

ABSTRACT
The liver is frequently affected in patients with active brucellosis. The present study demonstrates that Brucella abortus infection induces the activation of the autophagic pathway in hepatic stellate cells to create a microenvironment that promotes a profibrogenic phenotype through the induction of transforming growth factor-β1 (TGF-β1), collagen deposition, and inhibition of matrix metalloproteinase-9 (MMP-9) secretion. Autophagy was revealed by upregulation of the LC3II/LC3I ratio and Beclin-1 expression as well as inhibition of p62 expression in infected cells. The above-described findings were dependent on the type IV secretion system (VirB) and the secreted BPE005 protein, which were partially corroborated using the pharmacological inhibitors wortmannin, a phosphatidyl inositol 3-kinase inhibitor, and leupeptin plus E64 (inhibitors of lysosomal proteases). Activation of the autophagic pathway in hepatic stellate cells during Brucella infection could have an important contribution to attenuating inflammatory hepatic injury by inducing fibrosis. However, with time, B. abortus infection induced Beclin-1 cleavage with concomitant cleavage of caspase-3, indicating the onset of apoptosis of LX-2 cells, as was confirmed by the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay and Hoechst staining. These results demonstrate that the cross talk of LX-2 cells and B. abortus induces autophagy and fibrosis with concomitant apoptosis of LX-2 cells, which may explain some potential mechanisms of liver damage observed in human brucellosis.
KEYWORDS: Brucella, liver
INTRODUCTION
Human brucellosis, a zoonotic infection caused by Brucella species, is an inflammatory disease, with inflammation being present both in the acute and chronic phases of the disease and in virtually all of the organs affected (1–3). The liver is frequently affected in patients with active brucellosis, and although numerous studies have focused on brucellar liver histopathology (1, 2, 4–6), the pathogenic mechanisms of Brucella liver disease have not been completely investigated at the molecular and cellular levels.
Liver fibrosis is a wound-healing response to chronic hepatic injury (7, 8). An early event in the development of liver fibrosis is the activation of hepatic stellate cells (HSCs), the major cell type responsible for increased synthesis of extracellular matrix proteins (9). Increased transforming growth factor β1 (TGF-β1) levels are also observed in the damaged liver, and this has a close correlation with fibrogenic changes in HSCs and liver tissue (10–12).
It has been demonstrated that autophagy is involved in the fibrotic response to chronic hepatic injury caused by alcohol abuse, hepatitis virus infection, and nonalcoholic steatohepatitis (13). Autophagy is a catabolic intracellular pathway that targets defective or excessive organelles to the lysosomes for degradation into amino acids, free fatty acids, or other small molecules used for material recycling or energy harvest (14). Autophagy, usually stimulated by energy restriction, stress, or inflammation, is regarded as a survival mechanism that plays a critical role in maintaining cellular homeostasis, which is involved in many human disorders, including fibrotic disease (15). During fibrosis, autophagy is mostly a cell survival mechanism that attenuates hepatic inflammatory injury and ultimately induces liver fibrosis (14).
Previously, we have demonstrated that upon infection of hepatic stellate cells, B. abortus triggers a profibrogenic response characterized by the inhibition of matrix metalloproteinase-9 (MMP-9) secretion, collagen deposition, and TGF-β1 secretion. This involves a functional type 4 secretion system (T4SS) and its effector protein, BPE005 of B. abortus. The TGF-β1 pathway is a classical signaling pathway activated in liver fibrosis that also induces autophagy (16). This suggests that autophagy participates in fibrosis during Brucella infection in a way that depends on TGF-β expression. Thus, we hypothesized that Brucella infection creates a microenvironment that promotes a profibrogenic phenotype and induces the activation of the autophagic pathway, which could have an important contribution in attenuating hepatic injury in the liver of patients with Brucella infection.
RESULTS
B. abortus infection induces LC3-II and Beclin-1 expression in LX-2 cells.
It has been demonstrated that autophagy participates in HSC activation (17). We have previously demonstrated that B. abortus infection induces HSC activation, leading to a profibrogenic phenotype (18, 19). To determine if B. abortus infection induces the activation of the autophagic pathway in HSCs, we evaluated by Western blotting at 24 h postinfection the expression of LC3 II, the lipidated form of LC3 I, and the only known protein that specifically associates with autophagosomes (20), the autophagy regulator Beclin-1 (21), as well as p62, which participates in the autophagic clearance of ubiquitinated proteins (22). B. abortus infection induced an increase in the LC3 II/LC3 I ratio, an increase in Beclin-1 expression, and the inhibition of p62 expression (Fig. 1). These results indicate that autophagy was induced by B. abortus in LX-2 cells.
FIG 1.

B. abortus infection induces autophagy in LX-2 cells. (A) LX-2 cells were infected with B. abortus at an MOI of 100 and 1,000, cell lysates obtained at 24 h postinfection were used to determine LC3 I and II, Beclin-1, and p62 production by Western blotting (vertical line, spliced image to eliminate redundant results at an MOI of 500). (B to D) Densitometric analysis of results from three independent experiments performed as described for panel A: LC3 II/LC3 I ratio (B) and Beclin-1 (C) and p62 (D) production. Data are given as the means ± SD from at least three individual experiments. *, P < 0.05; **, P < 0.01 versus noninfected cells (N.I.).
B. abortus induction of a profibrogenic phenotype on LX-2 cells depends on PI3-kinase and lysosomal proteases.
Acquisition of a fibrogenic phenotype by resident HSCs is a critical event of the liver's response to injury. Increasing evidence supports the notion that autophagy participates in the pathophysiology of hepatic fibrosis (14). A key cytokine involved in fibrosis is TGF-β1 (23). We have demonstrated that B. abortus infection induced fibrosis in vitro and in vivo, as revealed by collagen deposition and MMP-9 downmodulation in a mechanism that is dependent on TGF-β1 (19). Thus, experiments were conducted to determine whether fibrosis induced by B. abortus infection is dependent on autophagy activation. To this end, we examined markers of a fibrogenic phenotype, including the secretion of TGF-β1, the deposition of collagen, and the downmodulation of MMP-9 in LX-2 cells during B. abortus infection in the presence of wortmannin (a phosphatidylinositol 3-kinase [PI3-kinase] inhibitor) and leupeptin plus E64 (inhibitors of lysosomal proteases). Both wortmannin and leupeptin plus E64 reversed B. abortus-induced MMP-9 downmodulation, collagen deposition, and TGF-β1 secretion (Fig. 2A to C). Accordingly, when we performed the infection experiment in the presence of rapamycin, an autophagy inductor, we observed an increase of the fibrotic phenotype with respect to infected but untreated cells, corroborating the association between autophagy and fibrosis (Fig. 2D and E). These results indicate that autophagy is involved in the induction of the fibrotic phenotype in B. abortus-infected LX-2 cells.
FIG 2.

B. abortus induction of a fibrotic phenotype depends on PI3-kinase and lysosomal proteases. (A) Effect of wortmannin (wort) and E64 plus leupeptin (E64+leupeptin) in the inhibition of MMP-9 secretion induced by B. abortus infection by zymography 24 h postinfection. (B) wort and E64+leupeptin effect on the induction of collagen deposition induced by B. abortus infection, as determined by quantification of Sirius red staining 7 days after infection. (C) Effect of wort and E64+leupeptin on TGF-β1 secretion during B. abortus infection. Effect of rapamycin during B. abortus infection on MMP-9 secretion by zymography 24 h postinfection (D) and collagen deposition determined by quantification of Sirius red staining 7 days after infection (E). Data are given as the means ± SD from at least three individual experiments. *, P < 0.05; ***, P < 0.001 versus infected and untreated cells.
B. abortus induces autophagy in LX-2 cells in a VirB-dependent manner.
The type IV secretion system (T4SS) VirB is vital for Brucella to establish an intracellular replication niche (24), and it also has been involved in the induction of inflammatory responses during infection (11, 25–27). In addition, we have previously demonstrated that the fibrotic phenotype induced by B. abortus in LX-2 cells is dependent on a functional T4SS (18). As such, we tested whether VirB was involved in the activation of the autophagy signaling pathway. Our results demonstrate that B. abortus wild-type infection increased the expression of LC3 II and Beclin-1. However, when LX-2 cells were infected with the B. abortus virB10 mutant, LC3 II and Beclin-1 levels did not differ significantly from those of uninfected cells (Fig. 3). These results indicate that the activation of the autophagy signaling pathway depends on a functional T4SS.
FIG 3.

B. abortus induces autophagy in LX-2 cells in a VirB-dependent manner. (A) LX-2 cells were infected with B. abortus (B.a.) and its virB10 isogenic mutant (ΔvirB10) at an MOI of 100 and 1,000, and cells were stained at 24 h postinfection to determine LC3 and Beclin-1 levels by immunofluorescence. (B, C, and D) Quantitative analysis of experiments presented in panel A. Data are given as the means ± SD from at least three individual experiments. *, P < 0.05; **, P < 0.0; ***, P < 0.001 versus noninfected cells (N.I.).
VirB-dependent effector protein BPE005 is involved in the autophagic phenotype induced by B. abortus in LX-2 cells.
Recently, B. abortus-secreted effectors have been identified to require a functional VirB system to be translocated into the host cells (27–35).
Our previous results indicated that the secreted protein BPE005 is involved in the induction of a fibrogenic phenotype in LX-2 cells (18). Thus, we decided to determine whether BPE005 is also involved in the induction of autophagy. To this end, we evaluated if the B. abortus bpe005 mutant was able to induce the expression of LC3 II, the only protein known to specifically associate with autophagosomes. As occurred with the B. abortus virB mutant, when LX-2 cells were infected with the B. abortus bpe005 mutant, there was no change in the levels of LC3 II with respect to LC3 I. In addition, the complemented bpe005 mutant restored the ability to induce autophagy observed in the wild-type strain (Fig. 4).
FIG 4.
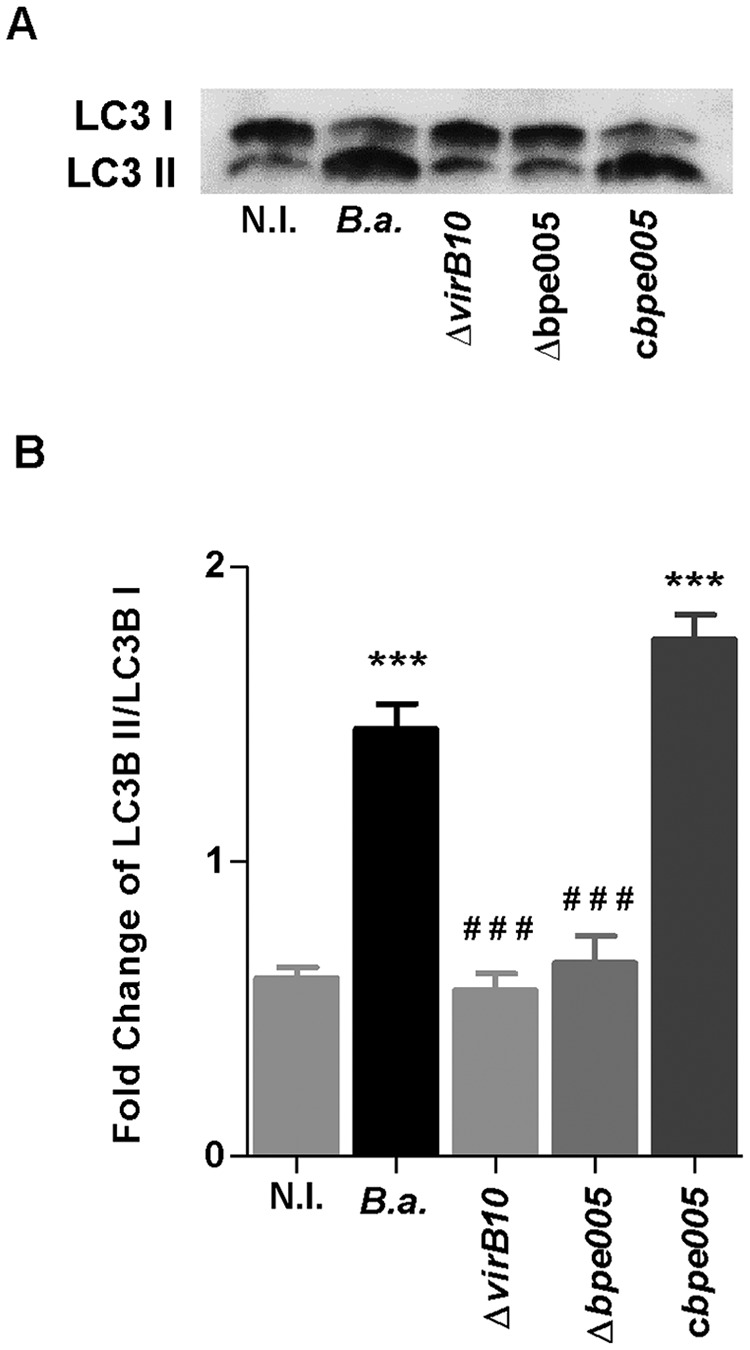
VirB-dependent effector protein BPE005 is responsible for the autophagy phenotype. (A) LX-2 cells were infected with B. abortus (B.a.), its virB10 (ΔvirB10) and bpe005 (Δbpe005) isogenic mutants, and the complemented bpe005 mutant (cbpe005) at an MOI of 1,000, and cell lysates obtained at 24 h postinfection were used to determine LC3 I/II ratios by Western blotting. (B) Densitometric analysis of results from two independent experiments performed as described for panel A. Data are given as the means ± SD from at least three individual experiments. ***, P < 0.001 versus noninfected cells (N.I.); ###, P < 0.001 versus B. abortus.
In addition, and taking into account that Brucella could use some components from the autophagy pathway to establish its replicative niche (36, 37), experiments were performed to corroborate the association between autophagy and the fibrotic phenotype induced by BPE005 in LX-2 cells. To this end, LX-2 cells were transfected with a eukaryotic expression vector harboring the bpe005 gene, and the levels of MMP-9 activity, collagen deposition, and TGF-β1 secretion were determined. Expression of BPE005 protein in LX-2 cells was able to inhibit MMP-9 activity and to induce collagen deposition and TGF-β1 secretion, and these phenomena were reversed when experiments were performed in the presence of wortmannin (a PI3-kinase inhibitor) and leupeptin plus E64 (inhibitors of lysosomal proteases), bafilomycin, or chloroquine (two inhibitors of autophagosome-lysosome fusion) (Fig. 5).
FIG 5.
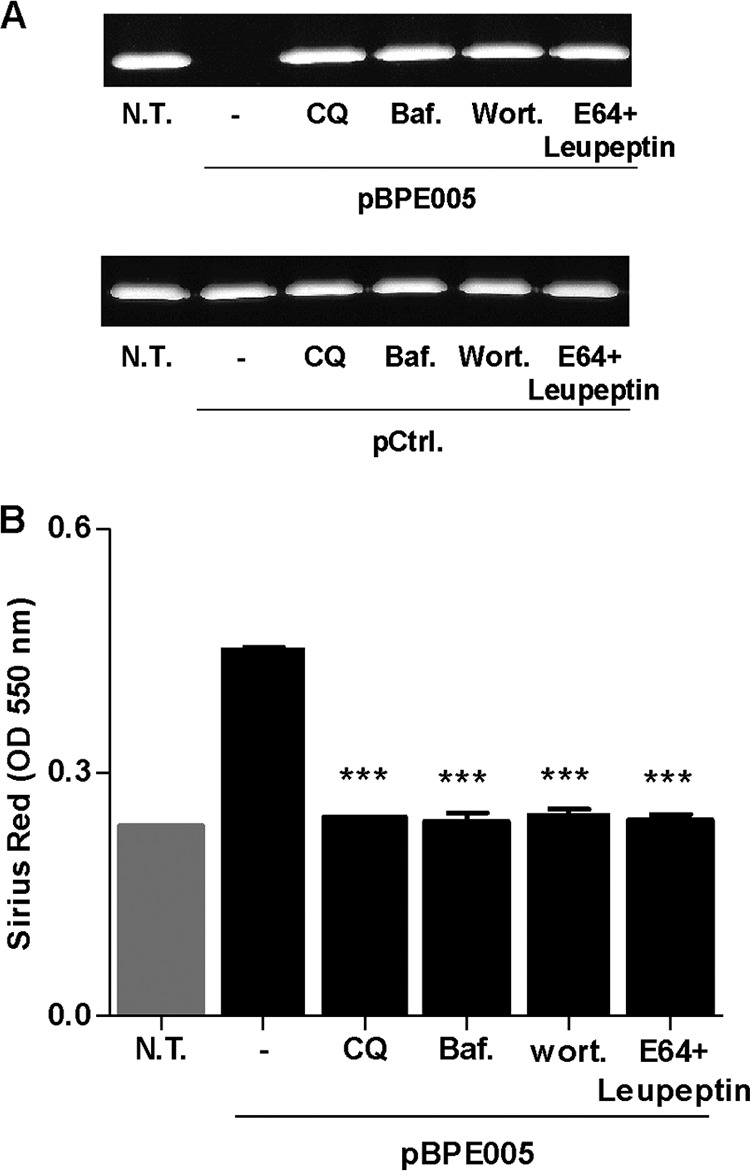
BPE005 protein induction of a fibrotic phenotype depends on PI3-kinase, lysosomal proteases, and autophagosome-lysosome fusion. (A) LX-2 cells were transfected with BPE005 plasmid DNA (pBPE005) or with pcDNA3-c-myc as a control (pCtrl.), and the effect of chloroquine (CQ), bafilomycin (Baf.), wortmannin (Wort.), and E64 plus leupeptin (E64+leupeptin) on the inhibition of MMP-9 secretion was determined by zymography at 24 h after transfection. (B) CQ, Baf., Wort., and E64+leupeptin effect on the induction of collagen deposition was determined by quantification of Sirius red staining 7 days after transfection. Data are given as the means ± SD from at least three individual experiments. ***, P < 0.001 versus transfected and untreated cells. N.T., nontreated.
Taken together, these results indicate that B. abortus induces autophagy pathway activation with a mechanism that is dependent on the presence of T4SS and its secreted protein, BPE005, with concomitant fibrosis dependent on PI3-kinase, lysosomal proteases, and autophagosome-lysosome fusion.
B. abortus infection induces cleavage of Beclin-1 with concomitant apoptosis.
In spite of the ability of Brucella infection to induce a fibrotic phenotype in HSCs (18, 19), liver cirrhosis is a debatable issue in Brucella-infected humans (5), indicating a possible balance between fibrotic and antifibrotic factors involved in infection. Increasing HSC death is a possible explanation for limiting liver fibrosis.
Beclin-1 is a dual regulator of both autophagy and apoptosis. When we evaluated Beclin-1 expression at 48 h postinfection, our results indicated that B. abortus infection induces Beclin-1 cleavage (Fig. 6). Caspase-3-mediated cleavage of Beclin-1 promotes cross talk between apoptosis and autophagy (38). Experiments then were conducted to determine if B. abortus infection induces apoptosis in LX-2 cells at 48 h postinfection. To this end, LX-2 cells were infected with B. abortus, and the presence of apoptotic cells was determined by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay, cleaved caspase-3 expression, and Hoechst 33342 staining. Our results indicate that B. abortus-infected LX-2 cells exhibited an increase of TUNEL-positive cells, cleaved caspase-3 expression, and apoptotic nuclei, as revealed by Hoechst 33342 staining, compared to uninfected controls (Fig. 7), indicating a proapoptotic effect of Brucella infection. Apoptosis depended on the expression of a functional T4SS, since the percentage of apoptotic cells did not differ significantly between LX-2 cells infected with the B. abortus virB mutant and uninfected controls. These results indicated that after 48 h postinfection, B. abortus infection induces apoptosis of LX-2 cells.
FIG 6.

B. abortus infection induces cleavage of Beclin-1. (A) LX-2 cells were infected with B. abortus at an MOI of 100 and 1,000, and cell lysates obtained at 48 h postinfection were used to determine LC3 I and II ratios and Beclin-1 and p62 production by Western blotting (vertical line, spliced image to eliminate redundant results at an MOI of 500). (B to E) Densitometric analysis of results from two independent experiments performed as described for panel A: LC3 II/LC3 I ratio (B) and Beclin-1 (C), cleaved Beclin-1 (D), and p62 (E) production. Data are given as the means ± SD from at least three individual experiments. *, P < 0.05; **, P < 0.01 versus noninfected cells (N.I.).
FIG 7.

B. abortus infection induces apoptosis of LX-2 cells. (A) LX-2 cells were infected with B. abortus (B.a.) and its virB10 isogenic mutant (ΔvirB10) at an MOI of 100 and 1,000, and apoptosis was evaluated at 48 h postinfection by TUNEL assay, cleaved caspase-3 expression, and Hoechst staining by fluorescence microscopy. (B to D) Quantitative analysis of experiments presented in panel A: TUNEL assay (B), cleaved caspase-3 expression (C), and Hoechst 33342 staining (D). Data are given as the means ± SD from at least three individual experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus noninfected cells (N.I.).
PI3-kinase and lysosomal proteases are necessary to induce apoptosis of B. abortus-infected LX-2 cells.
Experiments were conducted to study if the PI3-kinase signaling pathway, lysosomal protein degradation, and apoptosis are related phenomena during B. abortus infection. To determine if these phenomena are necessary to induce apoptosis, infection experiments were performed in the presence of wortmannin and leupeptin plus E64, and the presence of apoptotic cells was determined by TUNEL assay, cleaved caspase-3 expression, and Hoechst 33342 staining. B. abortus-infected LX-2 cells exhibited an increase of TUNEL-positive cells, cleaved caspase-3, and apoptotic nuclei, as revealed by Hoechst 33342 staining, compared to levels for uninfected controls (Fig. 8). Apoptosis depended on the PI3-kinase signaling pathway and lysosomal protein degradation, since the percentage of apoptotic cells was significantly reduced when infection experiments were performed in the presence of the mentioned inhibitors (Fig. 8). These results indicated that PI3-kinase and lysosomal proteases are necessary to induce apoptosis in B. abortus-infected hepatic stellate cells.
FIG 8.
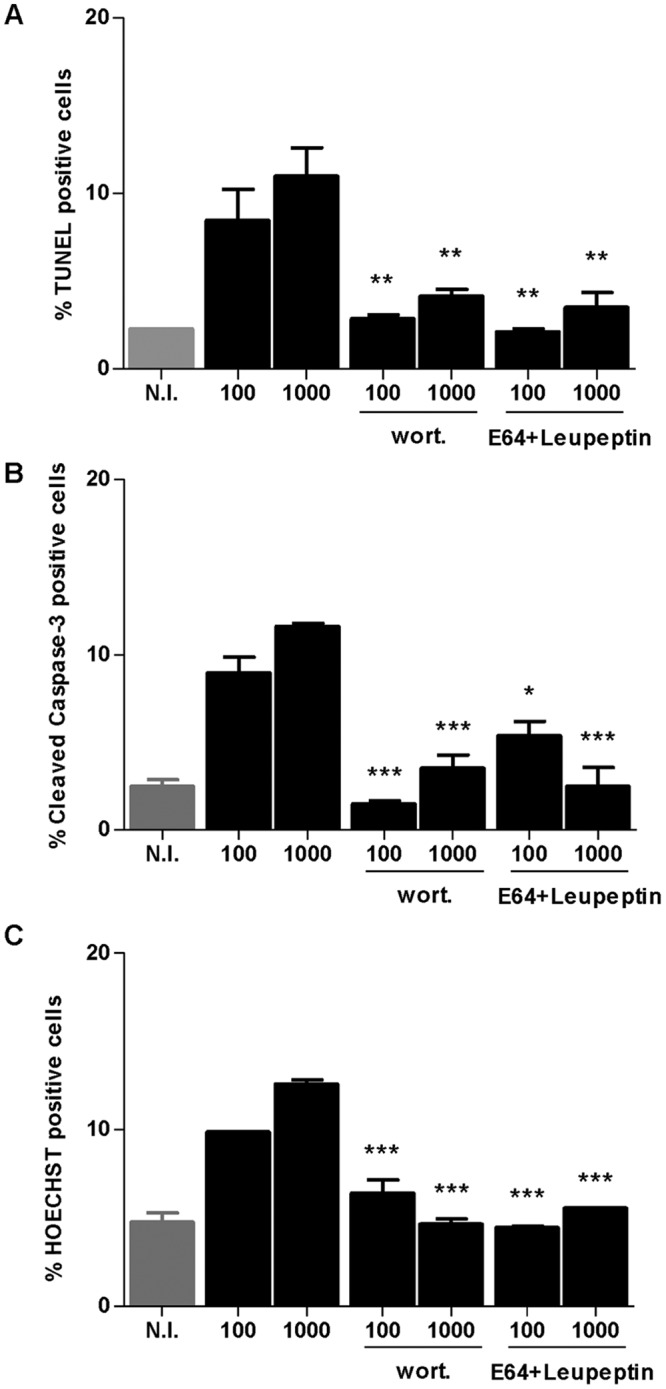
B. abortus infection induces apoptosis of LX-2 cells that is dependent on PI3-kinase and lysosomal proteases. Effect of wortmannin (wort) and E64 plus leupeptin (E64+leupeptin) in the apoptosis induced by B. abortus infection at 48 h postinfection. The presence of apoptotic cells were determined and quantified by TUNEL assay (A), cleaved caspase-3 expression (B), and Hoechst 33342 staining (C). Data are given as the means ± SD from at least three individual experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus infected and untreated cells.
DISCUSSION
Brucellosis is a worldwide zoonosis characterized by hepatomegaly, splenomegaly, and peripheral lymphadenopathy. It is a chronic and debilitating infection caused by Gram-negative facultative intracellular bacteria that infect domestic and wild animals and can be transmitted to humans (2, 5). However, although numerous studies have focused on brucellar liver histopathology (1, 4–6), the pathogenic mechanisms of liver disease caused by Brucella have not been completely elucidated.
In humans, the liver is frequently affected during active brucellosis. The frequency of liver involvement in active brucellosis ranges from 5% to 52% or more (5). In any circumstance, the persistence of an infectious stimulus might drive liver fibrosis because its presence could induce marked alterations in a variety of immune and structural cells, leading to a healing phenotype which is characterized by the deposition of extracellular matrix (39). This occurs as a compensatory response to the tissue repairing process in a wide range of chronic liver injures. However, although numerous studies have focused on brucellar liver histopathology (4), the pathogenic mechanisms of liver disease caused by Brucella have not been completely elucidated.
Autophagy is a cellular pathway crucial for development, differentiation, survival, and homeostasis. Its implication in human diseases has been highlighted in recent years (13). In addition, several data show that autophagy is involved in major aspects of hepatology (40). In liver, autophagy may supply energy for activation of HSCs, and this activation also can induce some liver diseases, including hepatic fibrogenesis (13). Autophagy fuels activation of HSCs inducing type I collagen synthesis, as was documented during cellular activation both in vitro and in vivo in mice following either carbon tetrachloride (CCL4)- or thioacetamide (TAA)-induced liver injury and in human HSCs from hepatitis B-infected liver (7). Accordingly, our results indicate that upon infection of LX-2 cells, B. abortus triggers autophagy activation characterized by the upregulation of Beclin-1 expression, an increase in the LC3 II/LC3 I ratio, and the downmodulation of p62. This was at least in part associated with a profibrogenic response characterized by inhibition of MMP-9 secretion, induction of collagen deposition, and TGF-β1 secretion in a way that involved a functional T4SS and its BPE005 effector protein.
As was mentioned, liver fibrosis is the excessive accumulation of extracellular matrix proteins, including collagen, that occurs in most types of chronic liver diseases. Advanced liver fibrosis results in cirrhosis, liver failure, and portal hypertension. Human brucellosis has been reported to have a possible causal relationship between B. abortus infection and cirrhosis (4); however, the presence of liver cirrhosis is a debatable issue, since in the studies reported, viral hepatitis was not excluded by the authors (5, 41, 42). In addition, cirrhosis was not observed during Brucella infection in animal models, indicating that although Brucella organisms have the ability to induce a fibrotic phenotype in HSCs, other factors could be involved to reduce the high fibrosis necessary to induce hepatic cirrhosis (18). The reversion of fibrotic phenotype and the concomitant resolution of hepatic fibrosis play an important role in the resolution of the pathogenesis of hepatic fibrosis without induction of cirrhosis. Recent studies suggest that apoptosis becomes the overriding process with resulting net HSC loss from the liver. This clearance of activated HSCs by apoptosis paved the way for the recovery of hepatic fibrosis (43, 44). Therefore, our results indicate that B. abortus was able to induce fibrosis with concomitant autophagy pathway activation in LX-2 cells at 24 h postinfection. However, at 48 h postinfection, caspase-mediated cleavage of Beclin-1 inactivates autophagy mediated by Beclin-1 and enhances apoptosis with concomitant apoptotic cell death.
Autophagy and apoptosis are two important and interconnected stress response mechanisms. The phenomenon in which autophagy precedes apoptosis could control, at least in part, liver fibrosis and inhibit the development of cirrhosis. This could partly explain why the development of cirrhosis is rare during hepatic brucellosis (5).
Taken together, these results indicate that upon infection of HSCs, B. abortus triggers a profibrotic response coinciding with autophagic pathway activation and then apoptotic cell death of activated HSCs that modulates the fibrotic phenotype.
MATERIALS AND METHODS
Bacterial culture.
Brucella abortus S2308, its isogenic virB10 polar and bpe005 mutants, and a complemented strain of the bpe005 mutant (18) were grown overnight in 10 ml of tryptic soy broth (Merck, Buenos Aires, Argentina) with constant agitation at 37°C. Bacteria were harvested and the inocula were prepared as described previously (45). All live Brucella experiments were performed in biosafety level 3 facilities located at the Instituto de Investigaciones Biomédicas en Retrovirus y SIDA (Buenos Aires, Argentina).
Cell culture.
The LX-2 cell line, a spontaneously immortalized human hepatic stellate cell line, was a gift from Scott L. Friedman (Mount Sinai School of Medicine, New York, NY). LX-2 cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies-Invitrogen, Carlsbad, CA) and supplemented with 2 mmol/liter of l-glutamine, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 5% (vol/vol) fetal bovine serum (Gibco-Invitrogen, Carlsbad, CA). Cells were seeded at 5 × 104 cells/well in 24-well plates and were cultured at 37°C with 5% CO2.
Cellular infection.
LX-2 cells were infected with either B. abortus, its isogenic virB10 and bpe005 mutants, or the complemented bpe005 mutant strain at different multiplicities of infection (MOI). After the bacterial suspension was dispensed, the plates were centrifuged for 10 min at 2,000 rpm and then incubated for 2 h at 37°C in a 5% CO2 atmosphere. Infected cells were extensively washed with Dulbecco's modified Eagle's medium to remove extracellular bacteria and then incubated in medium supplemented with 100 μg/ml of gentamicin and 50 μg/ml of streptomycin to kill extracellular bacteria. LX-2 cells were harvested at different times of infection depending on the specific experiment to determine autophagy, apoptosis, and fibrosis markers.
Western blotting.
Infected LX-2 cells were lysed in ice-cold lysis buffer consisting of 1% Triton X-100 in 150 mM NaCl, 25 mM Tris-HCl (TBS), pH 7.4, and a protease inhibitor cocktail (Sigma-Aldrich). Lysates were incubated on ice for 10 min and cleared by centrifugation at 13,000 × g for 10 min. Protein concentrations were determined by the bicinchoninic acid method (Pierce, Rockford, IL, USA) using bovine serum albumin (BSA) as the standard. Equal amounts of proteins were loaded onto electrophoresis gels and after separation; proteins were transferred to a nitrocellulose membrane (GE Healthcare, Little Chalfont, United Kingdom) and blocked for 1 h with 5% milk protein–0.05% Tween 20. After blocking, membranes were incubated with rabbit anti-LC3B (Cell Signaling Technology, Danvers, MA), goat anti-BECN-1 (Santa Cruz Biotechnology), or rabbit anti-p62/SQSTM1 (R&D Systems) overnight at 4°C, followed by washing, and then incubated with a 1:1,000 dilution of peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) for 1 h. Protein bands were visualized on Hyperfilm ECL (GE Healthcare) by chemiluminescence. Equal loading was confirmed by Ponceau S, acid red staining, and incubation of the blots with an anti-β-actin antibody (clone C4; Santa Cruz Biotechnology).
Immunofluorescence.
LX-2 cells were infected with B. abortus and, after 24 or 48 h postinfection, were fixed in 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.3% Triton X-100 (Roche Diagnostics GmbH, Mannheim, Germany) for 10 min, and blocked with PBS containing 1% BSA for 1 h. Infected cells were stained with rabbit anti-LC3B (Cell Signaling Technology, Danvers, MA), goat anti-BECN-1 (Santa Cruz Biotechnology), or rabbit anti-cleaved caspase-3 (Cell Signaling Technology) diluted in 0.1% PBS–Tween 20 overnight at 4°C. Cells then were incubated with Alexa Fluor 488 anti-rabbit (Jackson ImmunoResearch Laboratories) or Alexa Fluor 488 anti-goat (Molecular Probes, Life Technologies) diluted in 0.1% PBS–Tween for 4 h at room temperature. 4′,6-Diamidino-2-phenylindole (DAPI) was used for nuclear staining, and cells were stained for 30 min at room temperature. After washing in PBS, cells were mounted and then analyzed by fluorescence microscopy.
Apoptosis assays.
LX-2 cells were infected with B. abortus or its isogenic virB10 polar mutant and were harvested 48 h later. Cells were washed, and the percentage of apoptotic cells was assessed by fluorescence microscopy after they were labeled by the terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay or by staining with the Hoechst 33342 dye. As a positive control, cells were treated with 200 μM hydrogen peroxide.
Inhibitors.
To study the potential involvement of molecules that participate in autophagy pathways in the induction of collagen deposition, increase of TGF-β secretion, and MMP-9 inhibition by B. abortus infection in LX-2, wortmannin or E64 plus leupeptin (46) (Sigma-Aldrich de Argentina S.A.) were added 2 h after infection. Wortmannin was used at a concentration of 10 μM, and E64 plus leupeptin were used at a concentration of 20 μM. To study the role of autophagy pathways in the induction of collagen deposition, increase of TGF-β secretion, and MMP-9 inhibition by BPE005 protein, LX-2 cells were transfected with BPE005 plasmid or pCDNA3-c-myc as a control of transfection efficiency, as previously described (18). After transfection, cells were treated with wortmannin or leupeptin plus E64, at the previously mentioned concentrations, or with bafilomycin A1 or chloroquine (46). Bafilomycin A1 (Sigma-Aldrich) was used at a concentration of 200 nM, and chloroquine (Sigma-Aldrich) was used at a concentration of 50 μM.
Zymography.
Gelatinase activity was assayed by the method of Hibbs et al., with modifications, as described previously (45, 47, 48).
Assessment of collagen deposition by Sirius red staining.
Collagen deposition was quantified using Sirius red (Sigma-Aldrich), a strong anionic dye that binds strongly to collagen molecules. Sirius red staining was performed as described previously (18, 19).
Measurement of cytokine concentrations.
TGF-β1 expression was determined in the culture supernatants by enzyme-linked immunosorbent assay (ELISA; BD Biosciences, San Jose, CA).
Statistical analysis.
Statistical analysis was performed with one-way analysis of variance (ANOVA), followed by post hoc Tukey's test using GraphPad Prism 5.0 software. Data are represented as means ± standard deviations (SD).
ACKNOWLEDGMENTS
We thank Scott Friedman for LX-2 cells and Horacio Salomón and the staff of the Instituto de Investigaciones Biomédicas en Retrovirus y SIDA (INBIRS) for their assistance with biosafety level 3 laboratory use.
P.C.A.B., A.I.P.V., and C.K.H. are recipients of a fellowship from CONICET. G.H.G., D.J.C., and M.V.D. are members of the Research Career of CONICET. The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Young EJ. 1989. Clinical manifestations of human brucellosis. In Young EJ, Corbel MJ (ed), Brucellosis: clinical and laboratory aspects. CRC Press, Boca Raton, FL. [Google Scholar]
- 2.Pappas G, Akritidis N, Bosilkovski M, Tsianos E. 2005. Brucellosis. N Engl J Med 352:2325–2336. doi: 10.1056/NEJMra050570. [DOI] [PubMed] [Google Scholar]
- 3.Colmenero JD, Reguera JM, Martos F, Sanchez-De-Mora D, Delgado M, Causse M, Martin-Farfan A, Juarez C. 1996. Complications associated with Brucella melitensis infection: a study of 530 cases. Medicine (Baltimore) 75:195–211. doi: 10.1097/00005792-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Akritidis N, Tzivras M, Delladetsima I, Stefanaki S, Moutsopoulos HM, Pappas G. 2007. The liver in brucellosis. Clin Gastroenterol Hepatol 5:1109–1112. doi: 10.1016/j.cgh.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Madkour MM. 2001. Osteoarthicular brucellosis. In Madkour MM. (ed), Madkour's brucellosis, 2nd ed Springer-Verlag, Berlin, Germany. [Google Scholar]
- 6.Young EJ, Hasanjani Roushan MR, Shafae S, Genta RM, Taylor SL. 2014. Liver histology of acute brucellosis caused by Brucella melitensis. Hum Pathol 45:2023–2028. doi: 10.1016/j.humpath.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 7.Hernandez-Gea V, Friedman SL. 2012. Autophagy fuels tissue fibrogenesis. Autophagy 8:849–850. doi: 10.4161/auto.19947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bataller R, Brenner DA. 2005. Liver fibrosis. J Clin Investig 115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeng GW, Wang CR, Liu ST, Su CC, Tsai RT, Yeh TS, Wen CL, Wu YQ, Lin CY, Lee GL, Chen MY, Liu MF, Chuang CY, Chen CY. 1997. Measurement of synovial tumor necrosis factor-alpha in diagnosing emergency patients with bacterial arthritis. Am J Emerg Med 15:626–629. doi: 10.1016/S0735-6757(97)90173-X. [DOI] [PubMed] [Google Scholar]
- 10.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. 1995. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Investig 96:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roux CM, Rolan HG, Santos RL, Beremand PD, Thomas TL, Adams LG, Tsolis RM. 2007. Brucella requires a functional type IV secretion system to elicit innate immune responses in mice. Cell Microbiol 9:1851–1869. doi: 10.1111/j.1462-5822.2007.00922.x. [DOI] [PubMed] [Google Scholar]
- 12.Tahashi Y, Matsuzaki K, Date M, Yoshida K, Furukawa F, Sugano Y, Matsushita M, Himeno Y, Inagaki Y, Inoue K. 2002. Differential regulation of TGF-beta signal in hepatic stellate cells between acute and chronic rat liver injury. Hepatology 35:49–61. doi: 10.1053/jhep.2002.30083. [DOI] [PubMed] [Google Scholar]
- 13.Song Y, Zhao Y, Wang F, Tao L, Xiao J, Yang C. 2014. Autophagy in hepatic fibrosis. Biomed Res Int 2014:436242. doi: 10.1155/2014/436242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao YQ, Fan XM. 2015. Autophagy: a new therapeutic target for liver fibrosis. World J Hepatol 7:1982–1986. doi: 10.4254/wjh.v7.i16.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin XM, Ding WX, Gao W. 2008. Autophagy in the liver. Hepatology 47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 16.Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, Sugimoto K, Miyazono K. 2009. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 69:8844–8852. doi: 10.1158/0008-5472.CAN-08-4401. [DOI] [PubMed] [Google Scholar]
- 17.Thoen LF, Guimaraes EL, Dolle L, Mannaerts I, Najimi M, Sokal E, van Grunsven LA. 2011. A role for autophagy during hepatic stellate cell activation. J Hepatol 55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Arriola Benitez PC, Rey Serantes D, Herrmann CK, Pesce Viglietti AI, Vanzulli S, Giambartolomei GH, Comerci DJ, Delpino MV. 2015. The effector protein BPE005 from Brucella abortus induces collagen deposition and matrix metalloproteinase 9 downmodulation via transforming growth factor beta1 in hepatic stellate cells. Infect Immun 84:598–606. doi: 10.1128/IAI.01227-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arriola Benitez PC, Scian R, Comerci DJ, Serantes DR, Vanzulli S, Fossati CA, Giambartolomei GH, Delpino MV. 2013. Brucella abortus induces collagen deposition and MMP-9 down-modulation in hepatic stellate cells via TGF-beta1 production. Am J Pathol 183:1918–1927. doi: 10.1016/j.ajpath.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Shintani T, Klionsky DJ. 2004. Autophagy in health and disease: a double-edged sword. Science 306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He C, Levine B. 2010. The Beclin 1 interactome. Curr Opin Cell Biol 22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lippai M, Low P. 2014. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int 2014:832704. doi: 10.1155/2014/832704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Border WA, Noble NA. 1994. Transforming growth factor beta in tissue fibrosis. N Engl J Med 331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 24.Sieira R, Comerci DJ, Sanchez DO, Ugalde RA. 2000. A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J Bacteriol 182:4849–4855. doi: 10.1128/JB.182.17.4849-4855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rolan HG, Tsolis RM. 2008. Inactivation of the type IV secretion system reduces the Th1 polarization of the immune response to Brucella abortus infection. Infect Immun 76:3207–3213. doi: 10.1128/IAI.00203-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, Zamboni DS, Oliveira SC. 2013. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J Immunol 190:3629–3638. doi: 10.4049/jimmunol.1202817. [DOI] [PubMed] [Google Scholar]
- 27.Marchesini MI, Herrmann CK, Salcedo SP, Gorvel JP, Comerci DJ. 2011. In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell Microbiol 13:1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myeni S, Child R, Ng TW, Kupko JJ III, Wehrly TD, Porcella SF, Knodler LA, Celli J. 2013. Brucella modulates secretory trafficking via multiple type IV secretion effector proteins. PLoS Pathog 9:e1003556. doi: 10.1371/journal.ppat.1003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Barsy M, Jamet A, Filopon D, Nicolas C, Laloux G, Rual JF, Muller A, Twizere JC, Nkengfac B, Vandenhaute J, Hill DE, Salcedo SP, Gorvel JP, Letesson JJ, De Bolle X. 2011. Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell Microbiol 13:1044–1058. doi: 10.1111/j.1462-5822.2011.01601.x. [DOI] [PubMed] [Google Scholar]
- 30.de Jong MF, Sun YH, den Hartigh AB, van Dijl JM, Tsolis RM. 2008. Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol Microbiol 70:1378–1396. doi: 10.1111/j.1365-2958.2008.06487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salcedo SP, Marchesini MI, Lelouard H, Fugier E, Jolly G, Balor S, Muller A, Lapaque N, Demaria O, Alexopoulou L, Comerci DJ, Ugalde RA, Pierre P, Gorvel JP. 2008. Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog 4:e21. doi: 10.1371/journal.ppat.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Barsy M, Mirabella A, Letesson JJ, De Bolle X. 2012. A Brucella abortus cstA mutant is defective for association with endoplasmic reticulum exit sites and displays altered trafficking in HeLa cells. Microbiology 158:2610–2618. doi: 10.1099/mic.0.060509-0. [DOI] [PubMed] [Google Scholar]
- 33.Radhakrishnan GK, Yu Q, Harms JS, Splitter GA. 2009. Brucella TIR domain-containing protein mimics properties of the Toll-like receptor adaptor protein TIRAP. J Biol Chem 284:9892–9898. doi: 10.1074/jbc.M805458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacerda TL, Salcedo SP, Gorvel JP. 2013. Brucella T4SS: the VIP pass inside host cells. Curr Opin Microbiol 16:45–51. doi: 10.1016/j.mib.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Dohmer PH, Valguarnera E, Czibener C, Ugalde JE. 2014. Identification of a type IV secretion substrate of Brucella abortus that participates in the early stages of intracellular survival. Cell Microbiol 16:396–410. doi: 10.1111/cmi.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamer I, Goffin E, De Bolle X, Letesson JJ, Jadot M. 2014. Replication of Brucella abortus and Brucella melitensis in fibroblasts does not require Atg5-dependent macroautophagy. BMC Microbiol 14:223. doi: 10.1186/s12866-014-0223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, Lopez-Otin C, Virgin HW, Celli J. 2012. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11:33–45. doi: 10.1016/j.chom.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryter SW, Mizumura K, Choi AM. 2014. The impact of autophagy on cell death modalities. Int J Cell Biol 2014:502676. doi: 10.1155/2014/502676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meneghin A, Hogaboam CM. 2007. Infectious disease, the innate immune response, and fibrosis. J Clin Investig 117:530–538. doi: 10.1172/JCI30595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rautou PE, Mansouri A, Lebrec D, Durand F, Valla D, Moreau R. 2010. Autophagy in liver diseases. J Hepatol 53:1123–1134. doi: 10.1016/j.jhep.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 41.McCullough CN, Eisele CW. 1951. Brucella hepatitis leading to cirrhosis of the liver. AMA Arch Intern Med 88:793–802. doi: 10.1001/archinte.1951.03810120094009. [DOI] [PubMed] [Google Scholar]
- 42.Spink WW. 1956. Brucellosis; epidemiology, clinical manifestations, diagnosis. Semin Int 5:15–17. [PubMed] [Google Scholar]
- 43.Kong D, Zhang F, Zhang Z, Lu Y, Zheng S. 2013. Clearance of activated stellate cells for hepatic fibrosis regression: molecular basis and translational potential. Biomed Pharmacother 67:246–250. doi: 10.1016/j.biopha.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 44.Elsharkawy AM, Oakley F, Mann DA. 2005. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 10:927–939. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]
- 45.Scian R, Barrionuevo P, Giambartolomei GH, Fossati CA, Baldi PC, Delpino MV. 2011. Granulocyte-macrophage colony-stimulating factor- and tumor necrosis factor alpha-mediated matrix metalloproteinase production by human osteoblasts and monocytes after infection with Brucella abortus. Infect Immun 79:192–202. doi: 10.1128/IAI.00934-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scian R, Barrionuevo P, Giambartolomei GH, De Simone EA, Vanzulli SI, Fossati CA, Baldi PC, Delpino MV. 2011. Potential role of fibroblast-like synoviocytes in joint damage induced by Brucella abortus infection through production and induction of matrix metalloproteinases. Infect Immun 79:3619–3632. doi: 10.1128/IAI.05408-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hibbs MS, Hasty KA, Seyer JM, Kang AH, Mainardi CL. 1985. Biochemical and immunological characterization of the secreted forms of human neutrophil gelatinase. J Biol Chem 260:2493–2500. [PubMed] [Google Scholar]