

Key Points
Partially HLA-matched third-party CMV-specific T cells provide long-term viral control in HSCT patients with resistant CMV infection.
Viral control occurs in the setting of recovery of CD8+ terminally differentiated effector T cells.
Abstract
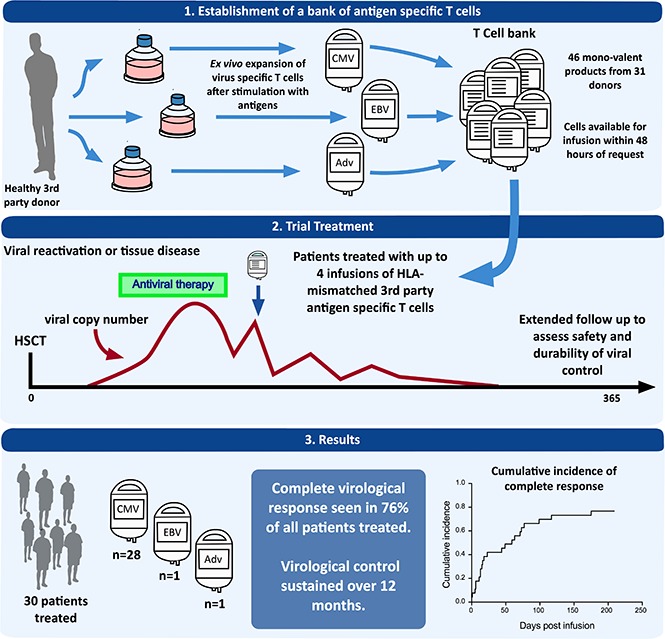
Donor-derived adoptive T-cell therapy is a safe and effective treatment of viral infection posttransplant, but it is limited by donor serostatus and availability and by its personalized nature. Off-the-shelf, third-party virus-specific T cells (VSTs) appear promising, but the long-term safety and durability of responses have yet to be established. We conducted a prospective study of 30 allogeneic hemopoietic stem cell transplant (HSCT) patients with persistent or recurrent cytomegalovirus (CMV) (n = 28), Epstein-Barr virus (n = 1), or adenovirus (n = 1) after standard therapy. Patients were treated with infusions of partially HLA-matched, third-party, ex vivo–expanded VSTs (total = 50 infusions) at a median of 75 days post-HSCT (range, 37 to 349 days). Safety, viral dynamics, and immune recovery were monitored for 12 months. Infusions were safe and well tolerated. Acute graft versus host disease occurred in 2 patients, despite a median HLA match between VSTs and the recipient of 2 of 6 antigens. At 12 months, the cumulative incidence of overall response was 93%. Virological control was durable in the majority of patients; the reintroduction of antiviral therapy after the final infusion occurred in 5 patients. CMV-specific T-cell immunity rose significantly and coincided with a rise in CD8+ terminal effector cells. PD-1 expression was elevated on CD8+ lymphocytes before the administration of third-party T cells and remained elevated at the time of viral control. Third-party VSTs show prolonged benefit, with virological control achieved in association with the recovery of CD8+ effector T cells possibly facilitated by VST infusion. This trial was registered at www.clinicaltrials.gov as #NCT02779439 and www.anzctr.org.au as #ACTRN12613000603718.
Visual Abstract
Introduction
Opportunistic infections are a significant cause of morbidity and mortality in patients who undergo allogeneic stem cell transplantation.1,2 Routine monitoring of the herpes viruses, Epstein-Barr virus (EBV) and cytomegalovirus (CMV), allows preemptive therapy that prevents progression to tissue disease. However, this approach has significant drawbacks. Antiviral medications are expensive, may require inpatient treatment, and are associated with significant toxicity. Furthermore, antivirals do not address the underlying immune defect and, consequently, many patients require prolonged or repeated treatment.3,4
Adoptive T-cell therapy with virus-specific T cells (VSTs) derived from the hemopoietic stem cell transplant (HSCT) donor offers an alternative approach. It appears safe and effective,5-11 but requires the HSCT donor to be seropositive for the virus of interest. Even when the donor is seropositive, a specific product must be made for each transplant recipient. In contrast, third-party VSTs can be used without full HLA matching, meaning that a single product can be stored and used for >1 transplant recipient. A recent trial using third-party cells from a cryopreserved bank demonstrated safety and short-term efficacy, with a 74% cumulative overall viral response rate at 6 weeks.12 Longer-term safety and efficacy was not examined. We generated a bank of cryopreserved VSTs and treated 30 patients with CMV, EBV, or adenovirus (ADV) infection or reactivation. We monitored adverse events, viral dynamics, use of antiviral therapy, and virus-specific immune recovery for 12 months after infusion. The intention of our study was to validate the previous short-term data on safety and viral control, and to assess longer-term questions of safety and response durability.
Methods
Study design and patients
The study was conducted as a multicenter, prospective, single-arm, phase I trial. Allogeneic HSCT patients with viral replication and/or tissue infection with CMV, ADV, or EBV that had failed standard therapy were eligible for recruitment. Standard therapy was defined as: ≥14 days of full-treatment dose of ganciclovir or foscarnet for CMV; therapy, which may include Cidofovir, for ADV; or immunosuppression reduction, rituximab, and/or cytotoxic chemotherapy for EBV or EBV posttransplant lymphoproliferative disorder (PTLD). Treatment failure was defined as persistent or recurrent CMV viremia at any level; or <50% reduction in ADV or EBV viral load or size of EBV lymphoma. Patients were excluded in the event of active acute graft versus host disease (GVHD), treatment with >1 mg/kg per day of prednisone or equivalent, treatment with anti-lymphocyte globulin, Eastern Cooperative Oncology Group performance status score >3, or deranged hepatic or renal function (detailed inclusion and exclusion criteria are provided in the supplemental Methods). The study was approved by the institutional research ethics committee at each site before recruitment. Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki. This study was registered on the Australian and New Zealand Clinical Trial Registry as #ACTRN12613000603718.
Third-party donors and T-cell generation
Venous blood or granulocyte colony-stimulating factor–primed apheresis product from healthy stem cell donors was used as the starting material for ex vivo T-cell expansion. Donors with common HLA types and positive CMV or EBV serostatus were recruited from Westmead Hospital and underwent standard assessment to confirm eligibility for allogeneic donation. HLA typing was performed by the Australian Red Cross Blood Service and was low resolution for class I loci and resolved to 4 digits at the DRB1 locus. Thirty-one donors were used to generate a bank of 46 monovalent (17 CMV, 14 EBV, and 15 ADV) VST products under good manufacturing practice conditions at the Sydney Cellular Therapies Laboratory, Westmead Hospital, as previously described.6,13 Donor monocyte-derived dendritic cells (moDCs) were pulsed with overlapping MACS GMP PepTivators (Miltenyi Biotec) peptide pools (15 mers overlapping by 11 peptides) for HCMV pp65, AdV5 Hexon, or EBV BZLF1/LMP2A/EBNA-1 proteins. Irradiated peptide-pulsed moDCs were used as stimulators in coculture with the monocyte-depleted fraction of granulocyte colony-stimulating factor–primed apheresis products or venous blood mononuclear cells isolated by Ficoll-Paque (GE Healthcare) gradient centrifugation. Cultures were restimulated with peptide-pulsed moDCs after 7 days and were continued for up to 21 days, with the addition of 20 U/mL interleukin-2 every 2 to 3 days, increasing to 50 U/mL of interleukin-2 from day 14 to 21. VST products were cryopreserved in multiple doses. One multivalent (CMV, EBV, ADV, and varicella-zoster virus) VST product was retained from a previous clinical study14 and was administered to patient 1. Standard VST product release criteria were applied (see supplemental Methods for criteria and VST shipping information).
Treatment and VST matching
Participants were treated with a dose of 2.0 × 107/m2 partially HLA-matched CMV-, EBV-, or ADV-specific T cells based on postthaw viability. VST-recipient matching required a minimum of 1 of 6 HLA antigens (HLA-A, -B, and -DRB1) shared between VSTs and the recipient. The VST-recipient matching algorithm incorporated the viral specificity of the product, the number of HLA matches with the recipient, the specific HLA antigen through which antiviral activity was mediated and demonstrated antiviral activity through a shared HLA antigen. VSTs were chosen based on the highest number of HLA matches with antiviral activity through the shared HLA antigen(s), with secondary preference given to products with the highest proportions of virus-specific major histocompatibility complex (MHC)–tetramer CD8+ cells or interferon-γ (IFN-γ) response. In the event of persistent viral replication detected ≥2 weeks after a previous infusion and continued fulfillment of eligibility criteria, patients were treated with ≤3 additional infusions of VSTs generated from the same or a different third-party donor. The VST dose could be increased up to 5.0 × 107/m2 cells if the patient had tolerated a previous VST dose at 2.0 × 107/m2 without VST toxicity. Antiviral therapy was administered according to local physician and institutional preference. A cycle of antiviral therapy was defined as the commencement or recommencement of full-dose antiviral therapy or a change to a new antiviral agent.
Outcomes and follow-up
Patients were monitored for 12 months from the final infusion of VSTs for evidence of clinical and virological response, toxicity, and immune cell recovery. Clinical review and peripheral blood sample collection were performed at regular time points, with resetting of the review cycle after each infusion. The primary end point of the trial was safety of the VST infusion. All adverse events were graded according to the National Cancer Institute’s common terminology criteria for adverse events, version 4.03. Secondary end points included the incidence of acute and chronic GVHD, viral reactivation, infection and organ damage, virus-specific immune reconstitution, and use of antiviral therapy. Virological response was defined as: complete response (CR) if the virus became undetectable by quantitative polymerase chain reaction (PCR) at any point post–VST infusion with resolution of any symptoms related to tissue or organ infection; and partial response (PR) if there was ≥50% reduction from the immediate preinfusion viral load. If the patient did not fulfill criteria for CR or PR, they were defined as having no response (NR). GVHD was graded according to standard criteria for acute and chronic GVHD (supplemental Tables 1-3).
Virological and immune monitoring
Viral load was measured by quantitative PCR at individual centers (for details of the CMV assays used, see supplemental Table 4). CMV drug resistance mutations were identified using PCR sequencing of the UL97 and UL54 genes.
Postinfusion immune monitoring was performed on batched peripheral blood mononuclear cells (PBMCs) to minimize interassay variability. Flow cytometry was performed on VST products or postinfusion PBMCs using monoclonal antibodies directed against CD3, CD4, CD8, CD14, CD19, CD56, CD62L, CD45RA, and PD-1 (BD Biosciences, San Jose, CA). Viability was assessed using 7-amino-actinomycin D (BD Biosciences) or hydroxystilbamidine (Life Technologies). PBMCs from otherwise healthy individuals undergoing venesection for hemochromatosis were assessed to provide a range for the percentage of PD-1 expression on CD8+ T cells. CD3+, CD4+, CD8+, and T memory subset cell counts were calculated by multiplication of the subset percentage and the lymphocyte count measured on the day of sample collection. Viral antigen specificity and HLA-restricted epitope recognition on VST products were assessed using phycoerythrin-conjugated virus-specific iTAg MHC class I human tetramers (Beckman Coulter, Brea, CA) or by intracellular cytokine flow cytometry after stimulation of VSTs with overlapping peptide mix or with individual epitopes, as previously described.13 Flow cytometry data were acquired on a FACSCanto II or LSRFortessa (BD Biosciences) and analyzed with FlowJo software (version 10.0.8r1; Tree Star, Inc., Ashland, OR). CMV-specific T-cell immune recovery was monitored by IFN-γ enzyme-linked immunospot (ELIspot) assay, as described previously.13
Statistical analysis
The cumulative incidence function was used to estimate the cumulative overall (CR and PR) and CR response rate to VSTs at the end of follow-up, with death considered a competing risk. The peak CMV pp65-specific T-cell responses, as measured by IFN-γ ELIspot assay before and after VST infusion, were compared using the Wilcoxon matched-pairs signed rank test to determine significance. A comparison of the percentage expression of PD-1 on CD8+ T cells between healthy individuals and trial patients before VST infusion was performed using a Mann-Whitney test to determine significance. Linear regression and Pearson correlation were performed to determine the significance of the relationship between the time to CR and the timing of the first VST infusion post-HSCT. Statistical analysis was performed using Prism 7 for Mac (GraphPad Software, Inc., La Jolla, CA). Cumulative incidence data were generated using the Analysis of Censored and Correlated Data software, version 8.4.8 (Boffin Software, Sydney, NSW, Australia).
Results
Participant Characteristics
Thirty-one patients were recruited at 7 centers across Australia and New Zealand (see supplemental Appendix) between February 2013 and August 2016. One patient with CMV reactivation did not receive treatment because no suitably HLA-matched product was available. Of the remaining 30 patients, 28 patients were treated for CMV reactivation, 1 for ADV reactivation, and 1 for EBV reactivation. The participant characteristics are described in Table 1. Of the 22 adults and 8 pediatric patients, 27 patients were transplanted for hematological malignancy and 3 pediatric patients were transplanted for immune deficiency syndromes. The majority of transplants were performed with in vivo T-cell depletion (22 of 30 patients). Two-thirds of CMV-seropositive recipients were transplanted from CMV-seronegative donors. Of the 28 patients with CMV reactivation, 3 patients had a history of previous CMV disease, and 3 patients had current CMV disease (2 patients with CMV enteritis and 1 patient with CMV pneumonitis). Patients were heavily pretreated for viral infection having received a median of 31 days (range, 14 to 113 days) of antiviral therapy before the initial VST infusion. Eighteen patients had ≥2 cycles of treatment, and 12 patients had been exposed to ≥2 different classes of antiviral therapy before the first VST infusion. Mutations conferring resistance to CMV drug therapy were found in 5 of 11 patients tested before first VST infusion and in 1 patient tested on day 7 after administration of CMV-specific T cells. Mutations in CMV UL54 were more frequent (5 of 6 patients) than UL97 kinase mutations (2 of 6 patients), as detailed in supplemental Table 5.
Table 1.
Participant characteristics
| Patient | Age, y | Sex | Transplant indication | Conditioning HSCT type, HLA match “mismatched antigen” (cell source other than PBSCs) | T-cell depletion | Target viral infection (tissue involvement) | R/D viral serostatus | Immunosuppression at first VST infusion | Previous antiviral therapy, d | Previous cycles of therapy, n | Previous antiviral agents, n | CMV drug resistance mutations | Peak viral load before first VST infusion, cp/ml |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 63 | M | AML/MDS | RIC MUD | ATG | CMV | Pos/Neg | Pred, TAC | 55 | 4 | 2 | UL54/97 | 84 600 |
| 2 | 41 | M | T-NHL | MAC MUD | Anti-CD52 | CMV | Pos/Neg | CSA | 27 | 1 | 1 | NT | 3990 |
| 3 | 64 | M | AML | RIC MUD | ATG | CMV | Pos/Neg | CSA | 23 | 1 | 1 | NT | 44 700 |
| 4 | 60 | M | AML | RIC MUD | ATG | CMV (active gastritis/enteritis) | Pos/Neg | CSA | 19 | 1 | 1 | NT | 30 000 |
| 5 | 53 | M | AML | RIC MSD | ATG | CMV (active enterocolitis) | Pos/Neg | CSA | 32 | 2 | 2 | UL54 | 9170 |
| 6 | 36 | F | MDS | RIC MSD | None | CMV | Pos/Pos | Pred, MMF, TAC | 22 | 2 | 2 | ND | 24 800 |
| 7 | 58 | F | ALL | RIC MSD | ATG | CMV | Pos/Neg | None | 44 | 2 | 1 | NT | 43 700 |
| 8 | 59 | F | AML | RIC MUD | ATG | CMV | Pos/Neg | CSA | 31 | 1 | 1 | NT | 39 300 |
| 9 | 49 | M | B-NHL | RIC HREL | None | CMV (previous colitis) | Neg/Pos | Pred, MMF, TAC | 22 | 2 | 2 | NT | 124 000 |
| 10 | 63 | M | SM-AHNMD (AML) | RIC HREL | None | CMV | Pos/Neg | TAC | 14 | 2 | 2 | ND | 277 000 |
| 11 | 33 | M | AML | MAC MMUD, 9/10 'B' | ATG | CMV | Pos/Pos | Pred, MMF | 14 | 1 | 1 | UL54* | 78 600 |
| 12 | 63 | F | MDS | RIC HREL | None | CMV | Pos/Pos | MMF, TAC | 18 | 2 | 2 | UL54 | 46 100 |
| 13 | 46 | F | ALL | MAC MUD | ATG | CMV | Pos/Neg | CSA | 14 | 2 | 2 | UL54 | 16 100 |
| 14 | 69 | M | AML | RIC MUD | ATG | CMV | Pos/Neg | CSA | 21 | 1 | 1 | ND | 67 800 |
| 15 | 45 | F | AML | MAC MSD | None | CMV | Pos/Neg | Pred, CSA | 113 | 2 | 1 | UL97 | 7 300 000 |
| 16 | 17 | M | ALL | MAC MUD | ATG | CMV | Pos/Neg | CSA | 94 | 2 | 1 | ND | 61 100 |
| 17 | 34 | F | AML | RIC MUD | ATG | CMV (previous pneumonitis) | Pos/Neg | None | 46 | 2 | 1 | NT | 12 400 |
| 18 | 67 | M | T-LBL | RIC HREL (BM) | None | CMV | Pos/Neg | None | 27 | 1 | 1 | NT | 4250 |
| 19 | 52 | M | T-NHL | RIC MSD | None | CMV (previous enterocolitis) | Pos/Pos | Pred, MMF, CSA | 88 | 3 | 1 | NT | 32 100 |
| 20 | 63 | M | AML/MDS | RIC MUD (BM) | ATG | CMV | Pos/Pos | CSA | 68 | 2 | 1 | NT | 42 000 |
| 21 | 59 | F | AML | RIC MUD | ATG | CMV | Pos/Neg | CSA | 50 | 1 | 1 | NT | 51 700 |
| 22 | 35 | M | ALL | MAC MUD | ATG | CMV (active pneumonitis) | Pos/Pos | CSA | 39 | 2 | 2 + CMV IVIG | NT | 62 088 |
| 23 | 3 | M | FA (SAA) | RIC MUD | ATG + TCR-αβ depletion | CMV | Pos/Pos | CSA | 15 | 1 | 1 | NT | 21 000 |
| 24 | 13 | M | ALL | MAC MMUD, 9/10 'DR' (BM) | ATG | CMV | Pos/Neg | Pred, CSA | 31 | 2 | 2 | NT | 39 000 |
| 25 | 12 | M | AML | MAC MUD (cord) | ATG | CMV | Pos/Pos | None | 60 | 1 | 1 | ND | 8 000 |
| 26 | 5 | M | Hyper-IgM | MAC MSD (BM) | ATG | CMV | Pos/Neg | MMF, CSA | 35 | 2 | 2 | NT | 45 000 |
| 27 | 3 | M | CD40-L deficiency | MAC MUD (BM) | Anti-CD52 | CMV | Pos/Neg | CSA | 51 | 2 | 2 | ND | 79 433 |
| 28 | 5 | F | FA (MDS) | RIC MUD | ATG + CD34 selection | CMV | Pos/Pos | CSA | 29 | 2 | 2 + CMV IVIG | NT | 21 700 |
| 29 | 1 | M | SCID | MAC MUD | Anti-CD52 | ADV | UNK | MMF, CSA | 30 | 1 | 1 | NA | 2 089 296 |
| 30 | 44 | M | NK/T-NHL,HLH | RIC HREL | None | EBV | Pos/Pos | None | NA† | NA | 81 240 | ||
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ATG, anti-thymocyte globulin; BM, bone marrow; B-NHL, B-cell non-Hodgkin lymphoma; CSA, cyclosporine; D, donor; F, female; FA, Fanconi's anemia; HLH, hemophagocytic lymphohistiocytosis HREL, haploidentical related; IgM, immunoglobulin M; IVIG, IV immunoglobulin; L, ligand; M, male; MAC, myeloablative conditioning; MDS, myelodysplasia; MMF, mycophenolate mofetil; MMUD, mismatched unrelated donor; MSD, matched sibling donor; NA, not applicable; ND, not detected; Neg, negative; NK/T-NHL, NK/T-cell non-Hodgkin lymphoma; NT, not tested; PBSC, peripheral blood stem cell; Pos, positive; Pred, prednisolone; R, recipient; RIC, reduced-intensity conditioning; SAA, severe aplastic anemia; SCID, severe combined immunodeficiency; SM-AHNMD, systemic mastocytosis with associated clonal hematological non–mast cell lineage disease; TAC, tacrolimus; TCR, T-cell receptor; T-LBL, T-cell lymphoblastic leukemia/lymphoma; T-NHL, T-cell non-Hodgkin lymphoma; UNK, unknown.
CMV drug resistance mutation testing was performed on day 7 post–VST infusion.
Patient 30 had EBV-associated NK/T-cell non-Hodgkin lymphoma and was treated with a full course of rituximab before haploidentical stem cell transplantation. The patient had recurrent EBV reactivation posttransplant managed with reduction of immunosuppression.
Characteristics of the VST products
Fifty VST infusions from 15 of 31 available donors were administered during the trial. Products from 8 donors were used in multiple patients (Table 2). The most frequently shared HLA types were A2 and A1, followed by B8 and B7 and DRB1 01:01 and 03:01. VST products were predominantly CD3+ (median, 98%; range, 92%-99%). The median percentage of CD3+ cells that were CD3+8+ was 58%, (range, 14%-84%) and CD3+4+ was 39% (range, 15%-85%); 9 of 15 products were predominantly CD3+8+. The CD3+8+ subset consisted predominantly of CD45RA–CD62L– effector memory T cells. Viral epitope-specific MHC class I tetramers were available for 10 products. The median percentage of total cells that were tetramer positive was 24% (range, 6%-47%).
Table 2.
Viral response and outcomes
| Patient | Viral load immediately before first VST, cp/mL | Day post- HSCT at first VST infusion | VST infusions, n | VST product infused | HLA match (VSTs to patient) | HLA antigen/allele(s) matched between VST and patient* | Best response | Post–VST course and outcome after best response | Rehospitalization post–VST infusion, d | Status, cause of death (days post–first VST infusion) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 55 000 | 171 | 3 | 2011-18, RPT | 4/6 | (A1, A1, B8, DR 03:01)1,2 | PR | Progressive viremia. Additional VST. Antivirals after final VST infusion. Persistent viremia. | 69 | Death, presumed CMV disease (209) |
| 13-0008 | 3/6 | (A1, B8, DR 03:01)3 | ||||||||
| 2 | <150 | 83 | 4 | 12-0022, RPT, RPT | 2/6 | (B7, DR 15:01)1-3 | CR | Quantifiable reactivation. Additional VSTs/antivirals. No antivirals restarted after final VST infusion, despite persisting viremia. | NA | Alive |
| 13-0008† | 3/6 | (A1, B8, DR 03:01)4 | ||||||||
| 3 | <150 | 63 | 2 | 12-0022, | 4/6 | (A2, A2, B7, DR 07:01)1 | CR‡ | Reactivation (BLQ), no additional VSTs/antivirals. | NA | Alive |
| 13-0006 | 2/6 | (A2, B7)2 | ||||||||
| 4 | 1440 | 52 | 1 | 12-0022 | 4/6 | A2, A2, B7, DR 15:01 | CR | Antivirals after final VST infusion. CR attained. Quantifiable reactivation. No additional antivirals. | NA | Alive |
| 5 | <150 | 81 | 1 | 13-0015 | 3/6 | B35, DR 01:01, DR 04:01 | CR | Reactivation (BLQ). No additional VSTs/antivirals. | 6 | Alive |
| 6 | 363 | 69 | 2 | 13-0019§, RPT§ | 3/6 | A2, A2, DR 11:01 | CR | Reactivation (BLQ). No additional VSTs/antivirals. | 14 | Alive |
| 7 | <150 | 122 | 1 | 14-0004 | 4/6 | A1, B8, DR 07:01, DR 07:01 | CR | Quantifiable reactivation. No additional VSTs/antivirals. | 3 | Alive |
| 8 | 270 | 66 | 2 | 14-0006, RPT† | 3/6 | A2, B44, DR 04:01 | CR | Sustained CR. | NA | Alive |
| 9 | 4680 | 67 | 1 | 13-0015 | 2/6 | B35||, DR 01:01|| | PR | Progressive viremia and CMV colitis. Not fit for additional VSTs/antivirals. | 20 | Death, bacterial infection (71) |
| 10 | 5000 | 69 | 1 | 13-0015 | 2/6 | A2, B35 | CR | Reactivation (BLQ). No additional VSTs/antivirals. | NA | Alive |
| 11 | 12 900 | 62 | 1 | 14-0008 | 2/6 | A24, B40¶ | CR | Antivirals after final VST infusion. CR attained. No additional antivirals. | Did not leave hospital | Death, gut GVHD + disseminated adenoviral infection (35) |
| 12 | 660 | 67 | 1 | 14-0007 | 2/6 | B8||, DR 03:01|| | CR | Reactivation (BLQ). Antivirals restarted after final VST infusion. | 30 | Alive |
| 13 | 1740 | 62 | 1 | 14-0007 | 3/6 | A1, B8, DR 03:01 | PR | Decreasing viral load. No additional VSTs/antivirals. | Did not leave hospital | Death, disseminated toxoplasmosis (33) |
| 14 | 281 | 69 | 2 | 13-0015 | 2/6 | (A2, DR 04:01)1 | NR | Stable viremia. No antivirals after final VST infusion. | 6 | Alive |
| 14-0010 | 1/6 | (A2)2 | ||||||||
| 15 | 11 900 | 161 | 2 | 13-0015 | 3/6 | (A2, B35, DR 01:01)1 | CR | Sustained CR. | 9 | Death, relapse (190) |
| 13-0019 | 2/6 | (A2, A2)2 | ||||||||
| 16 | 60 300 | 126 | 1 | 14-0003 | 1/6 | B7 | CR | Quantifiable reactivation. No additional VSTs/antivirals. | NA | Alive |
| 17 | 9800 | 104 | 2 | 14-0004, RPT | 3/6 | A1, DR 07:01, DR 07:01 | PR | Progressive viremia. Additional VSTs, no additional antivirals. | 15 | Death, bacterial infection (37) |
| 18 | 386 | 153 | 1 | 14-0004 | 2/6 | A1, B8 | CR | Quantifiable reactivation. No additional VSTs/antivirals. | 8 | Alive |
| 19 | 1940 | 349 | 2 | 13-0015, RPT† | 3/6 | A2, B35, DR 01:01 | CR | CMV enteritis post–first VST infusion, treated with additional VSTs. CR attained. Quantifiable reactivation. No additional VSTs/antivirals. | 5 | Alive |
| 20 | 3260 | 118 | 4 | 14-0010, RPT† | 1/6 | (A2)1,2 | CR | Quantifiable reactivation. Additional VSTs/antivirals. No antivirals restarted after final VST infusion. | 40 | Alive/Relapse |
| 13-0006§ | 2/6 | (A2, DR 04:01)3 | ||||||||
| 13-0019 | 2/6 | (A2, A2)4 | ||||||||
| 21 | 51700 | 132 | 3 | 14-0007, RPT† | 3/6 | (A1, B8, DR 03:01)all | CR | Quantifiable reactivation. Additional VSTs/antivirals. No antivirals restarted after final VST infusion. | NA | Alive |
| 13-0008 | 3/6 | |||||||||
| 22 | 897 | 89 | 1 | 14-0007 | 3/6 | A1, A31, B40 | CR | Sustained CR. | NA | Alive |
| 23 | 6100 | 37 | 1 | 13-0015 | 2/6 | B35, DR 01:01 | CR | Sustained CR. | 3 | Alive |
| 24 | 8500 | 124 | 2 | 14-0003, RPT | 2/6 | B7, DR 14:54|| | CR | Antivirals after final VST infusion. CR attained. No additional antivirals. | 59 | Death, CNS EBV PTLD (80) |
| 25 | 2300 | 102 | 1 | 14-0004§ | 3/6 | A1, DR 07:01, DR 07:01 | CR | Reactivation (BLQ). No additional VSTs/antivirals. | NA | Alive |
| 26 | 150 | 57 | 2 | 14-0007, RPT† | 1/6 | DR 01:01 | CR | Quantifiable reactivation. Additional VSTs, no antivirals. No antivirals after final VST infusion. | 11 | Alive |
| 27 | 1260 | 68 | 2 | 13-0005 | 1/6 | (A1)all | CR | Reactivation (BLQ). No additional VSTs/antivirals. | NA | Alive |
| 13-0008 | 1/6 | |||||||||
| 28 | 483 | 47 | 1 | 13-0019 | 3/6 | A2, A2, B40 | PR | Decreasing viral load. Off study 10 days post–VST infusion. | NA | Death, relapse (58) |
| 29 | 389 045 | 42 | 1 | 14-0002 | 1/6 | DR 01:01 | CR | Reactivation (BLQ). No additional VSTs/antivirals. | NA | Alive |
| 30 | 81 240 | 133 | 1 | 13-0029 | 1/6 | A24 | NR | Progressive viremia. Not fit for additional VSTs/antivirals. | Did not leave hospital | Death, relapse (14) |
BLQ, below level of quantitation; CNS, central nervous system; cp, copies; NA, not applicable; RPT, repeat infusion of the same VST product.
In column 7, HLA match was low resolution for HLA-A and B and allelic for DRB1. Antigens or alleles with HLA-restricted viral activity are marked in bold. Superscript outside parenthesis refers to the VST infusion number (e.g. 1 indicates first infusion).
Infusion dose >2.0 × 107 cells/m2 (range, 3.1 to 5.0 × 107/m2).
CR achieved before donor lymphocyte infusions administered for relapse prophylaxis on day 117 and 178 post–first VST infusion.
Infusion dose <2.0 × 107 cells/m2 (range, 1.37 to 1.65 ×107/m2).
HLA allele of product matched to HLA allele of recipient (not transplant donor).
HLA allele of product matched to HLA allele of transplant donor (not recipient).
Administration of VSTs and VST matching
The median day of first infusion was day 75 (range, 37 to 349 days) post-HSCT. The details of VST treatment and characteristics are described in Table 2. A single infusion was administered to 17 patients, 9 patients received 2 infusions, 2 patients received 3 infusions, and 2 patients received 4 infusions. Eight patients were administered VST products from >1 donor. The median VST cell dose was 2.0 × 107cells/m2 with a range of 1.37 to 5.0 × 107cells/m2 (6 patients received increased cell doses after an incomplete response to a previous dose of 2.0 × 107cells/m2). The median HLA match of product to recipient was 2 of 6 HLA antigens; 11 VST infusions were based on a single antigen or allele match. Sixteen of the 50 infusions were matched at either a class I or class II HLA locus, whereas the remainder were matched at both.
Safety and toxicity of VSTs
There were no immediate infusion-related toxicities attributed to VSTs. One patient was hospitalized within 24 hours of infusion due to fever and was found to have central line–related bacteremia.
Patients were followed up for a median of 12 months. No adverse event was attributed to VST infusion. The majority of adverse events were related to infections. All serious adverse events (SAEs) are detailed in Table 3. All non–VST-targeted infections are described in supplemental Table 6. Noninfective SAE included posterior reversible encephalomyelitis syndrome in 2 patients (both on calcineurin inhibitors), Stevens-Johnson syndrome attributed to ganciclovir (n = 1), respiratory failure due to fluid overload (n = 1), and respiratory failure with relapsed malignancy (n = 1). Other adverse events included transient paraproteinemia in 2 patients, 1 case of self-limited asymptomatic thyrotoxicosis, 1 case of Bell’s palsy, 1 case of drug-related skin eruption, 1 case of idiopathic facial edema, and 1 case of steroid responsive thrombocytopenia
Table 3.
Serious adverse events
| Patient | Days post–first VST infusion | Event |
|---|---|---|
| 1 | 108 | Rhinovirus RTI |
| 180 | Escherichia coli UTI | |
| 5 | 354 | Respiratory syncytial virus RTI |
| 6 | 314 | Streptococcus pneumoniae pneumonia/sepsis |
| 7 | 251 | RTI (no organism identified) |
| 9 | 45 | RTI (no organism identified) |
| 54 | Rhinovirus and Influenza A(H3) RTI* | |
| 54 | Pseudomonas putida pneumonia/sepsis† | |
| 56 | Enterococcus faecium pneumonia/sepsis† | |
| 11 | 8 | Disseminated ADV infection† (urine, blood, stool, GIT) |
| 21 | Grade IV gut GVHD with hemorrhage† | |
| 12 | 29 | Stevens-Johnson syndrome (skin, ocular, oral, and genital mucosa) |
| 41 | Posterior reversible encephalopathy syndrome | |
| 13 | 9 | Disseminated Toxoplasma gondii† (cerebral, CSF, blood, and BMAT) |
| 14 | 50 | Serratia marcescens bacteremia (line-related) |
| 15 | 190 | Presumed Pneumocystis jirovecii pneumonia† |
| 16 | 19 | Respiratory failure due to fluid overload |
| 32 | Posterior reversible encephalopathy syndrome | |
| 17 | 23 | Human metapneumovirus RTI |
| 31 | Enterococcus faecium pneumonia/empyema† | |
| 18 | 1, 16 | Pseudomonas aeruginosa RTI |
| 16 | Rhinovirus RTI | |
| 19 | 80 | Rotavirus diarrhea |
| 20 | 126 | RLL pneumonia, no organism identified |
| 23 | 291 | Enterococcus faecalis and Enterobacter cloacae UTI |
| 24 | 7 | Respiratory syncytial virus RTI |
| 24 | Orbital cellulitis, no organism identified | |
| 52 | Pseudomonas aeruginosa bacteremia | |
| 53 | Cerebral EBV PTLD† | |
| 65 | Respiratory failure, Candida sp., Corynebacterium sp., CMV PCR+, EBV PCR+ (BAL)† | |
| 26 | 11 | Febrile, no organism identified |
BAL, bronchoalveolar lavage; BMAT, bone marrow aspirate and trephine; CSF, cerebral spinal fluid; GIT, gastrointestinal; RLL, right lower lobe; RTI, respiratory tract infection; sp., species; UTI, urinary tract infection.
*Rhinovirus and Influenza A(H3) RTI was initially diagnosed on day 34 post–VST infusion.
Contributed to death.
A total of 9 deaths occurred during follow-up (Table 2). Overall unadjusted survival at 12 months post–first VST infusion was 69%, with median survival not reached. One patient died of presumed CMV disease and is discussed below. Three patients died of relapsed malignancy, including 1 patient who was given EBV-specific T cells for EBV reactivation in the context of EBV-associated NK/T lymphoma. Two patients died of nonviral infections. Patient 24 died of presumed cerebral EBV PTLD 80 days post–CMV-specific VST infusion. He was not eligible for EBV-specific VSTs on trial at the time of the PTLD diagnosis due to poor performance status.
Acute de novo GVHD occurred in 2 patients within 2 weeks of VST infusion. Patient 9 had steroid responsive grade II acute GVHD (stage 3 skin, stage 1 gut) that resolved before death due to infection 71 days after VST infusion. Patient 11 received a CMV-specific VST infusion 62 days after an HLA-B antigen–mismatched matched unrelated donor (MUD) transplant. He died 35 days postinfusion from refractory grade IV acute GVHD (gut) and disseminated ADV infection. He was not eligible for ADV-specific VSTs on trial due to active GVHD. Chronic GVHD (mild, n = 2; moderate, n = 1; and severe, n = 2) occurred in 5 patients.
Virological response and clinical outcomes
At 12 months, the cumulative incidence of overall response was 93%, and the cumulative incidence of CR was 76% (Figure 1). Virological response and outcomes for all patients are provided in Table 2.
Figure 1.

Cumulative incidence of response at 12 months post–VST infusion. (A) The cumulative time to best overall response (OR) (either CR or PR) for all patients. (B) The cumulative time to CR for all patients.
CRs
Twenty-three patients achieved a complete virological response at a median of 59 days (range, 1 to 175 days) post–first VST infusion. All 3 patients with active tissue infection achieved a CR after a single infusion. Figure 2 illustrates the CR seen in patient 5, in whom positive immunohistochemical stains for CMV colitis resolved coincident with a rise in the absolute lymphocyte count and an increase in CMV-specific T-cell response. The patient treated for ADV reactivation (patient 29) had a CR to a single infusion of VSTs.
Figure 2.

Outcome of CMV enterocolitis in patient 5 post–CMV-specific VST infusion. (A) Viral load, lymphocyte count, and colonic biopsy in patient 5, who was treated with a single VST infusion. The patient had persistent symptomatic biopsy–proven CMV enterocolitis after a month of antiviral therapy. Post–VST infusion, the symptoms resolved, the patient developed an absolute lymphocytosis, and viral load fluctuated between negative and below the level of quantitation (<150 cp/mL). (B) CMV pp65-specific T-cell response as measured by IFN-γ ELIspot. (C-D) Photomicrograph of colonic biopsy with CMV immunostain pre–VST (C) and post–VST (D) infusion (original magnification ×200, Olympus microscope, model BX43). LLQ, lower limit of quantitation.
Of the 23 patients who achieved a CR, 14 patients remained virus PCR-negative (n = 5) or below the level of quantitation (n = 9) for the duration of follow-up. An additional 5 patients had brief episodes of quantifiable reactivation, but were not administered additional VST or antiviral therapy after CR.
PRs
Five patients achieved a PR. Patient 1 achieved PR but had persistent CMV infection refractory to additional VSTs and multiple antiviral agents and died of presumed CMV disease. Of the 4 other patients that achieved PR, 3 died of subsequent non-viral infections and 1 from disease relapse.
NRs
Two patients did not meet criteria for a CR or PR. One patient had persisting low-level CMV in the blood 152 days post–final VST infusion. The second patient was treated with EBV VSTs for EBV reactivation, but died 2 weeks later with relapsed EBV-associated NK/T lymphoma.
Use of antivirals before and after VST infusion is illustrated in Figure 3. The median number of days since cessation of antiviral therapy at last follow-up for all patients was 326 days (range, 0 to 412 days). Five patients recommenced antiviral therapy after the final VST infusion. In 4 of these patients (patients 4, 11, 12, and 25), this occurred in the context of high-dose steroids administered for suspected or confirmed GVHD. Patient 1 received 44 days of antiviral therapy after reinitiation and died of presumed CMV disease. Figure 4 details the viral load after the final VST infusion for all patients treated for CMV.
Figure 3.

Antiviral therapy pre– and post–final VST infusion. The numbers to the left of the red bars refer to the patient number. The red bars on the left show the cumulative days of antiviral therapy administered before the final infusion (including therapy before the first VST infusion). The blue bars to the right show the duration and number of blocks of antiviral therapy administered over the course of follow-up after the final VST infusion.
Figure 4.

CMV response and reactivations post–final VST infusion. CMV viral load (cp/mL) after final VST infusion in the 28 patients treated for CMV reactivation.
The presence of drug resistance mutations did not appear to affect the virological response to VSTs with 4 of the 6 patients with UL54 or UL97 mutations achieving a CR and 5 of the 6 patients without mutations achieving a CR.
Immune reconstitution
Of the 30 patients treated with VSTs, 18 patients were lymphopenic preinfusion. The median lymphocyte count for all patients was 0.8 × 109/L (range, 0.2 to 14.1 × 109/L). Immunophenotyping performed in 26 of the 28 patients with CMV reactivation showed a rise in the CD3+ count after VST infusion. This was predominantly CD8+ T cells (preinfusion median, CD8+ 0.26 × 109/L, day 59 postinfusion median CD8+, 0.91 × 109/L). This rise coincided with the median time to CR (Figure 5A) and a shift in the composition of CD8+ T cells from effector memory CD45RA–62L– T cells to terminally differentiated effector CD45RA+62L– T cells (Figure 5B). In contrast, within the CD4+ T-cell memory subset, effector memory T cells were dominant throughout follow-up (Figure 5C). The percentage of CD8+ T cells expressing PD-1 before VST infusion was significantly higher than healthy individuals at steady state (preinfusion median, 28% [range, 5%-71%] of CD8+ T cells; healthy individuals’ median, 6% (range, 2%-10%] of CD8+ T cells; P = .005). Postinfusion PD-1 expression exhibited a downward trend over the course of follow-up, but did not completely normalize (Figure 5D). There was no correlation between the number of days post–stem cell transplant and the time to complete virological response (Figure 6).
Figure 5.

T-cell subset responses post–VST infusion. (A) Median cell number for CD3+, CD4+, and CD8+ T cells for time points post–first VST infusion (× 109/L). (B) Median cell number for CD8+ T-cell memory subsets defined as T terminal effector (CD45RA+62L–), T naive (CD45RA+62L+), T central memory (CD45RA–62L+), and T effector memory (CD45RA–62L–). (C) Median cell number for the CD4+ T-cell memory subsets. (D) Percentage of CD8+ T cells expressing PD-1. Bars and lower and upper whiskers represent the median, 25th, and 75th percentiles, respectively. The results for healthy individuals at steady state are indicated by the dashed line (median) and gray shading (range).
Figure 6.

Correlation between the time to CR with timing of first VST infusion post-HSCT. To assess whether virological responses may be explained by natural immune recovery post-HSCT, linear regression and Pearson’s correlation were performed to determine any significant relationship between the timing of the response and the timing of first VST infusion post-HSCT in the 23 patients who achieved CR. No correlation was observed.
Functional CMV-specific immunity was measured by ELIspot in 23 of the 28 patients treated for CMV. Preinfusion CMV pp65 T-cell responses were almost undetectable with a median of 4 spot forming cells (SFCs)/105 cells (range, 0-67 SFCs/105 cells). The median peak CMV pp65-specific immunity occurred at a median of 44 days (range, 0 to 309 days) after the first infusion and measured 49 SFCs/105 cells (range, 3-921 SFCs/105 cells; P < .0001; Figure 7).
Figure 7.

Peak CMV pp65-specific T-cell responses pre– and post–VST infusion. Peak SFC count as measured by IFN-γ ELIspot after the first VST infusion in 23 patients. Bars and lower and upper whiskers represent the median, 25th, and 75th percentiles, respectively. Significance was determined by the Wilcoxon matched-pairs signed rank test.
Discussion
Donor-derived VSTs have beneficial clinical effects when administered prophylactically and therapeutically to allogeneic HSCT recipients.5,6,8,9,11,15,16 Banked third-party, partially HLA-matched VSTs have been explored as an alternative to minimize the inconvenience and delay involved in generating specific donor-derived products for each recipient. Several studies have suggested that this approach leads to clinical benefit in the short term,5,7,8,11,12 although long-term effects on viral control and overall immune reconstitution have not been reported.
In this multicenter study of 30 patients, we used partially HLA-matched third-party VSTs to treat viral infections that persisted or recurred after an initial period of standard antiviral treatment. We followed patients for a median of 12 months and studied viral control and requirement for additional antiviral therapy as well as parameters of immune reconstitution. As in other studies, we observed no immediate infusion-related toxicities. Rates of GVHD and SAEs during the 12 months of follow-up were within expectations for the studied population. A lack of GVHD occurred despite the fact that all infusions came from unrelated third-party donors, 25 of the 50 infusions matched at 1 or 2 of 6 HLA antigens or alleles, 8 patients received products from ≥2 different donors, and infusions contained T-cell doses of up to 5.0 × 107/m2.
The overall virological response rate (CR and PR) for all patients at 12 months was 93%, and 76% of patients achieved a CR. One patient who achieved a PR subsequently died of unconfirmed CMV and thus could be considered a long-term NR. These high response rates were achieved in a heavily pretreated population in which all patients had standard antiviral therapy failure and drug resistance mutations were frequently detected. Our findings corroborate those of Leen et al12 who noted an almost identical CR rate in a similar patient group. Complete virological responses were seen irrespective of drug mutation status, suggesting that mutations affecting the action of traditional antivirals are not relevant when using cell therapy to enhance immune reconstitution.
Late CMV disease post-HSCT is the result of defective CMV immune function. The patients treated for CMV reactivation in our study were at high risk for recurrent CMV reactivations and late CMV disease.3,4,17,18 Despite this, we observed excellent control of CMV over 12 months post–final T-cell infusion. Only 5 patients reinitiated antiviral treatment, and in 4 patients, this occurred in the context of steroid administration for presumed or proven GVHD.
The virological response coincided with a rise in the numbers of CD3+ cells, and specifically CD8+ terminal effector memory cells, in the blood and the time of peak CMV-specific T-cell immunity. CD8+ T-cell reconstitution, in particular the CD8+ terminal effector subset (CD45RA+62L– or CD45RA+CCR7–),19-21 is known to be important in virological control post-HSCT.5,10 The recovery of CD8+ T cells did not correlate with time posttransplant, suggesting that VSTs were not acting purely as a bridge to coincidental hemopoietic recovery. Rather, CD8+ T-cell recovery was related to recent VST administration irrespective of time after transplantation, implicating VSTs in facilitating the recovery process, although confirmation of this hypothesis requires additional study.
Before VST administration, the percentage of CD8+ T cells expressing PD-1 in the study population was high compared with healthy individuals, and it remained elevated even at the time of viral control. The significance of this finding is currently unclear. Chronic antigenic stimulation (viral or alloreactive) may be responsible for PD-1 upregulation and T-cell dysfunction. The fact that viral control was observed without downregulation of PD-1 might imply that immune checkpoint inhibitor therapy would be of limited value as an adjunct to this form of cell therapy. Recent reports implicating PD-1 inhibitors in severe GVHD post-HSCT also suggest caution in combining cell therapy and immune checkpoint inhibition after allogeneic stem cell transplantation.22,23 More detailed assessment of immunoinhibitory markers is warranted.
Our results validate previous reports of the safety and short-term efficacy of third-party VST infusions. The novelty of our data lies in the demonstration of long-term virus control in a group of heavily pretreated patients, some with life-threatening tissue infections at the time of cell administration. This efficacy is particularly notable given the convenience and flexibility of third-party VST infusion using products from cryopreserved cell banks to treat recipients of multiple types of allogeneic HSCT. Over 95% of our patients were treated within 3 days of request, mainly in the outpatient setting. The HLA mismatching of third-party VSTs that can be tolerated means that VST products derived from 15 donors were adequate to meet the diverse HLA needs of our cohort. In many instances, the use of VSTs provided an alternative to further prolonged antiviral therapy and allowed the limitation of toxicity, especially the myelosuppression present at study entry. Our results may have relevance for treating multiple other viral and nonviral opportunistic pathogens occurring after allogeneic transplant. The establishment of third-party virus-specific cell banks could reduce the cost and complexity associated with personalized donor-derived cell therapy while retaining its efficacy and enhancing its practicality in routine transplantation.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank all the patients, investigators, and laboratory and clinical trial staff from all participating centers. The authors thank Gillian Huang, Ashita Chunilal, Yi Qiu Sun, and Ming Qiao for their assistance. Flow cytometry was performed in the Flow Cytometry Core Facility, which is supported by the Westmead Research Hub, Cancer Institute of New South Wales, and National Health and Medical Research Council.
This work was supported by project grant PG1032431 from the National Health and Medical Research Council. B.W. is a doctoral candidate at the University of Sydney and was provided conference support from the Cancer Institute of New South Wales, Sydney West Translational Cancer Research Centre. B.W., E.B., M.-C.D., D.B., C.K.K.M., and K.P.M. are currently or have been previously supported by the Leukaemia Foundation of Australia. E.B. is a National Health and Medical Research Council postdoctoral fellow.
Authorship
Contribution: B.W. recruited, treated, and followed up patients taking part in the study, coordinated the collection of clinical follow-up data from all centers, performed postinfusion immunophenotyping and some ELIspot assays, analyzed the data, and wrote the manuscript; E.B. assisted with the study design, wrote the clinical trial protocol, recruited, treated, and followed up patients taking part in the study, and supervised data analysis and writing of the manuscript; L.E.C. supervised the running of the clean room facilities, manufacture of the T-cell products for therapeutic use, and performance of assays for product release and performed searches of the bank for VST selection; A.Y. and C.F. supervised the recruitment, treatment, and follow-up of patients; J.B., R.S., and R.B. manufactured cell products for therapeutic use and assisted with postinfusion monitoring experiments; D.K. and G.S. assisted with data collection of patients on the study; M.-C.D., D.B., and C.K.K.M assisted with recruitment, follow-up, and data collection of patients in the study; P.J.S. supervised the recruitment, treatment, and follow-up of patients and provided guidance on issues specific to the pediatric setting; K.P.M. recruited, treated, and followed up patients in the study and assisted with study design; D.J.G. provided overall academic leadership, treated and followed up patients, designed the study, and supervised data analysis and preparation of the manuscript; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David J. Gottlieb, Department of Medicine, Westmead Hospital, Corner Hawkesbury Rd and Darcy Rd, Sydney, NSW 2145, Australia; e-mail: david.gottlieb@sydney.edu.au.
References
- 1.Hiwarkar P, Gaspar HB, Gilmour K, et al. . Impact of viral reactivations in the era of pre-emptive antiviral drug therapy following allogeneic haematopoietic SCT in paediatric recipients. Bone Marrow Transplant. 2013;48(6):803-808. [DOI] [PubMed] [Google Scholar]
- 2.Hill JA, Mayer BT, Xie H, et al. . The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood. 2017;129(16):2316-2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almyroudis NG, Jakubowski A, Jaffe D, et al. . Predictors for persistent cytomegalovirus reactivation after T-cell-depleted allogeneic hematopoietic stem cell transplantation. Transpl Infect Dis. 2007;9(4):286-294. [DOI] [PubMed] [Google Scholar]
- 4.Servais S, Dumontier N, Biard L, et al. . Response to antiviral therapy in haematopoietic stem cell transplant recipients with cytomegalovirus (CMV) reactivation according to the donor CMV serological status. Clin Microbiol Infect. 2016;22(3):289.e1-289.e7. [DOI] [PubMed] [Google Scholar]
- 5.Neuenhahn M, Albrecht J, Odendahl M, et al. . Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia. 2017;31(10):2161-2171. [DOI] [PubMed] [Google Scholar]
- 6.Blyth E, Clancy L, Simms R, et al. . Donor-derived CMV-specific T cells reduce the requirement for CMV-directed pharmacotherapy after allogeneic stem cell transplantation. Blood. 2013;121(18):3745-3758. [DOI] [PubMed] [Google Scholar]
- 7.Uhlin M, Gertow J, Uzunel M, et al. . Rapid salvage treatment with virus-specific T cells for therapy-resistant disease. Clin Infect Dis. 2012;55(8):1064-1073. [DOI] [PubMed] [Google Scholar]
- 8.Doubrovina E, Oflaz-Sozmen B, Prockop SE, et al. . Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119(11):2644-2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heslop HE, Slobod KS, Pule MA, et al. . Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925-935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peggs KS, Verfuerth S, Pizzey A, Chow SL, Thomson K, Mackinnon S. Cytomegalovirus-specific T cell immunotherapy promotes restoration of durable functional antiviral immunity following allogeneic stem cell transplantation. Clin Infect Dis. 2009;49(12):1851-1860. [DOI] [PubMed] [Google Scholar]
- 11.Feuchtinger T, Opherk K, Bethge WA, et al. . Adoptive transfer of pp65-specific T cells for the treatment of chemorefractory cytomegalovirus disease or reactivation after haploidentical and matched unrelated stem cell transplantation. Blood. 2010;116(20):4360-4367. [DOI] [PubMed] [Google Scholar]
- 12.Leen AM, Bollard CM, Mendizabal AM, et al. . Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121(26):5113-5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clancy LE, Blyth E, Simms RM, et al. . Cytomegalovirus-specific cytotoxic T lymphocytes can be efficiently expanded from granulocyte colony-stimulating factor-mobilized hemopoietic progenitor cell products ex vivo and safely transferred to stem cell transplantation recipients to facilitate immune reconstitution. Biol Blood Marrow Transplant. 2013;19(5):725-734. [DOI] [PubMed] [Google Scholar]
- 14.Ma CK, Blyth E, Clancy L, et al. . Addition of varicella zoster virus-specific T cells to cytomegalovirus, Epstein-Barr virus and adenovirus tri-specific T cells as adoptive immunotherapy in patients undergoing allogeneic hematopoietic stem cell transplantation. Cytotherapy. 2015;17(10):1406-1420. [DOI] [PubMed] [Google Scholar]
- 15.Peggs KS, Thomson K, Samuel E, et al. . Directly selected cytomegalovirus-reactive donor T cells confer rapid and safe systemic reconstitution of virus-specific immunity following stem cell transplantation. Clin Infect Dis. 2011;52(1):49-57. [DOI] [PubMed] [Google Scholar]
- 16.Barker JN, Doubrovina E, Sauter C, et al. . Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. 2010;116(23):5045-5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Özdemir E, Saliba RM, Champlin RE, et al. . Risk factors associated with late cytomegalovirus reactivation after allogeneic stem cell transplantation for hematological malignancies. Bone Marrow Transplant. 2007;40(2):125-136. [DOI] [PubMed] [Google Scholar]
- 18.Boeckh M, Leisenring W, Riddell SR, et al. . Late cytomegalovirus disease and mortality in recipients of allogeneic hematopoietic stem cell transplants: importance of viral load and T-cell immunity. Blood. 2003;101(2):407-414. [DOI] [PubMed] [Google Scholar]
- 19.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78(11):5535-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moins-Teisserenc H, Busson M, Scieux C, et al. . Patterns of cytomegalovirus reactivation are associated with distinct evolutive profiles of immune reconstitution after allogeneic hematopoietic stem cell transplantation. J Infect Dis. 2008;198(6):818-826. [DOI] [PubMed] [Google Scholar]
- 21.Ganepola S, Gentilini C, Hilbers U, et al. . Patients at high risk for CMV infection and disease show delayed CD8+ T-cell immune recovery after allogeneic stem cell transplantation. Bone Marrow Transplant. 2007;39(5):293-299. [DOI] [PubMed] [Google Scholar]
- 22.Haverkos BM, Abbott D, Hamadani M, et al. . PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood. 2017;130(2):221-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh AK, Porrata LF, Aljitawi O, et al. . Fatal GvHD induced by PD-1 inhibitor pembrolizumab in a patient with Hodgkin’s lymphoma. Bone Marrow Transplant. 2016;51(9):1268-1270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.