

Abstract
Aims
The aims of the present study were to assess the safety, tolerability and pharmacokinetics of radavirsen following single ascending doses and multiple doses given as intravenous infusions in healthy adults.
Methods
A phase I safety and pharmacokinetic study of radavirsen was performed in healthy volunteers. The study was divided into two parts. The first was a single‐ascending‐dose study of five cohorts of eight subjects each, randomized 6:2 to receive single intravenous doses of radavirsen ranging from 0.5 mg kg–1 to 8 mg kg–1 or placebo. The second was a multiple‐dose study of 16 subjects randomized 12:4 to receive 8 mg kg–1 or placebo once daily for 5 days.
Results
A total of 66 subjects were screened, and 56 subjects were dosed between 2013 and 2015. At least one adverse event occurred in 31/42 (74%) who received radavirsen, and 13/14 (93%) receiving placebo. The most common adverse events were headache and proteinuria, and were similar in incidence and severity among those receiving radavirsen or placebo. Single‐dose pharmacokinetics demonstrated relatively linear and dose‐proportional increases in maximal concentration and in area under the concentration–time curve from zero to 24 h (AUC0–24). At 8 mg kg–1 in the multiple‐dose cohort, the day 4 geometric mean AUC0–24 was 57.9 μg*h ml–1.
Conclusion
Single infusions of radavirsen up to 8 mg kg–1, and multi‐dosing at 8 mg kg–1 once daily for 5 days, appear to be safe and well tolerated in healthy subjects. The multi‐dose day 4 AUC0–24 in the present study was comparable with that associated with protection from viral infection in a preclinical ferret influenza model. Further evaluation of radavirsen for the treatment of influenza infections is warranted.
Keywords: antiviral, phosphorodiamidate morpholino oligomer, therapeutic
What is Already Known about this Subject
New therapeutic agents for influenza are needed due to resistance and suboptimal efficacy of current therapies and preventative vaccines.
Radavirsen is an antisense oligomer that inhibits the translation of M1 and M2 protein in influenza A.
What this Study Adds
This is the first clinical report of an antisense oligomer for the possible treatment of influenza in humans.
These data demonstrate that radavirsen is safe and well tolerated.
At 8 mg kg–1, radavirsen has a mean AUC0–24 of 57.9 μg*h ml–1, which is an AUC expected to be effective in the treatment of influenza A in humans.
Introduction
Therapeutic interventions for influenza A include adamantanes (amantadine and rimantadine), which target the M2 transmembrane proton channel, and neuraminidase inhibitors (oseltamivir, zanamivir, peramivir, and laninamivir), which inhibit viral budding from infected respiratory epithelial cells by blocking neuraminidase activity.
New forms of treatment for influenza A are needed, based on: (i) the known propensity of this virus to undergo both continuous low‐level antigenic drift and less frequent but unpredictable major antigenic shift leading to pandemic disease; (ii) the clear failure of vaccination, even when strains are reasonably matched, to prevent influenza‐related illness in a significant proportion of vaccine recipients; and (iii) the increased frequency of resistance to approved forms of therapy for influenza [e.g. adamantane resistance in all strains (H1N1 and H3N2) since 2009, and oseltamivir resistance seen in the 2007–2009 A/H1N1 influenza].
Several decades ago, antisense oligonucleotides were found to inhibit influenza virus replication 1. Phosphorothioate oligodeoxynucleotides inhibit the replication of influenza A at very low concentrations 2. A more stable phosphorothioate formulation used in a mouse model led to 45% survival and a 1.5 log reduction in viral titre in the lung when 40 mg kg–1 doses were administered every 12 h 3. The liposomal formulation of various small interfering RNA compounds was demonstrated to be substantially more potent, with 3 mg kg–1 doses providing for a 0.8–1.8 log reduction in lung viral titre, prolongation of median survival time, and up to 45% survival in lethal challenge models 4. The peptide‐conjugated phosphorodiamidate morpholino oligomer (PMO) was the most effective inhibitor of viral titre, with doses as low as 0.15 mg kg–1, resulting in a 1.9–2.0 log reduction in lung viral titre and 50% survival in a lethal challenge mouse model 5, 6.
PMOs are synthetic molecules designed to mimic the natural nucleic acid structure. As antisense oligonucleotides, they bind to complementary sequences of RNA by standard nucleic acid base‐pairing. PMOs are similar to DNA in structure, in that they have standard nucleic acid bases; however, PMOs have morpholine rings instead of the deoxyribose rings of DNA, and are linked through phosphorodiamidate groups instead of phosphates. Radavirsen (previously AVI‐7100) is a PMO containing three positive charges in the form of piperazine residues at defined locations along the backbone (PMOplus®). Unlike classic siRNA or other antisense therapeutic approaches with mechanisms of action that ultimately degrade the target mRNA, radavirsen is designed to recognize and bind to specific mRNA sequences in the matrix segment of influenza A and physically block translation as well as processing of pre‐mRNA, by preventing the splicing of M1 and M2 transcripts. These proteins are involved in the complex viral replication, assembly and budding processes that enable the virus to reproduce in a host cell and infect other cells in the body. Inhibition of the M1 and M2 matrix proteins is intended to disrupt the viral life‐cycle and stop or slow the spread of the disease to other cells in the body. Radavirsen contains CpG motifs, but does not result in immune activation or immune suppression. The viral genomic sequence bound by radavirsen is highly conserved, suggesting no existing resistance potential. Preclinical data suggest that this mechanism may enable broad‐spectrum activity against multiple influenza viruses. Further, radavirsen acts synergistically with neuraminidase inhibitors in animal models of infection.
The present phase I study evaluated the safety, tolerability and pharmacokinetics of radavirsen following single and multiple intravenous infusions in healthy adults.
Methods
Study design
This was a phase I randomized, double‐blind, placebo‐controlled study. It was conducted in accordance with the applicable regulatory and International Conference on Harmonization – Good Clinical Practice requirements. The study protocol and consent were approved by the National Institute of Allergy and Infectious Diseases Institutional Review Board (Protocol 13‐I‐0029; ClinicalTrials.gov Identifier: NCT01747148). Written informed consent was obtained from potential subjects prior to screening.
Subjects were screened up to 28 days prior to dosing. On the first day of dosing (day 0), subjects were assigned sequentially into one of five cohorts, and randomized to receive radavirsen or normal saline placebo. In the single‐dose cohorts, vital signs were obtained 15, 30, 60, and 90 min after the start of each 120‐min infusion, at the end of the infusion, and approximately 0.5, 1, 2, 4, and 8 h after the end of the infusion. Pharmacokinetic (PK) blood samples were obtained at baseline, at the end of infusion and approximately 1, 2, 4, and 8 h after the end of the infusion. Subjects were then seen on days 1, 2, 4, 10 and 28 for safety and tolerability follow‐up visits (with PK samples drawn on days 1, 2 and 4).
For the multi‐dose cohort, subjects were dosed once daily on days 0–4. These subjects were monitored for 2 h after all doses (8 h after the first and last dose), with samples for PK analysis drawn preinfusion on days 0–4, and postinfusion at 1, 2, 4 and 8 h on days 1 and 4 only. Subjects were then seen on days 5, 8, 14 and 32 for follow‐up safety visits (with PK measurements drawn on days 5 and 8). All new symptoms or abnormal laboratory results were captured as adverse events.
Study treatment
Radavirsen was manufactured by Sarepta Therapeutics (Cambridge, MA, USA), and supplied in 100 mg ml–1 vials. For subjects randomized to receive radavirsen, the calculated dose of radavirsen was diluted into a 150 ml bag of normal saline. The bag was masked with an opaque cover, and administered to the subjects as a constant‐rate infusion over 2 h.
Based on the no observed adverse events level of 24 mg kg–1 in female rats (the human equivalent of 3.9 mg kg–1), a starting dose of 0.5 mg kg–1 was chosen. The highest dose of 8 mg kg–1 was chosen as the human equivalent dose that demonstrated efficacy in a preclinical ferret model. The single ascending dose (SAD) portion of the study had five cohorts of eight subjects each (six active, two placebo), dosed at 0.5, 1, 2.5, 5 and 8 mg kg–1, respectively. Based on the findings from the SAD cohorts, the safety of multiple doses was tested in one cohort of 16 subjects (12 active, four placebo) at 8 mg kg–1 once daily for 5 days.
Study population
Healthy adult male or female subjects aged 18–60 years, with a body mass index (BMI) of 19–32 kg m–2 and a creatinine clearance ≥90 ml min–1 (calculated using the Chronic Kidney Disease Epidemiology Collaboration formula) were eligible for the study. All subjects were required to have normal laboratory parameters at enrolment, and did not use any prescription or over‐the‐counter medications, with the exception of acetaminophen, vitamins, seasonal allergy medications and/or contraceptive medications. Females of child‐bearing potential were enrolled as long as they had a negative pregnancy test at enrolment and used two effective forms of contraception, at least one which was a barrier method.
Analytical methods
A liquid chromatographic–tandem mass spectrometric (LC–MS/MS) method was used for the quantitative determination of radavirsen in human plasma. An automated solid phase extraction method was used to extract plasma samples, and the resulting extracts were analysed using reversed phase LC–MS/MS with an electrospray interface and selected reaction monitoring in the positive ionization mode. This method was validated over the calibration curve range of 5–1000 ng ml–1 with a 1/x2 weighted quadratic regression. The within‐run and between‐run precision ranged from 1.5% to 10.9% relative standard deviation (RSD) and 0.0% to 5.2% RSD, respectively. The within‐run accuracy ranged from 85.9% to 105.0% of nominal, and the between‐run accuracy from 91.6% to 100.0% of nominal for all quality control (QC) concentrations, including the lower limit of quantification. For samples above 1000 ng ml–1, study samples were diluted along with QCs (DiQC) in the same batch, to make sure that the samples were properly diluted and that all the DiQCs were within the acceptance criteria. Extraction recoveries for radavirsen were between 46.8% and 47.5%, and long‐term matrix storage stability for radavirsen in human plasma was established at −70°C for up to 209 days.
Statistical methods
This study sample size was designed to detect an adverse event with a true rate of 20% with a probability of 0.74 in any given SAD cohort and 0.93 in the MD cohort, while rarer events with a true rate of 1% could be detected with a probability of 0.06 in any SAD cohort and 0.11 in the MD cohort.
Continuous data were described using descriptive statistics: number of observations (n), mean (arithmetic), median, standard deviation, minimum and maximum. Frequencies and percentages were used for summarizing categorical data. Missing data were not imputed. Data are summarized by treatment group [pooled placebo and dose level of radavirsen (0.5 mg kg–1, 1 mg kg–1, 2.5 mg kg–1, 5 mg kg–1, 8 mg kg–1, multi‐dose 8 mg kg–1)].
PK data were analysed via noncompartmental and compartmental analysis (Phoenix WinNonlin v 6.4, Certara, St Louis, MO, USA). Key parameters of interest, including the maximal observed concentration (Cmax), apparent elimination rate constant (λZ) determined by calculating the absolute value of the slope of the log‐linear regression plasma concentration–time plot, the elimination half‐life (t1/2) calculated as 0.693/λZ, and the area under the concentration vs. time curve from time zero to 24 h postdose (AUC0–24) and extrapolated to infinity (AUC0‐∞), were calculated using the ‘linear‐up, log‐down’ trapezoidal rule. Dose proportionality and accumulation at steady state were assessed by comparison of geometric mean ratios (GMRs) and 90% confidence intervals (CIs) between doses.
Results
Sixty‐six unique subjects were screened between April 2013 and August 2015. Of these, 56 subjects were randomized in the six cohorts. One subject enrolled in the single‐dose cohort was also enrolled in the multi‐dose cohort (as permitted per protocol) after appropriate washout. Of the 11 subjects screened but not randomized, six were ineligible, one subject moved away from the study site, two subjects did not return calls and two subjects were screened but not needed as all cohorts were fully enrolled. One subject in cohort 1 withdrew after day 2 owing to time constraints. Baseline demographic characteristics of the randomized subjects were comparable among all dose groups, including placebo (Table 1).
Table 1.
Baseline demographics
| Placebo (n = 14) | Cohort 1 0.5 mg kg–1 (n = 6) | Cohort 2 1.0 mg kg–1 (n = 6) | Cohort 3 2.5 mg kg–1 (n = 6) | Cohort 4 5.0 mg kg–1 (n = 6) | Cohort 5 8.0 mg kg–1 (n = 6) | Cohort 6 8.0 mg kg–1 (n = 12) | Total Radavirsen (n = 42) | |
|---|---|---|---|---|---|---|---|---|
| Age (years) | ||||||||
| Mean | 29.8 | 26.5 | 30.3 | 31.2 | 31 | 25.7 | 33.7 | 30.3 |
| SD | 9.3 | 3.5 | 12.6 | 10.7 | 11.9 | 3.9 | 9.1 | 9.2 |
| Median | 25 | 25 | 27.5 | 27.5 | 26.5 | 25 | 30.5 | 27 |
| Min, max | 19, 49 | 24, 33 | 19, 54 | 23, 51 | 24, 55 | 22, 33 | 20, 48 | 19, 55 |
| Race [no. (%)] | ||||||||
| Asian | 2 (14.3) | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.4) |
| Black | 4 (28.6) | 2 (33.3) | 0 | 0 | 1 (16.7) | 1 (16.7) | 6 (50) | 10 (23.8) |
| Hawaiian/Pacific Islander | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Multiple race | 1 (7.1) | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.4) |
| Unknown | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.4) |
| White | 6 (42.9) | 4 (66.7) | 5 (83.3) | 4 (66.7) | 5 (83.3) | 5 (83.3) | 6 (50) | 29 (69) |
| Gender [no. (%)] | ||||||||
| Male | 12 (85.7) | 3 (50) | 5 (83.3) | 6 (100) | 1 (16.7) | 2 (33.3) | 5 (41.7) | 22 (52.4) |
| Female | 2 (14.3) | 3 (50) | 1 (16.7) | 0 | 5 (83.3) | 4 (66.7) | 7 (58.3) | 20 (47.6) |
| BMI | ||||||||
| BMI (kg m –2 ) | 27.5 | 25.4 | 24.2 | 24.3 | 23.3 | 23.9 | 25.4 | 24.6 |
BMI, body mass index; SD, standard deviation
Safety
A total of 153 AEs, regardless of relatedness, were reported in the study: 116 for subjects receiving radavirsen and 37 for the subjects receiving placebo, roughly proportional to the 3:1 randomization of participants to active drug vs. placebo. At least one AE occurred in 44 of the 56 subjects: 31/42 (74%) in subjects who received radavirsen, and 13/14 (93%) in subjects receiving placebo. There were no serious AEs and no subjects discontinued the study because of an AE. There were two grade 3 AEs and two grade 4 AEs – two subjects had a grade 3 AE for increased aspartate aminotransferase (AST) and a grade 4 AE for increased creatine phosphokinase, which was temporally associated with vigorous exercise and judged not to be related to the study drug. Of the remaining 149 AEs, 29 were grade 2 and 120 were grade 1.
One subject in cohort 6 on day 3 had an episode of hypotension and hypoglycaemia (captured as AEs) 1 h and 35 min into the infusion. After several minutes, the symptoms and hypotension resolved, and the infusion was resumed and completed without recurrence of similar symptoms.
The most common AEs were headache [15 events in 12 subjects (28.57%) who received radavirsen and four events in four subjects (28.57%) who received placebo] and proteinuria [15 events in 12 subjects (28.57%) who received radavirsen and three events in three subjects (21.42%) who received placebo]. A drug‐related AE was defined as any AE with an investigator causality assessment of possible, probable or definite. All drug‐related AEs are presented in Table 2.
Table 2.
Number of drug‐related adverse events (AEs) (and percentage of subjects with the AE) by preferred term and cohort
| MedDRA AE term | Placebo (n = 14) | Radavirsen 0.5 mg kg –1 (n = 6) | Radavirsen 1 mg kg –1 (n = 6) | Radavirsen 2.5 mg kg –1 (n = 6) | Radavirsen 5 mg kg –1 (n = 6) | Radavirsen 8 mg kg –1 (n = 6) | Radavirsen 8 mg kg –1 multi‐dose (n = 12) |
|---|---|---|---|---|---|---|---|
| Acute gingivitis | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Alanine aminotransferase increased | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (8.3) |
| Albuminuria | 2 (14.3) | 1 (16.7) | 0 | 0 | 0 | 2 (33.3) | 0 |
| Arthralgia | 0 | 0 | 0 | 0 | 0 | 0 | 3 (8.3) |
| Aspartate aminotransferase increased | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 3 (16.7) |
| Urine beta‐2 microglobulin increased | 0 | 0 | 0 | 0 | 0 | 0 | 3 (16.7) |
| Blood bilirubin increased | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 |
| Chills | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Common cold | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
| Cough | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Diarrhoea | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Electrocardiogram PR interval prolonged | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 0 | 2 (16.7) |
| Headache | 2 (14.3) | 3 (50.0) | 2 (33.3) | 2 (16.7) | 2 (33.3) | 2 (33.3) | 1 (8.3) |
| Haematuria | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Hyperglycaemia | 3 (21.4) | 0 | 0 | 0 | 2 (33.3) | 2 (33.3) | 2 (16.7) |
| Hypertension | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 |
| Hypoalbuminaemia | 1 (7.1) | 1 (16.7) | 0 | 0 | 1 (16.7) | 0 | 0 |
| Hypoglycaemia | 2 (7.1) | 0 | 0 | 0 | 0 | 0 | 2 (16.7) |
| Hypomagnesaemia | 3 (7.1) | 1 (16.7) | 0 | 1 (16.7) | 2 (33.3) | 0 | 0 |
| Hyponatraemia | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Hypotension | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Increased appetite | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Indigestion | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 |
| Infusion site infiltration | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Injection site tenderness | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 |
| Light‐headedness | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (8.3) |
| Muscle soreness | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
| Palpitations | 0 | 0 | 0 | 0 | 0 | 0 | 1 (8.3) |
| Proteinuria | 3 (21.4) | 1 (16.7) | 0 | 2 (16.7) | 0 | 1 (16.7) | 10 (75.0) |
| Rash | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 2 (16.7) |
| Sore throat | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 |
| Systolic hypertension | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
| Vomiting | 1 (7.1) | 0 | 0 | 0 | 0 | 0 | 0 |
In the multi‐dose cohort, the rate of transaminase elevations increased slightly: increased AST levels were noted in five events in four subjects (33%) who received radavirsen (one of which was judged not to be related) vs. none in subjects receiving placebo, and increased ALT levels were noted for three events in three subjects (two of which were judged not to be related) who received radavirsen vs. none in subjects receiving placebo (Table 2). Proteinuria was more common in the multi‐dose cohort, being seen in 9/12 subjects (75%) for a total of 10 events. Proteinuria generally began on days 4–5 (range days 2–14), was detected only on 1–3 study days and peaked at a mean of 37 mg ml–1 (Figure 1). When adjusted for urine creatinine, this increase in urine protein persisted. A similar increase was seen in urine beta‐2 microglobulin. All proteinuria reversed by day 32.
Figure 1.
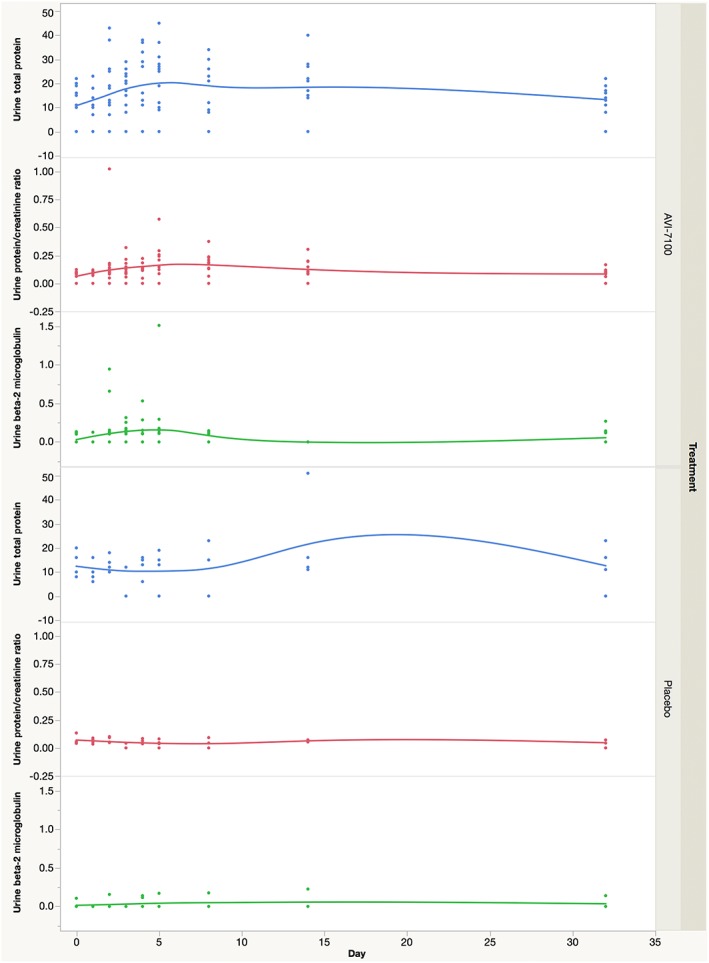
Urine protein, protein to creatinine ratio, and beta‐2 microglobulin over time in the multi‐dose cohort by treatment
Pharmacokinetics
Analysis of the SAD cohorts demonstrated that radavirsen demonstrated roughly linear and dose‐proportional increases in the PK parameters of Cmax and AUC0–24 over the range 0.5–8.0 mg kg–1 (Table 3). SAD data also showed that radavirsen appeared to follow a bi‐exponential decline/two‐compartmental PK model (Figure 2); however, analysis from the multiple‐dose cohort with extended PK sampling from 24–96 h postdose demonstrated that a third compartment is present in the concentration vs. time profile of radavirsen (Figure 3). SAD data indicated an apparent terminal elimination t1/2 of radavirsen of approximately 3 h (range 2.7–3.4 h), while the t1/2 calculated from the newly observed third compartment ranged from 47 h to 162 h. While this third compartment represents the true terminal elimination, it is likely not to be clinically relevant, as all concentrations measured after 24 h postdose throughout the study (including those obtained on day 4 of the multiple‐dose cohort) were less than 2% of the observed Cmax, and the median AUC percentage extrapolation from 24 h extrapolated to infinity was 1.3% in the multiple‐dose cohort. Furthermore, multiple‐dose data demonstrated no accumulation of radavirsen on day 4 of dosing, based on Cmax and AUC0–24 (Table 4). Geometric mean PK parameters using the two‐compartmental model were similar in both the SAD and multiple‐dose cohorts (Table 5). When using dose‐normalized Cmax and AUC0–24 to compare across dosing cohorts, there were no differences in any PK parameters based on gender or BMI (data not shown).
Table 3.
Pharmacokinetic parameters after a single dose of radavirsen
| Dose level | C max (μg ml –1 ) | AUC 0–∞ (μg*h ml –1 ) | Dose ratio a (vs. 0.5 mg kg –1 ) | C max b (vs. 0.5 mg kg –1 ) | AUC 0–∞ c (vs. 0.5 mg kg –1 ) |
|---|---|---|---|---|---|
| GM (CV%) | GMR (90% CI) | ||||
| 0.5 mg kg –1 | 1.4 (24.1) | 3.2 (28.0) | – | – | – |
| 1 mg kg –1 | 3.6 (9.5) | 8.7 (12.8) | 2 | 2.3 (1.7–3.0) | 2.4 (1.7–3.2) |
| 2.5 mg kg –1 | 7.2 (20.3) | 19.4 (19.3) | 5 | 4.7 (3.2–6.2) | 5.4 (3.8–7.1) |
| 5 mg kg –1 | 20.4 (44.9) | 48.2 (39.3) | 10 | 14.6 (7.2–22.0) | 15.2 (8.4–21.9) |
| 8 mg kg –1 | 21.3 (9.4) | 54.0 (15.0) | 16 | 15.2 (10.5–20.0) | 17.0 (11.4–22.5) |
AUC0–∞, area under the concentration–time curve from time 0 extrapolated to infinity; CI, confidence interval; Cmax, maximum concentration; CV%, coefficient of variation; GM, geometric mean; GMR, geometric mean ratio
Dose ratio comparing respective dose level vs. 0.5 mg kg–1 dosing level
GMR comparison of respective dose level vs. 0.5 mg kg–1 Cmax estimate
GMR comparison of respective dose level vs. 0.5 mg kg–1 AUC0–∞ estimate
Figure 2.
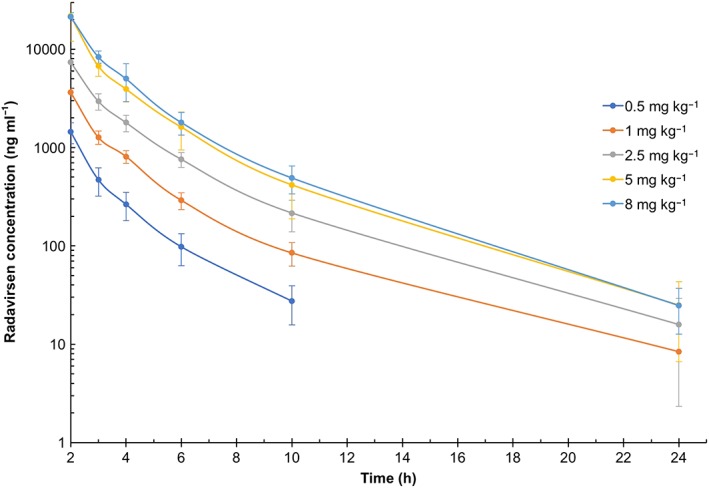
Plasma concentrations of radavirsen over time – Cohorts 1–5. Data are presented as arithmetic mean ± standard deviation
Figure 3.
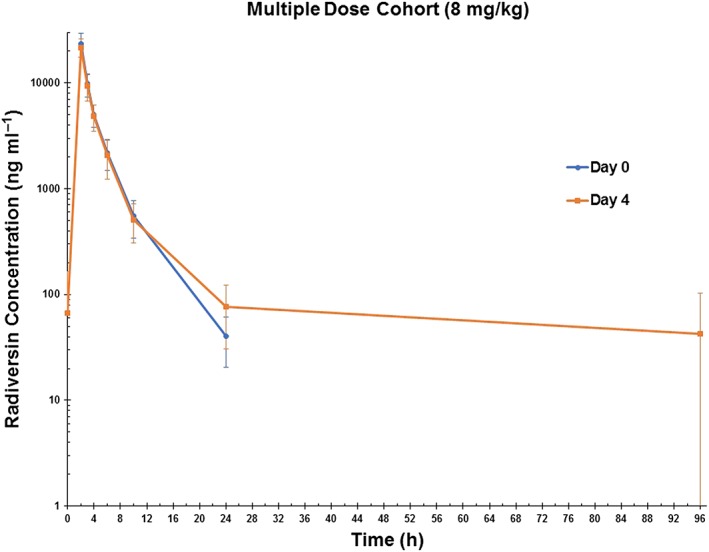
Plasma concentrations of radavirsen (ng ml–1) in the multiple‐dose cohort (cohort 6) after the first (day 0) and last (day 4) dose. Data are presented as arithmetic mean ± standard deviation
Table 4.
Multiple dosing accumulation test
| C max (μg ml –1 ) | AUC 0–24 (μg*h ml –1 ) | C max a | AUC 0–24 b | |
|---|---|---|---|---|
| GM (CV%) | GMR (90% CI) | |||
| Day 0 | 22.7 (25.5) | 58.2 (24.6) | 0.96 (0.88–1.03) | 1.00 (0.94–1.05) |
| Day 4 | 21.7 (19.6) | 57.9 (22.7) | ||
AUC0–24, area under the concentration–time curve from time 0 to 24 h; CI, confidence interval; Cmax, maximum concentration; CV%, coefficient of variation; GM, geometric mean; GMR, geometric mean ratio
GMR comparison of day 4 vs. day 0 Cmax estimates
GMR comparison of day 4 vs. day 0 AUC0–24 estimates
Table 5.
Geometric mean (CV%) pharmacokinetic parameters from the two‐compartmental model of single ascending and multiple‐dose cohorts
| CL 1 (ml h –1 kg –1 ) | CL 2 (ml h –1 kg –1 ) | V 1 (ml kg –1 ) | V 2 (ml kg –1 ) | α (1 h –1 ) | β (h –1 ) | |
|---|---|---|---|---|---|---|
| SAD | 115 (2.3) | 50.2 (23.0) | 110 (10.9) | 86.6 (12.4) | 1.77 (18.7) | 0.34 (12.1) |
| MD | 121 (4.1) | 40.6 (33.2) | 159 (10.5) | 130.9 (25.3) | 1.17 (17.9) | 0.20 (25.2) |
Data are presented as the geometric mean (CV%)
α, elimination rate from the central compartment; β, elimination rate from the peripheral compartment; CL1, clearance from the central compartment; CL2, clearance from the peripheral compartment; CV%, coefficient of variation; MD, multiple‐dose cohort; SAD, single ascending dose cohort; V1, volume of distribution of the central compartment; V2, volume of distribution of the peripheral compartment
Discussion
Radavirsen is a novel inhibitor of influenza A which is being developed for the treatment of influenza A. This putative drug candidate was developed within 7 days of acquisition of the 2009 novel swine‐origin influenza A virus H1N1 virus RNA sequence (Sarepta Therapeutics, data on file). The targeting of radavirsen only at influenza A, and not influenza B, is less than ideal in the current influenza drug development landscape. However, the ability of the platform quickly to generate a new putative drug candidate against an emerging influenza virus makes this class a potentially significant addition to the armamentarium of influenza therapeutics.
The results of the present phase I study suggest that, following single doses ranging up to 8 mg kg–1, and multiple doses at 8 mg kg–1 day–1 for 5 days, radavirsen appears to be safe and well tolerated. Proteinuria, while mild and reversible, was an unexpected finding. The fact that it was accompanied by elevated beta‐2 microglobulin suggests that it may represent glomerular injury. In preclinical toxicity studies with Sprague–Dawley (SD) rats treated for 1 week with radavirsen at 240 mg kg–1 day–1, there was a mildly higher urine gamma‐glutamyl transferase (UGGT) : urine creatinine (UCR) ratio (Sarepta Therapeutics, data on file). In a 2‐week toxicity study in SD rats receiving a dose of 240 mg kg–1 day–1, there were minimally higher urine total protein (UTP) : UCR, moderately higher UGGT : UCR and mildly higher urine cystatin C (UCYC) : urine creatinine ratios, suggestive of potential glomerular injury (Sarepta Therapeutics, data on file). In a 2‐week monkey toxicity study in which a dose of 12 mg kg–1 day–1 was used, there were mild‐to‐moderately increased UGGT : UCR ratios, but evaluation of UCYC, UCYC : UCR ratios, UTP concentration and UTP : UCR ratios did not detect any radavirsen‐related changes in glomerular function (Sarepta Therapeutics, data on file). Renal toxicities have been described in the preclinical testing of other antisense oligonucleotides 7. However, in an integrated safety database across multiple preclinical and clinical studies comprising 710 monkeys and 750 human subjects, there was no evidence of class effect renal toxicities 8. Furthermore, proteinuria was not reported in other human studies in which a PMO was employed – for example, in exon skipping in Duchenne muscular dystrophy 9, or with PMOplus® as an antiviral agent in haemorrhagic fever virus infection 10, 11. There were also no changes in urine beta‐2 microglobulin or other sensitive urinary markers of kidney injury, such as cystatin C and kidney injury molecule‐1, reported in these clinical studies (Sarepta Therapeutics, data on file).
Radavirsen exposure (Cmax and AUC0–24) increased relatively proportionately with dose, and did not change with repeated dosing. The 5 mg kg–1 dosing group appeared to deviate from dose proportionality, but this was likely to have been due to a single subject outlier with a high AUC0–24 estimate (89 998 ng*h ml–1). Dose proportionality was observed in the other dosing cohorts, including those receiving 8 mg kg–1 doses.
In preclinical efficacy studies, ferrets were infected intranasally with 5 × 105 plaque‐forming units of influenza A/Hong Kong/2369/09 H1N1 virus per ferret (which carries the H275Y oseltamivir resistance mutation) and administered radavirsen by the intraperitoneal route at 10 mg kg–1 or 30 mg kg–1 once a day, oseltamivir or saline controls. Ferrets treated with radavirsen 30 mg kg–1 (AUC 53.8 μg*h ml–1) had lower mean day 1 viral shedding (0.57 ± 0.49 log10 copies ml–1) compared with oseltamivir (2.75 ± 0.89 log10 copies ml–1) or saline (2.64 ± 1.18 log10 copies ml–1), lower cumulative AUC viral shedding (2.94 ± 0.86, 7.51 ± 3.09 and 6.14 ± 3.18 log10 copies*day ml–1) and low viral titre on bronchial alveolar lavage on day 7 (0.00 ± 0.00, 1.04 ± 1.49, and 1.14 ± 1.42 log10 copies ml–1), all of which had a P value of <0.05 vs. saline control by Dunnett's multiple comparisons test (Sarepta Therapeutics, data on file). The AUC at 30 mg kg–1 in a ferret is comparable with that achieved with the 8 mg kg–1 dose in humans (Table 3), suggesting that effective levels can be reached in treatment studies.
The third compartment observed in the PK analysis was consistent with a long intracellular t1/2. Similar results were described in other PMO studies that were analysed using a noncompartmental approach 10, 11. Plasma concentrations declined in a multi‐phasic manner with prolonged terminal elimination phases, particularly at higher doses, suggesting redistribution back into the plasma from a slowly diffusing tissue source (Sarepta Therapeutics, data on file). These PK observations are similar to those reported with other PMO derivatives under study as potential therapeutic or prophylactic interventions for Ebola 11 or Marburg 10 haemorrhagic virus infections.
Conclusion
This is the first demonstration of the safety, tolerability and pharmacokinetics of an antisense oligonucleotide for the treatment of influenza A. The data attained in the present study suggest that radavirsen is safe and well tolerated at relevant, potentially therapeutic exposures. Further clinical evaluation of radavirsen for the treatment of influenza A virus infections is warranted.
Competing Interests
J.H.B., J.V., P.M., P.K., K.M.B. and R.T.D. do not have any conflicts of interest to declare. J.Z. is an employee of Sarepta Therapeutics. P.I., A.H. and M.W. are prior employees of Sarepta Therapeutics.
The authors thank the volunteers who participated in this research study, without whom the study would not have been possible. We also thank the physicians, nurses and other healthcare personnel at the National Institutes of Health who provided invaluable services and support for this study. Study drug and analytical assays were provided by Sarepta Therapeutics. This project was funded, in part, with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. This project has been funded in part with federal funds from the Intramural Research Programs of the National Institute of Allergy and Infectious Diseases, National Institutes of Health. This project has also been funded in part with federal funds from the NIH Clinical Center, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Contributors
J.H.B., J.V., P.I., A.H. and R.T.D. were responsible for the initial study design, although all authors were involved with subsequent study amendments. J.H.B., J.V., P.M. and R.T.D. were responsible for study implementation, and enrolment of the participants. P.K. and K.M.B. were responsible for pharmacokinetic analysis. J.H.B., J.V., P.M. and R.T.D. analysed and interpreted the data and wrote the first draft of the report, although all authors had the opportunity to review the data and contributed to the editing of the final report.
Beigel, J. H. , Voell, J. , Muñoz, P. , Kumar, P. , Brooks, K. M. , Zhang, J. , Iversen, P. , Heald, A. , Wong, M. , and Davey, R. T. (2018) Safety, tolerability, and pharmacokinetics of radavirsen (AVI‐7100), an antisense oligonucleotide targeting influenza a M1/M2 translation. Br J Clin Pharmacol, 84: 25–34. doi: 10.1111/bcp.13405.
References
- 1. Kabanov AV, Vinogradov SV, Ovcharenko AV, Krivonos AV, Melik‐Nubarov NS, Kiselev VI, et al A new class of antivirals: antisense oligonucleotides combined with a hydrophobic substituent effectively inhibit influenza virus reproduction and synthesis of virus‐specific proteins in MDCK cells. FEBS Lett 1990; 259: 327–330. [DOI] [PubMed] [Google Scholar]
- 2. Zamecnik PC, Agrawal S. Oligodeoxynucleotide hybridization inhibition of HIV and influenza virus. Nucleic Acids Symp Ser 1991; 127–131. [PubMed] [Google Scholar]
- 3. Mizuta T, Fujiwara M, Abe T, Miyano‐Kurosaki N, Yokota T, Shigeta S, et al Inhibitory effects of an antisense oligonucleotide in an experimentally infected mouse model of influenza a virus. Biochem Biophys Res Commun 2000; 279: 158–161. [DOI] [PubMed] [Google Scholar]
- 4. Ge Q, Filip L, Bai A, Nguyen T, Eisen HN, Chen J. Inhibition of influenza virus production in virus‐infected mice by RNA interference. Proc Natl Acad Sci U S A 2004; 101: 8676–8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gabriel G, Nordmann A, Stein DA, Iversen PL, Klenk HD. Morpholino oligomers targeting the PB1 and NP genes enhance the survival of mice infected with highly pathogenic influenza a H7N7 virus. J Gen Virol 2008; 89: 939–948. [DOI] [PubMed] [Google Scholar]
- 6. Lupfer C, Stein DA, Mourich DV, Tepper SE, Iversen PL, Pastey M. Inhibition of influenza a H3N8 virus infections in mice by morpholino oligomers. Arch Virol 2008; 153: 929–937. [DOI] [PubMed] [Google Scholar]
- 7. Engelhardt JA. Comparative renal Toxicopathology of antisense oligonucleotides. Nucleic Acid Ther 2016; 26: 199–209. [DOI] [PubMed] [Google Scholar]
- 8. Crooke ST, Baker BF, Kwoh TJ, Cheng W, Schulz DJ, Xia S, et al Integrated safety assessment of 2′‐O‐methoxyethyl chimeric antisense oligonucleotides in nonhuman primates and healthy human volunteers. Mol Ther 2016; 24: 1771–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mendell JR, Goemans N, Lowes LP, Alfano LN, Berry K, Shao J, et al Telethon Foundation DMDIN . Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016; 79: 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heald AE, Charleston JS, Iversen PL, Warren TK, Saoud JB, Al‐Ibrahim M, et al AVI‐7288 for Marburg virus in nonhuman primates and humans. N Engl J Med 2015; 373: 339–348. [DOI] [PubMed] [Google Scholar]
- 11. Heald AE, Iversen PL, Saoud JB, Sazani P, Charleston JS, Axtelle T, et al Safety and pharmacokinetic profiles of phosphorodiamidate morpholino oligomers with activity against ebola virus and marburg virus: results of two single‐ascending‐dose studies. Antimicrob Agents Chemother 2014; 58: 6639–6647. [DOI] [PMC free article] [PubMed] [Google Scholar]