

Abstract
Aims
Population pharmacokinetic modelling has been widely used across many therapeutic areas to identify sources of variability, which are incorporated into models as covariate factors. Despite numerous publications on pharmacokinetic drug–drug interactions (DDIs) between antiepileptic drugs (AEDs), such data are not used to support the dose rationale for polytherapy in the treatment of epileptic seizures. Here we assess the impact of DDIs on plasma concentrations and evaluate the need for AED dose adjustment.
Methods
Models describing the pharmacokinetics of carbamazepine, clobazam, clonazepam, lamotrigine, levetiracetam, oxcarbazepine, phenobarbital, phenytoin, topiramate, valproic acid and zonisamide in adult and paediatric patients were collected from the published literature and implemented in NONMEM v7.2. Taking current clinical practice into account, we explore simulation scenarios to characterize AED exposure in virtual patients receiving mono‐ and polytherapy. Steady‐state, maximum and minimum concentrations were selected as parameters of interest for this analysis.
Results
Our simulations show that DDIs can cause major changes in AED concentrations both in adults and children. When more than one AED is used, even larger changes are observed in the concentrations of the primary drug, leading to significant differences in steady‐state concentration between mono‐ and polytherapy for most AEDs. These results suggest that currently recommended dosing algorithms and titration procedures do not ensure attainment of appropriate therapeutic concentrations.
Conclusions
The effect of DDIs on AED exposure cannot be overlooked. Clinical guidelines must consider such covariate effects and ensure appropriate dosing recommendations for adult and paediatric patients who require combination therapy.
Keywords: drug–drug interactions, epilepsy, modelling and simulations, personalized medicine, pharmacokinetics
What is Already Known about this Subject
First‐line and alternative first line antiepileptic drugs (AEDs) are often used in combination with second‐line line drugs (i.e. add‐on).
Many AED combinations lead to pharmacokinetic drug–drug interactions (DDIs), which may result in large variation in drug exposure.
The implications of such DDIs have not been considered in existing clinical guidelines.
What this Study Adds
We evaluate how demographic and clinical factors, including comedications (polytherapy), affect systemic exposure to AEDs in the target patient population. In addition, we demonstrate that AED dosing regimens can be optimized to ensure drug concentrations are maintained within a reference therapeutic range.
DDIs can lead to significant changes in systemic exposure and potentially alter the efficacy and safety profile of AEDs in adult and paediatric patients.
These results form the basis for a comprehensive review of clinical guidelines for the use of first and second line AEDs, including novel algorithms for dose adjustment.
Introduction
Epilepsy is a collection of syndromes characterized by the occurrence of paroxysmal seizures. Many patients require prolonged and often life‐long treatment with antiepileptic drugs (AEDs), which are developed and approved based primarily on the evidence of efficacy in specific seizure types. From a clinical perspective, this has led to treatment choices based on a classification system that discriminates AEDs into first and second‐line treatment. A first‐line treatment is tried first and usually used on its own. If first‐line treatment does not work, then another drug (i.e. an alternative first‐line treatment) may be tried on its own. First‐line treatment drugs may also be used as combinations (i.e. add‐on treatment) if seizure control is not achieved or a given regimen is not tolerated 1.
Approximately 20 AEDs are available, including first and second line treatment options. Different guidelines have been proposed to guide healthcare professionals and prescribing physicians on the use of AEDs, with special focus on the criteria for selection of newer drugs. In addition to providing recommendations for the treatment of specific populations such as women and patients infected with human immunodeficiency virus, attention is also given to the importance of dose titration and tapering procedures. Nevertheless, it has been shown that 10–20% of the patients whose target dose has been achieved, still show unresolved seizures and can benefit from subsequent dose‐adjustments 2, 3. Despite evidence on the role of pharmacoresistance and progression of the underlying pathological processes, the lack of response can be partly explained by interindividual variability in the pharmacokinetics (PK) of AEDs 4. The impact of such variability is particularly important in the paediatric population, where maturation processes and developmental growth are known to affect drug disposition 5, 6, 7. In addition, children who do not adequately respond to first‐line treatment are given multiple AEDs in combination, which can result in PK and pharmacodynamic drug–drug interactions (DDIs).
Population PK modelling has been widely used across many therapeutic areas to describe drug exposure and identify sources of variability, which are then incorporated into models as covariate factors 8, 9. Consequently, differences in drug exposure due to explanatory factors such as DDIs or demographic and clinical parameters can be predicted before treatment is initiated. The availability of such models also allows us to perform clinical trial simulations and not‐in‐trial simulations to explore the potential implication of covariate effects on individual patients or subgroups of the target patient population 3, 10. When performed in a systematic manner, the use of simulation scenarios becomes a powerful tool for the evaluation of the impact of multiple, concurrent factors on drug exposure, providing the rationale for dose adjustment purposes 11, 12. Here, we show how clinical trial simulations can be used to characterize PK DDIs for the most widely used AEDs at clinically relevant doses and regimens. Scenarios are evaluated that reflect the impact of titration steps, different maintenance doses and add‐on treatments. Bearing in mind current clinical practice, we aim to assess the impact of DDIs on the exposure to AEDs and establish the need for further dose adjustment. We anticipate that our analysis will assist the review of clinical guidelines, taking into account the role of covariate factors in future dosing recommendations. Most importantly, it will provide clinicians with further insight into the role of PK variability in the overall efficacy and safety profile of AEDs.
Methods
PK models and virtual patient demographics
Models describing the PK of carbamazepine (CBZ) 13, clobazam (CLBZ) 14, clonazepam (CLNZ) 15, lamotrigine (LMT) 16, 17, levetiracetam (LVT) 18, oxcarbazepine (OXC) 19, phenobarbital (PHB) 20, phenytoin (PHT) 21, topiramate (TPM) 22, valproic acid (VPA) 23, 24 and zonisamide (ZNS) 25 were collected from the published literature. Given the primary objective of our analysis, models were selected if covariate effects were identified for one or more AEDs and study population included >50 patients. In addition, whenever possible, preference was given to models based on PK data from both adult and paediatric patients. Furthermore, parameterization of the covariate effect (i.e. DDI) should be based on changes in clearance to allow easier differentiation between treatment conditions, i.e. the presence of the comedication. An overview of the model structure, including details on the parameterization of the covariate effects for each AED is presented in Table 1. Further information on the clinical protocols used to develop the PK models and identify the covariate effects is provided in the online supplemental material. As modelling codes were not available in the original publications, models were transcribed manually into standard control‐stream file format in NONMEM v7.2 26. For the sake of accuracy and quality, model transcription was assessed one by one before the implementation of the simulation scenarios by comparing model‐predicted concentrations for the original patient population to the reported results in the corresponding publications (see online supplemental material). If no deviations were observed during this initial quality check, the PK model code was subsequently transcribed into the appropriate format for simulation purposes in R v3.1.1 27. Simulation scenarios, comprising treatment conditions at different dose levels and DDIs were selected for both adult and paediatric patients. For each scenario, a population of 1000 virtual patients was simulated using the demographic baseline characteristics listed in Table 2. It was anticipated that spurious correlations between covariates would be negligible using random sampling for such a large number of patients. One exception was the correlation (colinearity) between weight and age in children, which is highly relevant for the characterization of PK in this population. This was particularly important for TPM, which had both weight and age as covariate factors in the model. In addition to demographic factors, other influential covariate factors such as genetic polymorphisms were also simulated if included in the original publication. To ensure accurate estimates of the covariate effects, demographic and other relevant clinical variables were sampled according to a uniform distribution.
Table 1a.
Overview of the population pharmacokinetic models used for the evaluation of drug–drug interactions for carbamazepine, clobazam, clonazepam, lamotrigine, levetiracetam and oxcarbazepine
| Model | Carbamazepine | Clobazam* | Clonazepam | Lamotrigine Adults | Lamotrigine Children | Levetiracetam | Oxcarbazepine |
|---|---|---|---|---|---|---|---|
| First author | Jiao 13 | Saruwatari 14 | Yukawa 15 | Rivas 16 | He 17 | Toublanc 18 | Park 19 |
| Population | Chinese | Japanese | Japanese | German, Spanish | Chinese | Japanese (model building), USA (validation) | Korean |
| Sample size (No.of patients) | 585 | 85 | 137 | 284 | 600 | 259 | 199 |
| Sample size (No.of patients) | 687 | 128 | 259 | 404 | 1699 | 1833 | 254 |
| Age (years) | 1.2–85.1 | 1–52 | 0.3–32.6 | 26.8–51.3 | 0.5–17 | 4–55 | 3–80 |
| Weight (kg) | 5–115 | 8–102 | 5–90 | 61.8–85 | 6–98 | 14–107 | 10–95 |
| Samples at | Trough | 0–10 h after dose | 2–6 h after dose | TDM | TDM | Random | TDM |
| Graphical representation |
|
|
|
|
|
|
|
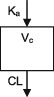
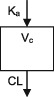
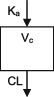
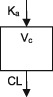
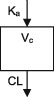
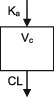
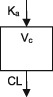
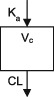
| Parameters | Ka, Vc, CL | Ka, Vc,CL | Ka, Vc, CL | Ka, Vc, CL | Ka, Vc, CL | Ka, Vc, CL | Ka, Vc, CL |
| Between‐subject variability | Vc, CL | Ka, Vc,CL | Vc, CL | CL | CL | Ka, Vc, CL | CL |
| Covariate effects on CL | WT, Dose, PHB, PHT, VPA, Elderly (>65) | WT, PHB, PHT, ZNS, CYP2C19 & POR*28 genotypes | WT, CBZ, VPA | WT, CBZ, PHB, VPA | WT, CBZ, PHB, VPA | WT, CBZ, PHB, PHT, VPA | WT, CBZ, PHB, PHT |
| Covariate effects on V | WT | WT | WT | WT | WT | WT | WT |
Simulations were performed only for the parent drug; the metabolite model not used CL, clearance; Ka, absorption rate constant; NA, not available; TDM, therapeutic drug monitoring; V, volume of distribution; Vc, volume of distribution of the central compartment; WT, weight
Table 2.
Patient baseline demographic characteristics used for the simulation scenarios, in which a virtual cohort of patients was treated with one or more antiepileptic drugs
| Population | adults | children |
|---|---|---|
| Age (years) | 18–65, uniformly distributed | 4–14, uniformly distributed |
| Mean weight (kg) | 75 (male) | (Age×3)+7a |
| 65 (female) | ||
| Coefficient of variation on weight | 16% | 10% |
| Dose interval (h) | 12 | 12 |
| Dose | mg day–1 | mg kg–1 day–1 |
| Number of simulated subjects per scenario | 1000 | 1000 |
Based on the weight‐by‐age formula proposed by Luscombe & Owens 51
Table 1b.
Overview of the population pharmacokinetic models used for the evaluation of drug–drug interactions of phenobarbital, phenytoin, topiramate, valproate and zonisamide
| Model | Phenobarbital | Phenytoin | Topiramate | Valproate Adults | Valproate Children | Zonisamide |
|---|---|---|---|---|---|---|
| First author | Goto 20 | Odani 21 | Girgis 22 | Blanco‐Serrano 23 | Blanco‐Serrano 24 | Okada 25 |
| Population | Japanese | Japanese | NA (Caucasian presumably) | Spanish | Spanish | Japanese |
| Sample size (No. of patients) | 79 | 116 | 1217 | 255 | 208 | 99 |
| Sample size (No. of patients) | 260 | 531 | 4640 | 770 | 534 | 282 |
| Age (y) | 0.8–44 | 1–37 | 2–85 | 14–95 | 0.1–14 | 1.36–39.24 |
| Weight (kg) | 8–80 | 42.4 ± 16.5 | NA | 4–74 | 27–100 | 10–117 |
| Samples at | TDM | Peak/Trough | NA | TDM | TDM | 4.3 ± 2.8 h after dose |
| Graphical representation |
|
|
|
|
|
|
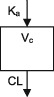
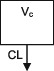
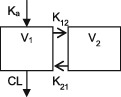
| Parameters | Ka, Vc, CL | Vc, CL (Vmax, Km) | Ka, Vss (V1 + V2), K12, K21, CL | Ka, Vc, CL | Ka, Vc, CL | Ka, Vc, CL |
| Between‐subject variability | Vc, CL | Vc, Vmax, Km | Ka, Vc, CL | CL | CL | CL |
| Covariate effects on CL | WT, PHT, VPA | WT, daily PHT dose, ZNS | Age, WT, Inducers (CBZ/PHB/PHT), VPA, NEMD, ZNS | WT, Dose, CBZ, PHT, PHB | WT, Dose, CBZ | WT, Dose, CYP2C19 genotype, CBZ, PHB, PHT |
| Covariate effects on V | ‐ | WT | WT | WT | WT | WT |
CL, clearance; Ka, absorption rate constant; Km, Michaelis‐Menten constant; K12/K21, distribution rate constants from and to central and peripheral compartments, respectively; NA, not available; TDM, therapeutic drug monitoring; V, volume of distribution; V1 / Vc, volume of distribution of the central compartment; V2, volume of distribution of the peripheral compartment, Vmax, maximum metabolic capacity; WT, weight
Simulation scenarios
One‐ and two‐compartment models were implemented in R according to equations (1) and (2.1)–(2.5), as described in the PFIM optimal design tool documentation 28. The concentration vs. time profiles of each AED were simulated at steady state for the typical adult and paediatric populations (Table 2), following the administration of a range of clinically relevant doses (Table 3). Given the objectives of the current investigation, we have decided not to apply bridging and extrapolation concepts to scale PK parameters from adults to children as a basis for the paediatric dose selection 29. Instead, paediatric doses were scaled by body weight on a mg kg–1 basis, as typically done by prescribing physicians in clinical practice. Secondary PK parameters were then derived, including average steady‐state (Css), peak (Cmax) and trough (Cmin) concentrations.
Table 3.
Simulated doses, comedications and corresponding reference therapeutic range for each antiepileptic drug. Reference antiepileptic drug concentration ranges were taken from Patsalos et al. 30. See main text for further details on supporting references. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA= valproic acid, ZNS= zonisamide
| Drug | Doses adults (mg day–1, * μg day–1) | Doses children (mg kg–1 day–1, * μg kg–1 day–1) | Add‐on medication simulated | Therapeutic window 30 (mg l–1, * μg l–1) |
|---|---|---|---|---|
| CBZ | 400, 800, 1200 | 10, 15, 20 | PHB ∨ PHT ∨ VPA | 4–12 |
| CLBZ | 10, 20, 30* | 0.2, 0.3, 0.4* | PHB ∨ PHT ∨ ZNS | 30–300* |
| CLNZ | 2, 5, 8* | 0.05, 0.075, 0.1* | VPA | 20–70* |
| LMT | 200, 300, 400 | 4, 6, 8 | [(CBZ ⊕ PHB ⊕ PHT) ⊕ INDc] ∨ VPA | 2.5–15 |
| LVT | 1000, 2000, 3000 | 20, 30, 40 | Inducersd | 12–46 |
| OXC | 600, 1200, 1800 | 15, 20, 25 | CBZ ⊕ PHB ⊕ PHT | 3–35 |
| PHB | 60, 150, 240 | 2, 4, 6 | PHT ∨ VPA | 10–40 |
| PHT | 200, 300, 400 | 5, 7.5, 10 | ZNS | 10–20 |
| TPM | 200, 300, 400 | 5, 7.5, 10 | Inducerse ∨ VPA | 5–20 |
| VPA | 400, 800, 1200 | 10, 20, 30 | CBZ ∨ PHB ∨ PHT | 50–100 |
| ZNS | 200, 300, 400 | 5, 7.5, 10 | CBZ ∨ PHB ∨ PHT | 10–40 |
∨ all combinations are possible, ⊕ only one combination is possible
Refers to the different units
For LMT, if more than 1 of CBZ, PHB, or PHT is added, only the effect indicated by IND (and/or VPA) affects LMT clearance
For LVT the original paper 17 mentions inducers such as carbamazepine
For TPM, clearance is induced by adding any of the following: CBZ, PHB and PHT, no distinction is made between adding one or more drugs
A key premise for the evaluation of the different simulation scenarios is the set of assumptions used, which include the following points:
Attainment and maintenance of AED exposure within a target range is desirable for optimal treatment response, irrespective of drug use as a single agent (monotherapy) or as combinations. The reference target concentration ranges published by Patsalos et al. 30 were considered relevant for both the adult and paediatric populations.
In addition, it was assumed that interindividual variability in pharmacodynamics, i.e. different sensitivity to individual drug effects are captured by the proposed target range, whereas resistance to treatment would impose exposure to higher drug concentrations, which are likely to be associated with poor tolerability.
Model misspecification was deemed to be minimal and parameter distributions to be precise and accurate to a sufficiently high degree to allow realistic simulations.
Covariate effects are reasonably well described by the models, despite the limited number of patients included for the development of the models (Table 1).
Bias in the estimates of the covariate effects is minimal even if DDIs are treated as discrete covariates in the available models. It is acknowledged, however, that discrete covariate effects may impair ability to adjust the dose, as variability in exposure or the use of different dose levels of the add‐on drug may alter the magnitude of the interaction. This is particularly important in the case of multiple DDIs.
Whereas discrete parameterization of DDIs may not fully capture the range of conditions or variation in clinical practice, it does provide a stronger basis for the dose rationale, as compared to scenarios where DDIs are completely overlooked.
Simulations were performed in two steps. First, we aimed to identify the dose or dose levels that maximized the fraction of virtual patients whose Css values remained within the target exposure range for each drug. Subsequently, the impact of DDIs on the systemic exposure of the first‐line or alternative first‐line AED was simulated (Table 3). In total, 76 scenarios were considered, taking into account the most clinically relevant dosing regimens and combinations. This resulted in a total of 33 scenarios for monotherapy and 43 scenarios for different AED combinations. As each scenario included 1000 virtual patients, our analysis comprises a population of 76 000 patients.
| (1) |
| (2.1) |
| (2.2) |
| (2.3) |
| (2.4) |
| (2.5) |
| (3) |
Equations (1)–(3). Ct: concentration at time t; τ: dosing interval; D: dose relative to the dose interval τ; DD: total daily dose; V or V1: central volume of distribution; ka: absorption rate constant; CL: clearance; t: time; tD: time of dose; Q: intercompartmental clearance; V2: peripheral volume of distribution. As none of the models included intravenous data, bioavailability estimates were not available; clearance and volume values used in the analysis were therefore based on apparent estimates.
Assessment of the impact of covariate effects on systemic drug exposure
The target Css value used for optimization was set to the drug concentration half‐way between the minimum and maximum values of the therapeutic window defined for each AED (Table 3). Details of the rationale for this approach are described in a previous publication by our group, where different dosing algorithms have been assessed for personalization of AED therapy 3. In brief, the ratio between predicted Css and target Css was calculated (ratio = predicted/target) and results were subsequently summarized in tabular and graphical format. Whisker‐box plots were generated separately for adults and children to describe the dispersion in drug exposure across the population, including the median and 95% prediction interval for each dose and DDI scenario. To facilitate the interpretation of the findings and visualize the impact of dosing titration and/or optimization procedures, the percentage of the population with concentrations outside the therapeutic range was also summarized numerically along with the whisker‐box plots. In addition, the percentage adjustment needed to bring the median Css values back to the target concentration was calculated and provided for each AED.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 31, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 32.
Results
A preliminary analysis of the PK models showed acceptable performance for the purposes of our investigation. Different dose and dosing regimens were simulated for each AED according to the scenarios shown in Table 3. For the sake of completeness, an overview of the concentration vs. time profiles for each AED in adult and paediatric patients is presented in the online supplemental material. These results are complemented by a summary of the procedures used for evaluation of model performance, including the results relative to the secondary PK parameters (Cmax, Cmin).
Monotherapy: impact of standard dosing regimens on systemic drug exposure
For most drugs, the simulated average Css fall within the reference values for a large fraction of the adult and paediatric populations. Notable exceptions were PHT and VPA, where a significant proportion of patients is at risk of achieving sub‐ or supratherapeutic drug concentrations (Figures 1 and 2). In fact, the deviations from the reference range are evident when considering the median estimates. Likewise, despite the use of dosing regimens in mg kg–1, PHT concentrations in children fall outside the therapeutic window in at least 50% of the patients. For VPA the situation is somewhat more favourable, with roughly 20% of the simulated population falling outside the reference therapeutic range. In the case of PHT, the deviation in exposure is compounded by the known nonlinearity and large interindividual variability in PK. There are important clinical implications for patients on PHT when plasma concentrations are >20 mg l–1. In reality, the evidence that a significant proportion of the population is exposed to drug concentrations above the therapeutic range may explain the incidence of adverse events.
Figure 1.
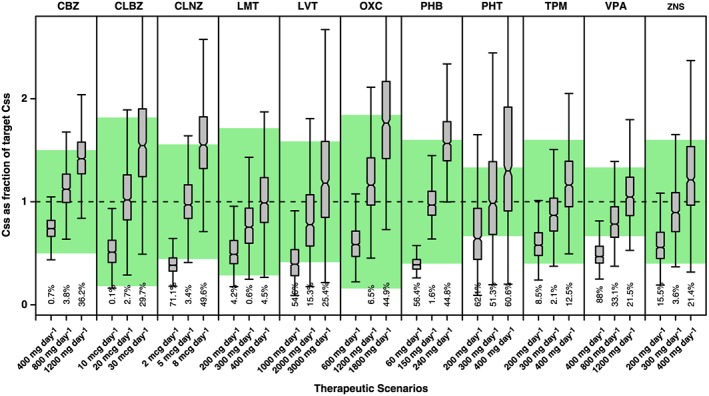
Whisker‐box plots show the median, quartiles and 95% prediction intervals for the steady‐state concentrations (Css) achieved in adults receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area depicts the reference therapeutic range; numbers shown below each bar are percentages of the population with Css values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure 2.
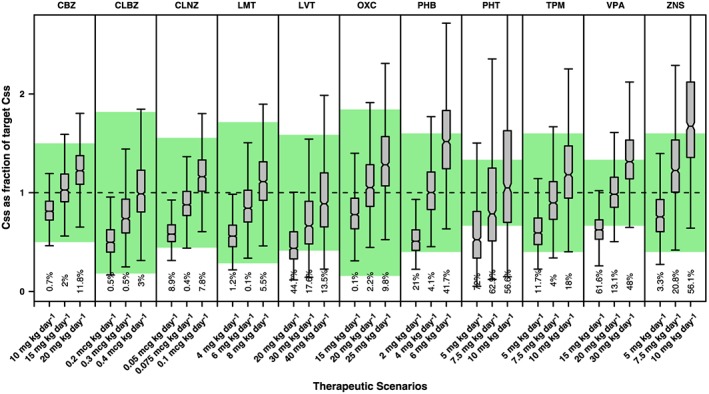
Whisker‐box plots show the median, quartiles and 95% prediction intervals for the steady‐state concentrations (Css) achieved in children receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area depicts the reference therapeutic range; numbers shown below the bars are percentages of the population with Css values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Polytherapy: impact of DDIs on systemic drug exposure
The use of simulations reveals that DDIs can cause major changes to AED concentrations both in adults and children (Figures 3 and 4). When more than one AED is added to the combination therapy, changes in the concentrations of the primary drug may be even larger. This contrasts with the results observed for monotherapy, where drug concentrations for the majority of the AEDs remained within the reference therapeutic range.
Figure 3.
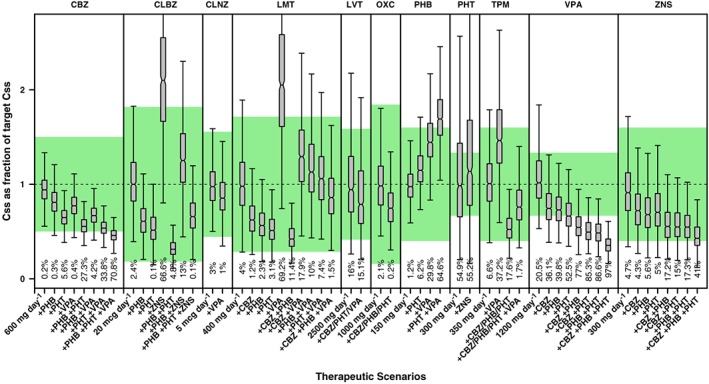
Whisker‐box plots show the median, quartiles and 95% prediction intervals for the steady‐state concentrations (Css) achieved in adults receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area depicts the reference therapeutic range; numbers shown below the bars are percentages of the population with Css values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure 4.
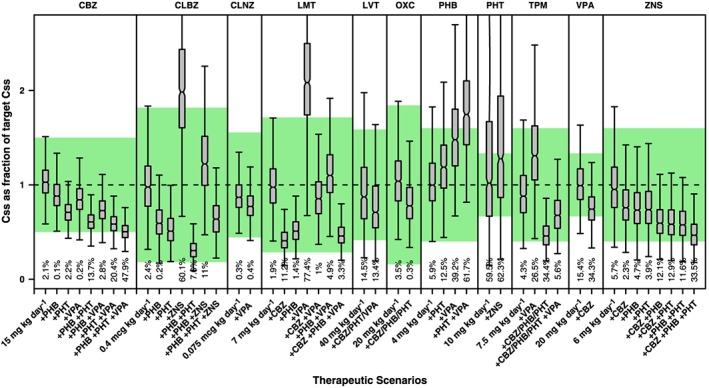
Whisker‐box plots show the median, quartiles and 95% prediction intervals for the steady‐state concentrations (Css) achieved in children receiving polytherapy. Different and drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area depicts the reference therapeutic range; numbers shown below the bars are percentages of the population with Css values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA= valproic acid, ZNS= zonisamide
In many cases, AED interaction results in median Css values that lie outside the reference therapeutic window. By contrast, in certain cases the interaction of multiple comedications may partially or completely counteract each other, resulting in no or minor net change in the exposure to the first line drug. An example of the latter is the interaction of LMT with combination therapy including PHT and VPA. A preliminary evaluation of the effect of DDIs suggests that the doses of the first line and possibly second line drugs used as add‐on treatment need to be adjusted, sometimes by even more than 200% (Table 4).
Table 4.
Predicted change in the dose of the backbone treatment, i.e. first‐line or alternative first‐line antiepileptic drug, as percentage required to ensure that systemic concentrations remain within the reference therapeutic range when add‐on treatment is initiated. (A) = adult patients; (P) = paediatric patients. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
| Add‐on drug(s) | Primary AED | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CBZ | CLBZ | CLNZ | LMT (A) | LMT (P) | LVT | OXC | PHB | PHT | TPM | VPA (A) | VPA (P) | ZNS | |
| +CBZ | – | – | +22 | +60 | +138 | +22 | +31 | – | – | +94 | +36 | +36 | +24 |
| +PHB | +17 | +66 | – | +80 | +88 | – | +31 | – | – | +94 | +40 | – | +29 |
| +PHT | +43 | +93 | – | +94 | – | +22 | +31 | −15 | – | +94 | +54 | – | +28 |
| +VPA | +21 | – | +14 | −51 | −53 | +22 | – | −32 | – | −31 | – | – | – |
| +ZNS | – | −52 | – | – | – | – | – | – | +16 | −37 | – | – | – |
| +CBZ&PHB | +17 | +66 | +22 | +22 | +349 | +22 | +31 | – | – | +94 | +90 | +36 | +60 |
| +CBZ&PHT | +45 | +93 | +22 | +32 | +138 | +22 | +31 | −15 | – | +94 | +110 | +36 | +60 |
| +CBZ&VPA | +21 | – | +39 | −22 | +12 | +22 | +31 | −32 | – | +33 | +36 | +36 | +24 |
| +CBZ&ZNS | – | −52 | +22 | +60 | +138 | +22 | +31 | – | +16 | +23 | +36 | +36 | +24 |
| +PHB&PHT | +70 | +220 | – | +49 | +99 | +22 | +31 | −15 | – | +94 | +115 | – | +65 |
| +PHB&VPA | +42 | +66 | +14 | −12 | −11 | +22 | +31 | −32 | – | +33 | +40 | – | +29 |
| +PHB&ZNS | +17 | −20 | +22 | +22 | +349 | +22 | +31 | – | +16 | +23 | +90 | +36 | +60 |
| +PHT&VPA | +75 | +93 | +14 | −5 | −43 | +22 | +31 | −42 | – | +33 | +54 | – | +28 |
| +PHT&ZNS | +45 | −7 | – | +94 | – | +22 | +31 | −15 | +16 | +23 | +54 | – | +28 |
| +VPA&ZNS | +21 | −52 | +14 | −51 | −43 | +22 | – | −32 | +16 | −56 | – | – | – |
| +CBZ&PHB&PHT | +70 | +220 | +22 | +137 | +349 | +22 | +31 | −15 | – | +94 | +193 | +36 | +105 |
| +CBZ&PHB&VPA | +42 | +66 | +39 | −40 | +111 | +22 | +31 | −32 | – | +33 | +90 | +36 | +60 |
| +CBZ&PHB&ZNS | +17 | −20 | +22 | +22 | +349 | +22 | +31 | – | +16 | +23 | +90 | +36 | +60 |
| +CBZ&PHT&VPA | +75 | +93 | +39 | −35 | +12 | +22 | +31 | −32 | – | +33 | +110 | +36 | +60 |
| +CBZ&PHT&ZNS | +45 | −7 | +22 | +32 | +139 | +22 | +31 | −15 | +16 | +23 | +110 | +36 | +60 |
| +CBZ&VPA&ZNS | +21 | −52 | +39 | −22 | +12 | +22 | +31 | −32 | +15 | −15 | +36 | +36 | +24 |
| +PHB&PHT&VPA | +105 | +220 | +14 | −27 | −11 | +22 | +31 | −32 | – | +33 | +115 | – | +65 |
| +PHB&PHT&ZNS | +70 | +55 | – | +49 | +99 | +22 | +31 | −15 | +16 | +23 | +115 | – | +65 |
| +PHB&VPA&ZNS | +41 | −20 | +14 | −12 | −11 | +22 | +31 | −32 | +16 | −15 | +40 | – | +29 |
| +PHT&VPA&ZNS | +75 | −7 | +14 | −5 | −53 | +22 | +31 | −42 | +16 | −15 | +54 | – | +28 |
Discussion
Given the incidence of epileptic seizures across a wide age range in the patient population, rational prescribing of AEDs requires not only an understanding of the drugs' pharmacodynamic properties, but also careful consideration of the factors known to affect drug disposition 6. Despite numerous publications in which demographic, clinical and genetic covariate factors have been identified, limited attention has been given to the magnitude and variability of such effects and their clinical implication. In most cases, covariate effects are assessed as part of a population PK analysis, where the main objective is the characterization of overall drug disposition properties, rather than the optimization of therapeutic interventions in a wider patient population 33, 34.
In a recent publication, we have shown how model‐based approaches can be used in conjunction with therapeutic drug monitoring to personalize AED therapy 3. The current investigation was aimed at exploring the implications of covariate effects on systemic exposure, with special focus on DDIs, i.e. when patients transition from monotherapy to combination treatment with alternative first line or second line therapy (polytherapy). We found that covariate effects on the disposition of LVT, PHT and VPA leads to considerable variation in drug exposure and consequently to a large proportion of patients reaching average steady‐state concentrations outside the therapeutic window (15%, 54% and 21%, respectively). The impact of covariate effects on the disposition of the other eight drugs included in the analysis appears to be less strong, resulting in a smaller proportion of patients outside the therapeutic window. By contrast, when DDIs come into play, exposure to most AEDs deviates from the reference therapeutic window (e.g. up to 98% in adults receiving VPA), most notably when more than one comedication was added. Moreover, there was no clear correlation between the mechanism of interaction and effect size 35.
Whilst the analysis and interpretation of the simulation results rely on a set of important assumptions regarding covariate effects, it is clear that the relevance of DDIs should not be overlooked in clinical practice, as first‐line treatments are often accompanied by second‐line drugs, which are combined as add‐on therapy in patients who fail to show acceptable clinical response on monotherapy. We have assumed that the models described the DDIs to a sufficiently accurate degree to explore their impact on exposure in the population. However, it should be highlighted that DDIs have been implemented as discrete covariates on clearance, i.e. clearance estimates change depending on whether a comedication was given or not. We cannot exclude the possibility that despite steady‐state concentrations the magnitude of such interactions may be dose‐dependent 36. To take into account the multiple interdependencies in the case of AED polytherapy, the application of more physiology‐based PK models may be required to accurately predict complex DDIs. On the other hand, one can assume that metabolic interactions [e.g. cytochrome P450 (CYP) enzymes] reflect high or maximum induction or inhibition when the comedication exposure is at therapeutically relevant concentrations. In this case, further variation in the dose or concentration does not affect the magnitude of the interaction any more. In this light, the predicted dose changes of first line drugs in Table 4 should be seen as typical values, based on commonly used dose levels of first‐line AEDs and comedication. These results do not exclude the fact that there may be additional variability, which is unaccounted for, depending on the dose of the comedication(s).
Currently, clinical guidelines do not consider the need to assess in a quantitative manner the contribution of covariate factors on drug exposure and consequently on the rationale for dose selection or titration algorithms 3. Whereas some product labels provide dosing recommendations for individuals with renal and hepatic impairment, no specific dose adjustment is proposed to account for other relevant factors. Often, DDIs are mentioned but no formal dosing recommendation is provided, which takes into account AED disposition and other relevant patient characteristics. This is particularly important in infants and children, in whom systemic clearance is often higher than adults after normalization for differences in body weight. This general pattern has been shown for various AEDs 37, 38. At the other extreme of age, in the elderly, systemic clearance is generally reduced compared with younger adults because of less efficient metabolism, reduced renal function, or both 37. Likewise, patient demographic characteristics, such as obesity, also lead to differences in drug disposition, with significant changes in hepatic blood flow and/or metabolic activity (e.g. increased CYP enzyme expression), which have not been considered in our analysis, as the weight range used for the simulations did not include obese patients 39. It should also be noted that despite known polymorphism in drug metabolism, there may be an interaction between genotype and extent of DDI that was not captured by the models in which CYP genotypes were identified as a covariate factor (Table 4). We have assumed that such a CYP genotype–DDI interaction may have a limited role in the overall shift from the target exposure when compared to the magnitude of the DDI itself. Additional data from in silico systems, such as SIMCYP would be required to explore phenotypical and genotypical differences in a systematic manner 40. Another potential factor leading to variability in systemic exposure, which has not been included in the current analysis, is plasma protein binding. In the presence of competing moieties, changes in unbound fraction may affect drug disposition and eventually treatment response, as has been described for VPA and PHT.
It should be highlighted that the lack of guidance regarding DDIs may be partly explained by the ongoing debate on the benefit of therapeutic drug monitoring, especially when performed in an empirical manner 7, 41, 42. Another point to consider is that clinicians tend to focus on age as the explanatory factor influencing the PK profile of AEDs. However, systemic exposure at any age may depend on different covariate factors, such as body weight, genetics, comorbidities, organ function and metabolic capacity. Clearly, in the presence of these multiple interacting factors, it may not be possible to disentangle the contribution of each one independently. Often, unless quantitative clinical pharmacology methods are implemented, such a situation prevents us from proposing dosing adjustment algorithms that correctly account for the effect of DDIs. This concept has been illustrated by the integration of therapeutic drug monitoring with Bayesian algorithms to support dose adjustment for CBZ and/or VPA 43, resulting in increased seizure control, better safety profile and reduced treatment costs.
Our investigation does not focus on the advantages of any specific approach. Rather, it draws attention to the fact that the characterization of covariate effects and variability in drug exposure is essential for dose optimization 44, 45, 46. However, we acknowledge that not all clinically relevant DDIs have been evaluated or parameterized accordingly (e.g. the effect of VPA coadministration on PHT PK) 47, 48.
We also recognize that even though many of the published models have been derived from limited clinical data and often lack a rigorous validation procedure in terms of parameter precision and predictive performance, some interesting lessons can be learnt from the simulation scenarios presented here. First, thanks to the identification of interindividual parameter variability, it is possible to select target (monotherapy) doses for most AEDs, which yield plasma concentrations that are within a reference therapeutic range, which can be deemed suitable for the majority of the population. This does not exclude the possibility that each patient may have an optimal target concentration and benefit from dose individualization 3. Second, DDIs can cause significant changes in the systemic exposure to first line drugs, and this also applies for many add‐on drugs in a combination 49, 50. In theory, this implies that the observed treatment response, or lack thereof, cannot be directly attributed to the add‐on drug. Instead, it may simply be the result of changes in exposure to the backbone drug in the combination. From a therapeutic perspective, one should envisage a scenario in which the systemic concentrations of the primary drug are not affected when patients are switched from monotherapy to combinations. Such a scenario provides the appropriate basis for titration of the add‐on drug.
In conclusion, we have explored the impact of DDIs on the systemic exposure to AEDs when they are used as add‐on in combination therapy. Whereas numerous factors may contribute to lack of efficacy and poor tolerability, the effect of interindividual PK variability and covariate factors on drug disposition cannot be ignored in clinical practice. Our analysis offers a strong basis for the review of clinical guidelines for the treatment of epileptic seizures with AEDs, taking into account the effect of DDIs on the dose rationale for adults and children.
Competing Interests
S.C.v.D. was funded by the Global Research in Paediatrics (GRIP) consortium, a FP7 Network of Excellence. O.D.P. is a member of GRiP and is also Senior Director Clinical Pharmacology at GlaxoSmithKline. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
The authors would like to thank Dr Saruwatari for sharing the NONMEM code for his PK model of clobazam.
The research leading to these results has received funding from the European Union Seventh Framework Programme FP7/2007–2013 under grant agreement no. 261 060.
Supporting information
Table S1 Population demographics used for the validation procedures for model implementation
Figure S1 Predicted vs. observed CBZ concentrations from the publication by Jiao et al. 13 (left) and distribution of simulated concentrations (right)
Figure S2 Predicted vs. observed CLBZ concentrations from the publication by Saruwatari et al. 14 (left) and distribution of simulated concentrations (right)
Figure S3 Predicted vs. observed CLNZ concentrations from the publication by Yukawa et al. 15 (left) and distribution of simulated concentrations (right)
Figure S4 Predicted vs. observed LMT concentrations from the publication by He et al. 17 (left) and distribution of simulated concentrations (right)
Figure S5 Predicted vs. observed LMT concentrations from the publication by Rivas et al. 16 (left) and distribution of simulated concentrations (right)
Figure S6 Observed LVT concentration vs. time profiles from the publication by Toublanc et al. 18 (left) and distribution of simulated concentrations (right)
Figure S7 Predicted vs. observed OXC (MHD) concentrations from the publication by Park et al. 19 (left) and distribution of simulated concentrations (right)
Figure S8 Predicted PHB concentration distribution from the summary statistics reported by Goto et al. 20 (left) and distribution of simulated concentrations (right)
Figure S9 Observed serum phenytoin concentration for different dosing regimens in the publication by Odani et al. 21 (left) and distribution of simulated concentrations (right)
Figure S10 TPM predicted minimum concentrations for different age groups in the publication by Girgis et al. 22 (left) and distribution of simulated concentrations (right)
Figure S11 Inferred VPA concentration distribution from the summary statistics reported by Blanco‐Serrano et al. 23 (left) and distribution of simulated concentrations (right)
Figure S12 Predicted vs. observed VPA concentrations from the publication by Blanco‐Serrano et al. 24 (left) and distribution of simulated concentrations (right)
Figure S13 Inferred ZNS concentration distribution from the summary statistics reported by Okada et al. 25 (left) and distribution of simulated concentrations (right)
Figure S14 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after doses of 400, 800, and 1200 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S15 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after doses of 600 mg day‐1 carbamazepine as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S16 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after doses of 10, 20, and 30 μg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S17 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after doses of 20 μg day‐1 clobazam as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S18 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after doses of 2, 5, and 8 μg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S19 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after a dose of 5 μg day‐1 clonazepam as monotherapy and in combination with valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S20 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S21 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after a dose of 400 mg day‐1 lamotrigine as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval time window of 12 hours
Figure S22 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after doses of 1000, 2000, and 3000 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S23 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after a dose of 2500 mg day‐1 levetiracetam as monotherapy and in combination with an inducer (carbamazepine (CBZ), or phenytoin (PHT), or valproic acid (VPA)). Each panel corresponds to a dosing interval of 12 hours
Figure S24 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state after doses of 600, 1200, and 1800 mg day‐1 oxcarbazepine. Each panel corresponds to a dosing interval of 12 hours
Figure S25 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state after a dose of 1000 mg day‐1 oxcarbazepine as monotherapy and in combination with an inducer (phenytoin (PHT), or phenobarbital (PHB), or carbamazepine (CBZ)). Each panel corresponds to a dosing interval of 12 hours
Figure S26 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after doses of 60, 150, and 240 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S27 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after a dose of 150 mg day‐1 phenobarbital as monotherapy and in combination with phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S28 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S29 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after a dose of 300 mg day‐1 phenytoin as monotherapy and in combination with zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S30 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S31 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after a dose of 350 mg day‐1 topiramate as monotherapy and in combination with valproic acid (VPA), and/or phenytoin (PHT) or phenobarbital (PHB) or carbamazepine (CBZ). Each panel corresponds to a dosing interval of 12 hours
Figure S32 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after doses of 400, 800, and 1200 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S33 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after a dose of 1200 mg day‐1 valproic acid as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT). Each panel corresponds to a dosing interval of 12 hours
Figure S34 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S35 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after a dose of 300 mg day‐1 zonisamide as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT). Each panel corresponds to a dosing interval of 12 hours
Figure S36 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after doses of 10, 15, and 20 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S37 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after a dose of 15 mg kg‐1 day‐1 carbamazepine as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S38 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after doses of 0.2, 0.3, and 0.4 μg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S39 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after a dose of 0.4 μg kg‐1 day‐1 clobazam as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S40 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after doses of 0.05, 0.075, and 0.1 μg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S41 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after a dose of 0.075 μg kg‐1 day‐1 clonazepam as monotherapy and in combination with valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S42 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after doses of 4, 6, and 8 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S43 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after a dose of 7 mg kg‐1 day‐1 lamotrigine as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S44 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after doses of 20, 30, and 40 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S45 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after a dose of 40 mg kg‐1 day‐1 levetiracetam as monotherapy and in combination with an inducer (carbamazepine (CBZ), or phenytoin (PHT), or valproic acid (VPA)). Each panel corresponds to a dosing interval of 12 hours
Figure S46 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state resulting from oxcarbazepine doses of 15, 20, and 25 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S47 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state after a dose of 20 mg kg‐1 day‐1 oxcarbazepine as monotherapy and in combination with an inducer (phenytoin (PHT), or phenobarbital (PHB), or carbamazepine (CBZ)). Each panel corresponds to a dosing interval of 12 hours
Figure S48 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after doses of 2, 4, and 6 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S49 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after a dose of 4 mg kg‐1 day‐1 phenobarbital as monotherapy and in combination with phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S50 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after doses of 5, 7.5, and 10 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S51 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after a dose of 10 mg kg‐1 day‐1 phenytoin as monotherapy and in combination with zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S52 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after doses of 5, 7.5, and 10 mg kg‐1 day‐1 Each panel corresponds to a dosing interval of 12 hours
Figure S53 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after a dose of 7.5 mg kg‐1 day‐1 topiramate as monotherapy and in combination with valproic acid (VPA), and/or phenytoin (PHT) or phenobarbital (PHB) or carbamazepine (CBZ). Each panel corresponds to a dosing interval of 12 hours
Figure S54 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after doses of 10, 20, and 30 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S55 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after a dose of 20 mg kg‐1 day‐1 valproic acid as monotherapy and in combination with carbamazepine (CBZ). Each panel corresponds to a dosing interval of 12 hours
Figure S56 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after doses of 5, 7.5, and 10 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S57 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after a dose of 6 mg kg‐1 day‐1 zonisamide as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT). Each panel corresponds to a dosing interval of 12 hours
Figure S58 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in adults receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S59 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in children receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S60 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in adults receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S61 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in children receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S62 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in adults receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S63 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in children receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S64 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in adults receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S65 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in children receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Supporting info item
van Dijkman, S. C. , Rauwé, W. M. , Danhof, M. , and Della Pasqua, O. (2018) Pharmacokinetic interactions and dosing rationale for antiepileptic drugs in adults and children. Br J Clin Pharmacol, 84: 97–111. doi: 10.1111/bcp.13400.
References
- 1. National Institute for Health and Care Excellence . Epilepsies: diagnosis and management (CG137). Available at http://www.nice.org.uk/guidance/cg137 (last accessed on 22 December 2016)
- 2. Kwan P, Brodie MJ. Epilepsy after the first drug fails: substitution or add‐on? Seizure 2000; 9: 464–468. [DOI] [PubMed] [Google Scholar]
- 3. van Dijkman SC, Wicha SG, Danhof M, Della Pasqua O. Individualised dosing algorithms and therapeutic monitoring for antiepileptic drugs. Clin Pharmacol Ther 2017. https://doi.org/10.1002/cpt.777. [DOI] [PubMed] [Google Scholar]
- 4. Piana C, de Jesus Antunes N, Della Pasqua O. Implications of pharmacogenetics for the therapeutic use of antiepileptic drugs. Expert Opin Drug Metab Toxicol 2014; 10: 341–358. [DOI] [PubMed] [Google Scholar]
- 5. Thomson AH, Brodie MJ. Pharmacokinetic optimisation of anticonvulsant therapy. Clin Pharmacokinet 1992; 23: 216–230. [DOI] [PubMed] [Google Scholar]
- 6. Italiano D, Perucca E. Clinical pharmacokinetics of new‐generation antiepileptic drugs at the extremes of age: an update. Clin Pharmacokinet 2013; 52: 627–645. [DOI] [PubMed] [Google Scholar]
- 7. van Dijkman SC, Alvarez‐Jimenez R, Danhof M, Della Pasqua O. Pharmacotherapy in pediatric epilepsy: from trial and error to rational drug and dose selection – a long way to go. Expert Opin Drug Metab Toxicol 2016; 12: 1143–1156. [DOI] [PubMed] [Google Scholar]
- 8. Piana C, Zhao W, Adkison K, Burger D, Jacqz‐Aigrain E, Danhof M, et al Covariate effects and population pharmacokinetics of lamivudine in HIV‐infected children. Br J Clin Pharmacol 2014; 77: 861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Castro FA, Piana C, Simões BP, Lanchote VL, Della Pasqua O. Busulfan dosing algorithm and sampling strategy in stem cell transplantation patients. Br J Clin Pharmacol 2015; 80: 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chain AS, Dieleman JP, van Noord C, Hofman A, Stricker BH, Danhof M, et al Not‐in‐trial simulation I: bridging cardiovascular risk from clinical trials to real‐life conditions. Br J Clin Pharmacol 2013; 76: 964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Musuamba FT, Teutonico D, Maas HJ, Facius A, Yang S, Danhof M, et al Prediction of disease progression, treatment response and dropout in chronic obstructive pulmonary disease (COPD). Pharm Res 2015; 32: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vegvari C, Cauët E, Hadjichrysanthou C, Lawrence E, Weverling GJ, De Wolf F, et al Using clinical trial simulators to analyse the sources of variance in clinical trials of novel therapies for acute viral infections. PLoS One 2016; 11: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiao Z, Shi XJ, Zhao ZG, Zhong MK. Population pharmacokinetic modeling of steady state clearance of carbamazepine and its epoxide metabolite from sparse routine clinical data. J Clin Pharm Ther 2004; 29: 247–256. [DOI] [PubMed] [Google Scholar]
- 14. Saruwatari J, Ogusu N, Shimomasuda M, Nakashima H, Seo T, Tanikawa K, et al Effects of CYP2C19 and P450 oxidoreductase polymorphisms on the population pharmacokinetics of clobazam and N‐desmethylclobazam in Japanese patients with epilepsy. 2014; 450:302–309. [DOI] [PubMed] [Google Scholar]
- 15. Yukawa E, Satou M, Nonaka T, Yukawa M, Ohdo S, Higuchi S, et al Influence of age and comedication on steady‐state clonazepam serum level‐dose ratios in Japanese epileptic patients. J Clin Pharm Ther 2001; 26: 375–379. [DOI] [PubMed] [Google Scholar]
- 16. Rivas N, Buelga DS, Elger CE, Santos‐Borbujo J, Otero MJ, Domínguez‐Gil A, et al Population pharmacokinetics of lamotrigine with data from therapeutic drug monitoring in German and Spanish patients with epilepsy. Ther Drug Monit 2008; 30: 483–489. [DOI] [PubMed] [Google Scholar]
- 17. He D, Wang L, Qin J, Zhang S, Lu W, Li L, et al Population pharmacokinetics of lamotrigine in Chinese children with epilepsy. Acta Pharmacol Sin 2012; 33: 1417–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Toublanc N, Sargentini‐Maier ML, Lacroix B, Jacqmin P, Stockis A. Retrospective population pharmacokinetic analysis of levetiracetam in children and adolescents with epilepsy – dosing recommendations. Clin Pharmacokinet 2008; 47: 333–341. [DOI] [PubMed] [Google Scholar]
- 19. Park KJ, Kim JR, Joo EY, Seo DW, Hong SB, Ko JW, et al Drug interaction and pharmacokinetic modeling of oxcarbazepine in Korean patients with epilepsy. Clin Neuropharmacol 2012; 35: 40–44. [DOI] [PubMed] [Google Scholar]
- 20. Goto S, Seo T, Murata T, Nakada N, Ueda N, Ishitsu T, et al Population estimation of the effects of cytochrome P450 2C9 and 2C19 polymorphisms on phenobarbital clearance in Japanese. Ther Drug Monit 2007; 29: 118–121. [DOI] [PubMed] [Google Scholar]
- 21. Odani A, Hashimoto Y, Takayanagi K, Otsuki Y, Koue T, Takano M, et al Population pharmacokinetics of phenytoin in Japanese patients with epilepsy: analysis with a dose‐dependent clearance model. Biol Pharm Bull 1996; 19: 444–448. [DOI] [PubMed] [Google Scholar]
- 22. Girgis IG, Nandy P, Nye JS, Ford L, Mohanty S, Wang S, et al Pharmacokinetic‐pharmacodynamic assessment of topiramate dosing regimens for children with epilepsy 2 to 10 years of age. Epilepsia 2010; 51: 1954–1962. [DOI] [PubMed] [Google Scholar]
- 23. Blanco‐serrano B, Otero MJ, Santos‐buelga D, García‐Sánchez MJ, Serrano J, Domínguez‐Gil A. Population estimation of valproic acid clearance in adult patients using routine clinical pharmacokinetic data. Biopharm Drug Dispos 1999; 20: 233–240. [DOI] [PubMed] [Google Scholar]
- 24. Blanco‐Serrano B, García Sánchez MJ, Otero MJ, Buelga DS, Serrano J, Domínguez‐Gil A. Valproate population pharmacokinetics in children. J Clin Pharm Ther 1999; 24: 73–80. [DOI] [PubMed] [Google Scholar]
- 25. Okada Y, Seo T, Ishitsu T, Wanibuchi A, Hashimoto N, Higa Y, et al Population estimation regarding the effects of cytochrome P450 2C19 and 3A5 polymorphisms on zonisamide clearance. Ther Drug Monit 2008; 30: 540–543. [DOI] [PubMed] [Google Scholar]
- 26. Beal SL, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user's guide. Icon Development Solutions, Ellicott City, Maryland, 2009. [Google Scholar]
- 27. R Core Team . R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2014. [Google Scholar]
- 28. Bazzoli C, Retout S, Mentré F. Design evaluation and optimisation in multiple response nonlinear mixed effect models: PFIM 3.0. Comput Methods Programs Biomed 2010; 98: 55–65. [DOI] [PubMed] [Google Scholar]
- 29. Harnisch L, Sheparp T, Pons G, Della Pasqua O. Modelling and simulation as a tool to bridge efficacy and safety data in special populations. CPT Pharmacometrics Syst Pharmacol 2013; 2: e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patsalos PN, Berry DJ, Bourgeois BFD, Cloyd JC, Glauser TA, Johannessen SI, et al Antiepileptic drugs – best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE commission on therapeutic strategies. Epilepsia 2008; 49: 1239–1276. [DOI] [PubMed] [Google Scholar]
- 31. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH, et al The IUPHAR/BPS guide to pharmacology in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2015/16. Voltage‐gated ion channels. Br J Pharmacol 2015; 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nielsen JC, Kowalski KG, Karim A, Patel M, Wesche DL, Tolbert D. Population pharmacokinetics analysis of vigabatrin in adults and children with epilepsy and children with infantile spasms. Clin Pharmacokinet 2014; 53: 1019–1031. [DOI] [PubMed] [Google Scholar]
- 34. Sugiyama I, Bouillon T, Yamaguchi M, Suzuki H, Hirota T. Population pharmacokinetic analysis for 10‐monohydroxy derivative of oxcarbazepine in pediatric epileptic patients shows no difference between Japanese and other ethnicities. Drug Metab Pharmacokinet 2014; 30: 160–167. [DOI] [PubMed] [Google Scholar]
- 35. Riva R, Albani F, Contin M, Baruzzi A. Pharmacokinetic interactions between antiepileptic drugs – clinical considerations. Clin Pharmacokinet 1996; 31: 470–493. [DOI] [PubMed] [Google Scholar]
- 36. Prostran M, Jovanović M, Sokić D, Grabnar I, Vovk T, Prostran M, et al Population pharmacokinetics of topiramate in adult patients with epilepsy using nonlinear mixed effects modelling. Eur J Pharm Sci 2013; 50: 282–289. [DOI] [PubMed] [Google Scholar]
- 37. Perucca E. Clinical pharmacokinetics of new‐generation antiepileptic drugs at the extremes of age. Clin Pharmacokinet 2006; 45: 351–363. [DOI] [PubMed] [Google Scholar]
- 38. Landmark CJ, Baftiu A, Tysse I, Valsø B, Larsson PG, Rytter E, et al Pharmacokinetic variability of four newer antiepileptic drugs, lamotrigine, levetiracetam, oxcarbazepine, and topiramate. Ther Drug Monit 2012; 34: 1. [DOI] [PubMed] [Google Scholar]
- 39. Ghobadi C, Johnson TN, Aarabi M, Almond LM, Allabi AC, Rowland‐Yeo K, et al Application of a systems approach to the bottom‐up assessment of pharmacokinetics in obese patients: expected variations in clearance. Clin Pharmacokinet 2011; 50: 809–822. [DOI] [PubMed] [Google Scholar]
- 40. Djebli N, Fabre D, Boulenc X, Fabre G, Sultan E, Hurbin F. Physiologically based pharmacokinetic modeling for sequential metabolism: effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab Dispos 2015; 43: 510–522. [DOI] [PubMed] [Google Scholar]
- 41. Jacob S, Nair AB. An updated overview on therapeutic drug monitoring of recent antiepileptic drugs. Drugs 2016; 16: 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patsalos PN. Drug interactions with the newer antiepileptic drugs (AEDs) – part 1: pharmacokinetic and pharmacodynamic interactions between AEDs. Clin Pharmacokinet 2013; 52: 927–966. [DOI] [PubMed] [Google Scholar]
- 43. Patsalos PN. Drug interactions with the newer antiepileptic drugs (AEDs) – part 2: pharmacokinetic and pharmacodynamic interactions between AEDs and drugs used to treat non‐epilepsy disorders. Clin Pharmacokinet 2013; 52: 1045–1061. [DOI] [PubMed] [Google Scholar]
- 44. Bondareva IB, Jelliffe RW, Andreeva OV, Bondareva KI. Predictability of individualized dosage regimens of carbamazepine and valproate mono‐ and combination therapy. J Clin Pharm Ther 2011; 36: 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bondareva IB, Sokolov AV, Tischenkova IF, Jelliffe RW. Population pharmacokinetic modelling of carbamazepine by using the iterative Bayesian (IT2B) and the nonparametric EM (NPEM) algorithms: implications for dosage. J Clin Pharm Ther 2001; 26: 213–223. [DOI] [PubMed] [Google Scholar]
- 46. Zhao W, Lopez E, Biran V, Durrmeyer X, Fakhoury M, Jacqz‐Aigrain E. Vancomycin continuous infusion in neonates: dosing optimisation and therapeutic drug monitoring. Arch Dis Child 2013; 98: 449–453. [DOI] [PubMed] [Google Scholar]
- 47. Yuen G, Taylor J. Predicting phenytoin dosages using Bayesian feedback: a comparison with other methods. Ther Drug Monit 1983; 5: 437–441. [DOI] [PubMed] [Google Scholar]
- 48. Tobler A, Mühlebach S. Intravenous phenytoin: a retrospective analysis of Bayesian forecasting versus conventional dosing in patients. Int J Clin Pharmacol 2013; 35: 790–797. [DOI] [PubMed] [Google Scholar]
- 49. Ohara M, Takahashi H, Lee MTM, Wen MS, Lee TH, Chuang HP, et al Determinants of the over‐anticoagulation response during warfarin initiation therapy in Asian patients based on population pharmacokinetic–pharmacodynamic analyses. PLoS One 2014; 9: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Siasos G, Oikonomou E, Zaromitidou M, Kioufis S, Kokkou E, Mourouzis K, et al Clopidogrel response variability is associated with endothelial dysfunction in coronary artery disease patients receiving dual antiplatelet therapy. Atherosclerosis 2015; 242: 102–108. [DOI] [PubMed] [Google Scholar]
- 51. Luscombe M, Owens B. Weight estimation in resuscitation: is the current formula still valid? Arch Dis Child 2007; 92: 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Population demographics used for the validation procedures for model implementation
Figure S1 Predicted vs. observed CBZ concentrations from the publication by Jiao et al. 13 (left) and distribution of simulated concentrations (right)
Figure S2 Predicted vs. observed CLBZ concentrations from the publication by Saruwatari et al. 14 (left) and distribution of simulated concentrations (right)
Figure S3 Predicted vs. observed CLNZ concentrations from the publication by Yukawa et al. 15 (left) and distribution of simulated concentrations (right)
Figure S4 Predicted vs. observed LMT concentrations from the publication by He et al. 17 (left) and distribution of simulated concentrations (right)
Figure S5 Predicted vs. observed LMT concentrations from the publication by Rivas et al. 16 (left) and distribution of simulated concentrations (right)
Figure S6 Observed LVT concentration vs. time profiles from the publication by Toublanc et al. 18 (left) and distribution of simulated concentrations (right)
Figure S7 Predicted vs. observed OXC (MHD) concentrations from the publication by Park et al. 19 (left) and distribution of simulated concentrations (right)
Figure S8 Predicted PHB concentration distribution from the summary statistics reported by Goto et al. 20 (left) and distribution of simulated concentrations (right)
Figure S9 Observed serum phenytoin concentration for different dosing regimens in the publication by Odani et al. 21 (left) and distribution of simulated concentrations (right)
Figure S10 TPM predicted minimum concentrations for different age groups in the publication by Girgis et al. 22 (left) and distribution of simulated concentrations (right)
Figure S11 Inferred VPA concentration distribution from the summary statistics reported by Blanco‐Serrano et al. 23 (left) and distribution of simulated concentrations (right)
Figure S12 Predicted vs. observed VPA concentrations from the publication by Blanco‐Serrano et al. 24 (left) and distribution of simulated concentrations (right)
Figure S13 Inferred ZNS concentration distribution from the summary statistics reported by Okada et al. 25 (left) and distribution of simulated concentrations (right)
Figure S14 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after doses of 400, 800, and 1200 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S15 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after doses of 600 mg day‐1 carbamazepine as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S16 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after doses of 10, 20, and 30 μg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S17 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after doses of 20 μg day‐1 clobazam as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S18 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after doses of 2, 5, and 8 μg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S19 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after a dose of 5 μg day‐1 clonazepam as monotherapy and in combination with valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S20 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S21 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after a dose of 400 mg day‐1 lamotrigine as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval time window of 12 hours
Figure S22 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after doses of 1000, 2000, and 3000 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S23 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after a dose of 2500 mg day‐1 levetiracetam as monotherapy and in combination with an inducer (carbamazepine (CBZ), or phenytoin (PHT), or valproic acid (VPA)). Each panel corresponds to a dosing interval of 12 hours
Figure S24 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state after doses of 600, 1200, and 1800 mg day‐1 oxcarbazepine. Each panel corresponds to a dosing interval of 12 hours
Figure S25 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state after a dose of 1000 mg day‐1 oxcarbazepine as monotherapy and in combination with an inducer (phenytoin (PHT), or phenobarbital (PHB), or carbamazepine (CBZ)). Each panel corresponds to a dosing interval of 12 hours
Figure S26 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after doses of 60, 150, and 240 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S27 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after a dose of 150 mg day‐1 phenobarbital as monotherapy and in combination with phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S28 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S29 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after a dose of 300 mg day‐1 phenytoin as monotherapy and in combination with zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S30 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S31 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after a dose of 350 mg day‐1 topiramate as monotherapy and in combination with valproic acid (VPA), and/or phenytoin (PHT) or phenobarbital (PHB) or carbamazepine (CBZ). Each panel corresponds to a dosing interval of 12 hours
Figure S32 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after doses of 400, 800, and 1200 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S33 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after a dose of 1200 mg day‐1 valproic acid as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT). Each panel corresponds to a dosing interval of 12 hours
Figure S34 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after doses of 200, 300, and 400 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S35 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after a dose of 300 mg day‐1 zonisamide as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT). Each panel corresponds to a dosing interval of 12 hours
Figure S36 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after doses of 10, 15, and 20 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S37 Median (red solid line) and 95% prediction interval (blue dashed lines) of carbamazepine concentrations at steady‐state after a dose of 15 mg kg‐1 day‐1 carbamazepine as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S38 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after doses of 0.2, 0.3, and 0.4 μg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S39 Median (red solid line) and 95% prediction interval (blue dashed lines) of clobazam concentrations at steady‐state after a dose of 0.4 μg kg‐1 day‐1 clobazam as monotherapy and in combination with phenobarbital (PHB), and/or phenytoin (PHT), and/or zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S40 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after doses of 0.05, 0.075, and 0.1 μg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S41 Median (red solid line) and 95% prediction interval (blue dashed lines) of clonazepam concentrations at steady‐state after a dose of 0.075 μg kg‐1 day‐1 clonazepam as monotherapy and in combination with valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S42 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after doses of 4, 6, and 8 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S43 Median (red solid line) and 95% prediction interval (blue dashed lines) of lamotrigine concentrations at steady‐state after a dose of 7 mg kg‐1 day‐1 lamotrigine as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S44 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after doses of 20, 30, and 40 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S45 Median (red solid line) and 95% prediction interval (blue dashed lines) of levetiracetam concentrations at steady‐state after a dose of 40 mg kg‐1 day‐1 levetiracetam as monotherapy and in combination with an inducer (carbamazepine (CBZ), or phenytoin (PHT), or valproic acid (VPA)). Each panel corresponds to a dosing interval of 12 hours
Figure S46 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state resulting from oxcarbazepine doses of 15, 20, and 25 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S47 Median (red solid line) and 95% prediction interval (blue dashed lines) of MHD concentrations at steady‐state after a dose of 20 mg kg‐1 day‐1 oxcarbazepine as monotherapy and in combination with an inducer (phenytoin (PHT), or phenobarbital (PHB), or carbamazepine (CBZ)). Each panel corresponds to a dosing interval of 12 hours
Figure S48 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after doses of 2, 4, and 6 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S49 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenobarbital concentrations at steady‐state after a dose of 4 mg kg‐1 day‐1 phenobarbital as monotherapy and in combination with phenytoin (PHT), and/or valproic acid (VPA). Each panel corresponds to a dosing interval of 12 hours
Figure S50 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after doses of 5, 7.5, and 10 mg day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S51 Median (red solid line) and 95% prediction interval (blue dashed lines) of phenytoin concentrations at steady‐state after a dose of 10 mg kg‐1 day‐1 phenytoin as monotherapy and in combination with zonisamide (ZNS). Each panel corresponds to a dosing interval of 12 hours
Figure S52 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after doses of 5, 7.5, and 10 mg kg‐1 day‐1 Each panel corresponds to a dosing interval of 12 hours
Figure S53 Median (red solid line) and 95% prediction interval (blue dashed lines) of topiramate concentrations at steady‐state after a dose of 7.5 mg kg‐1 day‐1 topiramate as monotherapy and in combination with valproic acid (VPA), and/or phenytoin (PHT) or phenobarbital (PHB) or carbamazepine (CBZ). Each panel corresponds to a dosing interval of 12 hours
Figure S54 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after doses of 10, 20, and 30 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S55 Median (red solid line) and 95% prediction interval (blue dashed lines) of valproic acid concentrations at steady‐state after a dose of 20 mg kg‐1 day‐1 valproic acid as monotherapy and in combination with carbamazepine (CBZ). Each panel corresponds to a dosing interval of 12 hours
Figure S56 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after doses of 5, 7.5, and 10 mg kg‐1 day‐1. Each panel corresponds to a dosing interval of 12 hours
Figure S57 Median (red solid line) and 95% prediction interval (blue dashed lines) of zonisamide concentrations at steady‐state after a dose of 6 mg kg‐1 day‐1 zonisamide as monotherapy and in combination with carbamazepine (CBZ), and/or phenobarbital (PHB), and/or phenytoin (PHT). Each panel corresponds to a dosing interval of 12 hours
Figure S58 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in adults receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S59 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in children receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S60 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in adults receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S61 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in children receiving different doses and dosing regimens of antiepileptic drugs administered as monotherapy. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S62 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in adults receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S63 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of peak drug concentrations (Cmax) in children receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmax values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S64 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in adults receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Figure S65 Whisker‐box plots show the median, quartiles, and 95% prediction intervals (bars) of trough concentrations (Cmin) in children receiving polytherapy. Different drug–drug interaction scenarios are presented for each antiepileptic drug based on commonly used combinations in clinical practice. Shaded area represents the reference therapeutic range, numbers listed below the bars are percentages of the population with Cmin values outside the reference therapeutic range. See text for details on the population PK models, covariate effects and simulation procedures. CBZ= carbamazepine, CLBZ= clobazam, CLNZ= clonazepam, LMT= lamotrigine, LVT= levetiracetam, OXC= oxcarbazepine, PHB= phenobarbital, PHT= phenytoin, TPM= topiramate, VPA+ valproic acid, ZNS= zonisamide
Supporting info item