

Key Points
More than 80% of patients with PMF harbor DNA variants/mutations other than JAK2/CALR/MPL.
Some of these variants/mutations adversely affect overall or leukemia-free survival independent of conventional risk stratification.
Abstract
A myeloid neoplasm–relevant 27-gene panel was used for next-generation sequencing of bone marrow or whole blood DNA in 182 patients with primary myelofibrosis (PMF). DNA sequence variants/mutations other than JAK2/CALR/MPL were detected in 147 patients (81%), with the most frequent being ASXL1 (36%), TET2 (18%), SRSF2 (18%), and U2AF1 (16%); furthermore, 35%, 26%, 10%, and 9% of the patients harbored 1, 2, 3, or 4 or more such variants/mutations, respectively. Adverse variants/mutations were identified by age-adjusted multivariable analysis of impact on overall survival or leukemia-free survival and included ASXL1, SRSF2, CBL, KIT, RUNX1, SH2B3, and CEBPA; their combined prevalence was 56%. Adverse variants/mutations were associated with inferior overall survival (median, 3.6 vs 8.5 years; P < .001) and leukemia-free survival (7-year risk, 25% vs 4%; P < .001), and the effect on survival was independent of both the Dynamic International Prognostic Scoring System Plus and JAK2/CALR/MPL mutational status, with respective hazard ratios of 2.0 (95% confidence interval [CI], 1.3-3.1) and 2.9 (95% CI, 1.9-4.4). Additional prognostic information was obtained by considering the number of adverse variants/mutations; median survivals in patients with zero (n = 80), 1 or 2 (n = 93), or 3 or more (n = 9) adverse variants/mutations were 8.5, 4, and 0.7 years, respectively (P < .001). Additional data were obtained on pattern of mutation co-segregation and phenotypic correlation, including significant associations between U2AF1 and JAK2 mutations (P = .04) and U2AF1 mutations and anemia (P = .003) and thrombocytopenia (P = .006). We conclude that DNA variants/mutations other than JAK2/CALR/MPL are prevalent in PMF and are qualitatively and quantitatively relevant in predicting overall and leukemia-free survival.
Visual Abstract
Introduction
Mutations in primary myelofibrosis (PMF) are operationally classified into 2 categories: drivers and others. The drivers include Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL). Others include additional sex combs such as transcriptional regulator 1 (ASXL1), serine/arginine-rich splicing factor 2 (SRSF2), isocitrate dehydrogenase 1 or 2 (IDH1/2), and enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) among others.1,2 Driver mutations in PMF are often mutually exclusive, and their frequencies are 50% to 60% for JAK2V617F, 20% to 25% for CALR, and 6% to 7% for MPL3,4; approximately 10% to 15% of patients with PMF do not express any of the 3 driver mutations and are referred to as being triple-negative.3,5 Recently, whole-exome sequencing has suggested the presence of somatic or germline MPL or JAK2 variants in some of the patients with triple-negative PMF.6 Regardless, driver mutations in PMF might be accompanied by other mutations whose pathogenetic relevance is even less clear; they include ASXL1, SRSF2, IDH1/2, EZH2, TET2, DNMT3A, and CBL, with respective frequencies of approximately 22%, 9%, 3%, 5%, 10%, 6%, and 4%.7
Driver mutations in myeloproliferative neoplasms (MPNs) have now been included in the World Health Organization diagnostic criteria for both prefibrotic and overtly fibrotic PMF.8,9 Furthermore, the World Health Organization system encourages screening for mutations other than JAK2/CALR/MPL to complement morphologic diagnosis in triple-negative cases.8 In terms of phenotypic correlations in PMF, JAK2 mutations are associated with older age, higher hemoglobin level, leukocytosis, and lower platelet count, and mutant CALR is associated with younger age, higher platelet count, and lower frequencies of anemia, leukocytosis, and spliceosome mutations.10,11 Furthermore, type 2 CALR mutations have been associated with higher risk category, circulating blast percentage, leukocyte count, and inferior survival.12 Genotype-phenotype correlations regarding other mutations include clustering of ASXL1 and SRSF2 mutations with older age and high-risk disease and clustering of ASXL1 mutations with leukocytosis, anemia, and constitutional symptoms.7 Most recently, the order of mutation acquisition was suggested as an additional determinant of phenotype in MPNs.13
In terms of disease prognostication in PMF, type 1 or type 1-like CALR mutations have been associated with superior survival,14,15 and ASXL1/SRSF2/EZH2 mutations have been associated with inferior survival7; in addition, leukemia-free survival was affected by IDH1/2 mutations.7 In a subsequent study,16 we showed that patients with 2 or more adverse mutations displayed significantly shorter survival compared with patients with 1 adverse mutation who in turn had worse survival than those with no adverse mutations. In this study, we applied a broader mutation screening strategy to identify the occurrence and prognostic value of additional mutations as well as their co-segregation patterns and phenotypic correlations.
Methods
This study was approved by the Mayo Clinic Institutional Review Board. Diagnoses and treatment approaches were in accordance with what was considered standard of care at the time of initial diagnosis.17 The study population was selected on the basis of the availability of sufficient DNA and was not otherwise biased by any other selection criteria. Targeted capture assays were carried out on bone marrow or whole blood DNA for the following genes: TET2 (exons 3, 9, 10, and 11), DNMT3A (exons 4, 8, 13, 15, 16, 18, 19, 20, 22, and 23), IDH1 (exon 4), IDH2 (exon 4), ASXL1 (exon 12), EZH2 (exons 8, 17, and 18), SUZ12 (all exons), SRSF2 (exon 1), SF3B1 (exons 13, 15, and 17), ZRSR2 (all exons), U2AF1 (exons 2 and 6), PTPN11 (all exons), TP53 (exons 5, 6, 7, and 8), SH2B3 (all exons), RUNX1 (exons 3, 4, and 8), CBL (exons 8 and 9), NRAS (exons 2 and 3), JAK2 (exons 12 and 14), CSF3R (exons 14 and 17), FLT3 (exons 14 and 20), KIT (all exons), CALR (all exons), MPL (exon 10), NPM1 (exon 11), CEBPA (exon 1), IKZF1 (all exons), and SETBP1 (exon 3).
Paired-end indexed libraries were prepared from individual patient DNA in the Mayo Genomic Sequencing Core Laboratory by using the NEBNext Ultra Library Prep protocol on the Agilent Bravo liquid handling platform (New England BioLabs; Ipswich, MA; Agilent Technologies Inc., Santa Clara, CA). Capture libraries were assembled according to NimbleGen standard library protocol (Roche NimbleGen, Inc., Basel, Switzerland). A panel including the regions of the aforementioned 27 heme-related genes was selected for custom target capture using Agilent SureSelect Target Enrichment Kit (see below for gene regions covered). Capture libraries were pooled at equimolar concentrations and loaded onto paired-end flow cells at concentrations of 7 to 8 pM to generate cluster densities of 600 000 to 800 000/mm2 following Illumina’s standard protocol using the Illumina cBot and HiSeq PE Cluster Kit Version 3 in batches of 48 samples per lane (Illumina, Inc., San Diego, CA). The flow cells were sequenced as 101 × 2 paired-end reads on an Illumina 2000 HiSequencing System using TruSeq SBS Sequencing Kit Version 3 and HiSeq Data Collection Version 2.0.12.0 software. Base-calling was performed by using Illumina’s Real-Time Analysis version 1.17.21.3.
GeneSifter software was used (PerkinElmer, Danvers, MA) to analyze targeted sequence data. Reads from the sequencing in fastq format were aligned by using Burrows-Wheeler Aligner against the genomic reference sequence for Homo sapiens according to Build 37.2 of the National Center for Biotechnology Information (Bethesda, MD). An additional alignment postprocessing set of tools was then used to perform local realignment, duplicate marking, and score recalibration to generate a final genomic aligned set of reads. Nucleotide variants were called by using the Genome Analysis Toolkit (Broad Institute, Cambridge, MA), which identified single nucleotide and small insertion/deletion events using default settings. AlamutVisual mutation analysis software (Interactive Biosoftware, Rouen, France) was used to help filter variations through public genomic databases. Variants were not further analyzed if they did not have a sequencing read depth of ≥50 reads and/or were present with ≤5% variant allele frequency. Minor allele frequency (MAF) was annotated by using both the National Center for Biotechnology Information Single Nucleotide Polymorphism public archive database (dbSNP), and the Exome Aggregation Consortium from the Broad Institute (Cambridge, MA). Remaining variants were further filtered through the Wellcome Trust Sanger Institute Catalogue of Somatic Mutations in Cancer (COSMIC) public database and characterized into 3 categories: VAR: variants not previously associated with a hematologic malignancy (by COSMIC) and either not present or present with ≤1% MAF in dbSNP; HEME: variants previously associated with a hematologic malignancy (by COSMIC or previously by the Mayo Clinic research laboratory; data not shown) and present with ≤1% MAF in dbSNP; and MUT: variants previously associated with a hematologic malignancy and also identified as being somatic (by COSMIC) and present with ≤1% MAF in dbSNP. Each case was assigned a unique patient number and corresponding variants/mutations can be viewed in supplemental Table 1. In cases with multiple variants/mutations in a single gene, the most significant type was illustrated in supplemental Table 1 (ie, MUT>HEME>VAR).
Prognostic evaluation of sequence variants/mutations considered both the number of sequence variants/mutations and the specific genes affected. Adverse sequence variants/mutations were identified by age-adjusted multivariable analysis of their impact on overall or leukemia-free survival. All statistical analyses considered clinical and laboratory parameters obtained at time of first referral at the Mayo Clinic. Differences in the distribution of continuous variables between categories were analyzed by either the Mann-Whitney U test or the Kruskal-Wallis test. Patient groups with nominal variables were compared by using the χ2 test. Survival analysis was considered from the date of referral to date of death or last contact. Leukemia-free survival calculations considered the transformation event as the uncensored variable. Survival curves were prepared by the Kaplan-Meier method and compared by the log-rank test. A Cox proportional hazards regression model was used for multivariable analysis. P values less than .05 were considered significant. All analyses were conducted by using StatView (SAS Institute, Cary, NC).
Results
A total of 182 Mayo Clinic patients with PMF were evaluated (median age, 63 years; range, 22-87 years; 65% males). Table 1 outlines the presenting clinical and laboratory details. Driver mutation distribution was 60% JAK2, 22% CALR, 6% MPL, and 12% triple negative. According to the Dynamic International Prognostic Scoring System Plus (DIPSS-Plus) model,18 survival risk distribution at time of referral was 31% high (n = 57), 38% intermediate-2 (n = 70), 17% intermediate-1 (n = 30), and 14% low (n = 25). Median follow-up was 4 years (range, 0.12-22.2 years). The numbers of documented deaths and leukemic transformations were 112 (62%) and 18 (10%), respectively.
Table 1.
Clinical and laboratory characteristics of 182 patients with PMF stratified by the presence or absence of adverse sequence variants/mutations
| Characteristic | Patients | P | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| All (n = 182) | With adverse variants (n = 102; 56%) | Without adverse variants (n =80; 44%) | ||||||||
| No. | % | Median (range) | No. | % | Median (range) | No. | % | Median (range) | ||
| Age, years | 63 (22-87) | 65 (37-84) | 61 (22-87) | .005 | ||||||
| Male sex | 118 | 65 | 71 | 70 | 47 | 59 | .13 | |||
| Hemoglobin, g/dL | 10.1 (5.8-16) | 10 (5.8-15.3) | 10.8 (6-16) | .05 | ||||||
| Leukocytes × 109/L | 10.5 (1.9-219) | 10 (2-219) | 10.7 (1.9-115.1) | .84 | ||||||
| Platelets × 109/L | 224 (11-1493) | 202 (11-692) | 263 (13-1493) | .03 | ||||||
| DIPSS-plus risk category | .002 | |||||||||
| Low | 25 | 14 | 9 | 9 | 16 | 20 | ||||
| Intermediate-1 | 30 | 17 | 12 | 12 | 18 | 22 | ||||
| Intermediate-2 | 70 | 38 | 39 | 38 | 31 | 39 | ||||
| High | 57 | 31 | 42 | 41 | 15 | 19 | ||||
| Karyotype | .54 | |||||||||
| Normal | 105/179 | 59 | 59/99 | 60 | 46/80 | 58 | ||||
| Unfavorable | 22/179 | 12 | 14/99 | 14 | 8/80 | 10 | ||||
| Driver mutation distribution | .26 | |||||||||
| JAK2 | 109 | 60 | 66 | 65 | 43 | 54 | ||||
| CALR | 40 | 22 | 17 | 17 | 23 | 29 | ||||
| MPL | 11 | 6 | 6 | 6 | 5 | 6 | ||||
| Triple-negative | 22 | 12 | 13 | 13 | 9 | 11 | ||||
| No. of variants/mutations other than JAK2/CALR/MPL | <.001 | |||||||||
| 0 | 35 | 19 | 0 | 0 | 35 | 44 | ||||
| 1 | 64 | 35 | 30 | 30 | 34 | 43 | ||||
| 2 | 48 | 26 | 39 | 39 | 9 | 11 | ||||
| 3 | 18 | 10 | 17 | 17 | 1 | 1 | ||||
| 4 | 10 | 6 | 9 | 9 | 1 | 1 | ||||
| 5 | 3 | 2 | 3 | 3 | 0 | 0 | ||||
| 6 | 3 | 2 | 3 | 3 | 0 | 0 | ||||
| 11 | 1 | <1 | 1 | 1 | 0 | 0 | ||||
Prevalence and co-segregation of sequence variants/mutations
DNA sequence variants/mutations other than JAK2/CALR/MPL were detected in 147 patients (81%), with no difference among driver mutational categories: 83% in JAK2, 73% CALR, 91% MPL, and 82% in triple-negative cases (P = .43) (Tables 1 and 2). Figure 1 depicts all sequence variants/mutations detected and their frequencies whereas supplemental Table 1 provides additional information on specific variants/mutations; the most frequent genes involved were ASXL1 (36%), TET2 (18%), SRSF2 (18%), and U2AF1 (16%). Approximately 35%, 26%, 10%, and 9% of the patients harbored 1, 2, 3, or 4 or more sequence variants/mutations other than JAK2/CALR/MPL, respectively (Table 1). The distribution in the number of mutations was similar among the different driver mutational categories (Table 2; P = .35); however, significant associations were noted for JAK2 with U2AF1 variants/mutations and infrequent occurrence of SRSF2 variants/mutations were noted in CALR-mutated cases (Table 2). Furthermore, among the most frequent sequence variants/mutations, significant associations were apparent for ASXL1 with U2AF1 (P = .002), CBL (P = .007), and SETBP1 (P = .04); for TET2 with CBL (P = .04), SUZ12 (P = .003), and PTPN11 (P < .001); for SRSF2 with IDH1/2 (P < .001) and MPL (P = .047); and for U2AF1 with JAK2 (P = .02), PTPN11 (P = .02), and ASXL1 (P = .002). Conversely, ASXL1 was less likely to coexist with DNMT3A (P = .03) and SRSF2 was less likely to coexist with U2AF1 (P = .006).
Table 2.
All and adverse DNA sequence variants/mutations among 182 patients with PMF
| Patients | P | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| All (n = 182) | JAK2 mutated (n = 109; 60%) | CALR mutated (n = 40; 22%) | MPL mutated (n = 11; 6%) | Triple negative (n = 22; 12%) | |||||||
| No. | % | No. | % | No. | % | No. | % | No. | % | ||
| No. of patients with DNA sequence variants/mutations other than JAK2/CALR/MPL | .24 | ||||||||||
| 0 | 35 | 19 | 19 | 17 | 11 | 27.5 | 1 | 9 | 4 | 18 | |
| 1 | 64 | 35 | 36 | 33 | 15 | 37.5 | 7 | 64 | 6 | 27 | |
| 2 | 48 | 27 | 30 | 28 | 11 | 27.5 | 2 | 18 | 5 | 23 | |
| 3 or more | 35 | 19 | 24 | 22 | 3 | 7.5 | 1 | 9 | 7 | 32 | |
| No. of patients with adverse DNA sequence variants/mutations other than JAK2/CALR/MPL | .3 | ||||||||||
| 0 | 80 | 44 | 43 | 39 | 23 | 57.5 | 5 | 45.5 | 9 | 41 | |
| 1 | 70 | 38 | 45 | 41 | 14 | 35 | 5 | 45.5 | 6 | 27 | |
| 2 | 23 | 13 | 16 | 15 | 2 | 5 | 0 | 5 | 23 | ||
| 3 or more | 9 | 5 | 5 | 5 | 1 | 2.5 | 1 | 9 | 2 | 9 | |
| Variants/mutations | |||||||||||
| ASXL1 | 65 | 36 | 38 | 35 | 14 | 35 | 3 | 27 | 10 | 45 | .73 |
| TET2 | 33 | 18 | 21 | 19 | 7 | 18 | 2 | 18 | 3 | 14 | .94 |
| SRSF2 | 32 | 18 | 21 | 19 | 2 | 5 | 4 | 36 | 5 | 23 | .05 |
| U2AF1 | 30 | 16 | 25 | 23 | 2 | 5 | 1 | 9 | 2 | 9 | .04 |
| CBL | 9 | 5 | 7 | 6 | 1 | 3 | 0 | 1 | 5 | .66 | |
| KIT | 2 | 1 | 1 | 1 | 0 | 0 | 1 | 5 | .39 | ||
| RUNX1 | 8 | 4 | 7 | 6 | 1 | 3 | 0 | 0 | .4 | ||
| CEBPA | 16 | 9 | 9 | 8 | 3 | 8 | 1 | 9 | 3 | 14 | .86 |
| SH2B3 | 11 | 6 | 9 | 8 | 0 | 0 | 2 | 9 | .21 | ||
Figure 1.

Twenty-seven-gene panel DNA sequence variants in Mayo Clinic patients with PMF (n = 182). (A) Co-segregation plot for individual variants/mutations in PMF. Each column represents 1 patient. Variants/mutations are depicted by representative colored bars. Red: variants previously associated with a hematologic malignancy, identified as being somatic and present with ≤1% MAF; Pink: variants previously associated with a hematologic malignancy and present with ≤1% MAF; Blue: variants not previously associated with a hematologic malignancy and present with ≤1% MAF. (B) Total variants/mutations in PMF ranked by gene and corresponding overall frequency percentage.
Phenotypic correlations of sequence variants/mutations
In general, older patients were more likely to display U2AF1, SF3B1, TET2, SETBP1, RUNX1, CEBPA, and PTPN11 variants/mutations, whereas female sex was associated with DNMT3A and male sex was associated with CSF3R variants/mutations (P < .05 for all). Additional phenotypic correlations were examined for sequence variants/mutations with recurrence rate of above 10% (Figure 1). Parameters examined for possible association included DIPSS-plus, karyotype, leukocyte count, hemoglobin level, platelet count, and splenomegaly. Anemia was associated with U2AF1 and SRSF2, thrombocytopenia with U2AF1, and constitutional symptoms and higher DIPSS-plus score with ASXL1 and SRSF2 variants/mutations (P < .05 for all, including P = .003 for U2AF1 and anemia association and P = .006 for U2AF1 and thrombocytopenia association). No association with karyotype, spleen size, or leukocyte count was evident.
Prognostic relevance of sequence variants/mutations
Age-adjusted univariable analysis identified ASXL1, SRSF2, IDH1/2, EZH2, CBL, KIT, and RUNX1 variants/mutations as being significantly associated with inferior survival; ASXL1, SRSF2, CBL, and KIT remained significant on age-adjusted multivariable analysis, with respective hazard ratios (HRs) of 2.1 (95% CI, 1.4-3.1), 2.0 (95% CI, 1.3-3.0), 3.0 (95% CI, 1.4-6.5), and 38.1 (95% CI, 8.6-168.2), whereas EZH2 and RUNX1 maintained borderline significance. For leukemia-free survival, univariable analysis showed significant associations with SRSF2, RUNX1, CEBPA, SH2B3, and IDH1/2; the first 4 remained significant during multivariable analysis with respective HRs of 4.9 (95% CI, 1.9-12.8), 8.7 (95% CI, 1.8-42.5), 5.4 (95% CI, 1.6-17.6), and 5.8 (95% CI, 1.6-21.7).
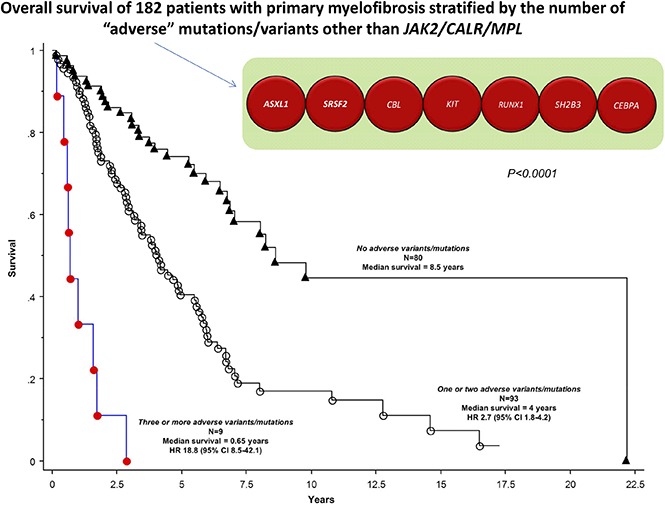
On the basis of their independent impact on overall or leukemia-free survival outlined above, ASXL1, SRSF2, CBL, KIT, RUNX1, CEBPA, and SH2B3 variants/mutations were identified as being adverse and at least 1 of them was present in 56% of the 182 study patients. Adverse variants/mutations were associated with inferior overall survival (median, 3.6 vs 8.5 years; P < .001) and leukemia-free survival (7-year risk, 25% vs 4%; P < .001), and the effect on survival was independent of both DIPSS-Plus and JAK2/CALR/MPL mutational status (HR, 2.0; 95% CI, 1.3-3.1). Figure 2 depicts survival data of patients with adverse vs nonadverse vs no detectable variants/mutations; the difference between the latter 2 became insignificant when adjusted for age (P = .15). Additional prognostic information was obtained by considering the number of adverse variants/mutations; median survivals in patients with zero (n = 80), 1 or 2 (n = 93), or 3 or more (n = 9) adverse variants/mutations were 8.5, 4, and 0.7 years, respectively (Figure 3; P < .001), and the difference was independent of DIPSS-Plus with HRs of 7.3 (95% CI, 3.2-16.8) for 3 or more and 1.9 (95% CI, 1.2-3.0) for 1 or 2 adverse variants/mutations.
Figure 2.

Overall survival curves in 182 patients with PMF, stratified by the presence of adverse (ASXL1, SRSF2, CBL, KIT, RUNX1, SH2B3, and CEBPA) vs nonadverse variants/mutations vs no DNA sequence variants/mutations.
Figure 3.

Overall survival curves in 182 patients with PMF, stratified by the number of adverse variants/mutations: 3 or more vs 1 or 2 vs no adverse variants/mutations.
Discussion
The frequent detection of multiple DNA sequence variants/mutations in PMF in this study is in line with that of a previous smaller study of 34 patients who underwent a 104-gene panel next-generation sequencing (NGS).19 The larger number of patients in this study has enabled us to examine the effect of driver mutational status on the particular phenomenon and its relevance to disease phenotype and prognostication. Frequencies of sequence variants/mutations were mostly similar among JAK2/CALR/MPL mutational categories, including triple-negative cases in which more than 80% of the patients displayed at least 1 sequence variant/mutation; however, significant clustering was noted between JAK2 and U2AF1 mutations, whereas SRSF2 mutations were infrequent in CALR-mutated patients (Table 2). In other words, multigene sequencing allows genetic confirmation of clonal myeloproliferation in virtually all patients with PMF, and it also discloses clustering of specific mutations that might signify pathogenetic relevance and provide an explanation for differences in phenotype and prognosis among patients with different driver mutations.
The phenotypic relevance of driver mutations in PMF is now well established and includes the association of JAK2 mutations with older age, higher hemoglobin level, leukocytosis, lower platelet count, and the association of CALR mutations with younger age, higher platelet count, and lower frequencies of anemia, leukocytosis, and spliceosome mutations.10 In this study, we show that older age was also associated with a number of other variants/mutations, including U2AF1, SF3B1, TET2, SETBP1, RUNX1, CEBPA, and PTPN11; the particular information underscores the need to adjust the analysis of survival impact for age. Additional phenotypic correlations included anemia with U2AF1 and SRSF2, thrombocytopenia with U2AF1, and constitutional symptoms and higher DIPSS-Plus score with ASXL1 and SRSF2 variants/mutations. Most of these associations were previously recognized7,20,21; the phenotypic effect of U2AF1 is particularly noteworthy because of its significant association with both JAK2 and ASXL1 and because of a recent mechanistic report on its potential contribution to abnormal hematopoiesis in patients with myelodysplastic syndromes.22
The key objective of this study was to identify variants/mutations with prognostic relevance. The need for such information is dire, considering the increasing use of targeted NGS in myeloid malignancies and its potential to facilitate refinement of current prognostic models. In a previous study7 that used mutation analysis of a limited number of genes, we had identified ASXL1, SRSF2, EZH2, and IDH1/2 mutations as risk factors for overall or leukemia-free survival in PMF.7 We also showed that the number of such prognostically relevant genes provided additional prognostic value; patients with 2 or more prognostically relevant mutations fared worse, in terms of both overall and leukemia-free survival, compared with those with 1 such mutation, which in turn displayed inferior survival compared with those without such mutations.16 In this study, we used NGS and a broader panel of genes, which enabled us to add CBL, KIT, RUNX1, SH2B3, and CEBPA variants/mutations to the unfavorable list; whether or not EZH2 and IDH1/2 mutations are thus no longer relevant in the presence of the newly identified adverse mutations requires additional studies for confirmation. This study also confirms the additional prognostic value of a number of adverse variants/mutations but, unlike our previous study,16 patients with 1 or 2 adverse variants/mutations displayed similar survival, which was inferior to that of patients without adverse variants/mutations and superior to that of those with 3 or more such mutations.
Additional studies are needed to validate and/or refine our observations. For example, some of the variants/mutations we labeled as being adverse have not yet been shown to affect protein function. Similarly, the distinction between rare germline variants and acquired somatic mutations was not fully addressed in this study. Thus, the observations from this study should be viewed with caution and regarded as being preliminary. Regardless, this study provides general information that might help interpret results from multigene sequencing studies that are being implemented with increasing frequency in routine clinical practice. The demonstration of additional (ie, independent) prognostic value of adverse variants/mutations to that of both DIPSS-Plus and driver mutational status suggests the practical value of including such molecular information in currently available prognostic models, which would also allow for a more appropriate use of such molecular information.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This study was supported in part by the Mayo Clinic Harvey-Yulman Charitable Foundation for Myelofibrosis Tissue Bank, by the Clinical Database of Molecular and Biological Abnormalities, and by the Henry J. Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN.
Authorship
Contribution: A.T. designed and sponsored the study, contributed patients, collected clinical data and patient samples, reviewed the molecular data, performed statistical analysis, and wrote the paper; T.L.L. performed and analyzed the molecular analysis; C.M.F. performed the molecular analysis; Y.E. collected clinical data; C.A.H. performed pathology review; R.P.K. performed cytogenetics review; N.G. contributed patients and collected clinical data; A.P. contributed patients and reviewed the molecular data; and all authors reviewed and approved the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ayalew Tefferi, Mayo Clinic College of Medicine, 200 First St SW, Rochester, MN 55905; e-mail: tefferi.ayalew@mayo.edu.
References
- 1.Tefferi A, Pardanani A. Myeloproliferative Neoplasms: A Contemporary Review. JAMA Oncol. 2015;1(1):97-105. [DOI] [PubMed] [Google Scholar]
- 2.Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;118(7):1723-1735. [DOI] [PubMed] [Google Scholar]
- 3.Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tefferi A, Lasho TL, Schwager SM, et al. The JAK2(V617F) tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specificity and clinical correlates. Br J Haematol. 2005;131(3):320-328. [DOI] [PubMed] [Google Scholar]
- 5.Vannucchi AM, Guglielmelli P. Molecular pathophysiology of Philadelphia-negative myeloproliferative disorders: beyond JAK2 and MPL mutations. Haematologica. 2008;93(7):972-976. [DOI] [PubMed] [Google Scholar]
- 6.Milosevic Feenstra JD, Nivarthi H, Gisslinger H, et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood. 2016;127(3):325-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861-1869. [DOI] [PubMed] [Google Scholar]
- 8.Barbui T, Thiele J, Vannucchi AM, Tefferi A. Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J. 2015;5:e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 10.Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia. 2014;28(7):1407-1413. [DOI] [PubMed] [Google Scholar]
- 11.Guglielmelli P, Nangalia J, Green AR, Vannucchi AM. CALR mutations in myeloproliferative neoplasms: hidden behind the reticulum. Am J Hematol. 2014;89(5):453-456. [DOI] [PubMed] [Google Scholar]
- 12.Tefferi A, Lasho TL, Finke C, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia. 2014;28(7):1568-1570. [DOI] [PubMed] [Google Scholar]
- 13.Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372(7):601-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. 2014;28(7):1472-1477. [DOI] [PubMed] [Google Scholar]
- 15.Tefferi A, Lasho TL, Tischer A, et al. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood. 2014;124(15):2465-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804-1810. [DOI] [PubMed] [Google Scholar]
- 17.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937-951. [DOI] [PubMed] [Google Scholar]
- 18.Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392-397. [DOI] [PubMed] [Google Scholar]
- 19.Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220-2228. [DOI] [PubMed] [Google Scholar]
- 20.Bartels S, Lehmann U, Büsche G, et al. SRSF2 and U2AF1 mutations in primary myelofibrosis are associated with JAK2 and MPL but not calreticulin mutation and may independently reoccur after allogeneic stem cell transplantation. Leukemia. 2015;29(1):253-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tefferi A, Finke CM, Lasho TL, et al. U2AF1 mutations in primary myelofibrosis are strongly associated with anemia and thrombocytopenia despite clustering with JAK2V617F and normal karyotype. Leukemia. 2014;28(2):431-433. [DOI] [PubMed] [Google Scholar]
- 22.Shirai CL, Ley JN, White BS, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell. 2015;27(5):631-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.