

ABSTRACT
Salmonella targets and enters epithelial cells at permissive entry sites: some cells are more likely to be infected than others. However, the parameters that lead to host cell heterogeneity are not known. Here, we quantitatively characterized host cell vulnerability to Salmonella infection based on imaged parameters. We performed successive infections of the same host cell population followed by automated high-throughput microscopy and observed that infected cells have a higher probability of being reinfected. Establishing a predictive model, we identified two combined origins of host cell vulnerability: pathogen-induced cellular vulnerability emerging from Salmonella uptake and persisting at later stages of the infection and host cell-inherent vulnerability. We linked the host cell-inherent vulnerability with its morphological attributes, such as local cell crowding, and with host cell cholesterol content. This showed that the probability of Salmonella infection success can be forecast from morphological or molecular host cell parameters.
KEYWORDS: Salmonella enterica serovar Typhimurium, cooperative behavior, cell vulnerability, single-cell heterogeneity, mathematical modeling
INTRODUCTION
Salmonella enterica serovar Typhimurium (S. Typhimurium) is a Gram-negative bacterium that causes enteric diseases in many vertebrates after ingestion of contaminated food or water. Salmonellosis is one of the most common causes of foodborne diseases in humans and is considered to be major public health and global economic problem (1). After oral uptake, more than 99% of S. Typhimurium bacteria are killed in the stomach or in the gut (2). The surviving bacteria reach the distal ileum where they invade nonphagocytic intestinal epithelial cells (3). In vitro experiments have shown that S. Typhimurium invasion of host cells occurs after a phase of bacterial near-surface swimming (NSS) on the epithelial layer. The bacteria scan the surface and eventually stop and dock at a selected host cell (4, 5). Docking is irreversible (6) and is followed by injection of Salmonella effectors into the host cell through a type 3 secretion system (T3SS), leading to the formation of ruffles that engulf the incoming bacterium (7, 8). Upon internalization S. Typhimurium either develops inside a Salmonella-containing vacuole (SCV), or it ruptures the SCV to escape into the cytoplasm where the pathogen replicates at a high rate, a phenomenon called hyperreplication (HR) (9, 10).
The mechanism by which S. Typhimurium targets specific host cellular sites for its entry remains open to debate. Santos and colleagues suggested that mitotic cells are selected due to increased cholesterol accumulation at the cell surface during metaphase (11). In contrast, Misselwitz and colleagues proposed that physical obstacles and forces that occur during the process of NSS lead to the targeting of topologically prominent sites, such as dividing cells or membrane ruffles (4). Finally, investigators have also reported that the invasion of S. Typhimurium at the ruffle site is a highly cooperative effort (6, 12). Indeed, coinfection of wild-type (WT) and noninvasive S. Typhimurium mutant strains resulted in the entry of both strains into the host cells; noninvasive S. Typhimurium mutants were trapped at ruffle sites and concomitantly internalized within the host cell following the active invasion by WT S. Typhimurium. However, the cooperative effect between intracellular and entering bacteria remains poorly understood at later stages of infection.
An increasing number of studies have highlighted the relevance of intrinsic cellular heterogeneity within eukaryotic monocultures. After seeding, cells display a dynamic range of variability in their morphologies depending on their local microenvironment, including the local density, and the peripheral or central positioning within cellular islets (13). This heterogeneity results in differences of transcription (14, 15), lipid composition (13, 15), and sensitivity to infections (13). Such cell-to-cell variations have been studied during viral infection, revealing that simian virus 40 (SV40) and mouse hepatitis virus (MHV) present a population-determined pattern of infection associated with differential, local cell crowding (13). In the context of bacterial infection, cell targeting has been related to bacterial cooperation at the entry site and evaluated at the whole-population level using CFU counting or flow cytometry analysis (12), but so far this has not been studied in situ at the single cell level.
Here, we investigated the susceptibility of epithelial host cells within the same cell population to become infected by S. Typhimurium. Our analysis revealed that some cells are more likely to be infected by Salmonella than others. We termed them vulnerable cells. Cell vulnerability was characterized in a quantitative manner by automated high-content imaging through double sequential infections with a delay of 1 to 3 h between the bacterial challenges. The number of intracellular bacteria per cell as well as the corresponding host cell parameters, such as cell perimeter, local density, and number of infected neighboring cells, was assessed. Using a mathematical model, we showed that host cell vulnerability can be induced by a first bacterial uptake but also arises from the cell's intrinsic morphological and microenvironmental characteristics.
RESULTS
Sequential infections allow studies of Salmonella cooperation at the single-cell level.
We carried out a microscopy-based double-infection assay to explore possible links between host cell vulnerability and successive bacterial infections of epithelial cells (Fig. 1). HeLa cells grown in 96-well plates were subjected to a first infection with green S. Typhimurium expressing the fluorescent protein (SLGFP) for 30 min, followed by elimination of the extracellular bacteria via gentamicin treatment and washing. The cells were then incubated for 1, 2, or 3 h before being subjected to a second wave of infection with red S. Typhimurium expressing the fluorescent protein dsRed (SLdsRed). Extracellular bacteria were again eliminated in the same way, and the host cells were stained with CellMask and 4′,6′-diamidino-2-phenylindole (DAPI) before automated image acquisition of entire culture wells (Fig. 1A). The obtained images were analyzed with CellProfiler, a widely used image analysis software program (16, 17) (Fig. 1B). The differently labeled bacteria and the stained host cells enabled us to distinguish and quantify distinct cellular populations, as follows: those cells infected during the first infection (I1) or not (noI1), those infected during the second infection (I2) or not (noI2), and the associated subpopulations (I1 & I2, noI1 & noI2, I1 & noI2, and noI1 & I2) (Fig. 1C). We based our analysis on comparing the probabilities of infection in these subpopulations.
FIG 1.

Double infections allow studies of Salmonella cooperation at the single-cell level. (A) An overview of the experimental workflow used in this study is shown. According to the sequential infection protocol, HeLa cells grown in 96-well plates for 24 h were subjected for 30 min to a first infection by SLGFP. This was followed by elimination of extracellular bacteria by gentamicin and incubation of the cells for 1, 2, or 3 h. The cells were subsequently challenged by a second infection with SLdsRed for 30 min. After removal of the extracellular bacteria, the samples were fixed. Nuclei were stained with DAPI, and cell membranes were stained with CellMask before microscopic acquisition of the entire wells. (B) Representative image of SLGFP and SLdsRed internalized in HeLa cells. Host cell nuclei are visualized through DAPI staining (blue), and cell membranes are visualized with CellMask (gray). Scale bar, 5 μm. (C) Scheme of our statistical analysis of different subpopulations. The following cellular populations can be distinguished: those cells infected during the first infection (I1) or not (noI1), those infected during the second infection (I2) or not (noI2), and the related subpopulations (I1 & I2; noI1 & noI2). This scheme maps the case of two independent infections. (D) Time distribution of the ruffle disappearance during Salmonella infection followed in actin-GFP transfected cells by time-lapse microscopy. (E and F) Comparison of the conditional probabilities of infection for two different populations during synchronous infection of SLGFP and SLdsRed in HeLa cells and Caco-2 cells. The MOIs were chosen to obtain an average of 30% of the cells infected and calculated after CFU counting (n ≥ 3). P values were obtained after a t test. (G) Comparison of an independent model (left) with the obtained data (right). The percentages are averaged from six independent experiments, represented in panel E, at an MOI of 30. **, P < 0.01; ***, P < 0.001; ns, not significant.
Salmonella cooperates for entry at ruffles.
In order to test the reliability of our method, we first examined whether we could detect ruffle-dependent cooperation between individual salmonellae during host cell entry, previously observed in infected HeLa and MDCK cells (4, 12). To do this, we determined first the time window during which ruffle-associated cooperation could potentially occur by performing time-lapse microscopy of Salmonella infection of HeLa cells transiently expressing GFP-tagged actin (Fig. 1D). Time series of 90 min at 3-min intervals provided image sequences of forming and disappearing ruffles. In most of the cases, we observed the uptake of one to two bacteria per ruffle, and we saw ruffle disappearance in less than 15 min (see Movie S1 in the supplemental material). We noticed that the greater the number of bacteria engulfed by the ruffles, the longer we could detect the presence of these ruffles. Therefore, newly arriving bacteria prompted additional growth of the ruffles (Movie S2). We quantified the ruffle lifetime by measuring the delay of ruffle disappearance after the entry of the last bacterium. The few cases of very high infection (>5 bacteria/ruffle) that could not be properly analyzed were excluded. Quantification revealed an average ruffle lifetime of 13 min and the complete disappearance of 90% of the ruffles after 24 min (Fig. 1D). Labeling Caco-2 cells with the membrane dye FM 4-64, we observed that the ruffle lifetime for infected Caco-2 cells was similar to that of infected HeLa cells.
We then challenged HeLa and Caco-2 cells with SLGFP and SLdsRed at the same time and compared the probability of SLdsRed infecting the same cell containing simultaneously SLGFP with that of infecting cells that did not contain SLGFP (Fig. 1E to G) (see Materials and Methods for details). The probability of SLdsRed infection was significantly higher in a cell infected by SLGFP than in a cell not infected by SLGFP for both HeLa (Fig. 1E) and Caco-2 (Fig. 1F) cells. The distribution of the different populations of infected cells shows a much larger overlap between the cells coinfected with SLGFP and SLdsRed than one would anticipate theoretically for two independent infections (Fig. 1G). Thus, the efficiency of Salmonella invasion of an individual epithelial cell depends on the concomitant invasion of the same cell by other salmonellae. Interestingly, increasing the multiplicity of infection (MOI) in HeLa cells (Fig. 1E) resulted in a significant increase of the SLdsRed infection in cells infected by SLGFP but not in cells not infected by SLGFP. This result confirmed that the direct effect of an MOI increase is a higher number of bacteria that infect certain cells rather than an increase in the overall number of cells that become infected. In addition to the previously reported Salmonella cooperative entry in HeLa and MDCK cells (4, 12), we showed here that this cooperation also takes place in Caco-2 cells, suggesting that this phenomenon is universal during Salmonella entry in epithelial cells. Taken together, these results validated that our system was operational.
The probability of being reinfected by Salmonella is higher for already-infected cells, even after the disappearance of the entry ruffles.
To study long-term and ruffle-unrelated cooperative events of Salmonella coinfections, we set up sequential infections with a delay of 1 h between the two infection waves, killing extracellular bacteria in between through gentamicin treatment. Scanning our time-lapse movies, we were ensured that this time lag led to the complete disappearance of any remaining entry ruffles from the first infection. In addition, we extended the delay between the two sequential infections to 2 h and 3 h (Fig. 1A). We compared the different populations of cells infected during the second infection (population I2), depending on whether they were already infected during the first wave of infection (population I2 ∣ I1) or not (population I2 ∣ noI1) for HeLa (Fig. 2A) and Caco-2 (Fig. 2B) cells. For both tested cell types, it was significantly more probable for a cell infected the first time to be reinfected the second time than for a cell not previously infected. We propose that such cells are somehow more vulnerable for future infection.
FIG 2.

The probability of being reinfected by Salmonella remains higher for already-infected cells after entry ruffle disappearance. (A and B) Conditional probability of infection for two different populations during sequential infection with a delay (Δt) of 2 h for HeLa cells and Caco-2 cells, as indicated. Results were obtained from three independent experiments, and P values were obtained after a paired t test. (C) The vulnerability score was plotted for infection with a 1-, 2-, or 3-h delay before the second infection in HeLa cells. The red line corresponds to P(I2 ∣ I1) = P(I2 ∣ noI1) = 1, indicating the independence of infections I2 and I1. Values above the red line correspond to P(I2 ∣ I1) > P(I2 ∣ noI1), indicating cooperation between infections. Values below the red line correspond to P(I2 ∣ I1) < P(I2 ∣ noI1), indicating competition between infections. Results were obtained from three independent experiments per time point, and P values were obtained after an unpaired t test. ***, P < 0.001; ns, not significant.
During all sequential infection experiments we also controlled the overall infection efficiencies of SLGFP and SLdsRed at all measured time points (first infection with SLGFP and second infection with SLdsRed, or in the reverse order) (Fig. S1). In all cases, the percentage of cells infected by each fluorescent Salmonella bacterium was similar to percentage of cells subjected to single (control) or sequential infections, underlining the observation that sequential infections did not change the overall infection efficiencies for the differently colored salmonellae. Nevertheless, we noticed a decrease in the number of infected cells between the early infection and that at later time points. This effect is most likely due to the technically unavoidable gentamicin treatment between infections. In addition, SLGFP showed a higher infectivity than SLdsRed for each condition, which is explained by the general deleterious effects of the heterologously overexpressed fluorescent proteins on Salmonella infectivity and by the partial loss of dsRed expression observed by us and others. Taking into account these issues, we took advantage of the observed consistency of the differences of infection efficiencies between the initial and the successive infections and between SLGFP and SLdsRed. This consistency allows comparative analyses of the ratio of the different infection probabilities, and it provided us with an analytical tool for precise quantification independently of the variances of the differently colored bacteria and technical hurdles of sequential infection.
We defined a vulnerability score as the conditional probability for a cell to be infected during the second infection after it had already been infected during the first one (I2 ∣ I1), divided by the conditional probability for a cell to be infected during the second infection when it had not been previously infected (I2 ∣ noI1) (described in detail in Materials and Methods). We also analyzed the changes of the vulnerability score over time by comparing cells subjected to sequential infections with 1-, 2-, and 3-h delays (Fig. 2B and S2 give a detailed representation of the conditional probability for each replicate). Surprisingly, the vulnerability score appeared unaltered. We obtained similar results when the order of the tested pathogens was reversed, infecting cells first with SLdsRed and then with SLGFP (Fig. S3). It was not possible to shorten the delay between infections to less than 1 h due to the ruffle influence, and we could not extend it beyond 3 h due to potential release into the extracellular medium of hyperreplicative (HR) bacteria from the first infection that could then reinfect new cells during the second wave of infection. Together, these results showed that, after ruffle disappearance, the infected cells remained more vulnerable to a new infection than the noninfected ones, and this vulnerability was stable over time.
Cell vulnerability to secondary infection can be predicted from the number of intracellular bacteria.
So far, we have considered only the characteristic “infected” or “noninfected” for each cell after SLGFP and SLdsRed infections that provides global trends on their interaction. To further exploit our data, we quantified the number of bacteria per host cell and related the obtained numbers to the previously extracted vulnerability scores. The distribution of intracellular bacteria inside infected cells at 2.5 h postinfection (p.i.) showed that most of the cells contained a few bacteria, and the proportion of cells decreased drastically when the number of intracellular bacteria increased. Overall, we were able to distinguish three groups of infected cells: the ones containing one to two intracellular bacteria (35% of the global population), the ones containing three to eight intracellular bacteria (39% of the global population), and the ones containing more than nine intracellular bacteria (26% of the global population), corresponding, respectively, to low, medium, and high infections (Fig. 3A).
FIG 3.

Cell vulnerability can be predicted from the number of bacteria previously internalized. (A) Distribution of the number of intracellular bacteria detected at 1.5 h p.i. in HeLa cells (average from three replicates). The infection efficiencies are clustered in three groups: low, medium, and high infection, corresponding, respectively, to 1 to 2, 3 to 8, or more than 9 bacteria per cell. (B) The vulnerability score is represented as a function of the number of intracellular bacteria resulting from the first infection in HeLa cells. (C) Probability of a cell to be highly infected during the second infection (I2 ≥ 9) as a function of the number of intracellular bacteria being internalized during the first infection in HeLa cells. Values in panels B and C represent the data merged from all the experiments (delay of 1, 2, and 3 h before the second infection). Infection efficiency groups are identical for experiments shown in all panels.
We compared the vulnerability scores of these three infection groups during sequential double infections (Fig. 3B). This analysis revealed that the greater the number of bacteria that had entered a given host cell during the first infection, the more likely it was that this cell became reinfected. Such tendencies still emerged when the bacteria were not grouped but analyzed individually, underlining the robustness of this result (Fig. S4).
Then, we investigated how the level of bacterial uptake during the second infection depends on the number of intracellular bacteria of the first infection. For this we quantified the probability for a cell to be highly infected during the second infection as a function of the efficiency of the first uptake (Fig. 3C). We found that the greater the number of intracellular bacteria that had been internalized during the first infection, the more likely the host cells were to engulf a large amount of new bacteria during the second infection. Therefore, we propose that cell vulnerability is maintained from the first to the second infection.
Cell vulnerability as an intrinsic or induced property.
The results from the sequential infections (Fig. 2 and 3) provided quantitative scores of cell vulnerability to Salmonella infection. We next investigated the origin of the observed cell vulnerability. Two possibilities can be anticipated: (i) the cellular vulnerability would be an intrinsic host cell attribute (hypothesis 1, intrinsic vulnerability), or (ii) it would be induced by bacterial uptake (hypothesis 2, induced vulnerability) (Fig. 4A). In theory, these hypotheses can be distinguished by the observable difference in the probabilities of the second wave of infection occurring in previously noninfected cells, P(I2 ∣ noI1), as depicted in the two schemes of Fig. 4B and described as follows: in the case of vulnerability as an intrinsic attribute, the probability of infection P(I2 ∣ noI1) would be lower than that for control cells [i.e., the infected cells among the population of cells exposed only to the second infection, or P(I2Ctr)] as the pool of vulnerable cells would have already been partially consumed during the first sequential infection, whereas it would remain conserved in the control (Fig. 4B, left). In the case of induced vulnerability, the probability of infection P(I2 ∣ noI1) would be similar to P(I2Ctr) as the cells would be considered to have equivalent vulnerabilities before their first infection (Fig. 4B, right). The experimental data obtained did not show a significant difference between P(I2 ∣ noI1) and P(I2Ctr) (t test, P value of >0.05) (Fig. 4C), suggesting that vulnerability may be induced by bacterial uptake (Fig. 4B, right). Taking into account the small percentage of cells belonging to the studied subpopulations, we caution that the absence of a statistically significant difference between these populations did not allow us to exclude the first hypothesis of host cell inherent vulnerability.
FIG 4.

Cell vulnerability examined as an intrinsic or an induced property. (A) Schemes of the two hypotheses for the origin of cell vulnerability. In hypothesis 1, cell vulnerability is inherent: some cells (in orange) are more vulnerable to infection than other cells (in yellow). In hypothesis 2, cell vulnerability is induced by bacterial uptake: before infection cells are equal regarding their vulnerability (in yellow), but after infection the infected cells turn progressively more vulnerable (in orange). (B) Graphic representation of the theoretical distribution of the different populations in the case of hypothesis 1 or hypothesis 2. (C) Probability of infection during sequential infection of HeLa cells with 1-, 2-, and 3-h delays for control cells (I2Ctr) and cells not infected during the first infection (noI1). P values were obtained after an unpaired t test [P(I2ctr) versus P(I2 ∣ noI1)]. ns, not significant.
Single-cell vulnerability to Salmonella infection is a combination of intrinsic and induced vulnerabilities.
Considering that the subpopulation comparison could not exclude an involvement of inherent vulnerability, we developed a mathematical model to evaluate the relative contribution of induced and inherent vulnerabilities to the overall cell vulnerability to Salmonella infection. To investigate the contribution of cell parameters at a single-cell level, we measured different intrinsic variables that could influence cellular vulnerability, namely, the cell morphology (cell perimeters and cell circularity), the local environment (local cell density and number of infected and noninfected neighboring cells), and the above-analyzed features of the Salmonella infection (delay between infections and load of intracellular bacteria per cell from I1) (Fig. 5A). We extracted all these elements using Icy, an image analysis software program (18) recently used for Salmonella infection studies in situ (19) (Fig. S5A gives an illustration of Icy cell segmentation).
FIG 5.

Single-cell vulnerability to Salmonella infection is a combination of intrinsic and induced vulnerability. (A) The depicted cellular parameters were determined for HeLa cells and Caco-2 cells as described in detail in Materials and Methods. An overlay of the distribution of some of these parameters in infected or noninfected cells is shown. (B) Quantification of the improvement of infection prediction by each cell parameter by subtracting the likelihood (log scale) of the model including all parameters from that of a model ignoring one parameter. Results are averaged over 100 training/testing circles for each model. P values were obtained after a paired t test. (C) Fold change of the probability of infection as a function of the intrinsic vulnerability and of a previous infection. (D) Increases or decreases in the probability of infection [P(inf)] when the listed cell parameters increase their values. *, P < 0.05; ***, P < 0.001; ns, not significant.
First, we analyzed the distribution of distinct cellular parameters in either infected or noninfected HeLa and Caco-2 cell populations (Fig. 5A). Caco-2 cells were cultured at high confluence so that the cells formed a continuous polarized monolayer (see Materials and Methods). For both cell types, the infected cells displayed distinct cellular features in comparison to the noninfected cells, such as higher local crowding reflected by a higher number of neighboring cells in direct contact. Comparing the relative correlations of the cellular parameters, we highlight the presence of strong links between many of them (Fig. S5B and C, S6, and S7). In particular, cell morphology is highly dependent on the local microenvironment, such as local cell density, which negatively correlates with the cell perimeter in HeLa and Caco-2 cells. Interestingly, cells that were infected during the second bacterial challenge are more likely to be nearby cells that were infected during the first bacterial challenge (infected neighbor cells) than by noninfected neighbor cells. Thus, Salmonella infection of one cell increases the probability that its neighboring cells will be subsequently infected.
To quantify the direct involvement of each studied parameter on overall cell vulnerability, we developed a statistical modeling approach adapted to our high-throughput microscopy data set on sequential Salmonella infection. This model is based on a logistic regression that is able to predict the infection efficiency at a single-cell level from cellular parameters. We measured the contribution of each parameter to the prediction by estimating how well the model predicts compared to a model that would ignore one parameter, as described in Materials and Methods (Fig. 5B). Taken separately, the load of intracellular bacteria resulting from I1 directly improved the prediction of cell vulnerability to subsequent infection (Fig. 5B). Thus, host cell vulnerability is induced by bacterial uptake, which is in line with our experimental data. In addition, the host cell parameters linked to cell morphology and local environment also significantly improved the model prediction of infection for HeLa and for Caco-2 cells (Tables S1 and S2 provide model details and the value of the coefficients). Together, our modeling approach revealed that single-host-cell vulnerability to Salmonella infection is a combination of intrinsic and bacterially induced vulnerability.
We quantified the relative involvement of the intrinsic and bacterially induced vulnerabilities by calculating the model-based fold change in the probability of infection of a cell not infected and having a low score of inherent vulnerability versus that of a cell infected and/or having a high score of inherent vulnerability (Fig. 5C). This showed that both induced and intrinsic vulnerabilities have a strong impact on overall cell vulnerability. Interestingly, the impact of induced vulnerability is more prevalent for Salmonella infection of HeLa cells (2.2-fold increase) than for infection of Caco-2 cells (1.3-fold increase), whereas inherent vulnerability plays a more prominent role for Caco-2 cell infections (2.6-fold increase) than for HeLa cells (1.6-fold increase). From these findings we conclude that the analyzed host cell parameters are differentially involved in relation to cell vulnerability to Salmonella infection depending on the cell type. In particular, the local cell density increases cell vulnerability for HeLa cells but reduces it for Caco-2 cells (Fig. 5D). This could be explained by the polarization of the Caco-2 cells at high confluence and highlights the specificity of each predicted model for a given cell type.
We also investigated whether the first infection affects the inherent host cell parameters; we compared the correlation between parameters that were identified as being either involved or not involved in the inherent vulnerability of the cell (Fig. S8). As their correlations were similar in infected and noninfected cells, we concluded that Salmonella infection did not impact the implications of the studied inherent cell parameters.
Reliability of the model-based prediction of infection.
To investigate the spatial distribution of cell vulnerability among the cell population, we generated vulnerability maps from the original images of the cell population after labeling each cell nucleus with a color corresponding to its probability of infection (Fig. 6A). Notably, we could confirm that, on average, the infected cells were properly assigned a higher prediction score to be infected than the noninfected ones (Fig. S9 shows the data for the predicted probabilities of infection). Based on our vulnerability maps, the predicted infected cells showed a very good overlap or were in the nearby vicinity of the experimentally infected cells (Fig. 6A). This illustrates the reliability of our approach in a qualitative way, and it also underlines the impact of local microenvironment on cell vulnerability. We went on to quantify the accuracy of the HeLa and Caco-2 cell-adapted models applied to 100 experimentally measured infected and 100 experimentally measured noninfected cells. For both cell types, models allowed a good prediction in the majority of the cases, with 62% for HeLa cells and 66% for Caco-2 cells (Fig. 6B). Taken together, these results attest that the probability of Salmonella infection success can be forecast at the near-single-cell level based on host cell parameters.
FIG 6.
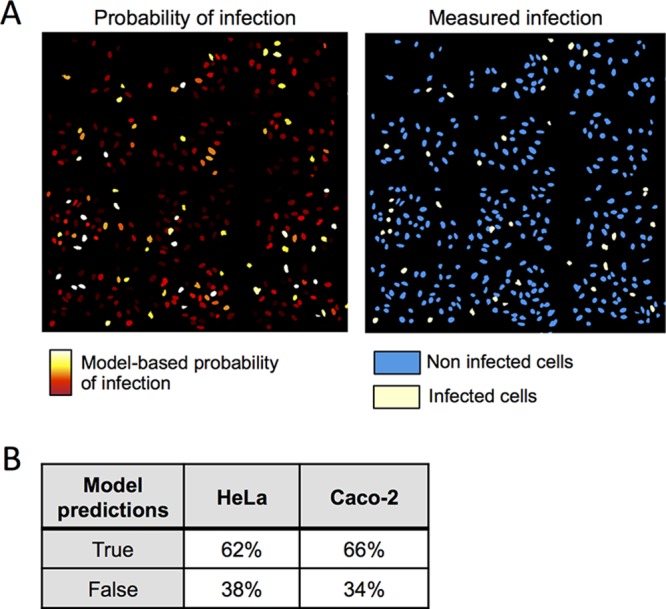
Comparison of model-predicted vulnerability of single-cell and measured infections. (A) Model-predicted probability of infection displayed on reproduced original images of HeLa cells (left panel). Colors are adapted for maximum contrast between the lowest (deep red) and highest (white) probability of infection. Measured infections from experiments are also shown (right panel). (B) Estimation of the reliability of the two (HeLa and Caco-2) models developed when tested on a total of 100 infected cells and 100 noninfected cells.
Involvement of cellular cholesterol level as an inherent vulnerability factor.
To investigate the molecular players that are linked to the inherent cell vulnerability to Salmonella infection, we analyzed plasma membrane composition as a main feature known to be relevant to Salmonella infection. We focused on cholesterol as the cells at low crowding present a larger amount of free cholesterol than the cells at high crowding (15). We monitored the relationship between global cellular cholesterol levels and host cell targeting by performing a Salmonella infection of HeLa cells for 30 min, followed by cholesterol labeling via filipin staining. Although filipin is the most commonly used tool to assess cholesterol content, it also displays very fast photobleaching properties (20). Thus, the automatic acquisition of an entire 96-well plate would introduce a strong bias due to the loss of filipin signal during the acquisition. To circumvent this technical issue, we carried out flow cytometry acquisition and analysis (Fig. 7). For each experiment, we binned the total cell population into five subpopulations corresponding to the increasing cellular levels of cholesterol that we classified as 1 to 5, with each subpopulation containing 20% of the total cells (Fig. S10 provides fluorescence-activated cell sorting [FACS] gating details). Comparing the number of infected cells in these different subpopulations with different amounts of cholesterol in HeLa (Fig. 7A) and Caco-2 (Fig. 7B) cells, we revealed that the probability of infection correlates in both cases with the cholesterol levels. Increasing the cholesterol level corresponds to a decrease in the probability of Salmonella infection in HeLa cells (Fig. 7A); however, it also corresponds to an increase in the probability of infection in Caco-2 cells (Fig. 7B). Thus, similarly to the cell density, the cholesterol level is a host cell parameter allowing us to estimate cell vulnerability to Salmonella infection in a cell-type-dependent manner.
FIG 7.

Probability of infection as a function of the single-cell cholesterol level. Variation in the probability of Salmonella infection at different levels of host cholesterol was measured by FACS as described in detail in the text. Cholesterol levels were binned in five categories at 20% steps from lowest to highest levels over the total cell population; each category contains 20% of the total cell population. (A) Results obtained for HeLa cells (n = 3). (B) Results obtained for Caco-2 cells (n = 3).
DISCUSSION
Cellular heterogeneity describes cases in which genetically identical cells present different behaviors and morphologies. This biological phenomenon is commonly present in an epithelial layer of an individual cell as well as within a monolayer of cultured cells. Despite the realization of the importance of cellular heterogeneity, its study has become feasible only during recent years, mainly thanks to the implementation of novel technologies such as imaging and computer-assisted analyses. In the context of pathogen infection, this heterogeneity produces cells unequally vulnerable or resistant, which impacts the overall infection.
We investigated the cell vulnerability of epithelial cells for S. Typhimurium infection. According to our results, infected cells display a strikingly higher probability of being reinfected with Salmonella, even after the disappearance of membrane ruffles. We obtained similar results in two relevant epithelial cell lines, HeLa and Caco-2, suggesting that this represents a conserved propensity toward Salmonella infection. The measured cellular vulnerability remained unaltered for all measured time points ranging from a delay of 1 h to 3 h between the infections. Assigning a vulnerability score to the challenged cells, we showed a higher vulnerability score in cells that had been previously infected, and we found that this score increased with the amount of intracellular bacteria contained by a given cell. This result raises the issue of the bacterial impact on cell vulnerability. Therefore, we aimed at distinguishing inherent cell vulnerability from vulnerability induced by bacterial uptake (Fig. 4A, hypotheses 1 and 2, respectively), exploiting the imaging data obtained via a high-content analytical pipeline. This allowed visualization of the infection in situ and provided a large number of associated cellular parameters. We quantified the implication of specific parameters associated with individual cells on cell vulnerability to Salmonella infection. It clearly appeared that the efficiency of early bacterial uptake during the first infection directly determined cell vulnerability. Thus, Salmonella induces an increase in cell vulnerability to subsequent infections.
While long-term cooperation among bacteria has been extensively studied for the communities of bacteria living in a common extracellular environment (21), little is known about the cooperation between intracellular and extracellular bacteria leading to increased bacterial uptake. Nevertheless, this phenomenon has been investigated more extensively for many viruses, including bacteriophages (22), influenza virus (23), poxviruses (24, 25), flaviviruses (26, 27), alphaviruses (28), and alphaherpesviruses (29). Generally, these studies have demonstrated that the first virus to infect a cell has the capacity to prevent coinfection of other viruses belonging to either the same strain or a more distantly related or unrelated strains. This is termed “superinfection exclusion” and may protect limited cellular resources and promote the replication and dissemination of the originally infecting virus. By analogy, the increased probability of cellular reinfection by Salmonella can be described as “superinfection promotion.” It remains to be clarified if such a process is relevant for all intracellular bacteria. For instance and in contrast to Salmonella infection, Jorgensen et al. reported that the Chlamydia effector protein CPAF secreted from bacteria within mature inclusions prevents bacteria that are still extracellular to invade (30). Thus, CPAF could be a factor mediating Chlamydia resistance to superinfection.
Our approach also allowed the relative quantification of the impact of different host cell parameters on the inherent vulnerability of the host cell to Salmonella infection. In particular, morphological attributes and local cell crowding are highly linked with this vulnerability. Cell crowding as a major determinant of the probability for infection has been proposed by Snijder and colleagues in the context of viral infection. They showed that during infections by the simian virus SV40 or the mouse hepatitis virus (MHV), the targeted cells have different localizations within cell islets (13). SV40 and MHV infect preferentially either peripheral or central cells, a phenomenon that is linked to the differential expression levels of focal adhesion kinase and the presence of sphingolipid GM1 at the plasma membrane of the challenged host cells. Thus, similarly to several viral infections, the probability of infection of a single cell by Salmonella is influenced by its local environment.
Our analytical tools will be useful for further studies on Salmonella and for other researchers working on different intracellular bacterial pathogens, such as Chlamydia, Listeria, or Shigella (see Materials and Methods). We revealed that some cells are indeed intrinsically more vulnerable to Salmonella and will be targeted by the bacteria first. Most of the tested parameters appeared to be relevant for model-based infection prediction but are differentially involved in the cell vulnerability depending on the cell type studied. Developing an adapted model based on host cell parameters, we could forecast the probability of Salmonella infection success at the near-single-cell level. Interestingly, the number of infected neighboring cells is highly increased in the population of infected cells. Cases of bacterial uptake impacting cells neighboring the infection (called bystander cells) have been previously reported for Shigella, which induces an interleukin-8 (IL-8) immune response after NF-κB activation that is detectable from 2 h p.i. in 70% of the bystander cells (31). However, it is not known whether the neighboring cells are also more susceptible to Shigella entry.
Because of our lack of knowledge of host factors that are involved in early attachment, such as potential entry receptors, it remains difficult to identify the molecular mechanisms that establish the differential vulnerability during Salmonella infection. Although receptors for direct recognition of Salmonella have been proposed, such as the cystic fibrosis transmembrane conductance regulator (CFTR) (32) and the epithelium growth factor receptor (EGFR) (33), many cell types infected by Salmonella do not express them (34). Therefore, it has been proposed that recognition mechanisms likely involve more ubiquitous factors (35). To explore the molecular cues involved in the inherent heterogeneity of host cell vulnerability, we decided to investigate membrane lipid composition, in particular, cellular cholesterol. We found that the cholesterol amount at the single-cell level in HeLa and Caco-2 cells correlates with the vulnerability of these cells to Salmonella infection. In HeLa cells Salmonella preferentially targets cells with small amounts of cholesterol. However, in Caco-2 cells, Salmonella preferentially targets cells with large amounts of cholesterol. Interestingly, these results with respect to a host cell molecule implicated in vulnerability are in agreement with the morphological feature of local density. Frechin and colleagues reported that cells at high density contain smaller amounts of cholesterol (15). Moreover, at high density HeLa and Caco-2 cells display an increase and a decrease in inherent cell vulnerability, respectively. This is in line with the correlation that we reported between the cholesterol level and host cell vulnerability. The molecular role of cholesterol during Salmonella infection is still under debate. Several studies have demonstrated that the Salmonella SipB effector and translocon component requires cholesterol for proper functioning (35, 36). In this context, it should be noted that the translocons operate in small cholesterol-rich microdomains at the plasma membrane and cannot be linked readily to the overall cholesterol levels. Furthermore, those studies were based on sterol-sequestering agents and biosynthesis inhibitors. Contrastingly, Gilk and colleagues, using an original mouse model, have shown that cholesterol is not essential for Salmonella invasion and intracellular replication inside host cells (37). In our study we highlighted that nontreated HeLa cells with a small amount of global cellular cholesterol are preferentially targeted by Salmonella, which does not exclude a potential involvement of cholesterol at the subcellular level. Santos and colleagues have also reported that the preferential invasion of hTERT-RPE1 and HeLa mitotic cells by Salmonella was SipB and cholesterol dependent (11). However, the small amount of mitotic cells in the whole population (<4%) may have a limited impact on the overall inherent vulnerability of the host cell population. Thus, our observation that the most vulnerable HeLa cells display a low cholesterol level is not in contradiction with previous publications on cholesterol involvement during the Salmonella infection process.
In conclusion, our study represents a first step in understanding Salmonella cell targeting and provides a path for the identification of cellular and bacterial factors involved in host cell vulnerability. Such factors could be targeted to render a cell more resistant to pathogen infections, allowing potential new therapeutic strategies. Together, the results of our study delineate in a quantitative manner the importance of vulnerable cell recognition and bacterial cooperation for cell targeting by S. Typhimurium.
MATERIALS AND METHODS
Bacterial strains.
The following S. Typhimurium strains were used: SL1344 (wild type), SL1344 pM965 (expressing GFP; SLGFP) described by Stecher et al. (38), and SL1344 pGG2 (expressing dsRed; SLdsRed) obtained after transformation of SL1344 with the pGG2 plasmid described by Lelouard et al. (39). Bacteria were grown in lysogeny broth (LB) medium supplemented with 0.3 M NaCl and ampicillin at 50 μg/ml at 37°C in an orbital shaker.
Cell culture.
All cell culture reagents were purchased from Invitrogen unless otherwise stated. Human epithelial HeLa cells (clone CCL-2 from ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% (vol/vol) fetal bovine serum (FBS), at 37°C in 5% CO2. HeLa cells were plated at a concentration of 1.5 × 104 cells/well in glass-bottom 96-well plates 24 h before infection so that they displayed about 80% of confluence on the infection day. Intestinal epithelial Caco-2 TC7 cells (kindly provided by P. Sansonsetti) were grown in DMEM supplemented with 10% FBS at 37°C in 10% CO2. Caco-2 cells were plated at a concentration of 3.5 × 104 cells/well in glass-bottom 96-well plates 48 h before infection so that they displayed a polarized (but not differentiated) continuous monolayer on the infection day. All infection assays were performed in EM buffer (120 mM NaCl, 7 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, 5 mM glucose, 25 mM HEPES, pH 7.4). HeLa cells were transfected with pEGFP-actin plasmid DNA (40) from a maxiprep, using X-tremeGENE 9 DNA transfection reagent (Roche), for 48 h.
Double-infection assays.
For invasion experiments, overnight bacterial cultures were subcultured at 1/20 and grown until late exponential/early stationary phase. Before infection, bacteria were gently washed and resuspended in EM buffer. Bacteria were added to the cells at an MOI of 30 corresponding to the CFU count and incubated for 30 min at 37°C in 5% or 10% CO2 for HeLa or Caco-2 cells, respectively. Noninternalized bacteria were eliminated by three washes with warm EM buffer and incubated for 1, 2, or 3 h at 37°C in 5% or 10% CO2 for HeLa or Caco-2 cells, respectively. Addition of EM buffer containing 100 μg/ml gentamicin for 1 h killed extracellular bacteria. The concentration of gentamicin was then decreased to 10 μg/ml, and 10% FBS was added to the medium. At the desired time points, the cells were washed again in EM buffer to eliminate the remaining gentamicin and reinfected with a fresh batch of subcultured bacteria according to the same protocol. After the extracellular bacteria were killed again by 1 h of incubation with EM buffer containing 100 μg/ml gentamicin, the cells were fixed with 4% paraformaldehyde at room temperature for immunofluorescence analysis.
Microscopy.
All image acquisitions were performed on a Nikon inverted widefield microscope using a 20× air objective (0.5 numerical aperture [NA]), an automatic programmable xy stage, and the Nikon perfect focus system. For sequential infections of HeLa and Caco-2 cells, 161 fields were imaged per well, and four channels per field were captured using a CoolSnap 2 camera (Roeper Scientific). Nuclei and cells were stained using DAPI (excitation and emission wavelengths of 350 and 470 nm, respectively), and the cell bodies were stained with CellMask DeepRed Plasma Membrane Stain (ThermoFisherScientific) (excitation and emission wavelengths of 640 and 670 nm, respectively). Caco-2 cells were stained with the FM 4-64 membrane dye (Invitrogen) before time-lapse imaging (excitation and emission wavelengths of 558 and 734 nm, respectively). Quantification of the ruffle timing was performed on the same microscope, using a 20× (0.5 NA) air objective and time intervals of 3 min for 90 min. Time-lapse imaging of ruffles was performed on a DeltaVision wide-field microscope using a 60× (1.42 NA) oil objective and z-stacks with a spacing of 500 nm. The images were subsequently deconvolved using DeltaVision Elite integrated software.
Cholesterol measurements.
HeLa and Caco-2 cells were challenged with SLGFP for 30 min before trypsinization, fixation with 4% paraformaldehyde at room temperature, and incubation with 16 μg/ml filipin complex from Streptomyces filipinensis (Sigma-Aldrich). This treatment was directly followed by FACS measurement on a BD FACSCanto cytometer using the excitation and emission wavelengths of 405 and 450 nm, respectively, and 488 and 530 nm, respectively, for filipin and GFP fluorescence detection, respectively. Infected and noninfected cells were distinguished using the green fluorescence emitted by SLGFP (see Fig. S10 in the supplemental material for gating details). Data were processed using FlowJo software.
Image analysis.
All images were analyzed with two open-source software programs: CellProfiler (http://cellprofiler.org/) and Icy (http://icy.bioimageanalysis.org/). CellProfiler was used to detect each single cell and the number of its intracellular salmonellae expressing either GFP or dsRed. The following modules were used during the analysis: IdentifyPrimaryObjects recognized nuclei and bacteria; IdentifySecondaryObjects identified cells (here the secondary objects) by extending the nuclear area previously recognized; RelateObjects assigned bacteria within individual cells. Icy was used for accurate detection of cell borders and cellular microenvironment analysis. We used a graphical environment within Icy, called Protocols, for the development of an analytical pipeline including the following plug-ins: HK-Means, which identifies nuclei by prefiltering the signal to identify objects within a size range; Spot Detector, which identifies bacteria; Active Contours, which identifies the edges of the plasma membrane by propagating the region of interest (ROI) detected for the nuclei; and Javascript, which relates each bacterium to the ROI of its host cell, measures local cell density, and distinguishes which neighboring cells are infected by which bacteria.
Probability.
P(I2 ∣ I1) is the probability of the second sequential infection, knowing that the cell has been infected in the first infection round, and is calculated as follows: P(I2 ∣ I1) = P(I1 & I2)/P(I1), where P(I1) is the number of cells in I1/total number of cells, and P(I1 & I2) is (the number of cells in I1 and I2)/total number of cells.
P(I2 ∣ noI1) is the probability of the second sequential infection, knowing that the cell has not been infected in the first round of infection and is calculated as follows: P(I2 ∣ noI1) = P(I2 & noI1)/P(noI1), where P(noI1) is the number of cells in noI1/total number of cells, and P(I2 & noI1) is (the number of cells in noI1 and I2)/total number of cells.
Model.
We modeled the influence of multiple parameters on the probability of a second infection. A Boolean variable Y represents the second infection; it is equal to 1 for infected cells and 0 otherwise. Its probability is predicted by the following seven parameters: load of infection (LOI) represents the number of infecting bacteria during the first infection, separated into four groups corresponding to no (0 bacteria), low (1 or 2), medium (3 to 8), or high (9+) infection. Delay is a categorical variable corresponding to the delay between the first and the second infections (1, 2, or 3 h). Infected neighbor cells (X1) refers to the number of cells in contact that had been infected during the first infection. Noninfected neighbor cells (X2) refers to the number of cells in contact which had not been infected during the first infection. Local cell density (X3) is the number of cells present in a vicinity of 100 μm. The distance is calculated between the centers of the nuclei. Cell perimeter (X4) is the length of the perimeter of the cell (in micrometers) obtained after segmentation. Circularity (X5) refers to the cell circularity, defined as (4π × area)/perimeter2. This parameter is higher for circular cells and lower for cells that are elongated or have complex shape but does not depend a priori on the cell size. In practice we used its square root. The conditional probability of Y during the second infection is modeled as follows: P(Y = 1 ∣ X1, … , Xn) = 1/[1 + exp(−(aLOI + aDelay + a1X1 + … a5X5))], where aLOI (respectively, aDelay) has a different value for each of the LOI categories (respectively, Delay categories), and a1, … ,a5 are constants. All parameters were learned by maximizing the likelihood of the model, e.g., the probability of the observed data as measured by the model. We used 115,000 and 327,000 cells to train and test the model for HeLa and Caco-2 cells, respectively. We divided the cell population into two random sets, the training set (9/10 of the cells per replicate) and testing set (1/10 of the cells), and computed the likelihood of infection observed in the testing set. The higher the likelihood was, the better the parameters of the model predicted infection. We repeated this procedure 100 times. To measure the improvement of infection prediction by taking into account each parameter, the likelihood of the complete model was compared (on a log scale) with the likelihood of seven models, each of which ignored one parameter. This difference in log-likelihood values is reported in Fig. 5B.
Quantification of the impact of a parameter to cell vulnerability was obtained by applying our statistical model to the 1st and the 3rd quantile values of a given parameter, while other parameters were kept equal at their median values. We obtained the probabilities of the second infection for these two sets and reported their ratio. In Fig. 5D, the upward- and downward-pointing arrows correspond to ratios above and under 1, respectively. The parameter and values corresponding to a low inherent vulnerability of HeLa and Caco-2 cells were the following: local cell density (1st quantile and 3rd quantile, respectively), cell perimeter (1st quantile), infected neighboring cells (median), noninfected neighboring cells (median), and circularity (median and 3rd quantile, respectively). The parameters and values corresponding to a high inherent vulnerability of HeLa and Caco-2 cells were the following: local cell density (3rd quantile and 1st quantile, respectively), cell perimeter (3rd quantile), infected neighboring cells (median), noninfected neighboring cells (median), and circularity (median and 1st quantile, respectively).
Model reliability was evaluated by using 100 infected and 100 noninfected cells and quantifying the amount of good predictions among those cells. We repeated this procedure 100 times and show the average. As a comparison, a random model would provide approximately 50% good predictions.
Statistical analysis.
The statistical analysis was performed using R and GraphPad Prism, and t tests were used to evaluate the significance of the results.
Availability of data.
The pipeline used on CellProfiler and on Icy, as well as the R code used to generate the model, can be provided by the authors.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jennifer Fredlund and Andrew Rutenberg for their help during the initial phase of the project, Adrien Sauvaget, Claude Loverdo, Kristine Schauer, and Uriel Hazan for productive discussions, Mariana Ferrari for her help with the FACS experiments, and all the members of the Dynamics of Host-Pathogen Interactions Unit and BioImage Analysis Group for helpful interactions.
V.S. was supported by a Ph.D. fellowship from the University Paris Diderot allocated by the ENS Cachan, Université Paris-Saclay. J.E. is a member of the LabEx consortia IBEID and MilieuInterieur. J.E. also acknowledges support from the ANR (grant StopBugEntry and AutoHostPath) and the ERC (CoG EndoSubvert).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00644-17.
REFERENCES
- 1.Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O'Brien SJ, Jones TF, Fazil A, Hoekstra RM. 2010. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis 50:882–889. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 2.Carter PB, Collins FM. 1974. The route of enteric infection in normal mice. J Exp Med 139:1189–1203. doi: 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson KG, Holden DW. 2010. Dynamics of growth and dissemination of Salmonella in vivo. Cell Microbiol 12:1389–1397. doi: 10.1111/j.1462-5822.2010.01511.x. [DOI] [PubMed] [Google Scholar]
- 4.Misselwitz B, Barrett N, Kreibich S, Vonaesch P, Andritschke D, Rout S, Weidner K, Sormaz M, Songhet P, Horvath P, Chabria M, Vogel V, Spori DM, Jenny P, Hardt WD. 2012. Near surface swimming of Salmonella Typhimurium explains target-site selection and cooperative invasion. PLoS Pathog 8:e1002810. doi: 10.1371/journal.ppat.1002810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vonaesch P, Cardini S, Sellin ME, Goud B, Hardt WD, Schauer K. 2013. Quantitative insights into actin rearrangements and bacterial target site selection from Salmonella Typhimurium infection of micropatterned cells. Cell Microbiol 15:1851–1865. doi: 10.1111/cmi.12154. [DOI] [PubMed] [Google Scholar]
- 6.Misselwitz B, Kreibich SK, Rout S, Stecher B, Periaswamy B, Hardt WD. 2011. Salmonella enterica serovar Typhimurium binds to HeLa cells via fim-mediated reversible adhesion and irreversible type three secretion system 1-mediated docking. Infect Immun 79:330–341. doi: 10.1128/IAI.00581-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haraga A, Ohlson MB, Miller SI. 2008. Salmonellae interplay with host cells. Nat Rev Microbiol 6:53–66. doi: 10.1038/nrmicro1788. [DOI] [PubMed] [Google Scholar]
- 8.LaRock DL, Chaudhary A, Miller SI. 2015. Salmonellae interactions with host processes. Nat Rev Microbiol 13:191–205. doi: 10.1038/nrmicro3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knodler LA. 2015. Salmonella enterica: living a double life in epithelial cells. Curr Opin Microbiol 23:23–31. doi: 10.1016/j.mib.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Santos JC, Enninga J. 2016. At the crossroads: communication of bacteria-containing vacuoles with host organelles. Cell Microbiol 18:330–339. doi: 10.1111/cmi.12567. [DOI] [PubMed] [Google Scholar]
- 11.Santos AJM, Meinecke M, Fessler MB, Holden DW, Boucrot E. 2013. Preferential invasion of mitotic cells by Salmonella reveals that cell surface cholesterol is maximal during metaphase. J Cell Sci 126:2990–2996. doi: 10.1242/jcs.115253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorkowski M, Felipe-López A, Danzer CA, Hansmeier N, Hensel M. 2014. Salmonella enterica invasion of polarized epithelial cells is a highly cooperative effort. Infect Immun 82:2657–2667. doi: 10.1128/IAI.00023-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snijder B, Sacher R, Rämö P, Damm E-M, Liberali P, Pelkmans L. 2009. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 461:520–523. doi: 10.1038/nature08282. [DOI] [PubMed] [Google Scholar]
- 14.Liberali P, Snijder B, Pelkmans L. 2015. Single-cell and multivariate approaches in genetic perturbation screens. Nat Rev Genet 16:18–32. doi: 10.1038/nrg3768. [DOI] [PubMed] [Google Scholar]
- 15.Frechin M, Stoeger T, Daetwyler S, Gehin C, Battich N, Damm E-M, Stergiou L, Riezman H, Pelkmans L. 2015. Cell-intrinsic adaptation of lipid composition to local crowding drives social behaviour. Nature 523:88–91. doi: 10.1038/nature14429. [DOI] [PubMed] [Google Scholar]
- 16.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin Da, Chang JH, Lindquist Ra, Moffat J, Golland P, Sabatini DM. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. 2011. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Chaumont F, Dallongeville S, Chenouard N, Hervé N, Pop S, Provoost T, Meas-Yedid V, Pankajakshan P, Lecomte T, Le Montagner Y, Lagache T, Dufour A, Olivo-Marin J-C. 2012. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods 9:690–696. doi: 10.1038/nmeth.2075. [DOI] [PubMed] [Google Scholar]
- 19.McQuate SE, Young AM, Silva-Herzog E, Bunker E, Hernandez M, de Chaumont F, Liu X, Detweiler CS, Palmer AE. 2017. Long-term live-cell imaging reveals new roles for Salmonella effector proteins SseG and SteA. Cell Microbiol 19:e12641. doi: 10.1111/cmi.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boutte Y, Men S, Grebe M. 2011. Fluorescent in situ visualization of sterols in Arabidopsis roots. Nat Protoc 6:446–456. doi: 10.1038/nprot.2011.323. [DOI] [PubMed] [Google Scholar]
- 21.Davey ME, O'Toole GA. 2000. Microbial biofilms: from ecology to molecular genetics. Microbiol Mol Biol Rev 64:847–867. doi: 10.1128/MMBR.64.4.847-867.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cumby N, Davidson AR, Maxwell KL. 2012. The moron comes of age. Bacteriophage 2:225–228. doi: 10.4161/bact.23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang IC, Li W, Sui J, Marasco W, Choe H, Farzan M. 2008. Influenza A virus neuraminidase limits viral superinfection. J Virol 82:4834–4843. doi: 10.1128/JVI.00079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876. doi: 10.1126/science.1183173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laliberte JP, Moss B. 2014. A novel mode of poxvirus superinfection exclusion that prevents fusion of the lipid bilayers of viral and cellular membranes. J Virol 88:9751–9768. doi: 10.1128/JVI.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zou G, Zhang B, Lim P-Y, Yuan Z, Bernard Ka, Shi P-Y. 2009. Exclusion of West Nile virus superinfection through RNA replication. J Virol 83:11765–11776. doi: 10.1128/JVI.01205-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol 81:4591–4603. doi: 10.1128/JVI.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karpf AR, Lenches E, Strauss EG, Strauss JH, Brown DT. 1997. Superinfection exclusion of alphaviruses in three mosquito cell lines persistently infected with Sindbis virus. J Virol 71:7119–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Criddle A, Thornburg T, Kochetkova I, DePartee M, Taylor MP. 2016. gD-Independent superinfection exclusion of alphaherpesviruses. J Virol 90:4049–4058. doi: 10.1128/JVI.00089-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jorgensen I, Bednar MM, Amin V, Davis BK, Ting JPY, McCafferty DG, Valdivia RH. 2011. The chlamydia protease CPAF regulates host and bacterial proteins to maintain pathogen vacuole integrity and promote virulence. Cell Host Microbe 10:21–32. doi: 10.1016/j.chom.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasper CA, Sorg I, Schmutz C, Tschon T, Wischnewski H, Kim ML, Arrieumerlou C. 2010. Cell-cell propagation of NF-κB transcription factor and MAP kinase activation amplifies innate immunity against bacterial infection. Immunity 33:804–816. doi: 10.1016/j.immuni.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, Ratcliff R, Evans MJ, Colledge WH. 1998. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature 393:79–82. doi: 10.1038/30006. [DOI] [PubMed] [Google Scholar]
- 33.Pace J, Hayman MJ, Galán JE. 1993. Signal transduction and invasion of epithelial cells by S. typhimurium. Cell 72:505–514. doi: 10.1016/0092-8674(93)90070-7. [DOI] [PubMed] [Google Scholar]
- 34.Jones BD, Paterson HF, Hall A, Falkow S. 1993. Salmonella typhimurium induces membrane ruffling by a growth factor-receptor-independent mechanism. Proc Natl Acad Sci U S A 90:10390–10394. doi: 10.1073/pnas.90.21.10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garner MJ, Hayward RD, Koronakis V. 2002. The Salmonella pathogenicity island 1 secretion system directs cellular cholesterol redistribution during mammalian cell entry and intracellular trafficking. Cell Microbiol 4:153–165. doi: 10.1046/j.1462-5822.2002.00181.x. [DOI] [PubMed] [Google Scholar]
- 36.Hayward RD, Cain RJ, McGhie EJ, Phillips N, Garner MJ, Koronakis V. 2005. Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells. Mol Microbiol 56:590–603. doi: 10.1111/j.1365-2958.2005.04568.x. [DOI] [PubMed] [Google Scholar]
- 37.Gilk SD, Cockrell DC, Luterbach C, Hansen B, Knodler LA, Ibarra JA, Steele-Mortimer O, Heinzen RA. 2013. Bacterial colonization of host cells in the absence of cholesterol. PLoS Pathog 9:e1003107. doi: 10.1371/journal.ppat.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stecher B, Hapfelmeier S, Müller C, Kremer M, Stallmach T, Hardt W-d, Mu C. 2004. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun 72:4138–4150. doi: 10.1128/IAI.72.7.4138-4150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lelouard H, Henri S, De Bovis Ba, Mugnier B, Chollat-Namy A, Malissen B, Meresse S, Gorvel JP. 2010. Pathogenic bacteria and dead cells are internalized by a unique subset of Peyer's patch dendritic cells that express lysozyme. Gastroenterology 138:173–184.e1–3. doi: 10.1053/j.gastro.2009.09.051. [DOI] [PubMed] [Google Scholar]
- 40.Ehsani S, Santos JC, Rodrigues CD, Henriques R, Audry L, Zimmer C, Sansonetti P, Van Nhieu GT, Enninga J. 2012. Hierarchies of host factor dynamics at the entry site of Shigella flexneri during host cell invasion. Infect Immun 80:2548–2557. doi: 10.1128/IAI.06391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.