

ABSTRACT
Salmonella enterica serovar Dublin is adapted to cattle but is able to infect humans with high invasiveness. An acute inflammatory response at the intestine helps to prevent Salmonella dissemination to systemic sites. Flagella contribute to this response by providing motility and FliC-mediated signaling through pattern recognition receptors. In a previous work, we reported a high frequency (11 out of 25) of S. Dublin isolates lacking flagella in a collection obtained from humans and cattle. The aflagellate strains were impaired in their proinflammatory properties in vitro and in vivo. The aim of this work was to elucidate the underlying cause of the absence of flagella in S. Dublin isolates. We report here that class 3 flagellar genes are repressed in the human aflagellate isolates, due to impaired secretion of FliA anti-sigma factor FlgM. This phenotype is due to an in-frame 42-nucleotide deletion in the fliE gene, which codes for a protein located in the flagellar basal body. The deletion is predicted to produce a protein lacking amino acids 18 to 31. The aflagellate phenotype was highly stable; revertants were obtained only when fliA was artificially overexpressed combined with several successive passages in motility agar. DNA sequence analysis revealed that motile revertants resulted from duplications of DNA sequences in fliE adjacent to the deleted region. These duplications produced a FliE protein of similar length to the wild type and demonstrate that amino acids 18 to 31 of FliE are not essential. The same deletion was detected in S. Dublin isolates obtained from cattle, indicating that this mutation circulates in nature.
KEYWORDS: FliE, proinflammatory capacity, Salmonella, flagella, serovar Dublin
INTRODUCTION
Nontyphoidal Salmonella enterica (NTS) is one of the main etiologic agents of foodborne diseases worldwide, with an estimated burden of 154 million cases per year (1–3; National Salmonella Centre [NSC] and Ministry of Health, Uruguay, unpublished data). NTS serovars normally cause gastroenteritis in healthy humans, but in immunocompromised hosts and children they can cause bloodstream infections, with high morbidity and mortality rates (4–7). It is estimated that 3.4 millon cases of invasive NTS (iNTS) diseases occur annually, with a case fatality rate of 20%, yielding 681,316 deaths per year (8). The highest incidence and number of cases are seen in sub-Saharan Africa, where iNTS is associated with HIV infection in adults and malaria, anemia, and malnutrition in children and is caused by strains of S. enterica serovars Typhimurium and Enteritidis with a genotype different than that of the classical strains that affect industrialized countries (9–11).
Salmonella enterica serovar Dublin is a nontyphoidal serovar adapted to cattle but is able to infect humans, in whom it is reported with an invasive index (the percentage of isolates obtained from blood to total isolates recovered for one serovar) as high as 71.4% (12–14). Among the serovars involved in iNTS infections, S. Dublin is among the most relevant serovars after Enteritidis and Typhimurium, the two serovars with the highest prevalence globally and ubiquitous in terms of host range (15–17; NSC, unpublished). However, when the invasive index is considered, S. Dublin shows a substantially higher invasive index than S. Enteritidis and S. Typhimurium globally (33% for S. Dublin and 1.8% and 1.6% for S. Enteritidis and S. Typhimurium, respectively) (14). The genetic bases for the high invasiveness exhibited by S. Dublin are not completely understood. Mohammed and Cormican postulated that two different type VI secretion systems encoded on Salmonella pathogenicity islands 6 and 19 (SPI-6 and SPI-19), a Gifsy-2-like prophage and a virulence plasmid, may contribute to the high capacity of S. Dublin to cause invasive disease in humans (18). In addition, it has been observed that Salmonella serovars adapted or restricted to a particular host show higher levels of genome degradation through genomic rearrangement and pseudogene formation and cause more severe disease than broad-host-range serovars (18–24).
For Salmonella, it is postulated that an acute inflammatory response at the intestine would contribute to maintaining the infection localized, preventing bacterial dissemination to systemic sites (25–27). Flagellin (encoded by fliC and by fljB in Salmonella diphasic serovars) is the main structural unit of the flagellum filament and the agonist of Toll-like receptor 5 (TLR5) and NAIP5/NLRC4 receptors (28, 29). It has also been demonstrated that flagellin is one of the main determinants of the inflammatory response at the intestine, by providing motility and activation of innate immune responses (30–34).
The flagellum is a macromolecular complex composed of three structures: a basal body containing the motor, a universal joint known as the hook, and a helical filament composed of up to 20,000 molecules of flagellin (35). Expression of flagellar genes follows a hierarchical cascade in which three classes of genes are distinguished: early, middle, and late genes (also named class 1, 2, and 3 genes, respectively), which code for regulators and proteins involved in the different stages of flagellar assembly (36, 37). FlhD4C2 (encoded by class 1 genes flhC and flhD) is the master regulator for transcription of all flagellar genes and drives expression of class 2 genes. Among them, genes coding for proteins composing the basal body and the hook are found, as well as fliA and flgM. fliA codes for the flagellum-specific sigma factor (σ28) responsible for expression of class 3 genes, whereas flgM codes for the FliA anti-sigma factor that inhibits FliA function in the early phase of flagellar assembly, through direct binding. When assembly of the hook-basal body (HBB) is completed, the type III flagellar protein export apparatus contained in it drives secretion of FlgM from the cell, resulting in release of FliA and the consequent transcription of class 3 genes (35, 38, 39). Among late genes, those coding for proteins forming the hook-filament junction, the filament (fliC), the stator complex (motAB), and the chemotaxis system are found.
Previously, we reported a high frequency of S. Dublin human isolates lacking flagella (4 out of 10), a phenotype due to significantly reduced levels of fliC mRNA compared to those in flagellated isolates, while fliA mRNA levels were not affected (40). Interestingly, the aflagellated isolates were impaired in their proinflammatory properties, as tested both in vivo and in vitro, and were all isolated from bloodstream infections, suggesting that a lack of flagella could promote systemic dissemination of S. Dublin through evasion of innate immune defenses. In this work, we sought to elucidate the mechanism underlying impaired fliC expression in S. Dublin isolates and evaluate its reversibility, in order to predict if it would revert under certain environmental conditions.
RESULTS
(i) Analysis of class 2 and 3 flagellar genes mRNA levels in S. Dublin isolates.
We previously reported that fliC mRNA levels were 2 orders of magnitude lower in nonmotile S. Dublin isolates than in motile ones, while fliA mRNA levels were not affected (40). In addition, the fliC coding sequence as well as adjacent regions (including promoter sequences) were identical between a nonmotile and a motile isolate, ruling out the possibility that impaired fliC mRNA levels were due to mutations in its regulatory regions leading to lower transcription efficiency or to differences in sequence leading to mRNA instability in the aflagellate isolates.
In order to evaluate if this impaired expression was specific for fliC or a general feature of late flagellar genes, we quantified mRNA levels for motB and cheB (class 3 genes) and fliN and flgB (class 2 genes [41]), using quantitative reverse transcription-PCR (qRT-PCR), in our collection of human S. Dublin isolates (Fig. 1A and B, respectively). Similar to the case with fliC, mRNA levels for motB and cheB were significantly lower in nonmotile isolates. However, no significant differences were observed in fliN and flgB mRNA levels between the two classes of isolates, suggesting that class 3 genes (controlled by FliA sigma factor), but not class 2 genes, were affected.
FIG 1.
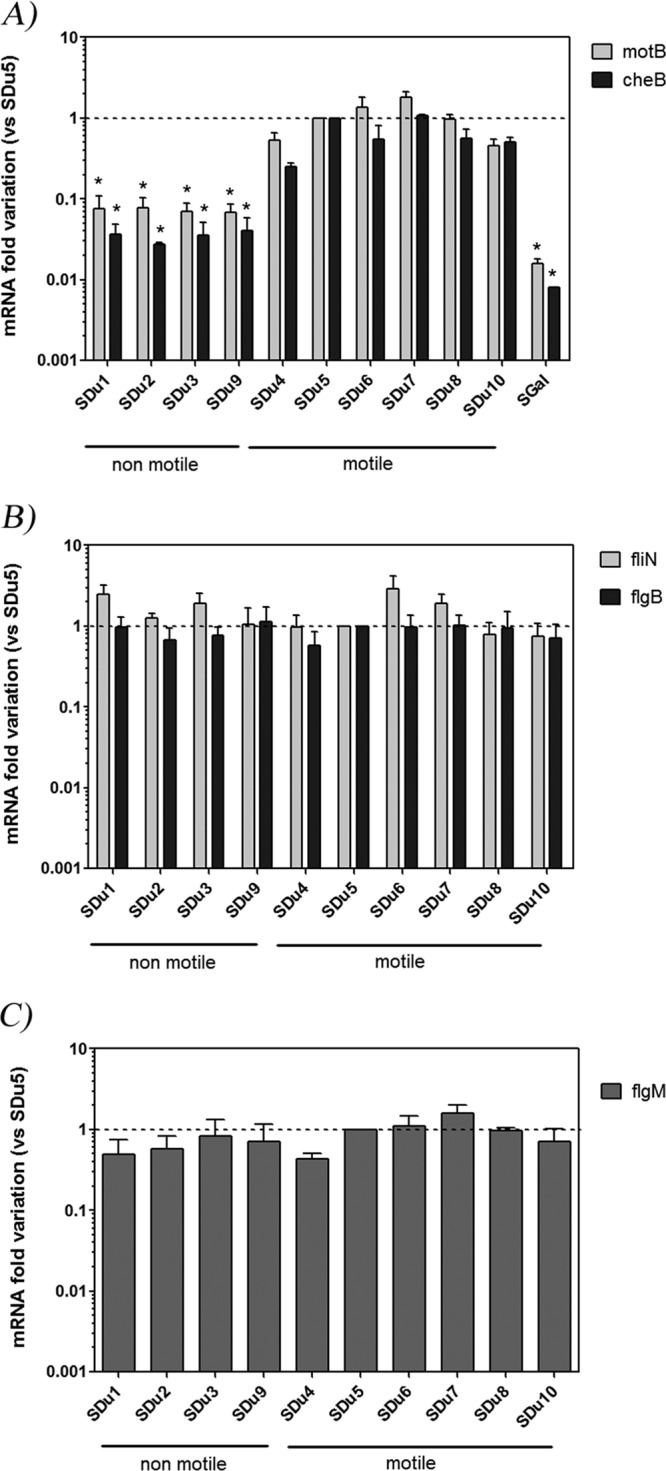
Flagellar gene mRNA levels quantification in S. Dublin isolates grown in LB medium to mid-log phase. Results (means and standard errors) from three independent experiments are shown. (A) motB and cheB (class 3); (B) fliN and flgB (class 2); (C) flgM (class 2 and 3). *, significant difference relative to SDu5 (P < 0.05). SGal is a strain of the aflagellate serovar S. Gallinarum. The dashed line indicates value 1, arbitrarily assigned to isolate SDu5.
In order to add support to this suggestion, we analyzed mRNA levels of one additional class 2 gene (fliF) and one additional class 3 gene (tsr) (41), and we verified that while fliF mRNA levels were not statistically different between the two groups of isolates, tsr mRNA levels were significantly lower in nonmotile than in motile ones (see Fig. S4 in the supplemental material).
As fliA mRNA levels were not affected in nonmotile isolates (40), and as FliA function depends on release of FlgM-dependent inhibition, we analyzed the mRNA levels of flgM, which is reported as a class 2 and 3 gene. flgM mRNA levels were not statistically different between the two groups of isolates (Fig. 1C).
(ii) Analysis of FlgM protein levels and subcellular localization.
FliA inhibition is released when FlgM is secreted outside the cell, a process that is initiated when HBB assembly is completed and there is a switch in secretion through the flagellar export apparatus from rod/hook-type to filament-type substrates (35, 42). Thus, we analyzed the cellular and secreted levels of FlgM and FliC proteins in our collection of S. Dublin clinical isolates (Fig. 2).
FIG 2.
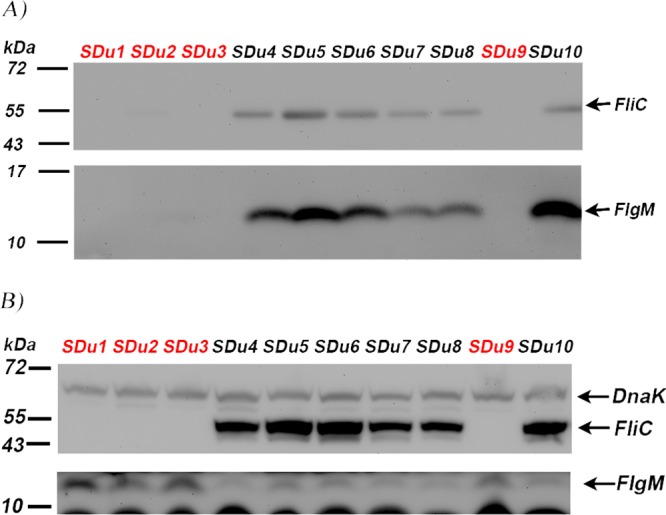
Western blot analysis of secreted (A) or cellular (B) protein extracts of S. Dublin isolates grown in LB medium to mid-log phase, using anti-FliC and anti-FlgM antisera. Detection of DnaK (69.1 kDa), a cytosolic protein, was used to verify equal loading quantities in cellular fractions (B) and the presence of protein in the secreted fraction due to cell lysis (A); as expected, it was not detected in secreted fractions (no signal is seen in panel A around 69.1 kDa). Sizes of molecular mass markers are indicated. FliC and FlgM are 53 kDa and 10.6 kDa, respectively. Nonmotile isolates are in red.
Consistent with our previously reported results (40), we found that the aflagellate isolates (SDu1 to -3 and SDu9) did not produce FliC in either the cellular or the secreted fraction. Surprisingly, FlgM was also not detected in the secreted fractions of aflagellate isolates (Fig. 2A), indicating that FlgM secretion is impaired in these isolates. Accordingly, FlgM was enriched in the cellular fractions of aflagellate isolates compared to flagellated ones (Fig. 2B), which would explain the lower levels of class 3 gene expression in the former.
(iii) In trans fliA overexpression in an aflagellate isolate.
In order to verify if motility impairment could be reversed by artificially increasing FliA levels, we transformed an aflagellate isolate (SDu3) with an expression plasmid harboring fliA under the control of an isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible promoter (PTac). The resulting strain (SDu3/pJfliA) was grown in the absence or presence of IPTG, and fliC and motB mRNA levels were quantified using qRT-PCR (Fig. 3A). In the presence of the inducer, fliC and motB mRNA levels increased significantly compared to those under the noninduced condition, even reaching higher levels than in a naturally motile strain. FliC protein levels were also evaluated by Western blotting, and this analysis revealed that it was being synthesized in SDu3/pJfliA in an IPTG-dependent manner (Fig. 3B). However, even if FliC was produced to high levels intracellularly in the IPTG-induced SDu3/pJfliA strain, it was almost undetectable in the secreted fraction (Fig. 3B). Moreover, FlgM intracellular levels were also increased in the IPTG-induced SDu3/pJfliA strain, probably due to fliA overexpression, but it was not detected in the secreted protein fraction (Fig. 3B).
FIG 3.

(A) fliC and motB mRNA level quantification in strain SDu3/pJfliA (SDu3pJA) grown in the absence (−) or presence (+) of the inducer. As controls, isolates SDu3 (not transformed) and SDu5 (naturally flagellated) were also evaluated. Results (means and standard errors) from two independent experiments are shown. *, P = 0.01 to 0.05; **, P = 0.001 to 0.01; ***, P < 0.001, compared to noninduced SDu5. (B) Western blot analysis with anti-FliC and anti-FlgM antisera of cellular or secreted proteins fractions of SDu3, SDu5, and SDu3 supplemented in trans with fliA (3pJA). A plus sign indicates growth in the presence of the inducer. Detection of DnaK was used to verify equal loading of samples in cellular protein fractions or absence of lysis in secreted protein fractions. Sizes of molecular mass markers are indicated. FliC and FlgM are 53 kDa and 10.6 kDa, respectively. Vertical lines between lanes of secreted protein fractions were included because the gel image was spliced for convenience (additional samples not related to this work were removed). (C) Image of 7th passage of SDu3/pJA in motility agar containing a gradient of IPTG concentrations. Each spot is subjected to a different inducer concentration, from 0 to 0.05 mM (see Materials and Methods). At this passage, the spot in the middle (corresponding to an IPTG concentration around 0.025 mM) showed a halo of motility. (D) Fluorescence labeling of flagellar filaments of isolate SDu3 (left) and SDu3/pJAmot+ grown in the absence (middle) or presence (right) of IPTG. Magnification, ×1,000.
These results indicate that in the aflagellate isolate, when fliA is overexpressed, the inhibition of transcription of class 3 flagellar genes is released and flagellin is synthesized, suggesting that silencing of late flagellar genes is a result of impaired secretion of FlgM that leads to permanent inhibition of FliA function. However, no motility was observed in SDu3/pJfliA strain at any IPTG concentration tested (data not shown), probably due to impaired FliC secretion. These results suggest that there is a general impairment of secretion of late flagellar products in the SDu3 isolate, so even if flagellin is produced at the cytosol level, it is not able to reach the extracellular location, where is assembled into the filament.
Finally, to force the strain SDu3/pJfliA to express the motility phenotype, we performed successive passages every 24 h in motility agar containing IPTG concentration gradients (from 0 to 0.05 mM). In three independent experiments, a motility halo was observed after 8 (1st experiment), 6 and 7 (2nd experiment), and 7 (3rd experiment) passages at intermediate IPTG concentrations but not at concentrations close to 0, indicating that induction of FliA synthesis was required to revert to the motile phenotype (Fig. 3C). It is important to note that in a previous work we reported that the SDu3 nonmotile phenotype was not reversible, at least after 12 daily passages in motility agar (40).
Fluorescent flagellar staining of an isolated revertant SDu3/pJfliA strain (named SDu3/pJAmot+) revealed a flagellated phenotype both in the presence and in the absence of the inducer, suggesting a capacity to assemble flagella independent of fliA overexpression (Fig. 3D).
(iv) Analysis of the nucleotide sequence of all flagellar operons in S. Dublin isolates.
We comparatively analyzed the nucleotide sequence of all flagellar operons (regions I, II, IIIa, and IIIb [36]) from nonmotile (SDu1 to -3 and SDu9) versus motile isolates (SDu4 to -8 and SDu10). The only difference found was a 42-nucleotide internal deletion in fliE (named fliEΔ42) (Fig. 4A), which codes for a protein postulated to be located in the junction zone between the MS ring and the rod in the flagellar basal body (43, 44). It was reported that secretion of FlgE (hook) and FlgD (hook-cap) through the flagellar export apparatus is impaired in fliE mutant strains (42, 45, 46). Interestingly, in fliEΔ42 the open reading frame (ORF) is not altered; thus, it is plausible that a protein lacking amino acids 18 to 31 could be synthesized. Indeed, qRT-PCR quantitation of fliE mRNA levels revealed no statistical differences between nonmotile and motile S. Dublin human isolates, although the transcripts were of different sizes (see Fig. S1 in the supplemental material).
FIG 4.

Sequence alignments of fliE genes from isolates described in this study (A) or FliE proteins from different Salmonella serovars (B) or different species from Enterobacteriaceae (C). (A) Sequence alignments of fliE genes from two Uruguayan isolates representative of each phenotype (SDu1, nonflagellated, and SDu5, flagellated). The nucleotide sequence of FliE from S. Dublin CT_02021853 (gi∣198241740) is included as a reference. In gray the region deleted from Uruguayan nonmotile isolates is shown. The sequences of the rest of the Uruguayan isolates are identical to those of the representative isolates shown, depending on the phenotype (SDu2, SDu3, and SDu9 are identical to SDu1; SDu4, SDu6 to -8, and SDu10 are identical to SDu5). Asterisks indicate sequence identity. (B and C) Amino acid sequence alignment of FliE from Salmonella enterica serovars and subspecies (B) and from diverse species of Enterobacteriaceae (C), including Shigella boydii, E. coli, Enterobacter cloacae, Yersinia pestis, Morganella morganii, and Proteus mirabilis. In gray the region deleted from Uruguayan nonmotile isolates is shown. Asterisks indicate fully conserved residues, colons indicate conservation between groups of residues of strongly similar properties, and periods indicate conservation between groups of weakly similar properties; the absence of a symbol indicates an absence of conservation. NCBI Protein database accession numbers for proteins in panel B: S. Heidelberg, KKE18834.1; S. Typhimurium strain LT2, NP_460921.1; S. Typhimurium D23580, CBG24957.1; S. Enteritidis strain P125109, B5R0Z0.1; S. Infantis, EHB41345.1; S. Dublin strain CT_02021853, B5FRW4.1; S. Paratyphi A strain ATCC 9150, AAV76882.1; S. Newport strain SL254, YP_002041232.1; S. Choleraesuis strain SC-B67, Q57N33.1; S. Lubbock, KIV43438.1; S. Typhi strain CT18, NP_456529.1; S. enterica subsp. arizonae, serovar 62:z4,z23:-, A9MML1.1; Salmonella enterica subsp. houtenae strain ATCC BAA-1581, EHY70991.1; and Salmonella enterica subsp. indica serovar 6,14,25:z10:1,(2),7, ESE86582.1. NCBI Protein database accession numbers for proteins in panel C: S. Weltevreden strain 2007-60-3289-1, CBY95195.1; S. Gallinarum strain SG9, EGE33731.1; S. Dublin strain CT_02021853, B5FRW4.1; S. Typhimurium strain 14028S, ACY88841.1; S. Typhi strain CT18, NP_456529.1; Shigella boydii, P95713.3; Escherichia coli O104:H4 strain C227-11, EGT69930.1; Shigella boydii Sb227, ABB65717.1; Enterobacter cloacae subsp. cloacae strain ATCC 13047, ADF62754.1; Yersinia pestis strain CO92, CAL20472.1; Morganella morganii subsp. morganii KT, AGG30849.1; and Proteus mirabilis strain HI4320, CAR43400.1.
The deletion corresponds to a less conserved region among FliE proteins from different serovars of Salmonella enterica subsp. enterica and from different Salmonella enterica subspecies (Fig. 4B). This region was also less conserved among FliE proteins from other species in the Enterobacteriaceae family (Fig. 4C).
(v) In trans complementation of an aflagellate isolate with wild-type fliE rescues the nonmotile phenotype and recovers proinflammatory properties in vivo.
To verify that the cause of the aflagellate phenotype in S. Dublin isolates studied in this work resides in the mutant fliE gene, a complementation assay was performed. Strain SDu3 was transformed with a plasmid carrying fliE from SDu5 cloned under the control of an arabinose-inducible promoter (PBAD). The resulting strain, designated SDu3/pBfliE, displayed motility dependent on the arabinose concentration (Fig. 5), reaching motility levels similar to those of a naturally motile isolate at medium arabinose concentrations. Western blotting analyses revealed that in strain SDu3/pBfliE induced with arabinose, FlgM was secreted and FliC was synthesized to levels similar to those in the naturally motile isolates (see Fig. S2 in the supplemental material). This indicates that mutation fliEΔ42 is the cause of the aflagellate phenotype in SDu3 and presumably also in SDu1, SDu2, and SDu9, which harbor the same deletion.
FIG 5.

Motility assay of strains SDu3/pBfliE (3BE), SDu3/pBAD22 (SDu3/BAD), and SDu5/pBAD22 (SDu5/BAD) at increasing arabinose concentrations. The diameter of the halo of growth after 6 h at 37°C is shown.
In a previous study, we demonstrated that the aflagellate isolate SDu3 had impaired proinflammatory properties in vivo at the cecum level, in streptomycin-pretreated C57BL/6 mice, in comparison with the naturally motile SDu5 isolate (40). In order to test if the fliE-complemented SDu3 strain that recovered motility was able to trigger a proinflammatory response at the intestine similar to that of SDu5, we tested them in the same in vivo model. For this purpose, streptomycin-resistant SDu3 was transformed with a plasmid carrying wild-type fliE (from SDu5) under the control of the PTac promoter, which exhibits higher basal leakage than PBAD. The resulting strain, named SDu3/pJfliE, showed IPTG-dependent motility, with approximately 70% of SDu5 motility in the absence of the inducer (see Fig. S3). The stability of the pJfliE plasmid was tested by growing SDu3/pJfliE in Luria-Bertani (LB) medium without antibiotic selection for 23 generations, with no apparent plasmid loss (data not shown).
The proinflammatory response in the ceca of streptomycin-pretreated mice 24 h after oral infection with SDu3, SDu5, and SDu3/pJfliE was evaluated in comparison with that in mock-infected animals (Fig. 6).
FIG 6.

mRNA variation at 24 h p.i. in the cecal mucosae of mice infected with S. Dublin isolates, as evaluated by qRT-PCR. Values are expressed as fold changes of mRNA levels for the indicated genes in infected mice relative to that in mock-infected animals (n/i). Means and standard errors from two independent experiments combined are shown. (A) mRNA quantification of genes coding for proinflammatory cytokines and chemokines. (B) mRNA quantification of genes coding for antimicrobial proteins. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, not statistically significant.
Cecal response at 24 h postinfection (p.i.) was chosen for analysis because it was previously determined that severe pathological changes upon Salmonella oral infection are restricted to this organ, and at early time points the differences in proinflammatory responses against aflagellate versus flagellate strains are more pronounced (31, 47). The genes selected for evaluation were those coding for proinflammatory cytokines (interleukin 17 [IL-17], tumor necrosis factor alpha [TNF-α], and gamma interferon [IFN-γ]), a chemokine involved in neutrophil recruitment (CXCL-1), and antimicrobial proteins involved in iron and zinc deprivation (lipocalin 2, encoded by Lcn-2, and calprotectin, one of its subunits encoded by S100.a9, respectively), which were reported previously to be induced in the murine intestine upon S. Typhimurium infection (32, 48).
All three isolates colonized efficiently the ceca of infected mice at 24 h p.i.; the only statistical difference in Salmonella counts in cecum content was found between SDu3 and SDu3/pJfliE (SDu3, 2.1 × 109 ± 3.9 × 108 CFU/g; SDu5, 1.3 × 109 ± 3.6 × 108 CFU/g; and SDu3/pJfliE, 0.84 × 109 ± 1.9 × 108 CFU/g; values are means ± standard errors of the means [SEMs]). However, the cecal mRNA levels of genes coding for proinflammatory cytokines, chemokines, and antimicrobial proteins in mice infected with the SDu3 isolate were similar to those in mock-infected animals and significantly lower than in SDu5-infected animals, as expected (Fig. 6A and B). Mice infected with the SDu3 fliE-complemented strain showed cecal mRNA levels for all six genes evaluated significantly higher than in mock-infected mice and similar to the levels observed in SDu5-infected mice, indicating that the SDu3 isolate harboring wild-type fliE recovered not only motility but also proinflammatory capacity in the mouse intestine. The analysis of motility and ampicillin resistance in isolates recovered from ceca and spleens revealed that isolate SDu3 was still nonmotile after passage through the host, whereas isolate SDu3/pJfliE was still motile and ampicillin resistant, indicating plasmid maintenance (data not shown).
(vi) Analysis of fliE nucleotide sequences in revertants SDu3/pJAmot+.
The elucidation of the molecular basis for the nonmotile phenotype in S. Dublin clinical isolates prompted us to analyze fliE sequences in SDu3 revertant strains (SDu3/pJAmot+). We selected four revertants obtained in three independent experiments (SDu3/pJAmot + 6, 7, 8, and 9; see Table 3), for which the fliE gene was PCR amplified and sequenced (Fig. 7A and B).
TABLE 3.
Non-Salmonella Dublin strains, bacterial constructions, and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| Escherichia coli MC1061 | F− Δ(ara-leu)7697[araD139]B/rΔ(codB-lacI)3 galK16 galE15 λ− e14−mcrA0 relA1 rpsL150 (Strr) spoT1 mcrB1 hsdR2(r− m+) | 63 |
| Escherichia coli DH5α | F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80dlacZΔM15 Δ(lacZYA-argF)U16 hsdR17(rK− mK+), λ− | 64 |
| Salmonella enterica serovar Typhimurium SL5338 | galE r− m+ | 65 |
| MC1061/pGEXM | E. coli MC1061 containing flgM cloned into pGEX4T-1 for protein production | This work |
| SDu3/pJfliA | SDu3 containing fliA cloned into pJF119EH | This work |
| SDu3/pJAmot + 6 | SDu3/pJfliA motility revertant after 6 successive passages (2nd expt) | This work |
| SDu3/pJAmot + 7 | SDu3/pJfliA motility revertant after 7 successive passages (2nd expt) | This work |
| SDu3/pJAmot + 8 | SDu3/pJfliA motility revertant after 8 successive passages (1st expt) | This work |
| SDu3/pJAmot + 9 | SDu3/pJfliA motility revertant after 7 successive passages (3rd expt) | This work |
| SDu3/pBAD | SDu3 containing empty vector pBAD22 | This work |
| SDu5/pBAD | SDu5 containing empty vector pBAD22 | This work |
| SDu3/pBfliE | SDu3 containing fliE cloned into pBAD22 | This work |
| SDu3 StrR/pJfliE | SDu3 containing pJfliE and aadA (Strr) | This work |
| SDu3 StrR | SDu3 containing aadA (Strr) | 40 |
| SDu5StrR | SDu5 containing aadA (Strr) | 40 |
| Plasmids | ||
| pBAD22 | Expression vector containing the PBAD promoter of the arabinose operon and its regulatory gene; araC Ampr | 66 |
| pGEX4T-1 | Expression vector with GST ORF under the control of Ptac promoter and lacIq; Ampr | 67 |
| pJF119EH | Expression vector containing the PTAC promoter and lacIq; Ampr | 68 |
| pBfliE | pBAD22 containing wild-type fliE under the control of the PBAD promoter and araC; Ampr | This work |
| pBfliEmot + 6 | pBAD22 containing fliE from revertant SDu3/pJAmot + 6 under the control of the PBAD promoter and araC; Ampr | This work |
| pBfliEmot + 7 | pBAD22 containing fliE from revertant SDu3/pJAmot + 7 under the control of the PBAD promoter and araC; Ampr | This work |
| pBfliEmot + 9 | pBAD22 containing fliE from revertant SDu3/pJAmot + 9 under the control of the PBAD promoter and araC; Ampr | This work |
| pGEXM | pGEX4T-1 containing flgM fused to GST under the control of the PTac promoter and lacIq; Ampr | This work |
| pJfliA | pJF119EH containing fliA under the control of the PTac promoter and lacIq; Ampr | This work |
| pJfliE | pJF119EH containing fliE under the control of the PTac promoter and lacIq; Ampr | This work |
FIG 7.

Nucleotide (A) and amino acid (B) sequence alignment of fliE/FliE from SDu3 revertant strains. In both alignments, the region highlighted in yellow corresponds to the region deleted in fliEΔ42 (42 bp, 14 amino acids), in sky blue duplicated region I, in orange duplicated region II, in green duplicated region III, and in yellow the deleted region in S. Dublin aflagellate isolates. Only the first 117 nucleotides of fliE are shown in panel A. Red arrows indicate the beginning of the deletion. Sequences of SDu5 (flagellated) and SDu3 (nonfagellated) are included as references. (C) Motility assay of strain SDu3 complemented in trans with fliE from SDu3 isolated revertants. Overnight cultures of SDu3/pBfliE (3BE), SDu3/pBAD22 (SDu3/BAD), SDu5/pBAD22 (SDu5/BAD), SDu3/pBfliEmot + 6 (3BEmot + 6), SDu3/pBfliEmot + 7 (3BEmot + 7), and SDu3/pBfliEmot + 9 (3BEmot + 9) were spotted in motility agar plates containing increasing arabinose concentrations, and the diameter of the halo of growth after 6 h at 37°C was measured.
The results revealed that in the four revertants, the deletion was filled up with duplications of adjacent regions that did not alter the coding frame, thus rendering proteins with sizes similar to that of the wild type (107 or 100 amino acids [aa], compared to 104 for the wild type and 90 for the protein with the deletion). However, the identities of residues 18 to 31 in the revertants were completely different from those in the wild type.
Revertants 6 and 8 have identical fliE sequences, in spite of being obtained in independent experiments. In these strains, two regions were duplicated: one of 21 bp, named region I, and one of 30 bp, named region II. Region I comprises nucleotides 7 to 27 and codes for 7 residues, AIQGIEG (highlighted in sky blue in Fig. 7A and B). Region II spans from nucleotides 37 to 66 and codes for 10 amino acids, QLQATVSFAG (highlighted in orange in Fig. 7A and B). Therefore, the resulting protein is 17 residues longer than the originally protein with the deletion (107 aa versus 90 aa) and 3 residues longer than the wild-type protein (104 aa). In revertant 7, one duplication occurred corresponding to region II, rendering a protein of 100 amino acids, 4 residues shorter than the wild type. Finally, in revertant 9, one 30-bp duplication occurred, named region III, which codes for 10 residues (ATVSFAGQLH, in green in Fig. 7A and B), rendering a protein 100 residues long. The fact that these four revertants recovered motility to normal levels suggests that the identities of amino acid residues 18 to 31 are not essential for function, but instead a protein of similar size to the wild type is required.
In order to demonstrate that the identity of the region from residues 18 to 31 of FliE is dispensable for its function and discard the possibility that extragenic suppressor mutations had occurred in the isolated revertants, we cloned fliE from revertants 6, 7, and 9 in pBAD22. The resulting plasmids, named pBfliEmot + 6, 7, and 9, were transformed into SDu3 and transformants were subjected to motility assays at increasing arabinose concentrations. fliE genes from all three revertants rescued the nonmotile phenotype in an arabinose-dependent manner, similarly to the pBAD-cloned wild-type fliE gene (Fig. 7C). Note that in all cases, at higher inducer concentrations motility was reduced, probably due to an excess of FliE synthesis that interferes with growth rate. This result indicates that FliE proteins from the revertants are functional and suggests that no additional genetic changes occurred in the isolated revertants.
(vii) fliEΔ42 screening in a collection of S. Dublin isolates obtained from cattle.
In a previous study we detected 10 S. Dublin isolates devoid of motility in a collection of 15 isolates obtained from cattle, the natural host of this Salmonella serovar. In 7 of the nonmotile isolates we also observed a lack of FliC in their total protein extracts (40). To investigate if in these isolates the deletion in fliE was responsible for the aflagellate phenotype, a PCR screening was applied, using primers designed to hybridize at deletion-flanking regions. A deletion with a size similar size to that of fliEΔ42 was observed in 4 of the 10 nonmotile cattle strains (SDuG2, SDuG3, SDuG6, and SDuG7), coincidently with a lack of FliC synthesis (Fig. 8). Analysis of the fliE nucleotide sequence in isolate SDuG6 indicated that this isolate harbors exactly the same deletion as observed in human isolates. Interestingly, in addition to these four isolates that displayed the fliEΔ42 mutation, there are three other isolates from cattle (SDuG5, SDuG8, and SDuG9) that lack FliC in their total protein extracts but do not harbor the deletion in fliE. Moreover, isolates SDuG10, SDuG12, and SDuG13 showed increased FliC levels in their protein extracts but no motility at all (40).
FIG 8.
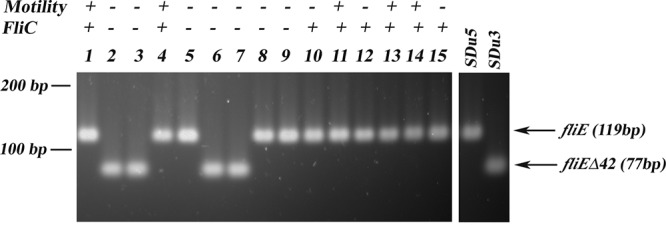
fliEΔ42 PCR screening of S. Dublin isolates from cattle. Lanes 1 to 15 show strains SDuG1 to SDuG15, respectively; control strains SDu5 (fliE+) and SDu3 (fliEΔ42) are also shown for reference. Motility and the absence or presence of FliC in total protein extracts analyzed by Western blotting (from reference 40) are indicated.
(viii) Analysis of fliE sequence in S. Dublin genomes available at Enterobase.
In order to investigate whether fliEΔ42 mutation is a frequent event worldwide in this Salmonella serovar, we analyzed fliE sequence in 1,230 S. Dublin genomes available at Enterobase (http://enterobase.warwick.ac.uk). These were all the strains assigned as serovar Dublin by Enterobase according to whole-genome sequencing (20 August 2017). We did not find the fliEΔ42 deletion in any of the genomes analyzed. However, we found one strain (SAL_DA4720AA) that has no identity to fliE in its genome and another strain (SAL_NA6662AA_AS) that harbors a 4-nucleotide insertion at positions 115 to 118 of fliE that generates a stop codon rendering a truncated FliE protein of 46 amino acids. No evidence of reversion of the fliEΔ42 deletion as described in the present study was found in the S. Dublin genomes from Enterobase.
(ix) Whole-genome phylogenetic analysis of Uruguayan S. Dublin isolates.
The availability of the complete genomic sequences of 14 S. Dublin Uruguayan isolates (5 out of 14 carrying the fliEΔ42 deletion) allowed us to evaluate the phylogenetic relationship among them, in order to investigate if the aflagellate strains described in this study are clonal or the deletion emerged several times as independent events.
In silico 7-gene multilocus sequence typing (MLST) analysis revealed that all 14 isolates belong to sequence type 10 (ST10), the main ST of serovar Dublin strains in Enterobase. Then, a whole-genome comparative single nucleotide polymorphism (SNP) analysis considering homologous regions (coding and noncoding regions) was performed. We identified a range of 114 to 306 SNPs of difference between all these isolates, and then we used the obtained SNP matrix to infer the phylogenetic relatedness of the strains, using the CT_02021853 S. Dublin strain as an outgroup and isolate SDu10 as a reference (see Fig. S5 in the supplemental material). Different phylogenetic methods (maximum likelihood and neighbor joining) and evolutionary models (HKY and Kimura 2P) were applied to test if the mutation emerged once or several times in different lineages. From the results of these analyses, it was not possible to establish if the strains having the deletion belong to a unique lineage, mainly because in all cases the key nodes showed very low or null statistical support (branch node support, bootstrap, and SH-like test; see Fig. S5).
DISCUSSION
In this work, we elucidated the molecular basis of the lack of flagella in isolates of S. enterica serovar Dublin causing invasive infections in humans. Dublin is a nontyphoidal serovar adapted to cattle, in which it usually causes systemic infections and abortion (19). In humans, S. Dublin frequently causes severe invasive infections, which may be fatal (49). As it is generally accepted that an acute inflammatory response at the intestine helps to prevent systemic dissemination of Salmonella, and as flagellin is one of the main determinants triggering this response, it is reasonable to speculate that the lack of flagella might contribute to the high invasiveness displayed by Salmonella Dublin. In fact, it has been reported that Salmonella Typhi, a human-restricted serovar that causes a severe systemic disease, represses fliC expression when interacting with the intestinal epithelium (25). In addition, the avian-restricted serovar Gallinarum, responsible for invasive typhoid-like disease in chickens, is nonflagellated, and a strain of S. Gallinarum engineered to express flagella causes overt intestinal pathology and is less pathogenic than the wild-type strain (34). In line with this, it was previously shown that typhoidal serovars (Typhi, Paratyphi A, and Sendai) are significantly less motile than S. Typhimurium, which in the case of serovar Paratyphi A was demonstrated to be due to a lower expression of the entire flagellar regulon than in S. Typhimurium (50–52). Moreover, the Salmonella Typhimurium variant ST313, which emerged in sub-Saharan Africa as an unusually invasive genotype in humans, was reported to show lower flagellin expression and lower proinflammatory capacity than S. Typhimurium ST19 (51, 52).
In this work we demonstrate that in human S. Dublin aflagellate isolates, there is a general silencing of expression of class 3 flagellar genes, which results in the absence of the flagellar appendage (Fig. 1A). We identified a 42-nucleotide deletion in fliE, coding for a protein located in the flagellar basal body, as the underlying cause of impaired FlgM secretion resulting in permanent FliA inhibition. Thus, trans-complementation of an aflagellate isolate with wild-type fliE recovered motility immediately, as well as secretion of FlgM and consequently expression of class 3 genes (Fig. 5; see also Fig. S2 in the supplemental material). The fact that the fliE mutants resulted from the identical 42-base in-frame deletions and appear in several isolates from different sources and periods of time suggests that this mutation was present in the strains before their isolation.
Our results show that the fliEΔ42 deletion is harbored by 4 out of 10 isolates of human origin (SDu1 to -3 and SDu9, all four nonmotile isolates from this source) and 4 out of 15 isolates from cattle (SDuG2, SDuG3, SDuG6, and SDuG7). In spite of the small sample size, this result suggests a high prevalence of this mutation in S. Dublin isolates from our country. As this deletion causes a general silencing of class 3 genes, the significant metabolic cost of synthesizing the flagellar filament would be saved. Thus, it is tempting to speculate that this mutation could provide an advantage over the flagellated wild type in the different environments that Salmonella encounters. The deletion in fliE seems not to be a rare trait but rather to be circulating in our country, as strains harboring this mutation were isolated from different sources (human and animals) and during an extended period (from 1995 to 2011). However, the fliEΔ42 deletion was not found in any other S. Dublin genome available in Enterobase. This result, together with the fact that the phylogenetic trees constructed with genomic data are not conclusive with regard to a clonal or independent origin of the fliEΔ42-harboring strains (see Fig. S5 in the supplemental material), suggests that this deletion emerged once in a common ancestor of all of them and thereafter persisted and disseminated through different hosts. However, the deletion in fliE is only one mechanism leading to the aflagellate phenotype, and many others may exist. Indeed, 3 additional cattle isolates (SDuG5, SDuG8, and SDuG9) showed an absence of FliC synthesis but did not harbor the deletion in fliE (Fig. 8), indicating the existence of distinct mechanisms leading to nonflagellated strains in this serovar and convergent functional evolution. In addition, the existence of important numbers of Salmonella isolates belonging to serovar Dublin devoid of flagella was previously reported in the United States (53) and Sweden (54). It would be very interesting to elucidate the mechanisms responsible for this phenotype in those isolates.
In a previous study, we showed significantly lower levels of fliC expression in aflagellate S. Dublin human isolates than in flagellate ones (40). In this work, we demonstrated that this silencing is not specific to fliC but instead seems to be general for class 3 genes, because motB, cheB, and tsr were also silenced in all nonmotile human isolates (Fig. 1A; see also Fig. S4). Conversely, mRNA levels of class 2 genes fliN, flgB, fliE, and fliF were unaffected (Fig. 1B; see also Fig. S4), indicating impairment of FliA function and not of the master regulator FlhD4C2. We then demonstrated that the impairment of FliA function is due to impaired FlgM secretion to the extracellular compartment (Fig. 2A and B). Accordingly, fliA overexpression in an aflagellate isolate resulted in titration of FlgM cytosolic levels and release of FliA inhibition, leading to increased expression of class 3 flagellar genes (Fig. 3A); however, no motility was observed. This could be explained by a defect in secretion of filament-type substrates through the flagellar export apparatus in the presence of the FliE protein with the deletion (designated FliEΔ18-31); thus, both FlgM and FliC secretions were impaired in the fliA-induced aflagellate isolate (Fig. 3B). Our results indicate that FliEΔ18-31 is a lack-of-function mutant and are in agreement with results reported previously for the spirochete Borrelia burgdorferi, for which a complete absence of the rod, hook, and filament in a fliE null mutant was demonstrated by cryo-electron tomography, whereas the rotor, stator, and export apparatus were normally assembled (44). Thus, as no hook is assembled, the switch in secretion from rod/hook-type to filament-type substrates (where FlgM and FliC are included) is impaired. Remarkably, a central channel visualized in the MS ring was closed in the B. burgdorferi ΔfliE mutant compared to the same channel open in the wild-type strain, providing a molecular explanation for the absence of secretion of substrates. The authors postulate that upon secretion of FliE and FlgB, the central channel adopts an open conformation and serves as the template for assembly of the rest of rod components (44). Moreover, work done with Salmonella Typhimurium by Macnab and others postulated FliE as an structural adaptor between the MS ring and the rod and demonstrated that FliE assembly is required for export of other substrates, such as the hook-capping protein FlgD and the hook protein FlgE (43, 46, 55). Thus, we can hypothesize that in the S. Dublin nonflagellated isolates studied in this work, FliEΔ18-31 is not assembled on the MS ring or that it is assembled but does not support assembly of the rod proteins. Minamino et al. also reported a direct interaction between FliE and proximal rod protein FlgB by affinity blotting and demonstrated that a FliE-V99G mutant (lying close to the C terminus of this 104-aa protein) that showed extremely poor flagellation and swarming motility reverted its phenotype due to extragenic suppressor mutations in flgB (43). Minamino et al. tentatively suggested that the N-terminal region of FliE would be important for its own export and its C-terminal region would be important for structural interactions with the proximal rod (43). Based on this suggestion, the FliE mutant protein reported here that harbors the deletion close to its N-terminal end would be impaired in its own secretion and thus would not be able to reach its location in the flagellar basal body. This is supported by the fact that FliEΔ18-31 showed no negative dominant effect over the wild-type protein, as a fliEΔ42 isolate was readily trans-complemented by wild-type fliE even under noninducing conditions (note that SDu3/pJfliE showed approximately 70% of SDu5 motility in the absence of IPTG [see Fig. S3 in the supplemental material]). Osorio-Valeriano et al. (56) reported the in silico prediction of FliE tertiary structure from Rhodobacter sphaeroides, suggesting that this protein folds into three packed α-helices, similar to the predicted structure of the inner rod component (PscI) of the injectisome type III secretion system from Pseudomonas aeruginosa (57). According to this prediction, the deletion described here would shorten the first α-helix of the protein. Interestingly, both FliE and PscI were reported to polymerize in vitro when purified, into long and flexible fibers with regular twists (57, 58). In the case of PscI, this polymerization was dependent on the integrity of the C-terminal α-helix, but the role of the N-terminal α-helix in polymerization or function of these proteins is unknown.
Remarkably, the aflagellate phenotype described here was hardly reversible; motile revertants of a naturally aflagellate isolate were obtained only after overexpression of fliA (and the class 3 genes controlled by it) with concomitant several passages in motility agar (both conditions imposing pressure in favor of the motility phenotype).
Interestingly, the revertants obtained in this work filled up the deletion with adjacent DNA sequences, rendering FliE proteins with a size similar to that of the wild type but with completely different amino acids at positions 18 to 31 (Fig. 7A and B). The revertant proteins (107, 100, and 100 residues long, compared to 104 residues for the wild type) rescued the nonmotile phenotype of a naturally aflagellate isolate similarly to the wild-type protein, indicating that these “patched” proteins are functional. This result indicates that the identities of residues 18 to 31 are not essential for functionality but instead a specific length of the N-terminal region is required, suggesting a structural role of this region for FliE functionality. Indeed, alignment of amino acid sequences of FliE proteins from diverse Salmonella serotypes and from different bacterial species among Enterobacteriaceae revealed that this region displays relatively low homology compared to the C-terminal moiety (Fig. 4B and C).
The aflagellate isolates reported here constitutively and irreversibly lack not only flagella but also flagellin production, meaning that they will not be able to activate extracellular innate immune receptor TLR5 (which leads to NF-κB translocation to the nucleus) or intracellular NAIP5/NLRC4 receptors (which lead to caspase 1 inflammasome activation). Thus, even if there are other pathogen-associated molecular patterns (PAMPs) able to activate these responses, the innate immune response mounted against these isolates would be dampened. Indeed, our results show that the aflagellate isolate SDu3 triggered significantly lower proinflammatory responses in murine ceca than did a flagellated strain, and this was fully reverted when SDu3 was complemented with wild-type fliE (Fig. 6A and B). This is in contrast to results obtained by our group with serovar Enteritidis, in which we found mutations in motor genes that render nonmotile but fully flagellate strains, which maintain their proinflammatory capacities (59). In addition, our results confirm previous results obtained by Reed et al., who reported that an S. Typhimurium mutant with a Tn10 insertion in fliE was unable to secrete flagellin, assemble flagella, or induce IL-8 secretion by cultured epithelial cells (60).
Together, the results presented here indicate that the absence of flagella is not unusual in strains belonging to serovar Dublin, suggesting that this feature is not essential for its biological cycle. This can be seen as another example of genomic degradation in a host-adapted Salmonella serovar that could contribute to its high invasive capacity.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
S. Dublin Uruguayan clinical isolates were obtained from the NSC and the Bacteriology Unit of the Ministry of Health collections (Table 1). These were all the human isolates of this serovar available in both collections in the period of study (1995 to 2011) and included seven isolated from blood (considered invasive) and three from other sources (feces or urine). An isolate obtained from urine was not considered invasive because we lacked the clinical data to distinguish between invasive infection and colonization or contamination of urine. S. Dublin cattle isolates (Table 2) were obtained from the NSC and were all S. Dublin isolates available from cattle in the period of study. All isolates had already been analyzed for motility (40).
TABLE 1.
S. Dublin human isolates used in this study
| Isolate | Yr of isolation | Source | Patient information | Motilitya |
|---|---|---|---|---|
| SDu1 | 1995 | Blood | NDb | − |
| SDu2 | 2004 | Blood | ND | − |
| SDu3 | 2006 | Blood | Female, 41 yrs old, asthmatic, diabetic | − |
| SDu4 | 2008 | Blood | Young man, drug addict | + |
| SDu5 | 2000 | Feces | ND | + |
| SDu6 | 2005 | Feces | Male, 40 yrs old | + |
| SDu7 | 2008 | Blood | Male, 1 yr old | + |
| SDu8 | 2011 | Blood | Male, 76 yrs old | + |
| SDu9 | 2011 | Blood | Female, 50 yrs old | − |
| SDu10 | 2011 | Urine | Male | + |
From reference 40.
ND, no data were available.
TABLE 2.
S. Dublin cattle isolates used in this study
| Isolatea | Date of isolation (mo/yr) | Source | Animal identification no.b | Motilityc |
|---|---|---|---|---|
| SDuG1 (75/95) | 06/1995 | NDd | 0341 | + |
| SDuG2 (56/96) | 08/1996 | Calf lymph | 0467 | − |
| SDuG3 (57/96) | 08/1996 | Calf lung | 0467 | − |
| SDuG4 (69/96) | 10/1996 | Calf | ND | + |
| SDuG5 (72/96) | 12/1996 | Calf | 0645 | − |
| SDuG6 (73/96) | 12/1996 | Bile | 7110 | − |
| SDuG7 (74/96) | 12/1996 | Liver | 7110 | − |
| SDuG8 (75/96) | 12/1996 | Calf | 0645 | − |
| SDuG9 (76/96) | 12/1996 | Calf | 0645 | − |
| SDuG10 (79/98) | 07/1998 | ND | 0847 | − |
| SDuG11 (210/00) | 09/2000 | Calf | ND | + |
| SDuG12 (73/04) | 06/2004 | Calf | calf104 | − |
| SDuG13 (74/04) | 06/2004 | Calf | calf107 | + |
| SDuG14 (75/04) | 06/2004 | Calf | calf108 | + |
| SDuG15 (99/04) | 07/2004 | Fetus | fetus018 | − |
Luria-Bertani (LB) agar (Sigma) and LB broth were used for routine cultures of Salmonella and Escherichia coli at 37°C; when grown in liquid medium, cultures were incubated at 200 rpm in an orbital shaking incubator. Isolates were stored in replicates at −80°C in LB broth containing 16.6% glycerol. Ampicillin and streptomycin were used at 100 μg/ml and 50 μg/ml, respectively.
Non-Salmonella Dublin strains, plasmids, and bacterial constructions are listed in Table 3.
For animal studies, strains were made streptomycin resistant by P22 phage transduction of the aadA gene from the streptomycin-resistant serovar Typhimurium strain SL1344 (http://www.sanger.ac.uk/Projects/Microbes/). The resulting transductants were able to grow in 500 μg/ml of streptomycin (although they were routinely grown in 50 μg/ml of this antibiotic) and were verified by anti-O antigen serum agglutination and motility testing. In addition, growth curves were performed to verify that genetic manipulation did not affect the growth properties of the original isolates.
For mouse infection experiments, bacteria were grown overnight (o/n) at 200 rpm at 37°C in LB broth containing 50 μg/ml of streptomycin; the o/n cultures were diluted 1:20 in the same medium plus 0.3 M NaCl and subcultured for 4 h with mild aeration (100 rpm).
Plasmid constructs and DNA manipulation.
Plasmids pBAD22, pJF119EH, and pGEX4T were used as cloning vectors in this study (Table 3). E. coli strain DH5α was used as a host for cloning, and E. coli strain MC1061 was used as a host for protein production for purification.
For fliA supplementation in trans into a nonmotile strain, fliA from a motile strain (SDu5) was cloned into pJF119EH. Genomic DNA was purified using a DNeasy blood and tissue kit (Qiagen) and used as the template for fliA PCR amplification using primers fliA3-F and fliA3-R (see Table S1 in the supplemental material). Final concentrations in a 50-μl mixture were 2 mM MgCl2, 0.3 mM deoxynucleoside triphosphates (dNTPs), 0.5 μM each primer, 0.3 μl of a mixture of Taq polymerase and Pfu polymerase (10 U and 1 U, respectively), and 2 μl of DNA. The cycling program was 5 min at 94°C and 30 cycles of 30 s at 94°C, 30 s at 50°C, and 90 s at 72°C. The PCR product was purified using a QIAquick PCR purification kit (Qiagen), digested with MfeI and HindIII, and ligated into pJF119EH previously cut with EcoRI and HindIII (note that MfeI produces cohesive ends compatible with those generated by EcoRI). The resulting recombinant plasmid contained fliA under the control of the PTac promoter and lacIq and was named pJfliA.
For fliE complementation of a nonmotile strain, fliE from SDu5 was amplified using primers fliE2-F and fliE2-R (see Table S1). Final concentrations in a 50-μl mixture were 1.75 mM MgCl2, 0.3 mM dNTPs, 0.5 μM each primer, 0.4 μl of a mixture of Taq polymerase and Pfu polymerase (10 U and 1 U, respectively), and 2.5 μl of DNA. The cycling program was 5 min at 95°C and 30 cycles of 30 s at 95°C, 30 s at 48°C, and 30 s at 72°C. The PCR product was purified, digested with EcoRI and HindIII, and ligated into pBAD22 previously cut with the same enzymes. The resulting recombinant plasmid contained fliE under the control of a PB promoter and araC and was named pBfliE. Cloning of fliE from motile revertants 6, 7, and 9 of isolate SDu3 into pBAD22 (resulting in plasmids named pBfliEmot + 6, 7, and 9, respectively) was done by following the same strategy.
For fliE complementation of a nonmotile strain for mouse infection experiments, fliE was cloned into pJF119EH, using exactly the same strategy as used for cloning into pBAD22. The resulting construct contained wild-type fliE under the control of the PTac promoter and lacIq and was named pJfliE.
Plasmid DNA extracted from E. coli DH5α was first modified by transformation into S. Typhimurium strain SL5338 (r− m+) and then electroporated into S. Dublin isolates.
For FlgM protein production and purification, SDu5 gene flgM was cloned into pGEX4T-1, which allows the expression of N-terminal fusions to glutathione S-transferase (GST) protein, controlled by the PTac promoter and lacIq. flgM was PCR amplified using primers flgM3-F/flgM3-R (see Table S1). Concentrations and cycling programs were the same as used in fliA amplification. The flgM PCR product was digested with EcoRI and XhoI and ligated into pGEX4T-1 previously cut with EcoRI and XhoI. The resulting recombinant plasmid was named pGEXM.
All recombinant plasmids were verified by restriction analysis and insertion sequencing to discard the presence of mutations in cloned genes.
Motility tests.
For motility tests, motility agar (LB medium containing 0.3% agar) was used. Two microliters of an overnight culture in LB medium were spotted in the center of a motility agar plate and incubated at 37°C. Values are expressed as the diameter of growth (in millimeters) obtained after 6 h of incubation at 37°C.
To obtain motile revertants of SDu3, successive passages of SDu3/pJfliA were done on motility agar plates containing IPTG concentration gradients (from 0 to 0.05 mM). Briefly, 10 ml of motility agar containing 0.05 mM IPTG was poured on a petri dish and kept at an incline. After solidification, 10 ml of motility agar was poured carefully on the plate and the plate was kept horizontal. Overnight culture was diluted 1/50 in LB broth containing 0.01 mM IPTG. After 2 h at 37°C and orbital shaking (200 rpm), 5 drops of 2 μl were spotted along the IPTG gradient with equal distance between each other and incubated for 24 h at 37°C. Every 24 h, a suspension of each spot was done in 50 μl of LB and 2 μl of this suspension was immediately spotted in the same place of a new IPTG gradient motility agar plate. In this way, we intended to maintain constant pressure conditions in favor of expression of a motility phenotype for bacteria in each spot. We used an IPTG gradient because a priori we did not know which IPTG concentration would be optimal for motility (too much fliA expression may be detrimental for growth, while expression at too low a level may be insufficient for FlgM titration and therefore expression of class 3 genes).
Motility of SDu3/pBfliE (and SDu3 complemented with a revertant's fliE gene cloned into pBAD22) was tested in individual plates of motility agar containing 0.005%, 0.02%, 0.05%, 0.1%, and 0% arabinose, and the diameter of the halo of growth was measured after 6 h at 37°C.
Production of specific polyclonal anti-FlgM antibodies.
For FlgM protein production and purification, overnight cultures of MC1061/pGEXM in LB broth with ampicillin (100 μg/ml) were diluted 1/100 in the same medium and grown for 2 h until they reached an optical density at 600 nm (OD600) of ∼0.4 to 0.6. Then 0.5 mM IPTG was added and growth continued for 2 h (to an OD600 of ∼1). Induced cultures were centrifuged and resuspended in phosphate-buffered saline (PBS) with 0.15 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM EDTA, and 0.1 mg/ml of lysozyme. After sonication, Triton X-100 was added to a 0.5% final concentration, and the extracts were centrifuged. The supernatants (soluble extracts) were incubated with glutathione agarose (Sigma), flow through separated, and column washed with PBS containing Triton X-100 at 0.5%. Finally, the purified proteins were treated in column with 20 U of thrombin (Sigma) for 2 h at room temperature and elution fractions were collected. Purified protein fractions were analyzed by polyacrylamide gel electrophoresis (SDS-PAGE) and the band corresponding to FlgM (10.6 kDa) was excised from the gel.
For mouse immunizations, each gel band piece was pulverized with liquid nitrogen, resuspended in 700 μl of PBS (∼0.4 μg/μl) and 233 μl of alumina, and inoculated in 4 female 8- to 10-week-old BALB/c mice (2 subcutaneously and 2 intraperitoneally, 150 μl each) every 7 days. After the 3rd inoculation, sera were positive for specific recognition of FlgM in Western blotting analyses.
Analysis of FliC and FlgM protein levels and subcellular localization.
Total protein extracts and secreted proteins were obtained as described by Yim et al. (59). The protein extracts were quantified by Bradford assay (Sigma).
For identification of FliC by Western blot analysis, 50 μg of total protein extracts or 30 μg of secreted protein fractions were loaded onto a 12% SDS-PAGE gel and analyzed using mouse anti-flagellar antigen Hg antibody (Bio-stat, UK) and ECL Prime Western blotting detection reagent (GE Healthcare). As an internal control, anti-DnaK monoclonal antibody was used (Abcam, UK). For identification of FlgM, 16% Tricine gels were used as described by Schägger (69) and revealed with antiserum anti-FlgM obtained in this work plus ECL Prime Western blotting detection reagent.
Flagellar staining.
For detection of flagella in live cells, we performed a previously described method using Alexa Fluor 594 carboxylic acid succinimidyl ester (Molecular Probes), an amino-specific fluorescent dye (70) with the modifications described in reference 59.
Sequencing and analysis of nucleotide sequences.
To obtain the nucleotide sequences of fliE in the motility revertants of SDu3, we purified genomic DNA from overnight bacterial cultures grown in LB using the DNeasy blood and tissue kit (Qiagen). One hundred nanograms of this DNA was used as the template to PCR amplify the complete gene, using primers fliE2-F and fliE2-R (see Table S1), and the resulting amplicon was sequenced at the Institut Pasteur of Montevideo Molecular Biology Unit (using Sanger technology) with primers fliE1-F and fliE1-R (Table S1). Before sequencing, all PCR products were purified using a QIAquick PCR purification kit (Qiagen).
The complete genome sequences of the 10 human isolates and 4 isolates from cattle (SDuG6, SDuG9, SDuG12, and SDuG14) were obtained thanks to collaboration with the Sanger Institute (Cambridge, UK). Genomes were sequenced as 50- to 76-bp paired-end runs on an Illumina Hiseq2000. High-quality reads were selected using sickle (available at https://github.com/najoshi/sickle), and then the quality was checked using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). De novo assembly was performed with Velvet (http://www.ebi.ac.uk/~zerbino/velvet/) and SPAdes (http://cab.spbu.ru/software/spades/).
All flagellar regions of these isolates and S. Dublin strain CT_02021853, available in the NCBI database (gi∣198241740), were compared using Bioedit software. Nucleotide and residues alignments were performed using Clustal 2.1 (61).
fliEΔ42 PCR screening.
For fliEΔ42 screening in S. Dublin isolates obtained from cattle, primers fliE1-F and fliE1-R were designed for PCR analysis, using genomic DNA of the corresponding strains as the template. Final concentrations were 2 mM MgCl2, 0.2 mM dNTPs, and 0.4 μM each primer. The cycling program was 5 min 95°C and 30 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C. PCR products were separated by electrophoresis in a 2.5% agarose gel.
Quantitative real-time PCR.
For bacterial mRNA quantifications, strains were grown to mid-log phase in LB medium and total RNA was extracted using the RNeasy minikit (Qiagen) with a pretreatment with the RNAprotect bacterial reagent (Qiagen). One microgram of this RNA was DNase treated (Invitrogen) and reverse transcribed using Moloney murine leukemia virus (MMLV) reverse transcriptase (Invitrogen) and random primers in a 20-μl reaction, and 2 μl of a 1/32 dilution of the resulting cDNA was used for real-time PCR using Sybr green (QuantiTect; Qiagen) in a Corbett thermocycler. Primer sequences used are in Table S1; the final concentration of each primer in the reaction was 0.3 μM. The cycling program for fliC and icdA quantification was as follows: 15 min at 95°C and 45 cycles of 15 s at 95°C, 30 s at 57°C, and 30 s at 72°C. For motB, cheB, fliN, flgB, fliE, tsr, and fliF the annealing temperature was 60°C. For analysis, we used the comparative threshold cycle (CT) method for relative mRNA quantitation, using icdA as the normalizing gene and an arbitrarily selected motile strain (SDu5) as the calibration condition (62). Each isolate was assayed in triplicate. Non-reverse-transcribed controls rendered no detectable CT values or were amplified at least 10 cycles later than the corresponding reverse-transcribed samples.
For fliC and motB mRNA level quantification in SDu3/pJfliA, the strain was grown in the absence or presence of 0.1 mM IPTG for 2 h until reaching an OD600 of ∼0.8, and RNA extraction, DNase treatment, reverse transcription, and qRT-PCR were performed as described above.
For mRNA quantification in the ceca of infected mice, fractions of the middle ceca were immediately removed after sacrifice, embedded in TRIzol (Invitrogen), and stored at −80°C. For total RNA extraction, the samples were homogenized using a TissueRuptor (Qiagen) and then the protocol indicated by the manufacturer was followed. One microgram of the resulting RNA was DNase treated and reverse transcribed using MMLV reverse transcriptase as described above. Two microliters of a 1/5 dilution of this reaction was used for real-time PCR using Sybr green (QuantiTect; Qiagen) in an ABI 7900HT thermocycler (Applied Biosystems). Primer sequences used are shown in Table S1; the final concentration of primers in the reaction was 0.9 μM. The cycling program was as follows: 15 min at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. We applied the CT method for relative mRNA quantitation, using the β-actin gene as the housekeeping gene, and the mRNA levels for each group of infected mice were compared to the levels obtained for the uninfected (streptomycin-pretreated) group.
Animal experiments.
Animal experiments were performed as described in reference 47. Briefly, groups of five 6- to 8-week-old female C57BL/6 mice (provided by the National Division of Veterinary Laboratories, Uruguay) were pretreated with 25 mg of streptomycin 24 h prior to infection with ∼5 × 107 CFU of the desired bacterial strain. Twenty-four hours postinfection (p.i.), mice were sacrificed, and fractions of the middle ceca were immediately removed, embedded in TRIzol (Invitrogen), and stored at −80°C for subsequent total RNA extraction and qRT-PCR analysis as described above. For bacterial count determination in organs, cecal contents and spleens were collected and weighed, resuspended in PBS containing 0.5% Tergitol, and homogenized for subsequent dilution and plating in MacConkey lactose agar plates (Oxoid) containing 50 μg/ml of streptomycin (ceca) or in LB plates (spleens).
Experiments with animals were performed according to national guidelines for animal experimentation that meet the guidelines in International Guiding Principles for Biomedical Research Involving Animals (https://grants.nih.gov/grants/olaw/guiding_principles_2012.pdf), and all protocols were approved by the University Ethics Committee.
Statistical analysis.
For analysis of differences in bacterial mRNA levels, the one-way analysis of variance (ANOVA) plus Dunnett's multiple-comparison posttest was used (GraphPad Prism 4.0 software), considering a P value of <0.05 to be statistically significant. For analysis of differences in the transcriptional response to the infection in mice, we used the Mann-Whitney U test (GraphPad Prism 4.0 software), considering a P value of <0.05 (two-tailed) to be statistically significant.
Accession number(s).
The complete genomic sequence data are freely available in the EMBL database (http://www.ebi.ac.uk) with the accession numbers ERR036125 to ERR036131 (SDu1 to SDu7, respectively), ERR405765 (SDu8), ERR405670 (SDu9), ERR405767 (SDu10), and ERR405672 to ERR405675 (SDuG6, SDuG9, SDuG12, and SDuG14, respectively).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Gordon Dougan and Dereck Pickard from the Sanger Institute (Cambridge, UK) for providing complete genome sequences of S. Dublin isolates. Gabriela Algorta and Teresa Camou are acknowledged for providing isolates from the NSC collection and MPH, respectively. Guy Tejedor and Fernando Silveira are acknowledged for invaluable help in working with animals.
This work was supported by Programa CSIC I+D (Universidad de la República, Uruguay).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00517-17.
REFERENCES
- 1.Kirk MD, Pires SM, Black RE, Caipo M, Crump JA, Devleesschauwer B, Dopfer D, Fazil A, Fischer-Walker CL, Hald T, Hall AJ, Keddy KH, Lake RJ, Lanata CF, Torgerson PR, Havelaar AH, Angulo FJ. 2015. World health organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med 12(12):e1001921. doi: 10.1371/journal.pmed.1001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang JY, Henao OL, Griffin PM, Vugia DJ, Cronquist AB, Hurd S, Tobin-D'Angelo M, Ryan P, Smith K, Lathrop S, Zansky S, Cieslak PR, Dunn J, Holt KG, Wolpert BJ, Patrick ME. 2016. Infection with pathogens transmitted commonly through food and the effect of increasing use of culture-independent diagnostic tests on surveillance—Foodborne Diseases Active Surveillance Network, 10 U.S. sites, 2012–2015. MMWR Morb Mortal Wkly Rep 65:368–371. [DOI] [PubMed] [Google Scholar]
- 3.Palacio R, Alfonso SAA, Legnani M, Da Silva A, Castro M, Camou T, Lopez D, Strozzi D, Rosa R. 2010. Surveillance of food-transmitted diseases in Uruguay, 2000–2009. In XX Congreso Latinoamericano de Microbiologia. Sociedad Uruguaya de Microbiología, Montevideo, Uruguay. [Google Scholar]
- 4.Crump JA, Sjolund-Karlsson M, Gordon MA, Parry CM. 2015. Epidemiology, clinical presentation, laboratory diagnosis, antimicrobial resistance, and antimicrobial management of invasive salmonella infections. Clin Microbiol Rev 28:901–937. doi: 10.1128/CMR.00002-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. 2012. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet 379:2489–2499. doi: 10.1016/S0140-6736(11)61752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barton Behravesh C, Jones TF, Vugia DJ, Long C, Marcus R, Smith K, Thomas S, Zansky S, Fullerton KE, Henao OL, Scallan E. 2011. Deaths associated with bacterial pathogens transmitted commonly through food: Foodborne Diseases Active Surveillance Network (FoodNet), 1996–2005. J Infect Dis 204:263–267. doi: 10.1093/infdis/jir263. [DOI] [PubMed] [Google Scholar]
- 7.Gordon MA. 2008. Salmonella infections in immunocompromised adults. J Infect 56:413–422. doi: 10.1016/j.jinf.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 8.Ao TT, Feasey NA, Gordon MA, Keddy KH, Angulo FJ, Crump JA. 2015. Global burden of invasive nontyphoidal Salmonella disease, 2010. Emerg Infect Dis 21:941–949. doi: 10.3201/eid2106.140999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, Gordon MA, Harris D, Clarke L, Whitehead S, Sangal V, Marsh K, Achtman M, Molyneux ME, Cormican M, Parkhill J, MacLennan CA, Heyderman RS, Dougan G. 2009. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res 19:2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feasey NA, Hadfield J, Keddy KH, Dallman TJ, Jacobs J, Deng X, Wigley P, Barquist Barquist L, Langridge GC, Feltwell T, Harris SR, Mather AE, Fookes M, Aslett M, Msefula C, Kariuki S, Maclennan CA, Onsare RS, Weill FX, Le Hello S, Smith AM, McClelland M, Desai P, Parry CM, Cheesbrough J, French N, Campos J, Chabalgoity JA, Betancor L, Hopkins KL, Nair S, Humphrey TJ, Lunguya O, Cogan TA, Tapia MD, Sow SO, Tennant SM, Bornstein K, Levine MM, Lacharme-Lora L, Everett DB, Kingsley RA, Parkhill J, Heyderman RS, Dougan G, Gordon MA, Thomson NR. 2016. Distinct Salmonella Enteritidis lineages associated with enterocolitis in high-income settings and invasive disease in low-income settings. Nat Genet 48:1211–1217. doi: 10.1038/ng.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uche IV, MacLennan CA, Saul A. 2017. A systematic review of the incidence, risk factors and case fatality rates of invasive nontyphoidal salmonella (iNTS) disease in Africa (1966 to 2014). PLoS Negl Trop Dis 11:e0005118. doi: 10.1371/journal.pntd.0005118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones TF, Ingram LA, Cieslak PR, Vugia DJ, Tobin-D'Angelo M, Hurd S, Medus C, Cronquist A, Angulo FJ. 2008. Salmonellosis outcomes differ substantially by serotype. J Infect Dis 198:109–114. doi: 10.1086/588823. [DOI] [PubMed] [Google Scholar]
- 13.Fierer J. 1983. Invasive Salmonella Dublin infections associated with drinking raw milk. West J Med 138:665–669. [PMC free article] [PubMed] [Google Scholar]
- 14.Langridge GC, Nair S, Wain J. 2009. Nontyphoidal Salmonella serovars cause different degrees of invasive disease globally. J Infect Dis 199:602–603. doi: 10.1086/596208. [DOI] [PubMed] [Google Scholar]
- 15.Al-Emran HM, Krumkamp R, Dekker DM, Eibach D, Aaby P, Adu-Sarkodie Y, Ali M, Rubach MP, Bjerregaard-Andersen M, Crump JA, Cruz Espinoza LM, Lofberg SV, Gassama Sow A, Hertz JT, Im J, Jaeger A, Kabore LP, Konings F, Meyer CG, Niang A, Pak GD, Panzner U, Park SE, Rabezanahary H, Rakotozandrindrainy R, Raminosoa TM, Razafindrabe TJ, Sampo E, Schutt-Gerowitt H, Sarpong N, Soura AB, Tall A, von Kalckreuth V, Wierzba TF, May J, Marks F. 2016. Validation and identification of invasive salmonella serotypes in Sub-Saharan Africa by multiplex polymerase chain reaction. Clin Infect Dis 62(Suppl 1):S80–S82. doi: 10.1093/cid/civ782. [DOI] [PubMed] [Google Scholar]
- 16.Tennant SM, Diallo S, Levy H, Livio S, Sow SO, Tapia M, Fields PI, Mikoleit M, Tamboura B, Kotloff KL, Nataro JP, Galen JE, Levine MM. 2010. Identification by PCR of non-typhoidal Salmonella enterica serovars associated with invasive infections among febrile patients in Mali. PLoS Negl Trop Dis 4:e621. doi: 10.1371/journal.pntd.0000621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendriksen RS, Vieira AR, Karlsmose S, Lo Fo Wong DM, Jensen AB, Wegener HC, Aarestrup FM. 2011. Global monitoring of Salmonella serovar distribution from the World Health Organization Global Foodborne Infections Network Country Data Bank: results of quality assured laboratories from 2001 to 2007. Foodborne Pathog Dis 8:887–900. doi: 10.1089/fpd.2010.0787. [DOI] [PubMed] [Google Scholar]
- 18.Mohammed M, Cormican M. 2016. Whole genome sequencing provides insights into the genetic determinants of invasiveness in Salmonella Dublin. Epidemiol Infect 144:2430–2439. doi: 10.1017/S0950268816000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uzzau S, Brown DJ, Wallis T, Rubino S, Leori G, Bernard S, Casadesus J, Platt DJ, Olsen JE. 2000. Host adapted serotypes of Salmonella enterica. Epidemiol Infect 125:229–255. doi: 10.1017/S0950268899004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langridge GC, Fookes M, Connor TR, Feltwell T, Feasey N, Parsons BN, Seth-Smith HM, Barquist L, Stedman A, Humphrey T, Wigley P, Peters SE, Maskell DJ, Corander J, Chabalgoity JA, Barrow P, Parkhill J, Dougan G, Thomson NR. 2015. Patterns of genome evolution that have accompanied host adaptation in Salmonella. Proc Natl Acad Sci U S A 112:863–868. doi: 10.1073/pnas.1416707112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betancor L, Yim L, Martinez A, Fookes M, Sasias S, Schelotto F, Thomson N, Maskell D, Chabalgoity JA. 2012. Genomic comparison of the closely related Salmonella enterica serovars Enteritidis and Dublin. Open Microbiol J 6:5–13. doi: 10.2174/1874285801206010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, Sebaihia M, Baker S, Basham D, Brooks K, Chillingworth T, Connerton P, Cronin A, Davis P, Davies RM, Dowd L, White N, Farrar J, Feltwell T, Hamlin N, Haque A, Hien TT, Holroyd S, Jagels K, Krogh A, Larsen TS, Leather S, Moule S, O'Gaora P, Parry C, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. 2001. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413:848–852. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 23.McClelland M, Sanderson KE, Clifton SW, Latreille P, Porwollik S, Sabo A, Meyer R, Bieri T, Ozersky P, McLellan M, Harkins CR, Wang C, Nguyen C, Berghoff A, Elliott G, Kohlberg S, Strong C, Du F, Carter J, Kremizki C, Layman D, Leonard S, Sun H, Fulton L, Nash W, Miner T, Minx P, Delehaunty K, Fronick C, Magrini V, Nhan M, Warren W, Florea L, Spieth J, Wilson RK. 2004. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet 36:1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- 24.Nuccio SP, Baumler AJ. 2014. Comparative analysis of salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. mBio 5(2):e00929-14. doi: 10.1128/mBio.00929-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winter SE, Winter MG, Godinez I, Yang HJ, Russmann H, Andrews-Polymenis HL, Baumler AJ. 2010. A rapid change in virulence gene expression during the transition from the intestinal lumen into tissue promotes systemic dissemination of Salmonella. PLoS Pathog 6(8):e1001060. doi: 10.1371/journal.ppat.1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raffatellu M, Santos RL, Verhoeven DE, George MD, Wilson RP, Winter SE, Godinez I, Sankaran S, Paixao TA, Gordon MA, Kolls JK, Dandekar S, Baumler AJ. 2008. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med 14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wangdi T, Winter SE, Baumler AJ. 2012. Typhoid fever: “you can't hit what you can't see.” Gut Microbes 3:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 29.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 30.Zeng H, Carlson AQ, Guo Y, Yu Y, Collier-Hyams LS, Madara JL, Gewirtz AT, Neish AS. 2003. Flagellin is the major proinflammatory determinant of enteropathogenic Salmonella. J Immunol 171:3668–3674. doi: 10.4049/jimmunol.171.7.3668. [DOI] [PubMed] [Google Scholar]
- 31.Stecher B, Hapfelmeier S, Muller C, Kremer M, Stallmach T, Hardt WD. 2004. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun 72:4138–4150. doi: 10.1128/IAI.72.7.4138-4150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winter SE, Thiennimitr P, Nuccio SP, Haneda T, Winter MG, Wilson RP, Russell JM, Henry T, Tran QT, Lawhon SD, Gomez G, Bevins CL, Russmann H, Monack DM, Adams LG, Baumler AJ. 2009. Contribution of flagellin pattern recognition to intestinal inflammation during Salmonella enterica serotype Typhimurium infection. Infect Immun 77:1904–1916. doi: 10.1128/IAI.01341-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iqbal M, Philbin VJ, Withanage GS, Wigley P, Beal RK, Goodchild MJ, Barrow P, McConnell I, Maskell DJ, Young J, Bumstead N, Boyd Y, Smith AL. 2005. Identification and functional characterization of chicken Toll-like receptor 5 reveals a fundamental role in the biology of infection with Salmonella enterica serovar Typhimurium. Infect Immun 73:2344–2350. doi: 10.1128/IAI.73.4.2344-2350.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Freitas Neto OC, Setta A, Imre A, Bukovinski A, Elazomi A, Kaiser P, Berchieri A Jr, Barrow P, Jones M. 2013. A flagellated motile Salmonella Gallinarum mutant (SG Fla+) elicits a pro-inflammatory response from avian epithelial cells and macrophages and is less virulent to chickens. Vet Microbiol 165(3-4):425–433. [DOI] [PubMed] [Google Scholar]
- 35.Chevance FF, Hughes KT. 2008. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol 6:455–465. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chilcott GS, Hughes KT. 2000. Coupling of flagellar gene expression to flagellar assembly in Salmonella enterica serovar Typhimurium and Escherichia coli. Microbiol Mol Biol Rev 64:694–708. doi: 10.1128/MMBR.64.4.694-708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Apel D, Surette MG. 2008. Bringing order to a complex molecular machine: the assembly of the bacterial flagella. Biochim Biophys Acta 1778:1851–1858. doi: 10.1016/j.bbamem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Karlinsey JE, Tanaka S, Bettenworth V, Yamaguchi S, Boos W, Aizawa SI, Hughes KT. 2000. Completion of the hook-basal body complex of the Salmonella typhimurium flagellum is coupled to FlgM secretion and fliC transcription. Mol Microbiol 37:1220–1231. doi: 10.1046/j.1365-2958.2000.02081.x. [DOI] [PubMed] [Google Scholar]
- 39.Ohnishi K, Kutsukake K, Suzuki H, Lino T. 1992. A novel transcriptional regulation mechanism in the flagellar regulon of Salmonella typhimurium: an antisigma factor inhibits the activity of the flagellum-specific sigma factor, sigma F. Mol Microbiol 6:3149–3157. [DOI] [PubMed] [Google Scholar]
- 40.Yim L, Sasias S, Martinez A, Betancor L, Estevez V, Scavone P, Bielli A, Sirok A, Chabalgoity JA. 2014. Repression of flagella is a common trait in field isolates of Salmonella enterica Serovar Dublin and is associated with invasive human infections. Infect Immun 82:1465–1476. doi: 10.1128/IAI.01336-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frye J, Karlinsey JE, Felise HR, Marzolf B, Dowidar N, McClelland M, Hughes KT. 2006. Identification of new flagellar genes of Salmonella enterica serovar Typhimurium. J Bacteriol 188:2233–2243. doi: 10.1128/JB.188.6.2233-2243.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirano T, Minamino T, Namba K, Macnab RM. 2003. Substrate specificity classes and the recognition signal for Salmonella type III flagellar export. J Bacteriol 185:2485–2492. doi: 10.1128/JB.185.8.2485-2492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minamino T, Yamaguchi S, Macnab RM. 2000. Interaction between FliE and FlgB, a proximal rod component of the flagellar basal body of Salmonella. J Bacteriol 182:3029–3036. doi: 10.1128/JB.182.11.3029-3036.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao X, Zhang K, Boquoi T, Hu B, Motaleb MA, Miller KA, James ME, Charon NW, Manson MD, Norris SJ, Li C, Liu J. 2013. Cryoelectron tomography reveals the sequential assembly of bacterial flagella in Borrelia burgdorferi. Proc Natl Acad Sci U S A 110:14390–14395. doi: 10.1073/pnas.1308306110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee HJ, Hughes KT. 2006. Posttranscriptional control of the Salmonella enterica flagellar hook protein FlgE. J Bacteriol 188:3308–3316. doi: 10.1128/JB.188.9.3308-3316.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minamino T, Macnab RM. 1999. Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol 181:1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. 2003. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun 71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raffatellu M, George MD, Akiyama Y, Hornsby MJ, Nuccio SP, Paixao TA, Butler BP, Chu H, Santos RL, Berger T, Mak TW, Tsolis RM, Bevins CL, Solnick JV, Dandekar S, Baumler AJ. 2009. Lipocalin-2 resistance confers an advantage to Salmonella enterica serotype Typhimurium for growth and survival in the inflamed intestine. Cell Host Microbe 5:476–486. doi: 10.1016/j.chom.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang FC, Fierer J. 1991. Human infection with Salmonella dublin. Medicine (Baltimore) 70:198–207. doi: 10.1097/00005792-199105000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Elhadad D, Desai P, Rahav G, McClelland M, Gal-Mor O. 2015. Flagellin is required for host cell invasion and normal Salmonella pathogenicity island 1 expression by Salmonella enterica serovar Paratyphi A. Infect Immun 83:3355–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramachandran G, Perkins DJ, Schmidlein PJ, Tulapurkar ME, Tennant SM. 2015. Invasive Salmonella Typhimurium ST313 with naturally attenuated flagellin elicits reduced inflammation and replicates within macrophages. PLoS Negl Trop Dis 9(1):e3394. doi: 10.1371/journal.pntd.0003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carden S, Okoro C, Dougan G, Monack D. 2015. Non-typhoidal Salmonella Typhimurium ST313 isolates that cause bacteremia in humans stimulate less inflammasome activation than ST19 isolates associated with gastroenteritis. Pathog Dis 73(4):ftu023. doi: 10.1093/femspd/ftu023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selander RK, Smith NH, Li J, Beltran P, Ferris KE, Kopecko DJ, Rubin FA. 1992. Molecular evolutionary genetics of the cattle-adapted serovar Salmonella Dublin. J Bacteriol 174:3587–3592. doi: 10.1128/jb.174.11.3587-3592.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franklin A, Linne T, Rehbinder V. 1990. Plasmid profile analysis and restriction enzyme fingerprinting of Salmonella DO-group strains. APMIS 98:665–668. doi: 10.1111/j.1699-0463.1990.tb04986.x. [DOI] [PubMed] [Google Scholar]
- 55.Müller V, Jones CJ, Kawagishi I, Aizawa S, Macnab RM. 1992. Characterization of the fliE genes of Escherichia coli and Salmonella typhimurium and identification of the FliE protein as a component of the flagellar hook-basal body complex. J Bacteriol 174:2298–2304. doi: 10.1128/jb.174.7.2298-2304.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Osorio-Valeriano M, de la Mora J, Camarena L, Dreyfus G. 2016. Biochemical characterization of the flagellar rod components of Rhodobacter sphaeroides: properties and interactions. J Bacteriol 198:544–552. doi: 10.1128/JB.00836-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monlezun L, Liebl D, Fenel D, Grandjean T, Berry A, Schoehn G, Dessein R, Faudry E, Attree I. 2015. PscI is a type III secretion needle anchoring protein with in vitro polymerization capacities. Mol Microbiol 96:419–436. doi: 10.1111/mmi.12947. [DOI] [PubMed] [Google Scholar]
- 58.Saijo-Hamano Y, Uchida N, Namba K, Oosawa K. 2004. In vitro characterization of FlgB, FlgC, FlgF, FlgG, and FliE, flagellar basal body proteins of Salmonella. J Mol Biol 339:423–435. doi: 10.1016/j.jmb.2004.03.070. [DOI] [PubMed] [Google Scholar]
- 59.Yim L, Betancor L, Martinez A, Bryant C, Maskell D, Chabalgoity JA. 2011. Naturally occurring motility-defective mutants of Salmonella enterica serovar Enteritidis isolated preferentially from nonhuman rather than human sources. Appl Environ Microbiol 77:7740–7748. doi: 10.1128/AEM.05318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reed KA, Hobert ME, Kolenda CE, Sands KA, Rathman M, O'Connor M, Lyons S, Gewirtz AT, Sansonetti PJ, Madara JL. 2002. The Salmonella typhimurium flagellar basal body protein FliE is required for flagellin production and to induce a proinflammatory response in epithelial cells. J Biol Chem 277:13346–13353. doi: 10.1074/jbc.M200149200. [DOI] [PubMed] [Google Scholar]
- 61.Li W, Cowley A, Uludag M, Gur T, McWilliam H, Squizzato S, Park YM, Buso N, Lopez R. 2015. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res 43(W1):W580–W584. doi: 10.1093/nar/gkv279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 63.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 64.Hanahan D. 1985. Techniques for transformation of E. coli, p 109–135. In Glover DM. (ed), DNA cloning. IRL Press, McLean, VA. [Google Scholar]
- 65.Bullas LR, Ryu JI. 1983. Salmonella typhimurium LT2 strains which are r− m+ for all three chromosomally located systems of DNA restriction and modification. J Bacteriol 156:471–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaelin WG Jr, Krek W, Sellers WR, DeCaprio JA, Ajchenbaum F, Fuchs CS, Chittenden T, Li Y, Farnham PJ, Blanar MA, Livingston DM, Flemington EK. 1992. Expression cloning of a cDNA encoding a retinoblastoma-binding protein with E2F-like properties. Cell 70:351–364. doi: 10.1016/0092-8674(92)90108-O. [DOI] [PubMed] [Google Scholar]
- 68.Fürste JP, Pansegrau W, Frank R, Blöcker H, Scholz P, Bagdasarian M, Lanka E. 1986. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 69.Schägger H. 2006. Tricine-SDS-PAGE. Nat Protoc 1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 70.Turner L, Ryu WS, Berg HC. 2000. Real-time imaging of fluorescent flagellar filaments. J Bacteriol 182:2793–2801. doi: 10.1128/JB.182.10.2793-2801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.