

Abstract
Parkinson's disease (PD) is now recognized as a neurodegenerative condition caused by a complex interplay of genetic and environmental influences. Chronic manganese (Mn) exposure has been implicated in the development of PD. Since mitochondrial dysfunction is associated with PD pathology as well as Mn neurotoxicity, we investigated whether Mn exposure augments mitochondrial dysfunction and neurodegeneration in the nigrostriatal dopaminergic system using a newly available mitochondrially defective transgenic mouse model of PD, the MitoPark mouse. This unique PD model recapitulates key features of the disease including progressive neurobehavioral changes and neuronal degeneration. We exposed MitoPark mice to a low dose of Mn (10 mg/kg, p.o.) daily for 4 weeks starting at age 8 wks and then determined the behavioral, neurochemical and histological changes. Mn exposure accelerated the rate of progression of motor deficits in MitoPark mice when compared to the untreated MitoPark group. Mn also worsened olfactory function in this model. Most importantly, Mn exposure intensified the depletion of striatal dopamine and nigral TH neuronal loss in MitoPark mice. The neurodegenerative changes were accompanied by enhanced oxidative damage in the striatum and substantia nigra (SN) of MitoPark mice treated with Mn. Furthermore, Mn-treated MitoPark mice had significantly more oligomeric protein and IBA-1-immunoreactive microglia cells, suggesting Mn augments neuroinflammatory processes in the nigrostriatal pathway. To further confirm the direct effect of Mn on impaired mitochondrial function, we also generated a mitochondrially defective dopaminergic cell model by knocking out the TFAM transcription factor by using a CRISPR-Cas9 gene editing method. Seahorse mitochondrial bioenergetic analysis revealed that Mn decreases mitochondrial basal and ATP-linked respiration in the TFAM KO cells. Collectively, our results reveal that Mn can augment mitochondrial dysfunction to exacerbate nigrostriatal neurodegeneration and PD related behavioral symptoms. Our study also demonstrates that the MitoPark mouse is an excellent model to study the gene-environment interactions associated with mitochondrial defects in the nigral dopaminergic system as well as to evaluate the contribution of potential environmental toxicant interactions in a slowly progressive model of Parkinsonism.
Keywords: Manganese, mitochondria, MitoPark, neuroinflammation, animal model, TFAM, dopamine, Parkinson's disease, gene-environment interaction
Graphical abstract
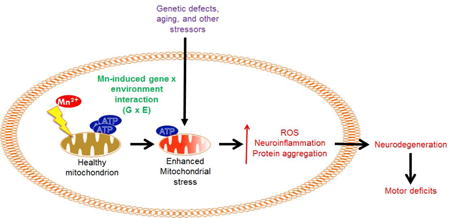
1. Introduction
Although manganese (Mn) is an essential cofactor needed for normal cellular functions, occupational and environmental exposures to the metal have been linked to increased risk for various neurological disorders including Parkinson's disease (PD) (Haynes et al., 2015; Lucchini et al., 2014; Sanders et al., 2015; Sikk and Taba, 2015). Mn exposure commonly occurs during the production of steel, batteries, fuel additives, fireworks, fungicides, welding, and ceramics (Mielke et al., 2002; Yabuuchi and Komaba, 2014). Given excessive exposure to Mn or failure to excrete it, the metal tends to accumulate in the basal ganglia, resulting in a movement disorder somewhat similar to PD called manganism (Bouabid et al., 2015; Peres et al., 2016b). Similar to PD, manganism manifests motor deficits such as rigidity and bradykinesia. However, in manganism patients, a distinctive “cock-walk” gait is observed and neuropsychiatric symptoms often present earlier rather than later in the disease progression (Bowler et al., 2006; Bowler et al., 1999; Kwakye et al., 2015). The neurons most affected in manganism occur in the globus pallidus, rather than the SN (Criswell et al., 2015; Perl and Olanow, 2007). Furthermore, manganism patients respond poorly to levodopa therapy, so instead chelation therapy has been used to treat the disorder (Discalzi et al., 2000; Ky et al., 1992).
Mn and other transition metals have long been implicated as risk factors in the etiology of PD, and a recent study suggests that Mn exposure dose-dependently increases UPDRS3 scores in welders (Gorell et al., 1999; Racette et al., 2016). Recent studies showed that welders exposed to Mn fumes display abnormal neurobehavioral changes that correlate with Mn accumulation in the basal ganglia as measured by magnetic resonance imaging (Lee et al., 2016; Lewis et al., 2016). Also, chronic exposure to metals and pesticides is associated with a younger age at onset of sporadic PD (Ratner et al., 2014). Other studies have revealed that genetic defects in Mn transporter proteins cause metal-induced Parkinsonism, including a form of childhood-onset Parkinsonism caused by an autosomal recessive Mn transporter defect. Patients having the homozygous mutation in SLC39A14 had excessive Mn accumulation and responded positively to chelation therapy (Tuschl et al., 2016). Furthermore, several mutations in SLC30A10, a Mn-specific efflux transporter thought to protect cells from Mn-induced toxicity, can cause familial Parkinsonism (Chen et al., 2015a; Leyva-Illades et al., 2014). The pathogenic mechanisms of Mn neurotoxicity are not completely understood, but evidence suggests that like classical Parkinsonian toxicants, Mn promotes protein aggregation and it also contributes to oxidative stress and mitochondrial dysfunction by inhibiting mitochondrial complexes I and II of the electron transport chain (Aschner et al., 2009; Carboni and Lingor, 2015; Liu et al., 2013; Peres et al., 2016a; Zheng et al., 1998). Furthermore, Mn can indirectly damage neurons by persistently triggering glial activation and neuroinflammation involving both microglia and astrocytes (Filipov and Dodd, 2012; Moreno et al., 2011; Moreno et al., 2009a; Streifel et al., 2012). Although striatal dopamine (DA) loss is a hallmark of PD models, contradictory results have been found in Mn-treated rodent studies (Moreno et al., 2009b; Witholt et al., 2000). These neurochemical changes are thought to contribute to the motor and neuropsychiatric symptoms present (Ferrer et al., 2012; Vermeiren and De Deyn, 2017).
Growing evidence has indicated that interactions between environmental exposures and genetic factors play a crucial role in the pathogenesis of PD. Mn and many genetic mutations associated with PD alter common biochemical pathways, allowing for a synergistic effect on the development of PD pathology (Bornhorst et al., 2014; Chen et al., 2015b; Roth, 2014). However, little direct evidence exists on Mn exposure exacerbating Parkinsonism in animal models. Such studies could provide much needed insight into how Mn hastens the pathophysiological processes involved in PD, and thus result in the development of better strategies to prevent or delay disease onset and progression. One study that used a toxin-based model to show effects of subchronic intraperitoneal Mn administration on DA-depleted rats reported that Mn potentiated neurobehavioral deficits but not the DA depletion (Witholt et al., 2000). Additionally, several PD-related genes such as parkin, LRRK2, and ATP13A2 are known to mediate Mn-induced toxicity in cell culture and animal models (Bornhorst et al., 2014; Higashi et al., 2004; Lovitt et al., 2010; Tan et al., 2011). However, none of the neurotoxin-based and genetic PD models faithfully recapitulate the chronic and progressive nature of the disease.
Mitochondrial impairment is well recognized as part of the normal aging process. Moreover, mitochondria serve as a key cellular target for PD pathology as well as for Mn neurotoxicity. However, we lack information on how Mn affects already comprised mitochondrial function as it relates to progression of the neurodegenerative process in PD and aging. Thus, in the present study, we evaluated the effect of a low-dose Mn exposure in the MitoPark mouse, a recently available, mitochondrially defective transgenic (Tg) mouse model of PD. MitoPark mice were generated by conditionally knocking out mitochondrial transcription factor A (TFAM) in DAergic neurons through the Cre-loxP system (Ekstrand et al., 2007). The MitoPark mouse is a unique PD model that recapitulates most of the hallmark behavioral symptoms and neuropathologies associated with PD, including progressive neurodegeneration and protein aggregates, representing a valuable model for studying the neurodegenerative process. Herein, we utilized the MitoPark mouse for a neurotoxicological study of Mn using a similar suite of behavioral, neurochemical, histological, and biochemical analyses routinely adopted in other animal models.
2. Materials and Methods
2.1 Chemicals
Dopamine hydrochloride, 3-4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 3,3′-diaminobenzidine (DAB), manganese chloride (MnCl2), and hydrogen peroxide were all purchased from Sigma (St Louis, MO). Halt protease and phosphatase inhibitor cocktail was obtained from Thermo Fisher (Waltham, MA). Bradford assay reagent and Western blotting buffers were purchased from Bio-Rad Laboratories (Hercules, CA). Anti-4-hydoxynonenal (4-HNE) antibody was purchased from R&D Systems (Minneapolis, MN). We purchased anti-IBA-1 antibodies from Wako Pure Chemical Industries (Richmond, VA) and Abcam (Cambridge, MA) for immunohistochemistry (IHC) and Western blot, respectively. Anti-tyrosine hydroxylase (TH) antibody was purchased from Millipore (Billerica, MA). Anti-oligomeric antibody (A11) and cell culture reagents were purchased from Invitrogen. The anti-mouse and anti-rabbit secondary antibodies (Alexa Fluor 680 conjugated anti-mouse IgG and IRDye 800 conjugated anti-rabbit IgG) were purchased from Invitrogen and Rockland Inc., respectively.
2.2 Cell lines
For in vitro mitochondrial function studies, the rat immortalized mesencephalic DAergic neuronal cell line (1RB3AN27, or N27) was cultured in RPMI 1640 containing 2 mM L-glutamine, 50 U/ml of penicillin and 50 μg/ml streptomycin with 0-10% FBS in incubators at 37°C and 5% CO2 as previously described by our lab (Charli et al., 2015). The lentivirus-based CRISPR/Cas9 TFAM knockout plasmid, pLV-U6gRNA-Ef1aPuroCas9GFP-TFAM, with the TFAM gRNA target sequence directed against the exon 1 sequence (CPR555e5e4099bf84.98), was purchased from Sigma-Aldrich. To make lentivirus, the lenti-CRISPR/Cas9 TFAM knockout plasmid was transfected into 293FT cells using the Mission Lentiviral Packaging Mix (Cat#SHP001, Sigma-Aldrich) according to manufacturer's instructions. For negative control lentivirus, Universal Negative Control Lentivirus from Sigma-Aldrich (U6-gRNA/CMV-Cas9-GFP) was similarly transfected into 293FT cells. The lentivirus was harvested 48 h post-transfection and titers were measured using the Lenti-X™ p24 Rapid Titer Kit (Cat # 632200, Takara Bio, Mountain View, CA). For stable knockout of TFAM in N27 cells, cells were plated at 0.1 × 106/well and lentivirus was added the following morning to the media at an MOI of 100. After 24 h, fresh media supplemented with puromycin (50 μg/mL) was added to the cells for stable cell selection.
2.3 Animal exposure
MitoPark mice were originally kindly provided by Dr. Nils-Goran Larson, currently at the Karolinska Institute in Stockholm, Sweden, who generated the mouse model in his laboratory at the Max Planck Institute for Biology of Ageing by conditionally knocking out TFAM in cells expressing dopamine transporter (DAT), as described in his publications (Ekstrand et al., 2007). MitoPark mice share many similarities with clinical PD including chronic, progressive neurodegeneration and motor deficits, protein aggregations, and adult onset of disease-related phenotypic changes (Ekstrand and Galter, 2009; Ekstrand et al., 2007). Upon L-DOPA administration, they recover motor function but will eventually develop dyskinesia (Galter et al., 2010; Gellhaar et al., 2015; Shan et al., 2015). Roles for neuroinflammation and oxidative stress were recently identified in the brain pathology of older-aged MitoPark mice (Ay et al., 2017; Langley et al., 2017). All mice for this study were bred, maintained, genotyped, and further characterized at Iowa State University. C57BL/6 mice (TFAM +/LoxP: Dat+/+ from MitoPark litter or from parental strain litters) and MitoPark mice (Dat +/Cre: TFAM LoxP/LoxP bred from C57BL/6 background) were fed ad libitum and housed in standard conditions approved and supervised by the Institutional Animal Care and Use Committee at Iowa State University. Eight-week-old mice (n=7-10/group, 3 males and 4 females for MitoPark treatment groups, and 5 males and 5 females for C57 treatment groups) received water or 10 mg/kg/day MnCl2 for 30 days by oral gavage. Mice were weighed and subjected to behavioral tests weekly. Neurochemical, biochemical, and histological studies were performed following sacrifice at 12 wks (Fig. 1A).
Figure 1. Mn accelerates and exacerbates progressive behavioral deficits in MitoPark mice.
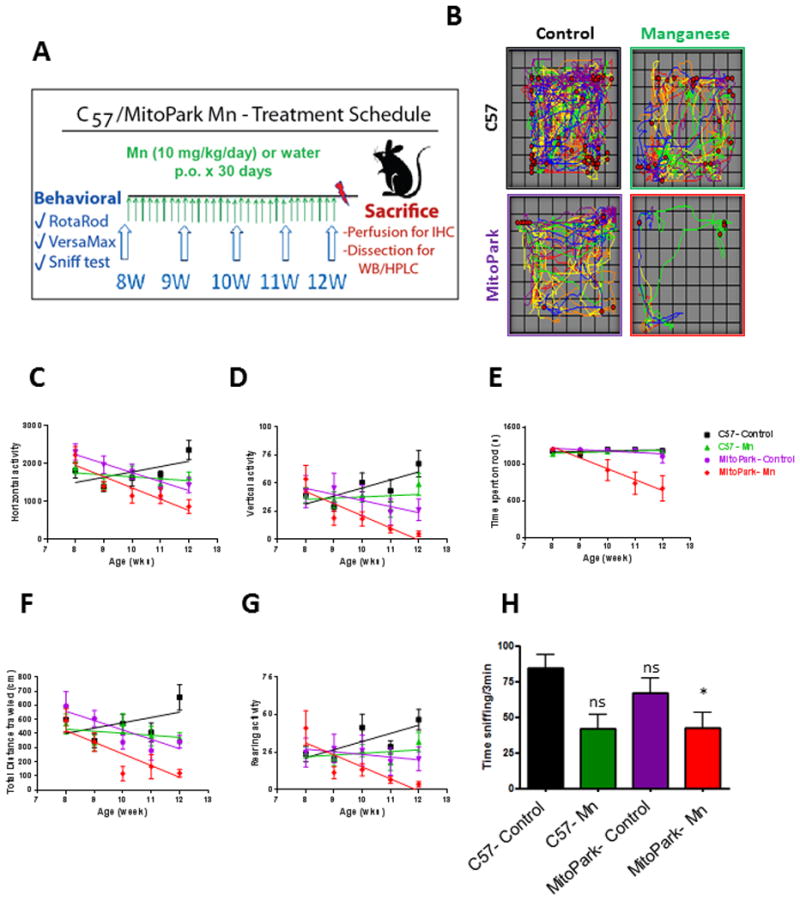
A, Exposure schedule showing C57 and MitoPark mice orally administered water or Mn (10 mg/kg) from ages 8-12 wks. B, VersaPlot showing horizontal activity (lines) and rearing activity (red dots) during a 10-min open-field test. Horizontal (C), vertical (D), and rearing (G) activities and distance traveled (F) during open-field test. E, Time spent on RotaRod. H, Time spent sniffing scented zone during 3-min social discrimination test. Graphical results represented as the mean±SEM (n=7-10 mice/group). *, p<0.05, and ns, p>0.05 versus water-treated C57 Control.
2.4 Motor function test
The VersaMax system (VersaMax monitor, model RXYZCM-16, and analyzer, model VMAUSB, AccuScan, Columbus, OH) was used for monitoring locomotor activity and the RotaRod (AccuScan) was used to test coordination of movement as previously described (Ghosh et al., 2012). For locomotor activity, mice acclimated 2 min prior to monitoring for 10 min using the VersaMax system during the daytime phase of the light cycle. Mouse order was randomized for each testing period. RotaRod speed was set to 20 rpm and time spent on rod was measured for 20 min maximum during a total of five trials.
2.5 Social discrimination test
To determine the olfactory function of control and MitoPark mice, we used a social discrimination test as previously described (Ngwa et al., 2014). However, this procedure was adapted to use ANY-maze tracking software (Stoelting, Wood Dale, IL) to determine time spent sniffing based on the animal's head being within a defined zone surrounding the bedding. Each mouse received the same amount (grams) of bedding from a cage housing 4 opposite-sex mice that had been changed one week prior to collecting the bedding. Total time spent sniffing the opposite sex's bedding was recorded during a 3-min interval.
2.6 High performance liquid chromatography (HPLC)
Striatum and olfactory bulb samples were prepared and processed for HPLC as described previously (Gordon et al., 2016a). Briefly, dissected brain regions were placed in a buffer containing 0.2 M perchloric acid, 0.05% Na2EDTA, 0.1% Na2S2O5, and isoproterenol (internal standard) to extract monoamine neurotransmitters. Monoamine lysates were placed in a refrigerated automatic sampler (model WPS-3000TSL) until being separated isocratically by a reversed-phase C18 column with a flow rate of 0.6 ml/min using a Dionex Ultimate 3000 HPLC system (pump ISO-3100SD, Thermo Scientific, Bannockburn, IL). Electrochemical detection was achieved using a CoulArray model 5600A coupled with an analytical cell (microdialysis cell 5014B) and a guard cell (model 5020). Data acquisition and analysis were performed using Chromeleon 7 and ESA CoulArray 3.10 HPLC Software and quantified data were normalized to wet tissue weight.
2.7 Western and slot blots
Protein lysates from the striatum and SN were prepared in RIPA buffer with protease and phosphatase inhibitors. For Western blot, equal amounts of protein lysates were run on a 12-15% SDS-PAGE as previously described (Jin et al., 2014) and transferred to a nitrocellulose membrane. For slot blot analysis, equal amounts of protein were loaded to each reservoir of the slot blot apparatus (Bio-Dot Microfiltration apparatus, Bio-Rad) and adsorbed to the nitrocellulose membrane as previously described (Harischandra et al., 2015). After blocking for 1 h, primary antibodies were incubated at 4°C overnight. The membranes were then incubated with secondary antibodies (Alexa Fluor 680 and Rockland IR800) at RT for one hour and images were captured via LI-COR Odyssey imager. Densitometric analysis was done using ImageJ software.
2.8 Immunohistochemistry
Brains perfused in 4% paraformaldehyde were cryoprotected with 30% sucrose the following day. Brains embedded in OCT (Sakura Finetek, Torrance, CA) at -80°C were cryosectioned to 30-μm sections, which were then stored in cryosolution (ethylene glycol and sucrose) until use. Fluoro-Jade C staining was done per manufacturer's instructions (Millipore). Immunostaining was performed as previously described (Gordon et al., 2016a). Briefly, following washing, antigen retrieval was achieved by keeping sections at 80°C for 30 min in sodium citrate (pH 8.5). After blocking in 2% BSA with 0.05% Tween-20 and 0.5% Triton X for one hour, sections were incubated with primary antibodies overnight at 4°C. For immunofluorescent staining, appropriate secondary antibodies (Alexa Fluor 488 and 555) were selected and incubated with sections for 1.5 h, followed by 7 min incubation with nuclear Hoechst stain (1:5000). For immunoperoxidase staining, a biotinylated secondary antibody followed by incubation with an avidin peroxidase solution (ABC Vectastain Kit, Vector laboratories, Burlingame, CA) to yield a brown stain after incubation with DAB solution. Dehydrated slides were mounted using DPX mountant and images were captured with an inverted Nikon TE2000-U microscope (Tokyo, Japan) attached to a SPOT digital camera with 2-40× lenses (Diagnostic Instruments, Sterling Heights, MI).
2.9 Statistics
All statistical analysis was done by repeated measures two-way ANOVA with Bonferroni post-test, two-way ANOVA with Holm-Sidak multiple comparisons post-test, or linear regression using GraphPad Prism. For behavioral analysis in Figures 1C-G and correlations in Figure 3, linear regression was used. Figures 1C-G were further analyzed by repeated measures two-way ANOVA as summarized in Table 1. The social discrimination test, HPLC, Western blots and Seahorse extracellular flux data were analyzed by two-way ANOVA with Holm-Sidak's multiple comparisons post-tests. Seahorse extracellular flux experiment data were also analyzed using Student's t-test for comparisons between control and treatment groups in each cell type. Differences with p-values ≤0.05 were considered significant.
Figure 3. Behavioral deficits correlate with neurochemical depletion in MitoPark mice.
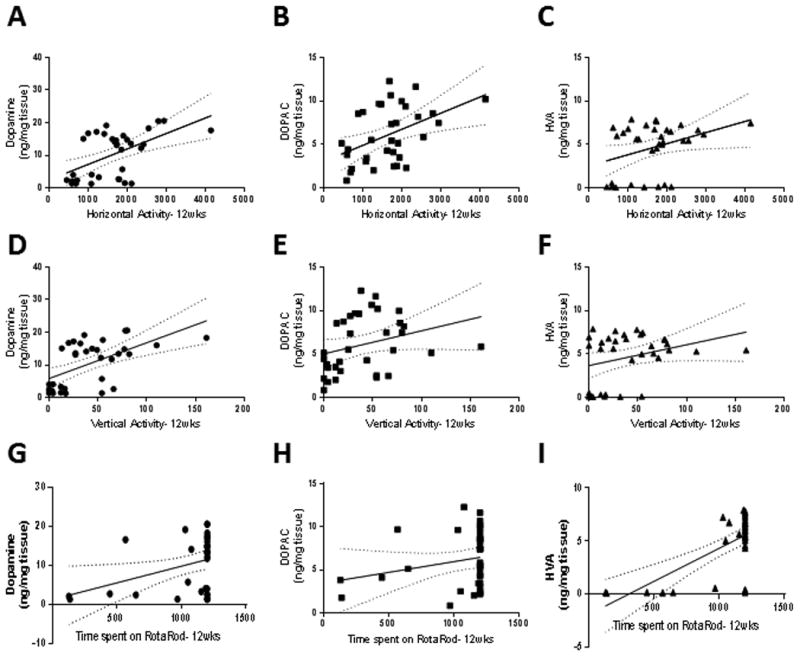
Correlation of horizontal activity with dopamine (A, R2= 0.29, F1,32=13.3, p=0.0010), DOPAC (B, R2=0.20, F1,32=8.2, p=0.0074), and HVA (C, R2=0.13, F1,32=4.6, p=0.040). Correlation of vertical activity with dopamine (D, R2=0.33, F1,32=16.2, p=0.0003), DOPAC (E, R2=0.088, F1,32=3.1, p=0.088), and HVA (F, R2=0.093, F1,32=3.3, p=0.079). Correlation of time spent on Rotarod with dopamine (G, R2=0.14, F1,32=5.2, p=0.030), DOPAC (H, R2=0.053, F1,32=1.8, p=0.19), and HVA (I, R2=0.43, F1,32=24.7, p<0.0001). Regression lines are displayed with the 95% confidence bands of possible regression lines.
Table 1.
Related to Figure 1. Summary of Bonferroni post-tests from repeated measures ANOVA for various behavioral parameters.
| Horizontal Activity | 8 | 9 | 10 | 11 | 12 wk |
|---|---|---|---|---|---|
| C57-Control vs. C57-Mn | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Control | ns | * | ns | ns | ns |
| C57-Control vs. MitoPark-Mn | ns | ns | ns | ns | *** |
| MitoPark-Control vs. MitoPark-Mn | ns | ns | ns | ns | ns |
| Vertical Activity | 8 | 9 | 10 | 11 | 12 wk |
| C57-Control vs. C57-Mn | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Control | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Mn | ns | ns | ns | * | ** |
| MitoPark-Control vs. MitoPark-Mn | ns | * | ns | ns | ns |
| RotaRod | 8 | 9 | 10 | 11 | 12 wk |
| C57-Control vs. C57-Mn | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Control | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Mn | ns | ns | ** | *** | *** |
| MitoPark-Control vs. MitoPark-Mn | ns | ns | ** | *** | *** |
| Total Distance Traveled | 8 | 9 | 10 | 11 | 12 wk |
| C57-Control vs. C57-Mn | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Control | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Mn | ns | ns | ns | ns | ns |
| MitoPark-Control vs. MitoPark-Mn | ns | ns | ns | ns | ns |
| Rearing Activity | 8 | 9 | 10 | 11 | 12 wk |
| C57-Control vs. C57-Mn | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Control | ns | ns | ns | ns | ns |
| C57-Control vs. MitoPark-Mn | ns | ns | ns | * | ** |
| MitoPark-Control vs. MitoPark-Mn | ns | ns | ns | ns | ns |
ns, not significant;
p<0.05;
p<0.01;
p<0.001.
3. Results
3.1 Low-dose Mn exposure accelerates the progressive behavioral deficits in MitoPark mice
MitoPark Tg mice display an adult-onset motor impairment, beginning around age 14 wks. Symptoms, including tremor and rigidity, progressively worsen by 20 wks (Ekstrand et al., 2007). Therefore, we began our study with mice at age 8 wks, prior to motor deficits in this model. Similarly, we chose a dose of Mn (10 mg/kg) that has been shown not to produce significant changes in behavioral parameters or striatal neurotransmitter levels in healthy adult mice (Moreno et al., 2009b). Therefore, we administered water or 10 mg/kg Mn by oral gavage daily to MitoPark and littermate control mice from ages 8-12 wks (Figure 1A). Behavioral tests were performed weekly, and biochemical and neurochemical analyses were performed on mice sacrificed at age 12 wks.
We first evaluated the locomotor activity and coordination of C57 and MitoPark mice using VersaMax and RotaRod tests, respectively. Representative activity plots obtained from open-field test at age 12 wks revealed a reduction in activity in Mn, MitoPark, and MitoPark-Mn groups when compared to C57 age-matched controls (Figure 1B). Linear regression of horizontal activity against age shows that C57 Control mice significantly increased their horizontal activity over time (R2=0.10, F1,48= 5.1, p=0.029). Mn-treated C57 mice did not significantly change their horizontal activity levels over time (R2=0.25, F1,48= 1.2, p=0.27) (Figure 1C). However, MitoPark mice showed a significant reduction in horizontal activity from 8 to 12 wks of age (R2=0.28, F1,33= 12.0, p=0.0015) and MitoPark mice treated with Mn had an even more significant reduction in horizontal activity over time (R2=0.41, F1,33= 22.9, p<0.0001).
Similarly, Mn-treated MitoPark mice experienced a significant decline in vertical activity (Figure 1D, R2=0.40, F1,33= 21.7, p<0.0001), which was not observed in MitoPark Controls (R2=0.06, F1,33= 2.0, p=0.17). C57 Controls had a significant increase in activity (R2=0.12, F1,48= 6.7, p=0.013), while Mn-treated C57 mice showed no significant change in vertical activity (R2=0.0044, F1,48= 0.2, p=0.65). Moreover, Mn-treated MitoPark mice were the only group that spent less time on the 20-rpm RotaRod (Figure 1E, R2=0.32, F1,33= 15.9, p=0.0004). C57 Controls (R2=0.013, F1,48= 0.6, p=0.42), C57 Mn (R2=0.052, F1,48= 2.7, p=0.11) and MitoPark Controls (R2=0.063, F1,33= 2.2, p=0.14) had no significant change in the time spent on Rotarod from 8 to 12 wks of age. Although both MitoPark Controls (R2=0.38, F1,33= 11.2, p=0.0020) and Mn-treated MitoParks (R2=0.55, F1,33= 22.0, p<0.0001) traveled less distance during the 10-min interval, the Mn-treated MitoParks showed an earlier and more severe deficit (Figure 1F). C57 Control (R2=0.047, F1,48= 2.3, p=0.13) and C57 Mn (R2=0.011, F1,48= 0.52, p=0.47) groups did not show a significant change in total distance traveled from 8 to 12 wks of age. C57 Control mice significantly increased rearing activity from 8 to 12 wks (R2=0.14, F1,48= 7.9, p=0.0073), however C57 mice treated with Mn showed no significant change (R2=0.011, F1,48=0.5, p=0.47). While MitoPark mice showed no significant change in rearing (R2=0.024, F1,33=0.39, p=0.54), Mn-treated MitoParks showed a significant reduction in rearing activity (R2=0.33, F1,33=16.0, p=0.0003). Overall, many locomotor and olfactory deficits present in MitoPark mice were exacerbated and accelerated by Mn exposure. Coordination and rearing activity were not yet impaired in MitoPark mice by age 12 wks, but were significantly affected in MitoPark mice exposed to Mn (Table 1).
Previous studies in our lab and others have indicated that olfactory dysfunction, which is a non-motor symptom of PD that can precede onset of motor symptoms by many years, is inherent in experimental models of PD and Mn neurotoxicity (Dranka et al., 2014; Neuner et al., 2014; Ngwa et al., 2014; Zhang et al., 2015). We therefore performed a social discrimination test to examine the effects of Mn exposure on olfaction. MitoPark mice did not show significantly decreased olfaction at age 12 wks, however, Mn-treated MitoParks spent significantly less time sniffing the opposite-sex bedding during a 3-min social discrimination test when compared to C57 Control (Figure 1H; MP, t=0.5, p=0.5; C57 Mn, t=2.1, p=0.084; MP Mn, t=2.8, p=0.028). Overall, the effect of treatment was significant, while genotype and interaction were not (Table 2). Only Mn-treated MitoParks were significantly different from C57 Controls, suggesting an additive effect of treatment and genotype is necessary to significantly reduce sniffing time. Total movement was not captured during this test, so the accompanying motor deficits may have influenced sniffing time in the scented zone during this behavioral task. Reduced interest in sniffing the bedding might also be affected by sexual changes in the MitoPark mice, therefore, breeding motivation and other factors should be explored in further studies.
Table 2.
| ANOVA | Factor | F (DFn, DFd) | P value |
|---|---|---|---|
| Social discrimination test | Interaction | F (1, 30) = 0.04 | P=0.83 |
| Treatment | F (1, 30) = 8.5 | P=0.0066 | |
| Genotype | F (1, 30) = 1.01 | P=0.32 | |
| Dopamine | Interaction | F (1, 30) = 0.006 | P=0.94 |
| Treatment | F (1, 30) = 3.05 | P=0.091 | |
| Genotype | F (1, 30) = 89.8 | P<0.0001 | |
| DOPAC | Interaction | F (1, 30) = 0.003 | P=0.95 |
| Treatment | F (1, 30) = 2.6 | P=0.11 | |
| Genotype | F (1, 30) = 29.4 | P<0.0001 | |
| HVA | Interaction | F (1, 30) = 0.2 | P=0.65 |
| Treatment | F (1, 30) = 1.3 | P=0.27 | |
| Genotype | F (1, 30) = 26.7 | P<0.0001 | |
| 4-HNE STR | Interaction | F (1, 10) = 1. 8 | P=0.21 |
| Treatment | F (1, 10) = 6.1 | P=0.034 | |
| Genotype | F (1, 10) = 9.3 | P=0.012 | |
| 4-HNE SN | Interaction | F (1, 10) = 2.9 | P=0.12 |
| Treatment | F (1, 10) = 3.3 | P=0.099 | |
| Genotype | F (1, 10) = 17.1 | P=0.0020 | |
| MTCO1 STR | Interaction | F (1, 10) = 0.2 | P=0.64 |
| Treatment | F (1, 10) = 4.2 | P=0.068 | |
| Genotype | F (1, 10) = 29.1 | P=0.0003 | |
| MTCO1 SN | Interaction | F (1, 10) = 0.3 | P=0.62 |
| Treatment | F (1, 10) = 1.8 | P=0.21 | |
| Genotype | F (1, 10) = 5.9 | P=0.035 | |
| IBA1 SN | Interaction | F (1, 10) = 2.8 | P=0.12 |
| Treatment | F (1, 10) = 0.1 | P=0.72 | |
| Genotype | F (1, 10) = 13.6 | P=0.0042 | |
| A11 SN | Interaction | F (1, 8) = 0.007 | P=0.93 |
| Treatment | F (1, 8) = 12.2 | P=0.0082 | |
| Genotype | F (1, 8) = 1.9 | P=0.20 | |
| Basal OCR | Interaction | F (1, 14) = 1.5 | P=0.24 |
| Treatment | F (1, 14) = 3.7 | P=0.073 | |
| Genotype | F (1, 14) = 70.3 | P<0.0001 | |
| ATP-linked | Interaction | F (1, 14) = 1.5 | P=0.25 |
| Treatment | F (1, 14) = 2.1 | P=0.17 | |
| Genotype | F (1, 14) = 45.8 | P<0.0001 | |
| Body weight | Interaction | F (1, 30) = 0.1 | P=0.71 |
| Treatment | F (1, 30) = 2.9 | P=0.10 | |
| Genotype | F (1, 30) = 1.2 | P=0.27 |
DFn= degrees of freedom, numerator; DFd= degrees of freedom, denominator.
3.2 Mn exacerbates striatal DA depletion and DAergic neuronal loss in MitoPark Tg mice
After establishing the behavioral effects of Mn in MitoPark mice, we next determined the effects on the nigrostriatal tract. In MitoPark mice, striatal DA loss is first observed at age 12 wks with only about 60% of TH+ neurons remaining at that time (Ekstrand et al., 2007). We first examined whether Mn exposure affects neurochemical deficits in the striatum by measuring DA, DOPAC and HVA by HPLC with electrochemical detection. Although MitoPark mice showed significantly less DA and metabolites than C57 controls, Mn exposure worsened the depletion of DA (Figure 2A; MP, t=6.6, p<0.0001; C57 Mn, t=1.3, p=0.24; MP Mn, t=7.9, p<0.0001), DOPAC (Figure 2B; MP, t=3.9, p=0.011; C57 Mn, t=1.3, p=0.20; MP Mn, t=5.0, p<0.0001), and HVA (Figure 2C; MP, t=3.3, p=0.005; C57 Mn, t=0.5, p=0.60; MP Mn, t=4.4, p=0.0003) in the striatum of MitoPark mice but not C57 mice. The effect of genotype was significant, while treatment and the interaction between treatment and genotype were not (Table 2). To further determine the effects of Mn on DAergic neuronal degeneration in the nigrostriatal tract, we performed TH IHC analyses on 30-μm sections of the striatal and nigral regions from 12-wk C57 and MitoPark mice treated with water or Mn. No significant reduction in TH-immunoreactivity was observed in vehicle- and Mn-treated non-Tg controls (Figure 2D). However, MitoPark mice did show less TH-immunoreactivity in nigral cell bodies and striatal terminals (Figure 2D). Importantly, Mn exposure exacerbated the loss of TH+ cells in the SN and immunostaining in the striatum in MitoPark mice (Figure 2D). Taken together, our neurochemical and histological data clearly demonstrate that Mn exposure exacerbates the neurodegenerative process in DAergic neurons.
Figure 2. Mn exacerbates striatal DA depletion and TH neuronal loss MitoPark mice.
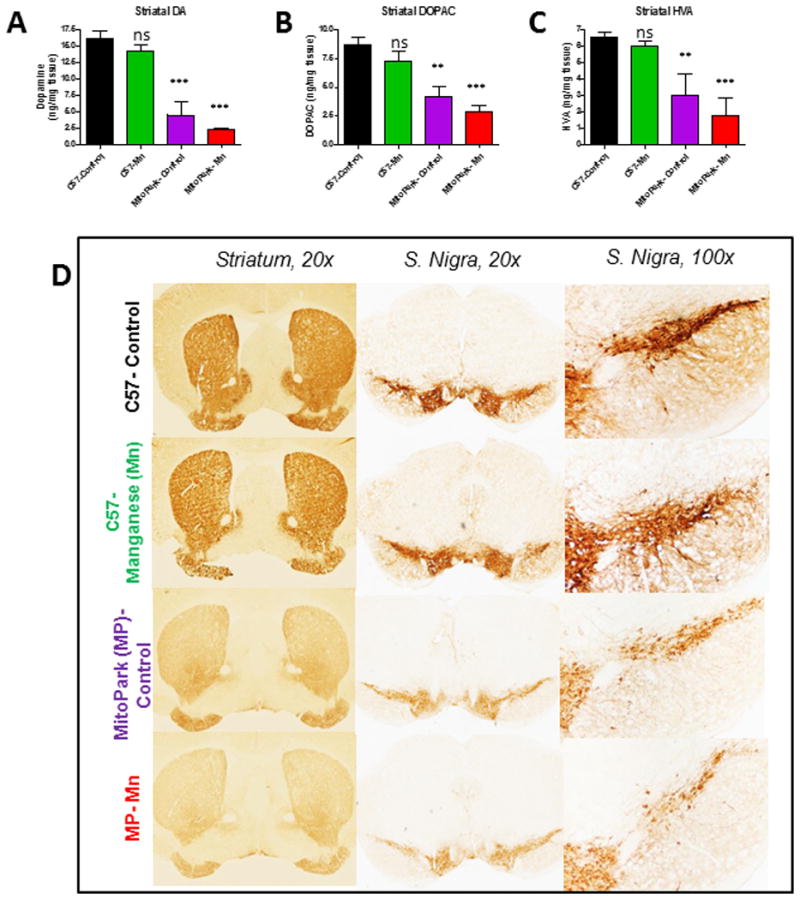
HPLC with electrochemical detection of the neurotransmitters dopamine (A), DOPAC (B), and HVA (C) in the striatum. DAB immunostaining (D) was performed in striatum and substantia nigra of vehicle-treated C57 (top), Mn-treated C57 (second row), vehicle-treated MitoPark (MP) (third row) and Mn-treated MP mice (bottom). Graphical results represented as the mean±SEM (n=7-10 mice/group). ns, p>0.05, **, p<0.01 and ***, p<0.001 versus water-treated C57 Control.
3.3 Behavioral deficits correlate with neurochemical depletion in MitoPark mice
Additionally, we show that dopamine and its metabolite levels correlated significantly with the behavioral tasks assessed. For example, horizontal activity (Figure 3A-C) significantly correlated with dopamine (R2= 0.29, F1,32=13.3, p=0.0010), DOPAC (R2=0.20, F1,32=8.2, p=0.007), and HVA (R2=0.13, F1,32=4.6, p=0.040). Vertical activity (Figure 3D-F) significantly correlated with dopamine (R2=0.33, F1,32=16.2, p=0.0003), but not DOPAC (R2=0.088, F1,32=3.1, p=0.088) or HVA (R2=0.093, F1,32=3.3, p=0.079). Rotarod coordination (Figure 3G-I) correlated with dopamine (R2=0.14, F1,32=5.2, p=0.030) and HVA (R2=0.43, F1,32=24.7, p<0.001), but not DOPAC (R2=0.053, F1,32=1.8, p=0.19).
3.4 Mn increases oxidative stress in the brains of MitoPark mice
Since Mn is known to induce oxidative damage, we assessed Mn-induced oxidative damage in MitoPark mice by measuring levels of 4-HNE, a lipid peroxidation product (Abdul-Muneer et al., 2013; Ghosh et al., 2016; Seo et al., 2016). As evidenced by Western blotting, 4-HNE was significantly increased in the SN (Figure 4A; MP, t=1.2, p=0.44; C57 Mn, t=0.7, p=0.47; MP Mn, t=3.9, p=0.0089) and striatum (Figure 4B; MP, t=1.7, p=0.22; C57 Mn, t=0.9, p=0.93; MP Mn, t=4.2, p=0.0054) of Mn-treated MitoPark mice when compared to C57 Controls. Consistently, double-IHC shows increased 4-HNE immunoreactivity (Figure 4C) in TH+ neurons in the SN of Mn-treated MitoPark mice, while no significant changes in 4-HNE were detectable in Mn-treated C57 or MitoPark-Control mice. Both treatment and genotype significantly affected 4-HNE levels in the striatum, while only the effect of genotype was significant in the SN (Table 2). In both regions, only the Mn-treated MitoPark mice significantly differed from the C57 Control group. These data suggest that low-dose Mn exposure and the inherent mitochondrial dysfunction in the MitoPark model combine to exacerbate oxidative stress in the nigrostriatal tract.
Figure 4. Mn increases oxidative stress in the brains of MitoPark mice.
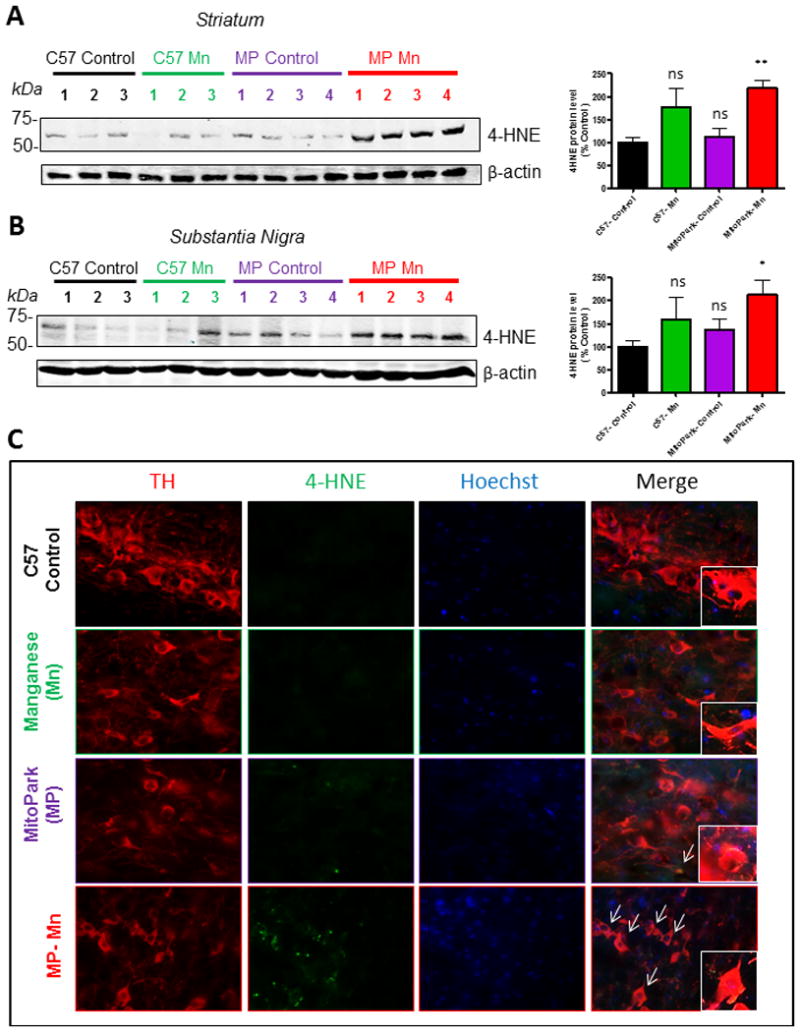
Representative Western blots and densitometric analysis of 4-HNE protein in the striatum (A) and substantia nigra (B). Immunohistochemistry of 12-wk mouse substantia nigra (C) reveals co-localization of TH and 4-HNE in dopaminergic neurons of Mn-treated MitoPark mice. Graphical results represented as the mean±SEM (n=3-4 mice/group). Ns, p>0.05, *, p<0.05, and **, p<0.01 versus water-treated C57 Control.
3.5 Mitochondrial dysfunction and neuronal cell death in Mn-treated MitoPark mice
MitoPark mice show decreased expression of the mtDNA encoded COX subunit I (MTCO1), indicating a severe reduction in mtDNA expression that results in respiratory chain deficiency in DAergic neurons (Ekstrand et al., 2007). We observed significantly reduced protein levels of MTCO1 in the striatum (Figure 5A) of MitoPark mice (t=4.1, p=0.0039) as well as Mn-treated MitoPark mice (t=5.3, p=0.0011). No change was observed in the Mn-treated C57 mice (t=1.7, 0.12). A similar reduction trend in MTCO1 protein levels, although not statistically significant, was observed in the SN (Figure 5B; MP, t=2.1, p=0.12; C57 Mn, t=1.2, p=0.25; MP Mn, t=2.7, p=0.070). As inherent mitochondrial dysfunction eventually results in the death of DAergic neurons in the MitoPark model, we next examined neuronal cell death by Fluoro-jade staining (Figure 5C), which revealed more degenerating neurons in the SN of MitoPark mice when compared to C57 Controls. However, a robust increase in Fluoro-Jade-labeled cells in the Mn-treated MitoPark mice suggests an enhanced cell death occurring in the Mn-treated MitoPark mice.
Figure 5. Mitochondrial dysfunction and neuronal cell death in Mn-treated MitoPark mice.
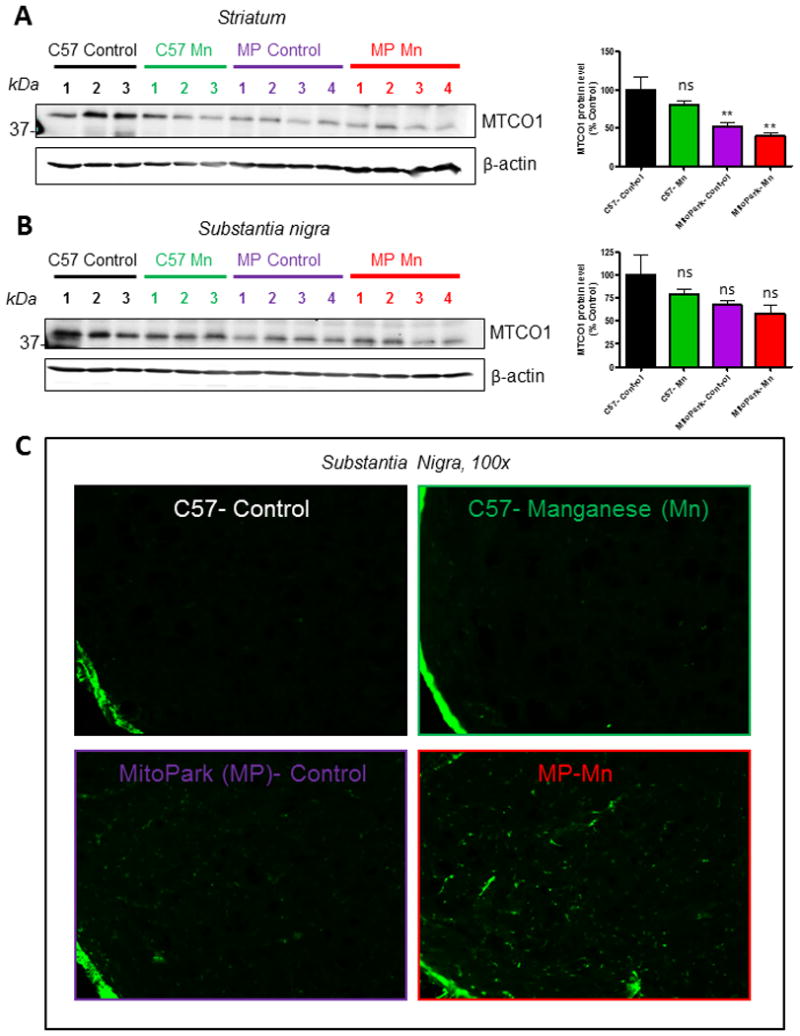
Representative Western blots and densitometric analysis of MTCO1 protein in the striatum (A) and substantia nigra (B). Fluoro-Jade C staining of 12-wk mouse substantia nigra (C) reveals increased neuronal cell death in Mn-treated MitoPark mice. Graphical results represented as the mean±SEM (n=3-4 mice/group). Ns, p>0.05, and **, p<0.01 versus water-treated C57 Control.
3.6 Mn induces neuroinflammatory response MitoPark mice
Neuroinflammation has been identified as an important contributor to neurodegenerative processes in PD. Although glial activation has not yet been characterized in the MitoPark model, it is understood that damaged neurons secrete factors which can activate glial cells (Block and Calderon-Garciduenas, 2009; Dhawan and Combs, 2012; Gordon et al., 2016b; Levesque et al., 2010; Panicker et al., 2015). Furthermore, Mn has been shown to promote microglia activation in various model systems (Kraft and Harry, 2011; Moreno et al., 2009a; Park and Chun, 2016). Western blotting revealed significantly more IBA-1 protein in the SN of Mn-treated MitoPark mice (t=2.9, p=0.049). However, neither Mn-treated C57 (t=0.9, p=0.40) or MitoPark-Controls (t=1.4, p= 0.34) had a significant elevation in IBA-1 (Figure 6A). These results are consistent with DAB-immunostaining showing more IBA-1+ cells in the SN of Mn-treated MitoParks (Figure 6B). Together, these results imply that Mn exposure in MitoPark mice can induce neuroinflammation in the SN, which may further contribute to neurodegeneration.
Figure 6. Neuroinflammatory changes in Mn-treated MitoPark mice.
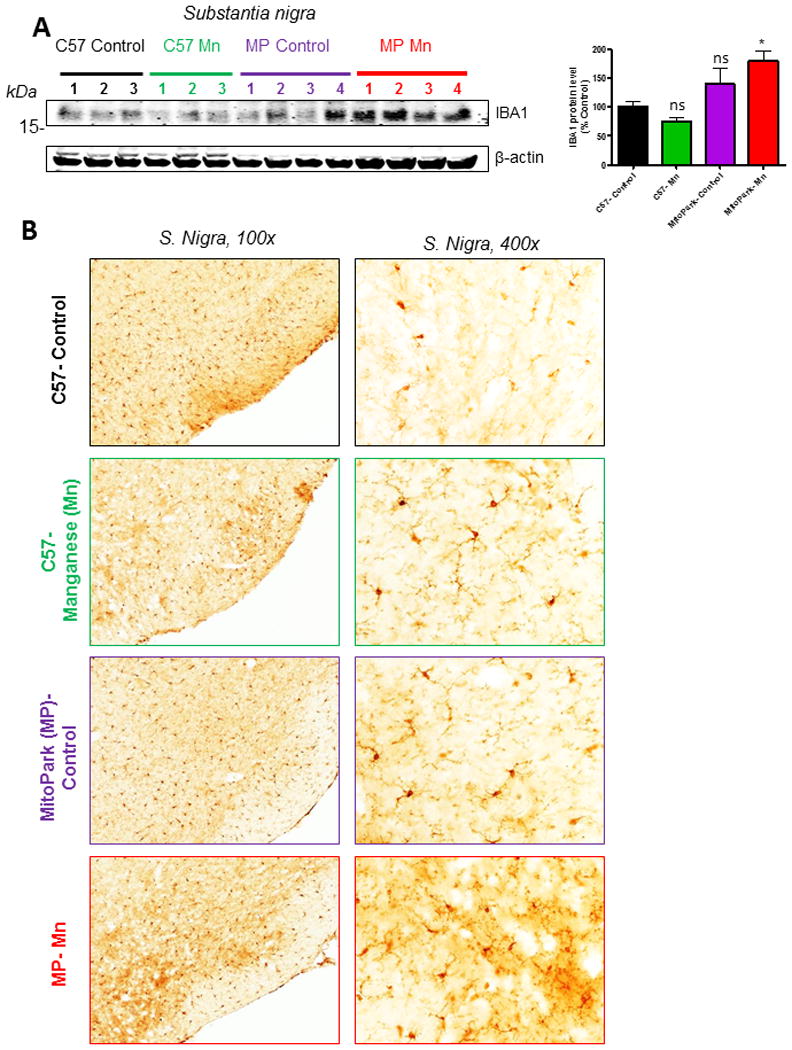
A, Western blot and corresponding densitometric analysis of IBA-1 protein in the SN. B, IBA-1 DAB immunostained sections from 12-wk mouse substantia nigra of vehicle-treated C57 (top), Mn-treated C57 (second row), vehicle-treated MitoPark (MP) (third row) and Mn-treated MP mice (bottom) show more IBA-1+ microglia in Mn-treated MitoPark mice when compared to vehicle-treated C57 Control mice. Graphical results represented as the mean±SEM (n=3-4 mice/group). Ns, p>0.05, and *, p<0.05 versus water-treated C57 Control.
3.7 Mn increases protein aggregation in MitoPark mice
Even prior to the symptomatic stage, MitoPark mice were found to contain protein aggregates in their DAergic neurons (Ekstrand et al., 2007). Although these aggregates did not contain α-synuclein, they did increase in size over time and partially co-localized with mitochondrial membrane proteins (Ekstrand et al., 2007). Our lab has shown that Mn enhances oligomerization of proteins in PD and prion models (Choi et al., 2006; Harischandra et al., 2015; Rokad et al., 2016). We therefore examined the levels of protein aggregation by slot blot analysis with an oligomeric-specific antibody, A11. The effect of treatment was significant; however, the effect of genotype and the interaction between treatment and genotype was not (Table 2). Although A11 modestly increased in C57-Mn (t=2.4, p=0.084) and MitoPark (t=0.9, p=0.38) groups, only the Mn-treated MitoPark (t=3.5, p=0.026) group showed significantly increased oligomeric protein levels when compared to C57 Control mice (Figure 7A). Overall, these results provide evidence that Mn enhances protein aggregation in MitoPark mice.
Figure 7. Mn increases protein aggregation in MitoPark mice.
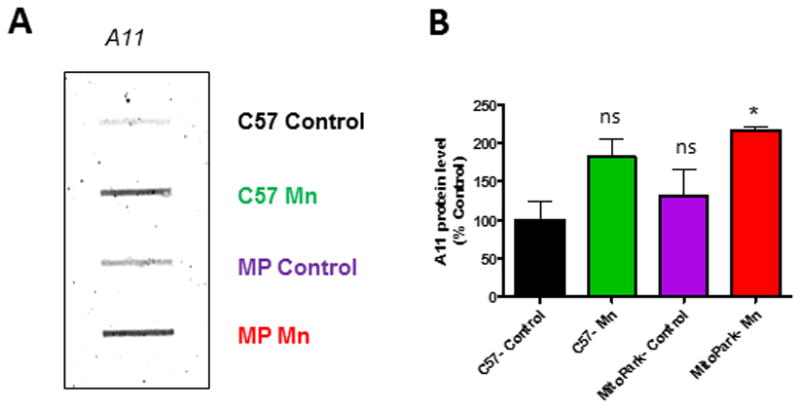
A, Representative slot blot analysis for oligomeric protein-specific antibody (A11) shows more oligomeric protein present in the substantia nigra of Mn-treated MitoPark mice. B, Quantification of A11 slot blot. Graphical results represented as the mean±SEM (n=3 mice/group). Ns, p>0.05, and *, p<0.05 versus water-treated C57 Control.
3.8 Mn potentiates mitochondrial deficits in a TFAM-KO DAergic neuronal cell model
Mitochondrial biogenesis is controlled by the transcription factor TFAM. Similar to MitoPark mice, knocking out TFAM produces defective mitochondria. To further confirm that Mn directly impairs mitochondrial dynamics in DAergic neuronal cells with defective mitochondria, we created a stable TFAM knockout (KO) N27 cell line using a CRISPR/Cas9-based lentiviral system. Our qRT-PCR analysis revealed more than an 80% loss of TFAM mRNA levels in TFAM-KO cells compared to CRISPR control cells (data not shown). Mitochondrial dynamics were then assessed by using a Seahorse extracellular flux analyzer. We exposed both TFAM-KO and control N27 neuronal cells to a low concentration of Mn (100 μM) for 24 h. This concentration of Mn alone did not result in mitochondrial deficits since both basal respiration rate (t=0.5, p=0.64) and ATP-linked respiration (t=0.1, p=0.88) did not significantly differ between vehicle- and Mn-treated control cells (Figure 7A-C). As expected, TFAM KO reduced basal respiration rate (t= 5.1, p=0.0003) and ATP-linked respiration (t=3.9; p=0.003) in untreated N27 cells. Interestingly, further reductions in basal respiration (t= 7.3, p<0.0001) were observed in Mn-treated TFAM-KO N27 cells (Figure 8A, B). Following oligomycin exposure, lower ATP-linked respiration was also observed in the Mn-treated TFAM-KO group (Figure 7C). The effect of TFAM-KO was significant; however, the effect of Mn treatment and the interaction between factors were not (Table 2). Taken together, these data suggest that Mn augments mitochondrial dysfunction by impairing both basal and ATP-linked respiration capacity in a DAergic neuronal cell model.
Figure 8. Mn exposure potentiated mitochondrial deficits in TFAM-KO neuronal cells.

A, CRISPR/Cas9-based TFAM-KO and Control N27 cells were treated with or without 100 μM Mn for 24 h and mitochondrial dynamics were measured using Seahorse XF24 analyzer. A, Quantification of basal respiration rate prior to MitoStressor injections. B, Quantification of ATP-linked respiration following oligomycin injection. Graphical results represented as the mean±SEM (n=3-4 /group). Ns, p>0.05, **, p<0.01, ***, p<0.001, ****, p<0.0001, #, p<0.05, versus untreated control from same cell type.
4. Discussion
In the present study, we systematically characterized neurobehavioral, neurochemical and biochemical changes contributing to nigral DAergic neurodegeneration in a transgenic, mitochondrially defective animal model (MitoPark mice) exposed to Mn. We demonstrated that orally administering low-dose Mn increased microglia activation and the formation of 4-HNE in MitoPark mice, suggesting that combining both genetic deficits and exposure to an environmental neurotoxic metal synergistically accentuates oxidative and inflammatory processes. Importantly, Mn exposure also accelerated and exacerbated motor deficits, the formation of aggregated proteins, and nigrostriatal DAergic degeneration in MitoPark mice. This is the first report, to our knowledge, describing a synergistic interaction between environmental Mn exposure and inherent mitochondrial dysfunction in accelerating the development of PD pathology.
Although neuropsychiatric symptoms of Mn at high doses are well researched, not much is known about low-dose Mn exposure and its neurobehavioral outcomes. A study by Moreno et al. (2009b) has shown that low-dose Mn alters anxiolytic behavior in male mice. Anxiolytic and depressive phenotyping were outside the scope of this paper, but would provide valuable insight and should be explored in future studies. In terms of non-motor deficits, olfactory deficits found in Mn-treated mice during this study (Figure 1H) could potentially be due to increased protein aggregation, reductions in neurogenesis, or changes in the olfactory epithelium or receptors (Chiu et al., 2015; Kurtenbach et al., 2013; Neuner et al., 2014; Postuma and Berg, 2016).
The pathophysiological mechanisms underlying Mn-induced exacerbation of DAergic neurons are not exactly clear. However, the increase in degenerating neurons and subsequent symptoms are likely a result of a combination of pathological pathways being further perturbed by Mn exposure. We have provided evidence that oxidative stress, protein aggregation, and neuroinflammation play a role in the Mn-mediated acceleration of disease progression in the MitoPark model. Microglia have previously been shown to become activated following Mn exposure, leading to secretion of proinflammatory factors, increased neuronal uptake of Mn, and neuronal cell death (Bade et al., 2013; Filipov and Dodd, 2012; Yin et al., 2017). In this report, we have shown that even a low dose of Mn is sufficient to induce neuroinflammatory changes in the presence of inherent mitochondrial dysfunction at 12 wks in MitoPark mice. Recently, we have shown that at 24 wks of age, MitoPark mice have increased microglial activation and express the proinflammatory enzymes NOX2 and iNOS in microglia (Langley et al., 2017). This suggests that Mn is accelerating the onset of neuroinflammatory changes in this model.
Furthermore, the effects that the interaction of these pathways could have on other processes should not be ignored. For example, the increased oxidative stress and protein aggregation observed could contribute to the gliosis found in Mn-treated MitoPark mice (Mosley et al., 2006; Zhang et al., 2005). Given the progressive phenotype, older-aged MitoPark mice should be characterized to determine when the development of reactive microgliosis first occurs in this model in the absence of Mn exposure. It would be interesting to decipher whether mitochondrial dysfunction in the MitoPark model was exacerbated by the Mn exposure or rather by an increased sensitivity to Mn toxicity in this model. Our results from TFAM-KO cells (Figure 7) suggest that Mn exposure directly impairs basal mitochondrial oxygen consumption rate and ATP-linked respiration. Also, the possibility of transport or excretion of Mn being compromised in this model cannot be ruled out. Further characterization of the biochemical mechanisms in MitoPark mice may help to address such questions.
Electrophysiological parameters in DAergic neurons were found to be affected in MitoPark mice, even prior to motor deficits (Branch et al., 2016). Because Mn enhances oxidative stress by catalyzing the auto-oxidation of DA and participating in Haber-Weiss reactions, this could explain the potent increase in lipid peroxidation (Figure 3) (Carboni and Lingor, 2015; Farina et al., 2013). Moreover, Mn is thought to preferentially accumulate in DAergic neurons due to increased calcium channel expression allowing for increased uptake of Mn (Carboni and Lingor, 2015). Branch and colleges (2016) revealed increased mRNA levels of Cav1.2 subunits in the 12-week-old MitoPark mouse, and more upregulation of ion channel subunit mRNAs associated with spontaneous firing at age 18-22 wks. In the mitochondria, Mn can inhibit mitochondrial aconitase and complexes I and II of the electron transport chain (Liu et al., 2013; Zheng et al., 1998). We previously reported that Mn exposure activates a mitochondria-dependent apoptotic cell death pathway in the DAergic system by a PKCδ-dependent proteolytic activation (Anantharam et al., 2004; Anantharam et al., 2002; Kitazawa et al., 2005; Kitazawa et al., 2002; Latchoumycandane et al., 2005). Thus, multiple mechanisms may be involved in regulating the Mn-induced sensitization of mitochondrial impairment. Further studies in in vitro and in vivo models of mitochondrial defects will provide additional mechanistic insights into Mn-induced neurodegeneration.
Gene-environment interactions are difficult to determine largely due to challenges in estimation of environmental exposures to toxicants and potential confounding factors (Polito et al., 2016; Thomas, 2010). A better understanding, however, could provide for novel biomarkers, therapeutic strategies, and personalized medicine. In the MitoPark mouse model, mitochondrial dysfunction is targeted to cells expressing DAT (Ekstrand et al., 2007). A number of studies have suggested that combining genetic variability in the DAT gene (SLC6A3) with pesticide exposure could interact synergistically to increase odds-ratios in PD patients (Kelada et al., 2006; Polito et al., 2016; Ritz et al., 2009; Ritz et al., 2016; Singh et al., 2008). Investigations of Mn and welding in relation to PD have led to controversial and opposing outcomes in various studies. Some studies report an increased PD risk, while others either do not report additional risk or emphasize the difference between PD and Mn-induced Parkinsonism (Guilarte and Gonzales, 2015; Nandipati and Litvan, 2016; Polito et al., 2016; Rentschler et al., 2012; Sriram et al., 2010). Multiple PARK genes have been shown to increase Mn susceptibility in model systems and epidemiological data (Bornhorst et al., 2014; Carboni and Lingor, 2015; Higashi et al., 2004; Lovitt et al., 2010; Rentschler et al., 2012; Tan et al., 2011). For instance, a study using induced pluripotent stem cell-derived neural progenitor cells showed significantly higher ROS generation in cells from a human subject with mutated PARK2 than in control subject-derived cells following Mn exposure, indicating increased sensitivity (Aboud et al., 2012). Other PARK genes are thought to function as Mn transporters (Carboni and Lingor, 2015; Tuschl et al., 2016). Exceptionally little data can be found linking epigenetic changes induced by Mn to effects on PD pathological mechanisms (Peng et al., 2015). Mn has also been implicated in gene-environment interactions in Huntington's and Alzheimer's diseases (Aboud et al., 2012; Bornhorst et al., 2014; Chin-Chan et al., 2015; Madison et al., 2012; Tong et al., 2014). Thus, continued efforts in studying the effect of environmental neurotoxicants in genetic defect models will shed light on gene-environment interactions in environmentally linked neurodegenerative diseases.
In conclusion, we show that low-dose Mn exposure significantly accelerates and exacerbates the motor deficits, striatal dopamine depletion and TH neuronal loss in MitoPark mice, demonstrating that Mn exposure can augment neurodegenerative processes in sensitive populations with mitochondrial deficiency, including the elderly community. Furthermore, our data demonstrate the utility of the MitoPark model for gene-environment interaction studies as well as for studying neurotoxic mechanisms in potentiating Parkinsonism in transgenic mouse models. The pathological role of Mn in other genetic models of PD and novel mitochondria-targeted therapeutic strategies for Mn-induced Parkinsonism should be further explored.
Highlights.
Mn accelerates and exacerbates progressive behavioral deficits in MitoPark mice
Mn exacerbates striatal DA depletion and TH neuronal loss in MitoPark mice
Mn increases oxidative stress in the brains of MitoPark mice
Mn induces neuroinflammation and protein aggregation in MitoPark mice brains
Mn accentuates mitochondrial impairment in MitoPark mice
Acknowledgments
We would like to thank Gary Zenitsky for his help in preparation of the manuscript. This work was supported by National Institutes of Health grants ES10586, ES26892, NS074443 and NS088206. The W. Eugene and Linda Lloyd Endowed Chair to AGK and Deans Professorship to AK are also acknowledged.
Footnotes
Conflict of interest: A.G.K. and V.A. are shareholders of PK Biosciences Corporation (Ames, IA). The company is interested in identifying novel biomarkers and potential therapeutic targets for PD. They do not have any direct interest in the present work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N, Haorah J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free radical biology & medicine. 2013;60:282–291. doi: 10.1016/j.freeradbiomed.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboud AA, Tidball AM, Kumar KK, Neely MD, Ess KC, Erikson KM, Bowman AB. Genetic risk for Parkinson's disease correlates with alterations in neuronal manganese sensitivity between two human subjects. Neurotoxicology. 2012;33(6):1443–1449. doi: 10.1016/j.neuro.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Blockade of PKCdelta proteolytic activation by loss of function mutants rescues mesencephalic dopaminergic neurons from methylcyclopentadienyl manganese tricarbonyl (MMT)-induced apoptotic cell death. Annals of the New York Academy of Sciences. 2004;1035:271–289. doi: 10.1196/annals.1332.017. [DOI] [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22(5):1738–1751. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Erikson KM, Herrero Hernandez E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. Neuromolecular medicine. 2009;11(4):252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay M, Luo J, Langley M, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. Molecular Mechanisms Underlying Protective Effects of Quercetin Against Mitochondrial Dysfunction and Progressive Dopaminergic Neurodegeneration in Cell Culture and MitoPark Transgenic Mouse Models of Parkinson's Disease. Journal of neurochemistry. 2017 doi: 10.1111/jnc.14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bade AN, Zhou B, Epstein AA, Gorantla S, Poluektova LY, Luo J, Gendelman HE, Boska MD, Liu Y. Improved visualization of neuronal injury following glial activation by manganese enhanced MRI. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2013;8(4):1027–1036. doi: 10.1007/s11481-013-9475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Calderon-Garciduenas L. Air pollution: mechanisms of neuroinflammation and CNS disease. Trends in neurosciences. 2009;32(9):506–516. doi: 10.1016/j.tins.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornhorst J, Chakraborty S, Meyer S, Lohren H, Brinkhaus SG, Knight AL, Caldwell KA, Caldwell GA, Karst U, Schwerdtle T, Bowman A, Aschner M. The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of alpha-synuclein in C. elegans. Metallomics : integrated biometal science. 2014;6(3):476–490. doi: 10.1039/c3mt00325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouabid S, Tinakoua A, Lakhdar-Ghazal N, Benazzouz A. Manganese Neurotoxicity: behavioral disorders associated with dysfunctions in the basal ganglia and neurochemical transmission. Journal of neurochemistry. 2015 doi: 10.1111/jnc.13442. [DOI] [PubMed] [Google Scholar]
- Bowler RM, Koller W, Schulz PE. Parkinsonism due to manganism in a welder: neurological and neuropsychological sequelae. Neurotoxicology. 2006;27(3):327–332. doi: 10.1016/j.neuro.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Bowler RM, Mergler D, Sassine MP, Larribe F, Hudnell K. Neuropsychiatric effects of manganese on mood. Neurotoxicology. 1999;20(2-3):367–378. [PubMed] [Google Scholar]
- Branch SY, Chen C, Sharma R, Lechleiter JD, Li S, Beckstead MJ. Dopaminergic Neurons Exhibit an Age-Dependent Decline in Electrophysiological Parameters in the MitoPark Mouse Model of Parkinson's Disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2016;36(14):4026–4037. doi: 10.1523/JNEUROSCI.1395-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni E, Lingor P. Insights on the interaction of alpha-synuclein and metals in the pathophysiology of Parkinson's disease. Metallomics : integrated biometal science. 2015;7(3):395–404. doi: 10.1039/c4mt00339j. [DOI] [PubMed] [Google Scholar]
- Charli A, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. Alterations in mitochondrial dynamics induced by tebufenpyrad and pyridaben in a dopaminergic neuronal cell culture model. Neurotoxicology. 2015 doi: 10.1016/j.neuro.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Bowman AB, Mukhopadhyay S, Aschner M. SLC30A10: A novel manganese transporter. Worm. 2015a;4(3):e1042648. doi: 10.1080/21624054.2015.1042648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, DeWitt MR, Bornhorst J, Soares FA, Mukhopadhyay S, Bowman AB, Aschner M. Age- and manganese-dependent modulation of dopaminergic phenotypes in a C. elegans DJ-1 genetic model of Parkinson's disease. Metallomics : integrated biometal science. 2015b;7(2):289–298. doi: 10.1039/c4mt00292j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Frontiers in cellular neuroscience. 2015;9:124. doi: 10.3389/fncel.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WH, Depboylu C, Hermanns G, Maurer L, Windolph A, Oertel WH, Ries V, Hoglinger GU. Long-term treatment with L-DOPA or pramipexole affects adult neurogenesis and corresponding non-motor behavior in a mouse model of Parkinson's disease. Neuropharmacology. 2015;95:367–376. doi: 10.1016/j.neuropharm.2015.03.020. [DOI] [PubMed] [Google Scholar]
- Choi CJ, Kanthasamy A, Anantharam V, Kanthasamy AG. Interaction of metals with prion protein: possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology. 2006;27(5):777–787. doi: 10.1016/j.neuro.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Criswell SR, Nelson G, Gonzalez-Cuyar LF, Huang J, Shimony JS, Checkoway H, Simpson CD, Dills R, Seixas NS, Racette BA. Ex vivo magnetic resonance imaging in South African manganese mine workers. Neurotoxicology. 2015;49:8–14. doi: 10.1016/j.neuro.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan G, Combs CK. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer's disease. Journal of neuroinflammation. 2012;9:117. doi: 10.1186/1742-2094-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discalzi G, Pira E, Herrero Hernandez E, Valentini C, Turbiglio M, Meliga F. Occupational Mn parkinsonism: magnetic resonance imaging and clinical patterns following CaNa2-EDTA chelation. Neurotoxicology. 2000;21(5):863–866. [PubMed] [Google Scholar]
- Dranka BP, Gifford A, McAllister D, Zielonka J, Joseph J, O'Hara CL, Stucky CL, Kanthasamy AG, Kalyanaraman B. A novel mitochondrially-targeted apocynin derivative prevents hyposmia and loss of motor function in the leucine-rich repeat kinase 2 (LRRK2(R1441G)) transgenic mouse model of Parkinson's disease. Neuroscience letters. 2014;583:159–164. doi: 10.1016/j.neulet.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrand MI, Galter D. The MitoPark Mouse - an animal model of Parkinson's disease with impaired respiratory chain function in dopamine neurons. Parkinsonism & related disorders. 2009;15(Suppl 3):S185–188. doi: 10.1016/S1353-8020(09)70811-9. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(4):1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M, Avila DS, da Rocha JB, Aschner M. Metals, oxidative stress and neurodegeneration: a focus on iron, manganese and mercury. Neurochemistry international. 2013;62(5):575–594. doi: 10.1016/j.neuint.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Lopez-Gonzalez I, Carmona M, Dalfo E, Pujol A, Martinez A. Neurochemistry and the non-motor aspects of PD. Neurobiology of disease. 2012;46(3):508–526. doi: 10.1016/j.nbd.2011.10.019. [DOI] [PubMed] [Google Scholar]
- Filipov NM, Dodd CA. Role of glial cells in manganese neurotoxicity. Journal of applied toxicology : JAT. 2012;32(5):310–317. doi: 10.1002/jat.1762. [DOI] [PubMed] [Google Scholar]
- Galter D, Pernold K, Yoshitake T, Lindqvist E, Hoffer B, Kehr J, Larsson NG, Olson L. MitoPark mice mirror the slow progression of key symptoms and L-DOPA response in Parkinson's disease. Genes, brain, and behavior. 2010;9(2):173–181. doi: 10.1111/j.1601-183X.2009.00542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellhaar S, Marcellino D, Abrams MB, Galter D. Chronic L-DOPA induces hyperactivity, normalization of gait and dyskinetic behavior in MitoPark mice. Genes, brain, and behavior. 2015;14(3):260–270. doi: 10.1111/gbb.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Kanthasamy A, Joseph J, Anantharam V, Srivastava P, Dranka BP, Kalyanaraman B, Kanthasamy AG. Anti-inflammatory and neuroprotective effects of an orally active apocynin derivative in pre-clinical models of Parkinson's disease. Journal of neuroinflammation. 2012;9:241. doi: 10.1186/1742-2094-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Langley MR, Harischandra DS, Neal ML, Jin H, Anantharam V, Joseph J, Brenza T, Narasimhan B, Kanthasamy A, Kalyanaraman B, Kanthasamy AG. Mitoapocynin Treatment Protects Against Neuroinflammation and Dopaminergic Neurodegeneration in a Preclinical Animal Model of Parkinson's Disease. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2016;11(2):259–278. doi: 10.1007/s11481-016-9650-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Neal ML, Luo J, Langley MR, Harischandra DS, Panicker N, Charli A, Jin H, Anantharam V, Woodruff TM, Zhou QY, Kanthasamy AG, Kanthasamy A. Prokineticin-2 upregulation during neuronal injury mediates a compensatory protective response against dopaminergic neuronal degeneration. Nature communications. 2016a;7:12932. doi: 10.1038/ncomms12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Singh N, Lawana V, Ghosh A, Harischandra DS, Jin H, Hogan C, Sarkar S, Rokad D, Panicker N, Anantharam V, Kanthasamy AG, Kanthasamy A. Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson's disease. Neurobiology of disease. 2016b;93:96–114. doi: 10.1016/j.nbd.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology. 1999;20(2-3):239–247. [PubMed] [Google Scholar]
- Guilarte TR, Gonzales KK. Manganese-Induced Parkinsonism Is Not Idiopathic Parkinson's Disease: Environmental and Genetic Evidence. Toxicological sciences : an official journal of the Society of Toxicology. 2015;146(2):204–212. doi: 10.1093/toxsci/kfv099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harischandra DS, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. alpha-Synuclein protects against manganese neurotoxic insult during the early stages of exposure in a dopaminergic cell model of Parkinson's disease. Toxicological sciences : an official journal of the Society of Toxicology. 2015;143(2):454–468. doi: 10.1093/toxsci/kfu247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes EN, Sucharew H, Kuhnell P, Alden J, Barnas M, Wright RO, Parsons PJ, Aldous KM, Praamsma ML, Beidler C, Dietrich KN. Manganese Exposure and Neurocognitive Outcomes in Rural School-Age Children: The Communities Actively Researching Exposure Study (Ohio, USA) Environmental health perspectives. 2015;123(10):1066–1071. doi: 10.1289/ehp.1408993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi Y, Asanuma M, Miyazaki I, Hattori N, Mizuno Y, Ogawa N. Parkin attenuates manganese-induced dopaminergic cell death. Journal of neurochemistry. 2004;89(6):1490–1497. doi: 10.1111/j.1471-4159.2004.02445.x. [DOI] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Harischandra DS, Kondru N, Ghosh A, Panicker N, Anantharam V, Rana A, Kanthasamy AG. Histone hyperacetylation up-regulates protein kinase Cdelta in dopaminergic neurons to induce cell death: relevance to epigenetic mechanisms of neurodegeneration in Parkinson disease. The Journal of biological chemistry. 2014;289(50):34743–34767. doi: 10.1074/jbc.M114.576702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelada SN, Checkoway H, Kardia SL, Carlson CS, Costa-Mallen P, Eaton DL, Firestone J, Powers KM, Swanson PD, Franklin GM, Longstreth WT, Jr, Weller TS, Afsharinejad Z, Costa LG. 5′ and 3′ region variability in the dopamine transporter gene (SLC6A3), pesticide exposure and Parkinson's disease risk: a hypothesis-generating study. Human molecular genetics. 2006;15(20):3055–3062. doi: 10.1093/hmg/ddl247. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Yang Y, Hirata Y, Kanthasamy A, Kanthasamy AG. Activation of protein kinase C delta by proteolytic cleavage contributes to manganese-induced apoptosis in dopaminergic cells: protective role of Bcl-2. Biochemical pharmacology. 2005;69(1):133–146. doi: 10.1016/j.bcp.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Wagner JR, Kirby ML, Anantharam V, Kanthasamy AG. Oxidative stress and mitochondrial-mediated apoptosis in dopaminergic cells exposed to methylcyclopentadienyl manganese tricarbonyl. The Journal of pharmacology and experimental therapeutics. 2002;302(1):26–35. doi: 10.1124/jpet.302.1.26. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Harry GJ. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. International journal of environmental research and public health. 2011;8(7):2980–3018. doi: 10.3390/ijerph8072980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtenbach S, Wewering S, Hatt H, Neuhaus EM, Lubbert H. Olfaction in three genetic and two MPTP-induced Parkinson's disease mouse models. PloS one. 2013;8(10):e77509. doi: 10.1371/journal.pone.0077509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwakye GF, Paoliello MM, Mukhopadhyay S, Bowman AB, Aschner M. Manganese-Induced Parkinsonism and Parkinson's Disease: Shared and Distinguishable Features. International journal of environmental research and public health. 2015;12(7):7519–7540. doi: 10.3390/ijerph120707519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ky SQ, Deng HS, Xie PY, Hu W. A report of two cases of chronic serious manganese poisoning treated with sodium para-aminosalicylic acid. British journal of industrial medicine. 1992;49(1):66–69. doi: 10.1136/oem.49.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley M, Ghosh A, Charli A, Sarkar S, Ay M, Luo J, Zielonka J, Brenza T, Bennett B, Jin H, Ghaisas S, Schlichtmann B, Kim D, Anantharam V, Kanthasamy A, Narasimhan B, Kalyanaraman B, Kanthasamy AG. Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxidants & redox signaling. 2017 doi: 10.1089/ars.2016.6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. The Journal of pharmacology and experimental therapeutics. 2005;313(1):46–55. doi: 10.1124/jpet.104.078469. [DOI] [PubMed] [Google Scholar]
- Lee EY, Eslinger PJ, Flynn MR, Wagner D, Du G, Lewis MM, Kong L, Mailman RB, Huang X. Association of neurobehavioral performance with R2* in the caudate nucleus of asymptomatic welders. Neurotoxicology. 2016;58:66–74. doi: 10.1016/j.neuro.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, Hong JS, Block ML. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain : a journal of neurology. 2010;133(Pt 3):808–821. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MM, Flynn MR, Lee EY, Van Buren S, Van Buren E, Du G, Fry RC, Herring AH, Kong L, Mailman RB, Huang X. Longitudinal T1 relaxation rate (R1) captures changes in short-term Mn exposure in welders. Neurotoxicology. 2016;57:39–44. doi: 10.1016/j.neuro.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34(42):14079–14095. doi: 10.1523/JNEUROSCI.2329-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Barber DS, Zhang P, Liu B. Complex II of the mitochondrial respiratory chain is the key mediator of divalent manganese-induced hydrogen peroxide production in microglia. Toxicological sciences : an official journal of the Society of Toxicology. 2013;132(2):298–306. doi: 10.1093/toxsci/kfs344. [DOI] [PubMed] [Google Scholar]
- Lovitt B, Vanderporten EC, Sheng Z, Zhu H, Drummond J, Liu Y. Differential effects of divalent manganese and magnesium on the kinase activity of leucine-rich repeat kinase 2 (LRRK2) Biochemistry. 2010;49(14):3092–3100. doi: 10.1021/bi901726c. [DOI] [PubMed] [Google Scholar]
- Lucchini RG, Guazzetti S, Zoni S, Benedetti C, Fedrighi C, Peli M, Donna F, Bontempi E, Borgese L, Micheletti S, Ferri R, Marchetti S, Smith DR. Neurofunctional dopaminergic impairment in elderly after lifetime exposure to manganese. Neurotoxicology. 2014;45:309–317. doi: 10.1016/j.neuro.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JL, Wegrzynowicz M, Aschner M, Bowman AB. Disease-toxicant interactions in manganese exposed Huntington disease mice: early changes in striatal neuron morphology and dopamine metabolism. PloS one. 2012;7(2):e31024. doi: 10.1371/journal.pone.0031024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielke HW, Gonzales CR, Powell E, Shah A, Mielke PW. Natural and anthropogenic processes that concentrate Mn in rural and urban environments of the lower Mississippi River delta. Environmental research. 2002;90(2):157–168. doi: 10.1006/enrs.2002.4382. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Streifel KM, Sullivan KA, Hanneman WH, Tjalkens RB. Manganese-induced NF-kappaB activation and nitrosative stress is decreased by estrogen in juvenile mice. Toxicological sciences : an official journal of the Society of Toxicology. 2011;122(1):121–133. doi: 10.1093/toxsci/kfr091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Streifel KM, Sullivan KA, Legare ME, Tjalkens RB. Developmental exposure to manganese increases adult susceptibility to inflammatory activation of glia and neuronal protein nitration. Toxicological sciences : an official journal of the Society of Toxicology. 2009a;112(2):405–415. doi: 10.1093/toxsci/kfp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Yeomans EC, Streifel KM, Brattin BL, Taylor RJ, Tjalkens RB. Age-dependent susceptibility to manganese-induced neurological dysfunction. Toxicological sciences : an official journal of the Society of Toxicology. 2009b;112(2):394–404. doi: 10.1093/toxsci/kfp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley RL, Benner EJ, Kadiu I, Thomas M, Boska MD, Hasan K, Laurie C, Gendelman HE. Neuroinflammation, Oxidative Stress and the Pathogenesis of Parkinson's Disease. Clinical neuroscience research. 2006;6(5):261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandipati S, Litvan I. Environmental Exposures and Parkinson's Disease. International journal of environmental research and public health. 2016;13(9) doi: 10.3390/ijerph13090881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuner J, Filser S, Michalakis S, Biel M, Herms J. A30P alpha-Synuclein interferes with the stable integration of adult-born neurons into the olfactory network. Scientific reports. 2014;4:3931. doi: 10.1038/srep03931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngwa HA, Kanthasamy A, Jin H, Anantharam V, Kanthasamy AG. Vanadium exposure induces olfactory dysfunction in an animal model of metal neurotoxicity. Neurotoxicology. 2014;43:73–81. doi: 10.1016/j.neuro.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker N, Saminathan H, Jin H, Neal M, Harischandra DS, Gordon R, Kanthasamy K, Lawana V, Sarkar S, Luo J, Anantharam V, Kanthasamy AG, Kanthasamy A. Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson's Disease. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35(27):10058–10077. doi: 10.1523/JNEUROSCI.0302-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Chun HS. Melatonin Attenuates Manganese and Lipopolysaccharide-Induced Inflammatory Activation of BV2 Microglia. Neurochemical research. 2016 doi: 10.1007/s11064-016-2122-7. [DOI] [PubMed] [Google Scholar]
- Peng C, Qiao Y, Ng JC. Epigenetics Involvement In Parkinson's Disease And Manganese-Induced Neurotoxicity. Journal of Clinical Epigenetics 2015 [Google Scholar]
- Peres TV, Parmalee NL, Martinez-Finley EJ, Aschner M. Untangling the Manganese-alpha-Synuclein Web. Frontiers in neuroscience. 2016a;10:364. doi: 10.3389/fnins.2016.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peres TV, Schettinger MR, Chen P, Carvalho F, Avila DS, Bowman AB, Aschner M. Manganese-induced neurotoxicity: a review of its behavioral consequences and neuroprotective strategies. BMC pharmacology & toxicology. 2016b;17(1):57. doi: 10.1186/s40360-016-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl DP, Olanow CW. The neuropathology of manganese-induced Parkinsonism. Journal of neuropathology and experimental neurology. 2007;66(8):675–682. doi: 10.1097/nen.0b013e31812503cf. [DOI] [PubMed] [Google Scholar]
- Polito L, Greco A, Seripa D. Genetic Profile, Environmental Exposure, and Their Interaction in Parkinson's Disease. Parkinson's disease. 2016;2016:6465793. doi: 10.1155/2016/6465793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postuma RB, Berg D. Advances in markers of prodromal Parkinson disease. Nature reviews. Neurology. 2016;12(11):622–634. doi: 10.1038/nrneurol.2016.152. [DOI] [PubMed] [Google Scholar]
- Racette BA, Searles Nielsen S, Criswell SR, Sheppard L, Seixas N, Warden MN, Checkoway H. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology. 2016 doi: 10.1212/WNL.0000000000003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner MH, Farb DH, Ozer J, Feldman RG, Durso R. Younger age at onset of sporadic Parkinson's disease among subjects occupationally exposed to metals and pesticides. Interdisciplinary toxicology. 2014;7(3):123–133. doi: 10.2478/intox-2014-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentschler G, Covolo L, Haddad AA, Lucchini RG, Zoni S, Broberg K. ATP13A2 (PARK9) polymorphisms influence the neurotoxic effects of manganese. Neurotoxicology. 2012;33(4):697–702. doi: 10.1016/j.neuro.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz BR, Manthripragada AD, Costello S, Lincoln SJ, Farrer MJ, Cockburn M, Bronstein J. Dopamine transporter genetic variants and pesticides in Parkinson's disease. Environmental health perspectives. 2009;117(6):964–969. doi: 10.1289/ehp.0800277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz BR, Paul KC, Bronstein JM. Of Pesticides and Men: a California Story of Genes and Environment in Parkinson's Disease. Current environmental health reports. 2016;3(1):40–52. doi: 10.1007/s40572-016-0083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokad D, Ghaisas S, Harischandra DS, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG. Role of neurotoxicants and traumatic brain injury in alpha-synuclein protein misfolding and aggregation. Brain research bulletin. 2016 doi: 10.1016/j.brainresbull.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JA. Correlation between the biochemical pathways altered by mutated parkinson-related genes and chronic exposure to manganese. Neurotoxicology. 2014;44:314–325. doi: 10.1016/j.neuro.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Sanders AP, Claus Henn B, Wright RO. Perinatal and Childhood Exposure to Cadmium, Manganese, and Metal Mixtures and Effects on Cognition and Behavior: A Review of Recent Literature. Current environmental health reports. 2015;2(3):284–294. doi: 10.1007/s40572-015-0058-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J, Singh NN, Ottesen EW, Sivanesan S, Shishimorova M, Singh RN. Oxidative Stress Triggers Body-Wide Skipping of Multiple Exons of the Spinal Muscular Atrophy Gene. PloS one. 2016;11(4):e0154390. doi: 10.1371/journal.pone.0154390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan L, Diaz O, Zhang Y, Ladenheim B, Cadet JL, Chiang YH, Olson L, Hoffer BJ, Backman CM. L-Dopa induced dyskinesias in Parkinsonian mice: Disease severity or L-Dopa history. Brain research. 2015;1618:261–269. doi: 10.1016/j.brainres.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikk K, Taba P. Methcathinone “Kitchen Chemistry” and Permanent Neurological Damage. International review of neurobiology. 2015;120:257–271. doi: 10.1016/bs.irn.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Singh M, Khan AJ, Shah PP, Shukla R, Khanna VK, Parmar D. Polymorphism in environment responsive genes and association with Parkinson disease. Molecular and cellular biochemistry. 2008;312(1-2):131–138. doi: 10.1007/s11010-008-9728-2. [DOI] [PubMed] [Google Scholar]
- Sriram K, Lin GX, Jefferson AM, Roberts JR, Wirth O, Hayashi Y, Krajnak KM, Soukup JM, Ghio AJ, Reynolds SH, Castranova V, Munson AE, Antonini JM. Mitochondrial dysfunction and loss of Parkinson's disease-linked proteins contribute to neurotoxicity of manganese-containing welding fumes. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24(12):4989–5002. doi: 10.1096/fj.10-163964. [DOI] [PubMed] [Google Scholar]
- Streifel KM, Moreno JA, Hanneman WH, Legare ME, Tjalkens RB. Gene deletion of nos2 protects against manganese-induced neurological dysfunction in juvenile mice. Toxicological sciences : an official journal of the Society of Toxicology. 2012;126(1):183–192. doi: 10.1093/toxsci/kfr335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Zhang T, Jiang L, Chi J, Hu D, Pan Q, Wang D, Zhang Z. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. The Journal of biological chemistry. 2011;286(34):29654–29662. doi: 10.1074/jbc.M111.233874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. Gene--environment-wide association studies: emerging approaches. Nature reviews. Genetics. 2010;11(4):259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Yang H, Tian X, Wang H, Zhou T, Zhang S, Yu J, Zhang T, Fan D, Guo X, Tabira T, Kong F, Chen Z, Xiao W, Chui D. High manganese, a risk for Alzheimer's disease: high manganese induces amyloid-beta related cognitive impairment. Journal of Alzheimer's disease : JAD. 2014;42(3):865–878. doi: 10.3233/JAD-140534. [DOI] [PubMed] [Google Scholar]
- Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, Hung CY, Simpson MA, Chong WK, Jacques TS, Woltjer RL, Eaton S, Gregory A, Sanford L, Kara E, Houlden H, Cuno SM, Prokisch H, Valletta L, Tiranti V, Younis R, Maher ER, Spencer J, Straatman-Iwanowska A, Gissen P, Selim LA, Pintos-Morell G, Coroleu-Lletget W, Mohammad SS, Yoganathan S, Dale RC, Thomas M, Rihel J, Bodamer OA, Enns CA, Hayflick SJ, Clayton PT, Mills PB, Kurian MA, Wilson SW. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nature communications. 2016;7:11601. doi: 10.1038/ncomms11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeiren Y, De Deyn PP. Targeting the norepinephrinergic system in Parkinson's disease and related disorders: The locus coeruleus story. Neurochemistry international. 2017;102:22–32. doi: 10.1016/j.neuint.2016.11.009. [DOI] [PubMed] [Google Scholar]
- Witholt R, Gwiazda RH, Smith DR. The neurobehavioral effects of subchronic manganese exposure in the presence and absence of pre-parkinsonism. Neurotoxicology and teratology. 2000;22(6):851–861. doi: 10.1016/s0892-0362(00)00108-2. [DOI] [PubMed] [Google Scholar]
- Yabuuchi N, Komaba S. Recent research progress on iron- and manganese-based positive electrode materials for rechargeable sodium batteries. Science and technology of advanced materials. 2014;15(4):043501. doi: 10.1088/1468-6996/15/4/043501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Dai Q, Jiang P, Zhu L, Dai H, Yao Z, Liu H, Ma X, Qu L, Jiang J. Manganese exposure facilitates microglial JAK2-STAT3 signaling and consequent secretion of TNF-a and IL-1beta to promote neuronal death. Neurotoxicology. 2017 doi: 10.1016/j.neuro.2017.04.001. [DOI] [PubMed] [Google Scholar]
- Zhang S, Xiao Q, Le W. Olfactory dysfunction and neurotransmitter disturbance in olfactory bulb of transgenic mice expressing human A53T mutant alpha-synuclein. PloS one. 2015;10(3):e0119928. doi: 10.1371/journal.pone.0119928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- Zheng W, Ren S, Graziano JH. Manganese inhibits mitochondrial aconitase: a mechanism of manganese neurotoxicity. Brain research. 1998;799(2):334–342. doi: 10.1016/s0006-8993(98)00481-8. [DOI] [PMC free article] [PubMed] [Google Scholar]