

Key Points
Combining phosphatidylinositol-3-kinase δ inhibition with rituximab, bendamustine, or both is feasible and active in relapsed iNHL.
The safety of novel combinations should be proven in phase 3 trials before adoption in clinical practice.
Abstract
Idelalisib, a first-in-class oral inhibitor of phosphatidylinositol-3-kinase δ, has shown considerable antitumor activity as a monotherapy in recurrent indolent non-Hodgkin lymphoma (iNHL). To evaluate the safety and activity of idelalisib in combination with immunotherapy, chemotherapy, or both, 79 patients with relapsed/refractory iNHL were enrolled based on investigator preference in 3 treatment groups. Patients received continuous idelalisib in combination with (1) rituximab (IR; 375 mg/m2 weekly × 8 doses), (2) bendamustine (IB; 90 mg/m2 per day × 2, for 6 cycles), or (3) both bendamustine and rituximab at aforementioned doses (IBR; monthly × 6 cycles). Patients had a median age of 61 years, a median of 3 prior therapies, and 46% had refractory disease. The overall response rate was 75% (22% complete response) for IR, 88% (36%) for IB, and 79% (43%) for IBR. The median progression-free survival was 37.1 months overall: 29.7 months for IR, 32.8 for IB, and 37.1 months for IBR. The median duration of response was 28.6 months in the IR group and has not been reached in the IB and IBR groups. The most common grade ≥3 adverse events and laboratory abnormalities were neutropenia (41%), pneumonia (19%), transaminase elevations (16%), diarrhea/colitis (15%), and rash (9%). The safety and efficacy reflected in these early data, however, stand in contrast with later observations of significant toxicity in subsequent phase 3 trials in frontline chronic lymphocytic leukemia and less heavily pretreated iNHL patients. Our findings highlight the limitations of phase 1 trial data in the assessment of new regimens. This trial was registered at www.clinicaltrials.gov as #NCT01088048 (an extension study was registered at www.clinicaltrials.gov as #NCT01090414).
Visual Abstract
Introduction
In 2015, approximately 20 000 people in the United States were diagnosed with indolent non-Hodgkin lymphomas (iNHL), and 7000 died of the disease.1,2 Treating recurrent iNHL continues to be challenging. Current therapies commonly include anti–cluster of differentiation 20 antibodies, such as rituximab3 (US approval in 1997) and the alkylator bendamustine4 (US approval in 2008), which have demonstrated activity and tolerability in combination with rituximab.5 Although initially effective for iNHL, standard chemotherapy and immunotherapy generally demonstrate decreasing efficacy with repeated administration.6-9 Thus, there is an unmet need for new treatments with different mechanisms of action, either as monotherapy or in combination with standard-of-care regimens, for patients with relapsed/refractory iNHL. With significant advances in the development of nonchemotherapeutic agents, “chemotherapy-free” regimens for iNHL are now an approach desired by many patients. However, there is also a need for more efficacious chemotherapy-containing regimens. The 20% of patients with follicular lymphoma (FL) treated with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone who progress within 2 years of diagnosis have a substantially increased risk of death within 5 years after diagnosis.10
Phosphatidylinositol 3-kinase (PI3K) is a lipid kinase that exists in 4 different isoforms: α, β, γ, and δ. In B lymphocytes, the δ isoform (PI3Kδ) plays a central role in normal B-cell development and function, transducing signals from the B-cell receptor and from receptors for various cytokines, chemokines, and integrins.11,12 Further, PI3Kδ signaling pathways are commonly hyperactive in B-cell malignancies.13,14 Idelalisib is a potent, small-molecule inhibitor of PI3K that is selective for the δ isoform.15 In lymphoid cell lines and primary patient samples, idelalisib inhibits PI3Kδ/AKT signaling and promotes apoptosis.16 A phase 1 trial demonstrated significant antitumor activity of idelalisib monotherapy in patients with relapsed iNHL with an overall response rate (ORR) of 48%, a median progression-free survival (PFS) of 7.6 months, and a median duration of response (DOR) of 18.5 months.17 A phase 2 study in patients with iNHL refractory to rituximab and an alkylating agent (n = 125) showed significant antitumor activity, with an ORR of 57%, including 6% complete responses (CRs), a median PFS of 11 months, and a median DOR of 12.5 months.18 Based on these data, the Food and Drug Administration granted idelalisib accelerated approval for treatment of patients with relapsed FL and small lymphocytic lymphoma (SLL) who had received at least 2 prior systemic therapies.
Based on single-agent activity in indolent lymphoma, we conducted a phase 1 trial evaluating idelalisib in combination with commonly used antilymphoma standard-of-care agents. Our objectives were to characterize the drug’s safety and clinical activity in combination with rituximab immunotherapy, bendamustine chemotherapy, or combined chemoimmunotherapy and to identify regimens to be tested in phase 3 studies.
Patients and methods
The present study describes the iNHL subgroup of a larger open-label study of idelalisib in patients with relapsed or refractory iNHL or chronic lymphocytic leukemia (CLL). The primary trial evaluated patients through 48 weeks of idelalisib treatment, followed by an extension study offering continued single-agent idelalisib therapy for patients who derived clinical benefit. The study protocol was approved by institutional review boards across the 11 participating study centers. All patients provided written informed consent before enrollment. These studies were conducted under a US Investigational New Drug Application in accordance with the International Conference on Harmonization guidelines for Good Clinical Practice and the original principles embodied in the Declaration of Helsinki. All authors had full access to study data and were involved in data interpretation, manuscript preparation, revision, and final approval.
Inclusion criteria
Previously treated patients with a confirmed diagnosis of FL (grades 1, 2, and 3a), SLL, or marginal zone lymphoma (MZL) were eligible.19 Patients must have been previously treated and relapsed or had refractory disease (defined as not responding to a standard regimen or progressing within 6 months of completing a standard regimen). Measurable disease consisting of ≥1 lesion of >2 cm in a single dimension by computed tomography (CT) scan was required. Patients were ≥18 years of age and had a World Health Organization performance status of ≤2.
Exclusion criteria
Key exclusion criteria included known active central nervous system lymphoma, active serious infection requiring systemic therapy, or prior allogeneic stem cell transplantation. Adequate bone marrow function was required and defined as no worse than mild neutropenia (absolute neutrophil count <1000/µL) or thrombocytopenia (<75 000/µL). Other reasons for exclusion were serum creatinine ≥2.0 mg/dL, serum bilirubin ≥2.0 mg/dL, serum transaminases ≥2 times the upper limit of normal (ULN), Child-Pugh class B or C hepatic impairment, serologic evidence of HIV, or active hepatitis B or hepatitis C. Female patients were excluded if they were pregnant or nursing.
Study design and treatments
The study was an open-label, unstratified, and nonrandomized phase 1 trial. Patients were treated in 1 of 3 cohorts based on investigator’s choice and received continuous oral therapy with idelalisib.4 The treatment groups received either rituximab (IV infusion of 375 mg/m2 weekly for 8 doses), bendamustine (30-minute IV infusion of 90 mg/m2 on days 1 and 2), or the combination of bendamustine and rituximab (bendamustine at 90 mg/m2 on days 1 and 2, then rituximab at 375 mg/m2 on day 1), and all groups received twice-daily idelalisib. Idelalisib was provided as 100- or 150-mg tablets, with 75- and 50-mg tablets available for dose reductions. The idelalisib dose was based on safety, efficacy, and pharmacokinetic data from phase 1 monotherapy studies in iNHL and CLL.17,20
The 3 main treatment groups are not comparable because they were not stratified or balanced in this trial. Patients in the first 2 cohorts were enrolled to the continuous idelalisib in combination with rituximab (IR; n = 8) and continuous idelalisib in combination with bendamustine (IB) groups (n = 8) and received idelalisib at 100 mg orally twice daily. After adequate safety was established, all subsequent patients in the following cohorts received 150 mg of idelalisib orally twice daily. In addition, a group was assigned to the combination of bendamustine, rituximab, and idelalisib (IBR; n = 14). In an attempt to decrease the incidence of transaminase elevations, subgroups with delayed initiation of idelalisib (28-day delay) were assessed, either with rituximab or with bendamustine (n = 12 each). Unless disease progression or intolerable toxicity occurred, patients could continue idelalisib through 48 weeks on the primary study (12 28-day cycles) and indefinitely on the extension study. The disposition of all treatment groups is summarized in Table 1.
Table 1.
Disposition of patients in the 7 subgroup-specific cohorts of the primary and extension studies
| Enrolled in primary study (n = 79) | Enrolled in extension study (n = 35) (%) | Ongoing (n = 17) (%) | |
|---|---|---|---|
| Idela + R | 32 | 14 (44) | 6 (19) |
| Idela 100 mg | 8 | 3 (38) | 3 (38) |
| Idela 150 mg | 12 | 8 (67) | 1 (8) |
| Delayed Idela 150 mg | 12 | 3 (25) | 2 (17) |
| Idela + B | 33 | 15 (45) | 8 (24) |
| Idela 100 mg | 8 | 1 (13) | 1 (13) |
| Idela 150 mg | 13 | 9 (69) | 6 (46) |
| Delayed Idela 150 mg | 12 | 5 (42) | 1 (8) |
| Idela + BR | |||
| Idela 150 mg | 14 | 6 (43) | 3 (21) |
B, bendamustine; Idela, idelalisib; R, rituximab.
Pretreatment evaluation
Screening assessments included documentation of iNHL diagnosis, staging (Ann Arbor system), prognostic factors (Follicular Lymphoma International Prognostic Index), B symptoms, World Health Organization performance status, physical examination, clinical laboratory tests, vital signs, 12-lead electrocardiogram, and disease status (including a CT scan and bone marrow biopsy if not already performed within 6 weeks before study initiation). At baseline, the following procedures were performed before study drug dosing: recording of adverse events (AEs); vital signs, electrocardiogram; and clinical laboratory tests, including serum immunoglobulin A (IgA), IgE, IgG, and IgM; and serum β2-microglobulin.
Safety assessments
All AEs were evaluated, and complete blood counts and standard serum chemistry tests were collected every 2 weeks for 12 weeks, then every 4 weeks for 12 weeks, and thereafter every 12 weeks. Serum immunoglobulins were measured at 12-week intervals through week 48. AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA), and the severity of AEs and laboratory abnormalities were graded using the Common Terminology Criteria for Adverse Events, version 4.02.21 Only serious AEs (SAEs) and grade ≥3 AEs were collected during the extension phase of the study.
Efficacy assessments
Patients were evaluated for disease response or progression every 8 weeks (weeks 8, 16, and 24) and then at 12-week intervals (week 36 onward). The ORR was derived from the best response measured from start of treatment until disease progression or early discontinuation. Nodal responses were calculated from the sum of the product of the greatest perpendicular diameters (SPD). CR, partial response (PR), stable disease (SD), and progressive disease (PD) were assessed using standard criteria.22 Bone marrow biopsies were performed as needed to confirm a CR and were required at week 24. Patients who had PD at any point stopped study drug treatment and discontinued the study.
Statistical analysis
Because this was a phase 1 study with the primary objective to assess safety, the sample size was not based on power calculations. The sample size of approximately 6 in cohorts 1a and 1b was based on the standard 3+3 design with a target observed dose-limiting toxicity threshold of 33%. The sample size of approximately 12 for the next cohorts was considered adequate for an initial assessment of safety and tolerability for subsequent evaluations. Unless otherwise noted, data from the primary and extension studies were analyzed together. All safety and efficacy analyses were based on the intent-to-treat analysis set, which included all patients who received at least 1 dose of study drug (idelalisib or combination therapy). The ORR was presented with a 2-sided 95% confidence interval (CI) using the exact method. PFS, DOR, and overall survival were summarized using the Kaplan-Meier (KM) method. Time to response (TTR) and DOR were analyzed in responding patients.
Results
Patient characteristics
Beginning in March 2010, 79 patients with iNHL were enrolled (Table 2). The primary 48-week study (Study 101-07) was completed in January 2013, and the extension study (Study 101-99) is ongoing; the present analysis is based on data up to October 2014. Demographics show that most (65.8%) patients were male and the median age was 61 years (range, 37-84). Lymphoma subtypes included FL (59 patients, 74.7%), SLL (15 patients, 19.0%), and MZL (5 patients, 6.3%). At baseline, patients frequently demonstrated elevated β2 microglobulin (59%), stage IV disease (57%), bulky adenopathy (>5 cm; 48%), anemia (hemoglobin <12 g/dL; 41%), and elevated lactate dehydrogenase (28%) in the context of a median of 3 prior therapies (range, 1-11). Most patients had received a rituximab-containing regimen, an alkylating agent, or an anthracycline (Table 2). Zero, 6%, and 14% of patients in the IR, IB, and IBR cohorts, respectively, received rituximab monotherapy as their only prior therapy. Approximately 46% of patients were refractory to their last prestudy therapy, and 58% of patients were refractory to rituximab (defined as lack of response or progression <6 months since completion of last regimen).
Table 2.
Patient characteristics and disposition (primary study, unless otherwise indicated)
| Idela + R (n = 32) | Idela + B (n = 33) | Idela + BR (n = 14) | All patients (n = 79) | |
|---|---|---|---|---|
| Age, median (range), y | 65 (40-84) | 59 (37-80) | 56 (48-76) | 61 (37-84) |
| Sex, male, n (%) | 22 (69) | 22 (67) | 8 (57) | 52 (66) |
| Disease subtype, n (%) | ||||
| FL | 23 (72) | 25 (76) | 11 (79) | 59 (75) |
| SLL | 7 (22) | 6 (18) | 2 (14) | 15 (19) |
| MZL | 2 (6) | 2 (6) | 1 (7) | 5 (6) |
| Prognostic factors, n (%) | ||||
| Performance status ≥1 | 12 (37) | 17 (52) | 4 (29) | 33 (41) |
| Stage IV | 20 (63) | 17 (52) | 8 (57) | 46 (57) |
| Bulky adenopathy (>5 cm) | 16 (50) | 18 (54) | 5 (36) | 39 (48) |
| Bone marrow involvement | 14 (44) | 14 (42) | 4 (29) | 32 (41) |
| β2 microglobulin (>ULN) | 18 (56) | 22 (67) | 7 (50) | 47 (59) |
| LDH (>ULN) | 9 (28) | 9 (27) | 4 (29) | 22 (28) |
| FLIPI score (FL patients) | ||||
| 0/1 | 2 (9) | 5 (20) | 4 (36) | 11 (19) |
| 2 | 10 (43) | 11 (44) | 2 (18) | 23 (39) |
| ≥3 | 8 (35) | 7 (28) | 5 (46) | 20 (34) |
| Missing | 3 (13) | 2 (8) | 0 | 5 (8) |
| Prior therapies, median, n (range) | 3 (1-9) | 3 (1-10) | 3 (1-8) | 3 (1-11) |
| Rituximab-containing regimen, % | 93 | 100 | 100 | 97 |
| Rituximab monotherapy as only prior treatment, % | 0 | 6 | 14 | 5 |
| Rituximab monotherapy as last prior treatment, % | 16 | 39 | 29 | 28 |
| Alkylating agent, % | 90 | 82 | 86 | 86 |
| Anthracycline, % | 50 | 49 | 57 | 51 |
| Bendamustine, % | 40 | 21 | 29 | 32 |
| Purine analog, % | 20 | 33 | 14 | 25 |
| Platinum agent, % | 16 | 18 | 7 | 15 |
| ASCT, % | 9 | 6 | 7 | 8 |
| Disease status, n (%) | ||||
| Relapsed | 19 (59) | 19 (58) | 5 (36) | 43 (56) |
| Refractory* | 13 (41) | 14 (42) | 9 (64) | 36 (46) |
| Idela dose received, n (%) | ||||
| 100 mg twice daily | 8 (25) | 8 (24) | 0 | 16 (20) |
| 150 mg twice daily | 24 (75) | 25 (76) | 14 (100) | 64 (81) |
| Duration of Idela therapy, median (range), mo | ||||
| Primary + extension study | 10.0 (0.5-33) | 9.9 (0.6-32.3) | 11.4 (0.9-25.3) | 10.1 (0.5-33) |
| Treatment disposition, n (%) | ||||
| Received study drug | 32 (100) | 33 (100) | 14 (100) | 79 (100) |
| Completed cycle 2 | 32 (100) | 32 (97) | 11 (79) | 75 (95) |
| Discontinued from primary study | 16 (50) | 18 (54) | 8 (57) | 42 (53) |
| Enrolled in extension study | 14 (44) | 15 (45) | 6 (43) | 35 (44) |
| Discontinued from extension study | 8 (57) | 7 (47) | 3 (50) | 18 (51) |
| Reasons for early discontinuation, primary study, n (%) | ||||
| AE | 6 (19) | 6 (18) | 4 (29) | 16 (20) |
| Disease progression | 4 (12) | 4 (12) | 1 (7) | 9 (11) |
| Patient/investigator request | 1 (3) | 4 (12) | 1 (7) | 6 (8) |
| Withdrew consent | 1 (3) | 1 (3) | 2 (14) | 4 (5) |
| Worsening disease/symptoms | 2 (6) | 1 (3) | 0 | 3 (4) |
| Death | 1 (3) | 1 (3) | 0 | 2 (3) |
| SCT | 1 (3) | 0 | 0 | 1 (1) |
| Reasons for discontinuation, extension study, n (%) | ||||
| Disease progression | 5 (36) | 2 (13) | 1 (17) | 8 (23) |
| AE | 2 (14) | 1 (7) | 2 (33) | 5 (14) |
| Death | 0 | 2 (13) | 0 | 2 (6) |
| Investigator request | 0 | 1 (7) | 0 | 1 (3) |
| SCT | 0 | 1 (7) | 0 | 1 (3) |
| Withdrew consent | 1 (7) | 0 | 0 | 1 (3) |
ASCT, allogeneic stem cell transplantation; FLIPI, Follicular Lymphoma International Prognostic Index; LDH, lactate dehydrogenase.
Refractory is defined as a lack of response or progression within 6 mo of completion of last therapy.
Patient disposition
The 3 main treatment groups contained a total of 7 subgroups as listed in Table 1: rituximab + 100 mg idelalisib, rituximab + idelalisib 150 mg, rituximab + 150 mg delayed idelalisib, bendamustine + 100 mg idelalisib, bendamustine + 150 mg idelalisib, bendamustine + 150 mg delayed idelalisib, and bendamustine/rituximab + 150 mg idelalisib. All regimens were administered twice daily. Analysis of safety and efficacy revealed no significant differences within the 3 IR subgroups and the 3 IB subgroups (see the supplemental Data); these subgroups were thus consolidated in subsequent analyses.
All patients in the intent-to-treat analysis set (N = 79) received idelalisib. The median duration of exposure for all patients in the study was 10.1 months (range, 0.5-32.7+). Specifics of disposition for all patients in the 3 main treatment groups are listed in Table 2. Thirty-seven patients (46.8%) completed the primary study and received at least 48 weeks of treatment. Forty-two patients discontinued the primary study. Reasons for discontinuation were AEs (16 patients, 20%), PD (9 patients, 11%), patient/investigator request (n = 6), withdrawal of consent (n = 4), worsening disease/symptoms not meeting criteria for progression (n = 3), death (n = 2), and stem cell transplantation (SCT; n = 1). Two additional patients completed the primary study but did not enroll in the extension study because of cholecystitis and investigator decision, respectively.
In the extension study, 35 patients continued with idelalisib treatment. Seventeen patients were on study at the data cutoff. Eighteen patients discontinued treatment in the extension study. The most common reasons were PD (8 patients), AEs (5 patients), death (2 patients), investigator request, SCT, and withdrawal of consent (1 patient each).
Safety profile
The incidences of treatment-emergent AEs (TEAEs) and laboratory abnormalities (regardless of attribution) are listed in Table 3. The table lists AEs of any grade (≥15% incidence), as well as associated rates of grade ≥3 AEs, by treatment cohort and total group. The most frequently reported nonlaboratory TEAEs of any grade were pyrexia (54%), nausea (44%), fatigue (43%), rash (38%), diarrhea or colitis (37%), cough (35%), insomnia (23%), pneumonia (22%), and upper respiratory infection (20%).
Table 3.
Incidence of TEAEs (≥15% of patients) and selected laboratory abnormalities by cohort type (combined primary and extension studies)
| Idela + R (n = 32) | Idela + B (n = 33) | Idela + BR (n = 14) | Total (n = 79) | |||||
|---|---|---|---|---|---|---|---|---|
| Grade | Grade | Grade | Grade | |||||
| Any | ≥3 | Any | ≥3 | Any | ≥3 | Any | ≥3 | |
| AE, n (%) | 32 (100) | 26 (81) | 33 (100) | 32 (97) | 14 (100) | 12 (86) | 79 (100) | 70 (89) |
| Pyrexia | 12 (37) | 0 | 21 (64) | 1 (3) | 10 (71) | 1 (7) | 43 (54) | 2 (2) |
| Nausea | 13 (41) | 0 | 13 (39) | 0 | 9 (64) | 0 | 35 (44) | 0 |
| Fatigue | 12 (37) | 1 (3) | 16 (48) | 2 (6) | 6 (43) | 0 | 34 (43) | 3 (4) |
| Rash | 8 (25) | 2 (6) | 13 (39) | 2 (6) | 9 (64) | 3 (21) | 30 (38) | 7 (9) |
| Diarrhea/colitis | 13 (41) | 6 (19) | 11 (33) | 3 (9) | 5 (36) | 3 (21) | 29 (37) | 12 (15) |
| Cough | 11 (34) | 0 | 13 (39) | 0 | 4 (29) | 0 | 28 (35) | 0 |
| Insomnia | 7 (22) | 0 | 10 (30) | 0 | 1 (7) | 0 | 18 (23) | 0 |
| Upper respiratory infection | 7 (22) | 0 | 8 (24) | 1 (3) | 1 (7) | 0 | 16 (20) | 1 (1) |
| Pneumonia | 4 (12) | 4 (12) | 10 (30) | 10 (30) | 3 (21) | 1 (7) | 17 (22) | 15 (19) |
| Chills | 4 (12) | 0 | 9 (27) | 0 | 1 (7) | 0 | 14 (18) | 0 |
| Vomiting | 3 (9) | 0 | 6 (18) | 1 (3) | 5 (36) | 0 | 14 (18) | 1 (1) |
| Constipation | 2 (6) | 0 | 7 (21) | 1 (3) | 3 (21) | 0 | 12 (15) | 1 (1) |
| Headache | 7 (22) | 0 | 4 (12) | 0 | 1 (7) | 0 | 12 (15) | 0 |
| Chemistry laboratory abnormality, n (%) | ||||||||
| ALT, increased | 13 (41) | 5 (16) | 19 (58) | 8 (24) | 4 (29) | 0 | 36 (46) | 13 (16) |
| AST, increased | 18 (56) | 4 (13) | 19 (58) | 5 (15) | 4 (29) | 0 | 41 (52) | 9 (11) |
| Alkaline phosphatase, increased | 12 (38) | 1 (3) | 20 (61) | 0 | 3 (21) | 0 | 35 (44) | 1 (1) |
| Bilirubin, increased | 7 (22) | 0 | 9 (27) | 0 | 3 (21) | 0 | 19 (24) | 0 |
| Creatinine, increased | 7 (22) | 0 | 3 (9) | 0 | 3 (21) | 0 | 13 (16) | 0 |
| Hematology laboratory abnormality, n (%) | ||||||||
| Lymphocytes, decreased | 18 (56) | 12 (38) | 28 (85) | 25 (76) | 13 (93) | 12 (86) | 59 (75) | 49 (62) |
| Neutrophils, decreased | 14 (44) | 11 (34) | 21 (64) | 15 (46) | 9 (64) | 6 (43) | 44 (56) | 32 (41) |
| Hemoglobin, decreased | 6 (19) | 2 (6) | 24 (73) | 5 (15) | 7 (50) | 1 (7) | 37 (47) | 8 (10) |
| Platelets, decreased | 8 (25) | 1 (3) | 19 (58) | 4 (12) | 6 (43) | 1 (7) | 33 (42) | 6 (8) |
TEAEs classified by PT using MedDRA, version 15.1. Patients who experienced multiple events within the same PT were counted once per PT.
PT, preferred term.
The most frequently reported (>1 patient) grade ≥3 TEAEs were pneumonia (19%), diarrhea/colitis (15%), rash (9%), fatigue (4%), and pyrexia (2%). The most common grade ≥3 laboratory abnormalities included lymphocytopenia (62%), neutropenia (41%), increased alanine aminotransferase (ALT; 16%), anemia (10%), and thrombocytopenia (8%). Grade ≥3 cytopenias were more common in the 2 bendamustine-containing cohorts. Grade ≥ 3 increased ALT occurred in 5/32 (16%) patients in the IR group and 8/33 (24%) in the IB group; no cases occurred in the IBR group.
AEs leading to discontinuation in the primary study included increased aspartate aminotransferase (AST/ALT; n = 3), rash (n = 3), diarrhea/colitis (n = 3), as well as anemia, myelodysplastic syndrome, thrombocytopenia, febrile neutropenia, fevers/weight loss, suicidal ideation, and vomiting (n = 1 each). AEs leading to discontinuation in the extension study (n = 5) included 2 patients with diarrhea/colitis and 1 patient each with cardiac arrest, esophageal cancer, and rash.
SAEs occurring in more than 1 patient are listed in Table 4. These included pneumonia (16%), pyrexia (14%), diarrhea/colitis (9%), rash (8%), febrile neutropenia (6%), sepsis (6%), acute renal failure (4%), and atrial fibrillation, cardiac arrest, herpes zoster infection, pneumonitis, and vomiting (3% each).
Table 4.
Incidence of SAEs (>1 patient) by cohort type (combined primary and extension studies)
| Idela + R (n = 32) | Idela + B (n = 33) | Idela + BR (n = 14) | Total (n = 79) | |
|---|---|---|---|---|
| SAE, n (%) | 14 (44) | 23 (70) | 11 (79) | 48 (61) |
| Pneumonia | 3 (9) | 9 (27) | 1 (7) | 13 (16) |
| Pyrexia | 0 | 7 (21) | 4 (29) | 11 (14) |
| Diarrhea/colitis | 2 (6) | 3 (9) | 2 (14) | 7 (9) |
| Febrile neutropenia | 0 | 5 (15) | 0 | 5 (6) |
| Rash | 1 (3) | 1 (3) | 4 (29) | 6 (8) |
| Renal failure, acute | 2 (6) | 1 (3) | 0 | 3 (4) |
| Sepsis | 0 | 4 (12) | 1 (7) | 5 (6) |
| Atrial fibrillation | 0 | 0 | 2 (14) | 2 (2.5) |
| Cardiac arrest | 2 (6) | 0 | 0 | 2 (2.5) |
| Herpes zoster | 1 (3) | 1 (3) | 0 | 2 (2.5) |
| Pneumonitis | 0 | 2 (6) | 0 | 2 (2.5) |
| Vomiting | 0 | 1 (3) | 1 (7) | 2 (2.5) |
SAEs classified by PT using MedDRA, version 15.1. Patients who experienced multiple events within the same PT were counted once per PT.
Two patients (3%) experienced a TEAE leading to death during the primary study. The causes, as determined by the investigator, were cardiac arrest (in the IR cohort) and sepsis (in the IB cohort). One additional patient (from the IB cohort) died of a lung infection 66 days after treatment ended. Two patients died during the extension study, 1 from a cerebrovascular accident and the other from sepsis, both in the IB cohort. All AEs leading to death were reported as unrelated to study drugs.
Grade ≥3 diarrhea or colitis was seen in 12 patients (15%), equally distributed across the 3 treatment groups. As in prior studies, symptoms typically consisted of painless, watery diarrhea, without blood or mucus, and had a median onset of 9 months (range, 2-37 months). Events were managed by supportive care measures as previously reported.23 Six patients were able to continue on study after drug interruption and improvement or resolution of the diarrhea/colitis (1 of the 6 received IV hydrocortisone). Two patients permanently discontinued treatment (1 treated with oral prednisolone and 1 treated with oral mesalazine) and 4 patients had previously discontinued idelalisib before the onset of the diarrhea/colitis and were not rechallenged.
Any-grade rash occurred in 38% of patients, including 7/79 (9%) grade ≥3 rash: 2/32 (6%) patients in the IR cohort, 2/33 (6%) in the IB cohort, and 3/14 (21%) in the IBR cohort. Four patients with grade ≥3 rash also had other AEs, including elevated ALT (n = 1), neutropenia (n = 1), pyrexia (n = 1), bronchitis (n = 1), dehydration (n = 1), supraventricular tachycardia (n = 1), and atrial fibrillation (n = 1). Four patients were rechallenged with idelalisib; 1 had a recurrence of grade 3 rash, and 3 patients were able to tolerate further idelalisib treatment.
For patients with suspected pneumonitis (eg, onset of cough, dyspnea, hypoxia, diffuse interstitial pattern or ground-glass opacities on chest imaging without obvious infectious etiology), idelalisib was interrupted. Pneumonitis occurred in 2 patients (at 4 and 5 months) in the IB treatment group; both cases were managed with drug interruption and antibiotic treatment (no steroids), and 1 patient was rechallenged without recurrence.
Efficacy
Of the 79 patients enrolled, 64 had a response, for an ORR of 81% (95% CI, 70.6-89.0) (Figure 1A). CRs were demonstrated in 25 patients (32%) and PRs in 39 patients (49%). In addition, 7 patients had SD (9%) and 4 patients had PD (5%) as best on-study response. Four patients were nonevaluable because they did not have follow-up CT scans.
Figure 1.

Response end points: response rate and changes in SPD. (A) ORR (gray) and CR (black) rates in the combined primary and extension studies. (B) Waterfall plot of best on-treatment changes in the SPD of measured lymph nodes, by evaluable patient. Criteria for response according to Cheson et al.22
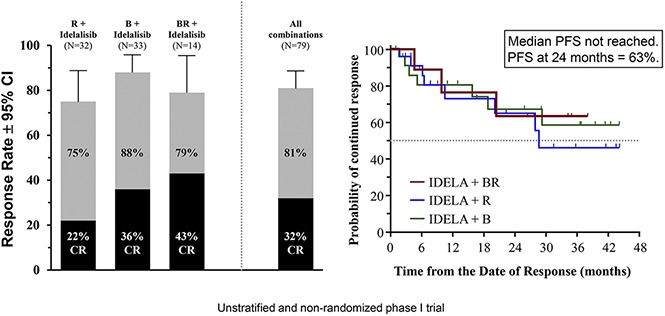
The groups are not comparable because they were not designed to be stratified or balanced in this trial. However, by treatment group, the ORR was 75% (95% CI, 57-89) for the IR group (n = 24/32), 88% (95% CI, 72-97) for the IB group (n = 29/33), and 79% (95% CI, 49-95) for the IBR group (n = 11/14) (Figure 1A). The CR rates were 22% (n = 7/32), 36% (n = 12/33), and 43% (n = 6/14), respectively. SD was noted in 4/32 patients (13%), 3/33 patients (9%), and 0/14 patients in the 3 groups, respectively. Nonevaluable patients included 2/32 in the IR group (needed radiotherapy, elevated liver function test) and 2/14 in the IBR group (intractable nausea/vomiting, withdrew consent). For patients with FL, the ORR (CR rate) was 74% (26%), 88% (44%), and 82% (45%) for the IR, IB, and IBR cohorts, respectively.
A waterfall plot of the best overall nodal response for all iNHL subtypes revealed that 73/75 (97%) of evaluable patients had some reduction in disease burden during treatment (Figure 1B). One patient progressed through therapy in the IB group. Responding patients experienced rapid reduction in lymphadenopathy; median TTR was 1.9 months (range, 1.0-16.6) (Figure 2A). Approximately 75% of responding patients demonstrated a response at their first CT scan at 8 weeks. However, some responses occurred later, and 2 patients converted from SD to PR in the extension study (1 each in the IR and IBR groups), both occurring at 16.6 months. The proportion of patients who responded and enrolled in the extension study was 42% in the IB group, 38% in the IR group, and 36% in the IBR group. Median time to CR was 8.2 months (range, 1.8-31.7 months). In addition, 3 patients converted from PR to CR in the extension study (2 in the IR group and 1 in the IB group). Although most responses occurred early, typically within weeks, some clinical responses occurred late, and some responses may have improved over time. The TTR by treatment group revealed minimal differences in response kinetics between the 3 groups (Figure 2B).
Figure 2.

Time-to-event end points: TTR, DOR, and PFS. (A) KM estimates for TTR (n = 79) and time to CR (n = 24). (B) TTR by the 3 treatment groups. (C) KM estimate for overall DOR. Median DOR not reached. PFS at 24 months was 70%. (D) KM estimate for DOR for the 3 treatment groups. KM estimate for overall PFS. Median PFS not reached. PFS at 24 months was 63%. (E) KM estimate for overall PFS. Median PFS not reached. PFS at 24 months was 63%. (F) KM estimate for PFS for the 3 treatment groups. IDELA, idelalisib.
Median DOR has not yet been reached; frequency of continued response was 66% at 24 months and 55% at 36 months (Figure 2C). Median DOR has been reached for the IR group (28.6 months), but has not yet been reached for the IB and IBR groups (Figure 2D). The median PFS for the overall population was 37.1 months (Figure 2E). The median PFS was 29.7, 32.8, and 37.1 months for the IR, IB, and IBR groups, respectively (Figure 2F). Median overall survival was not reached during the observation period (data not shown), and the KM estimate of the proportion of surviving patients at 24 months was 91.5%.
Discussion
This noncomparative, nonrandomized phase 1 study constitutes the first clinical assessment of IR and/or bendamustine for the treatment of iNHL. No new treatment-related toxicities were identified. The observed AE profile was consistent with previous trials of idelalisib, immunotherapy, or chemotherapy in the advanced-disease setting.
Similar to treatment with idelalisib monotherapy, 13/79 (16%) of patients had asymptomatic grade ≥3 transaminase elevations. Incidence in the prior trials was 25% in the dose-escalation phase 1 trial17 and 13% in the phase 2 trial.18 As in prior trials, these events occurred early, with a median onset of 6 weeks (range, 4-12 weeks) and were managed by temporary interruption. Most patients (9/12) were able to continue idelalisib after rechallenge at a 100 mg twice-daily dose, with subsequent dose reescalation. Two patients had grade 4 elevations, but both were successfully rechallenged and remain on the study. The ALT elevations observed with idelalisib do not appear to be exacerbated or worsened in the combination therapy setting.
The incidence of grade ≥3 diarrhea/colitis was comparable to a prior phase 2 monotherapy trial with a similar duration of idelalisib exposure, where it occurred in 17% of patients.18 Diarrhea/colitis was initially treated with antimotility agents, antibiotics, and/or study drug dose interruptions.23 After excluding infectious etiologies, steroid (prednisolone or budesonide) treatment could be initiated at the investigator’s discretion. The steroid schedule used was prednisolone 1 mg/kg or budesonide 9 mg, with a quick taper after response. Any-grade rash occurred in 38% of patients, with grade ≥3 rash in 7 patients (9%), which is a slightly higher incidence compared with prior idelalisib monotherapy studies.17,18 Bendamustine may have contributed to the increased frequency of rash.
Overall, median PFS was 37.1 months, and median DOR was not yet reached (with probability of continued response at 55% at 3 years). These results compare favorably with the idelalisib monotherapy trials, in which the median PFS and DOR were 7.6 and 18.4 months in the phase 1 trial17 and 11 and 12.5 months in the phase 2 trial.18 The study was designed to evaluate safety of combinations with idelalisib and not intended or powered to compare the different treatment arms; however, the response durations were largely overlapping for the 3 treatment groups and only diverged in the tail of the curves. The median response durations were 28.6 months for the IR group and had not yet been reached for the IB and IBR groups. PFS, DOR, and TTR did not significantly vary at different idelalisib doses or schedules within each treatment group. The primary combination treatments were only administered in the first 2 months (IR) or 6 months (IB or IBR combination), and rituximab maintenance was not part of the protocol, suggesting a role of idelalisib in the suppression of progression. The response durations appear to be longer than those observed in idelalisib monotherapy trials,17,18 or with rituximab,3,9 or bendamustine therapy studies in relapsed/refractory iNHL.24,25
There are no in vitro models for iNHL to test hypotheses about the mechanism of action of IR, bendamustine, or bendamustine and rituximab because all cell lines are derived from aggressive NHLs. In addition, the effects of inhibiting PI3K malignant B cells and their microenvironment on efficacy can only be tested in a clinical trial. The concept of combining idelalisib and rituximab was further supported by the full approval for IR for patients with relapsed CLL.20,26
In the context of these efficacy data associated with apparent acceptable toxicity, randomized phase 3 trials of IR (#NCT01732913) or bendamustine/rituximab (#NCT01732926) were initiated. Recently, an important safety signal of increased AE rates, including deaths, was seen in 6 phase 3 combination trials of idelalisib in patients with CLL/SLL/iNHL. Infectious issues were likely a contributing factor. These trials are undergoing detailed analyses by Gilead Sciences, Inc, and regulators.27,28 Although we cannot draw robust conclusions on similarities and differences between patient populations across studies and other factors that may account for different outcomes, 1 obvious difference between the 6 closed phase 3 trials and our study is the number of prior treatments. Patients in the randomized studies were treatment-naive or had a median of 1 prior regimen, whereas our study population and the CAL-101-09 study (US Food and Drug Administration approval of idelalisib in relapsed/refractory iNHL) had a median of 3 and 4 prior regimens, respectively. Lymphoma refractoriness, number of prior therapies, and state of the patients’ immune systems seem to affect relative risks and benefits of idelalisib combination regimens in specific disease contexts. Idelalisib administered to CLL patients frontline is associated with frequent immune-mediated hepatotoxicity. Lampson et al29 noted that as median age and number of prior therapies increased, the frequency of immune-mediated AEs with idelalisib decreased.
Our data provide an important example of phase 1 trial limitations and the value of conducting phase 3 studies before adopting new and potentially toxic regimens. Although idelalisib is useful for some patients with lymphoid malignancies, its role in combination therapies for relapsed iNHL remains to be determined. Further assessments of optimal patient selection, modified treatment dosing and schedules, supportive care, infection prophylaxis, and increased immune and infectious monitoring are warranted.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients for their dedication to this clinical trial and the clinical personnel at each of the study sites for diligence in caring for patients and collecting study data. The authors also acknowledge study team members at Novella Clinical, Inc (Columbus, OH) and INC Research (Raleigh, NC). Timothy DiChiara, medical writer at Gilead Sciences, and Impact Communication Partners, Inc, assisted in the preparation of the manuscript. Albert S. Yu of Gilead Sciences, Inc, designed the trial.
This clinical study was supported by research funding from Gilead Sciences, Inc (Foster City, CA) and Calistoga Pharmaceuticals, Inc (Seattle, WA), acquired by Gilead Sciences, Inc.
Authorship
Contribution: S.d.V., N.D.W.-J., S.E.C., I.W.F., M.T.S., N.H.F., J.P.S., R.V.B., J.C.B., K.R.R., T.E.B., R.R.F., and J.P.L. performed the research; W.R.G. and Y.K. analyzed the data; and S.d.V., W.R.G., and Y.K. wrote and/or edited the manuscript.
Conflict-of-interest disclosure: S.d.V., S.E.C., I.W.F., M.T.S., N.H.F., J.P.S., R.V.B., J.C.B., K.R.R., T.E.B., R.R.F., and J.P.L. received institutional research grants from Gilead Sciences; N.D.W.-J. received institutional research grants and honoraria from Gilead Sciences; Y.K. and W.R.G. are current or former employees of Gilead Sciences.
Correspondence: Sven de Vos, David Geffen School of Medicine at UCLA, UCLA Lymphoma Program, 10833 Le Conte Ave, Los Angeles, CA 90095; e-mail: devos@mednet.ucla.edu.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5-29. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute. Cancer Statistics Review, 1975-2012: Non-Hodgkin Lymphoma. http://seer.cancer.gov/csr/1975_2012/results_merged/sect_19_nhl.pdf. Bethesda, MD: National Institutes of Health; 2016. Accessed 8 February 2016.
- 3.Molina A. A decade of rituximab: improving survival outcomes in non-Hodgkin’s lymphoma. Annu Rev Med. 2008;59:237-250. [DOI] [PubMed] [Google Scholar]
- 4.Cheson BD, Rummel MJ. Bendamustine: rebirth of an old drug. J Clin Oncol. 2009;27(9):1492-1501. [DOI] [PubMed] [Google Scholar]
- 5.Rummel MJ, Niederle N, Maschmeyer G, et al. ; Study group indolent Lymphomas (StiL). Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203-1210. [DOI] [PubMed] [Google Scholar]
- 6.Lunning MA, Vose JM. Management of indolent lymphoma: where are we now and where are we going. Blood Rev. 2012;26(6):279-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gribben JG. How I treat indolent lymphoma. Blood. 2007;109(11):4617-4626. [DOI] [PubMed] [Google Scholar]
- 8.Zelenetz AD, Gordon LI, Wierda WG, et al. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Non-Hodgkin’s Lymphoma. Version 1.2016. National Comprehensive Cancer Network website. www.nccn.org/professionals/physician_gls/pdf/nhl.pdf. Accessed 3 February 2016.
- 9.Leonard JP, Gregory SA, Maloney DG, Vose JM, Younes A, Zelenetz AD. Optimizing the treatment of patients with rituximab-pretreated recurrent indolent non-Hodgkin lymphoma. Clin Adv Hematol Oncol. 2008;6(6):437-445. [PubMed] [Google Scholar]
- 10.Casulo C, Byrtek M, Dawson KL, et al. . Early relapse of follicular lymphoma after rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone defines patients at high risk for death: an analysis from the National LymphoCare Study. J Clin Oncol. 2015;33(23):2516-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195-203. [DOI] [PubMed] [Google Scholar]
- 12.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329-341. [DOI] [PubMed] [Google Scholar]
- 13.Durand CA, Hartvigsen K, Fogelstrand L, et al. . Phosphoinositide 3-kinase p110δ regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol. 2009;183(9):5673-5684. [DOI] [PubMed] [Google Scholar]
- 14.Bilancio A, Okkenhaug K, Camps M, et al. . Key role of the p110δ isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110δ function in B cells. Blood. 2006;107(2):642-650. [DOI] [PubMed] [Google Scholar]
- 15.Lannutti BJ, Meadows SA, Herman SE, et al. . CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoellenriegel J, Meadows SA, Sivina M, et al. . The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118(13):3603-3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flinn IW, Kahl BS, Leonard JP, et al. . Idelalisib, a selective inhibitor of phosphatidylinositol 3-kinase-δ, as therapy for previously treated indolent non-Hodgkin lymphoma. Blood. 2014;123(22):3406-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gopal AK, Kahl BS, de Vos S, et al. . PI3Kδ inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swerdlow SH, Campo E, Harris NL, et al. . WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. 4th ed. Geneva, Switzerland: WHO Press; 2008. [Google Scholar]
- 20.Brown JR, Byrd JC, Coutre SE, et al. . Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123(22):3390-3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). Version 4.02. https://evs.nci.nih.gov/ftp1/CTCAE/Archive/CTCAE_4.02_2009-09-15_QuickReference_8.5x11.pdf. Accessed 28 March 2016.
- 22.Cheson BD, Pfistner B, Juweid ME, et al. ; International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579-586. [DOI] [PubMed] [Google Scholar]
- 23.Coutré SE, Barrientos JC, Brown JR, et al. . Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;56(10):2779-2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedberg JW, Cohen P, Chen L, et al. . Bendamustine in patients with rituximab-refractory indolent and transformed non-Hodgkin’s lymphoma: results from a phase II multicenter, single-agent study. J Clin Oncol. 2008;26(2):204-210. [DOI] [PubMed] [Google Scholar]
- 25.Kahl BS, Bartlett NL, Leonard JP, et al. . Bendamustine is effective therapy in patients with rituximab-refractory, indolent B-cell non-Hodgkin lymphoma: results from a Multicenter Study. Cancer. 2010;116(1):106-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furman RR, Sharman JP, Coutre SE, et al. . Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.European Medicines Agency. EMA reviews cancer medicine Zydelig. www.ema.europa.eu/docs/en_GB/document_library/Press_release/2016/03/WC500203235.pdf. 11 March 2016. Accessed 25 April 2016.
- 28.U.S. Food and Drug Administration. FDA alerts healthcare professionals about clinical trials with Zydelig (idelalisib) in combination with other cancer medicines. http://www.fda.gov/Drugs/DrugSafety/ucm490618.htm. Updated 14 March 2016. Accessed 25 April 2016.
- 29.Lampson BL, Kasar SN, Matos TR, et al. . Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128(2):195-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.