

Key Points
In BP-CML multiple, nonhierarchically arranged immunophenotypic stem/progenitor populations have functional LSC activity.
BP-associated cytogenetic abnormalities are detected equally in all immunophenotypic stem/progenitor cells.
Abstract
Chronic myeloid leukemia (CML) is an excellent model of the multistep processes in cancer. Initiating BCR-ABL mutations are required for the initial phase of the disease (chronic phase, CP-CML). Some CP-CML patients acquire additional mutation(s) that transforms CP-CML to poor prognosis, hard to treat, acute myeloid or lymphoid leukemia or blast phase CML (BP-CML). It is unclear where in the hemopoietic hierarchy additional mutations are acquired in BP-CML, how the hemopoietic hierarchy is altered as a consequence, and the cellular identity of the resulting leukemia-propagating stem cell (LSC) populations. Here, we show that myeloid BP-CML is associated with expanded populations that have the immunophenotype of normal progenitor populations that vary between patients. Serial transplantation in immunodeficient mice demonstrated functional LSCs reside in multiple populations with the immunophenotype of normal progenitor as well as stem cells. Multicolor fluorescence in situ hybridization detected serial acquisition of cytogenetic abnormalities of chromosome 17, associated with transformation to BP-CML, that is detected with equal frequency in all functional LSC compartments. New effective myeloid BP-CML therapies will likely have to target all these LSC populations.
Visual Abstract
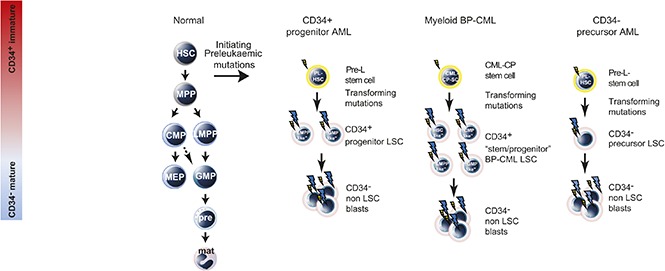
Introduction
Chronic phase (CP) chronic myeloid leukemia (CML), a clonal myeloproliferative disease, requires the constitutively active tyrosine kinase BCR-ABL. The majority of CP-CML patients achieve a durable complete cytogenetic response1 with tyrosine kinase inhibitors (TKIs; eg, imatinib, dasatinib, nilotinib). However, in the first few years after diagnosis, 1% to 1.5% of CP-CML patients per annum progress to a more aggressive acute leukemia, blast phase (BP)-CML. The rate of progression of CP-CML to BP-CML falls sharply when a major molecular response to TKI therapy is obtained. Less than 10% of patients present with de novo BP-CML, and two-thirds of these BP-CML patients have a myeloid immunophenotype. Response to TKIs in BP-CML is short-lived, and median survival following diagnosis of BP-CML is 6.5 to 11 months,2 with many patients developing additional mutations within the BCR-ABL kinase domain, leading to TKI resistance and rapid disease progression.3
Recent studies of de novo acute myeloid leukemia (AML)4-6 and myelodysplastic syndrome7 suggest AML-initiating mutations occur in a functional stem cell compartment that could either be long-term or short-term hemopoietic stem cell (ST-HSC/multipotent progenitor [MPP]) to create pre–leukemic stem cells (pre-LSCs) that expand by clonal advantage. However, pre-LSCs still differentiate completely, or almost completely, and can be associated with normal blood counts.8-10 Additional mutations, possibly acquired in pre-LSCs or downstream pre–leukemic progenitors, are required for full transformation. In fully transformed AML, most functional leukemia-propagating stem cell (LSC) populations have global gene expression profiles and immunophenotypes most similar to normal progenitor populations (“progenitor-like”)11 or normal myeloid precursors,12 suggesting that LSCs are arrested at progenitor or precursor stages. When LSCs are arrested at progenitor-like stages, patient samples contain a mixture of expanded functional LSC populations with global RNA expression profiles most similar to lymphoid-primed multipotent progenitors (LMPP) and granulocyte-macrophage progenitors (GMP).11 A smaller group of patients have expanded populations that share the same immunophenotype as ST-HSC/MPP and the common myeloid progenitor (CMP).11 However, it is unclear if, in AML, functional LSCs exist in the ST-HSC/MPP and CMP compartments. In comparison, in CP-CML, CML propagating stem cells are contained within immunophenotypic HSC populations, and progenitors do not have extended self-renewal capacity.13
In contrast, there have been limited studies of LSC populations in myeloid BP-CML. Jamieson et al identified a GMP-like population as potential LSCs in myeloid BP-CML.14 However, this study did not examine in vivo LSC activity and did not comprehensively assess all immunophenotypic stem/progenitor compartments for LSC function. Here, we demonstrate that in myeloid BP-CML functional LSC populations are present in multiple immunophenotypic compartments with marked interpatient variability of in vivo LSC characteristics.
Methods
Patient samples
Informed consent was obtained in accordance with the Declaration of Helsinki and with approval from UK Ethics Committees (Oxford 06\Q1606\110; Greater Glasgow and Clyde 10/S0704/2). Mononuclear cells (MNC) were isolated by Histopaque density gradient within 24 to 48 hours of collection. CD34+ cells were purified using the CD34 Microbead Kit/MACS separation columns (Miltenyi Biotec).
NSG xenograft assay
Experiments were performed in accordance with UK Government Home Office–approved Project License 30/2465. Eight- to 14-week-old female NSG mice were irradiated 100 to 125 cGy twice, 4 hours apart, followed 24 hours later by IV tail vein injection of myeloid BP-CML stem/progenitor cells. To abrogate antibody-mediated cell clearance, NSG mice were injected intraperitoneally with 200 mg of anti-CD122 antibody or IV immunoglobulin (1 mg/g body weight).11 Peripheral blood or bone marrow engraftment was monitored by blood sampling from 12 weeks onwards. Mice were culled for bone marrow harvesting between 16 and 22 weeks. Human myeloid (hCD45+CD33+CD19−) or B-lymphoid (hCD45+CD33−CD19+) engraftment was analyzed by fluorescence-activated cell sorting (FACS) and defined as ≥0.1% of live MNC gate. Leukemic engraftment was confirmed by karyotypic and BCR-ABL analysis.
Flow cytometric analysis and sorting
Different antibody panels were used for FACS purification of cells for injection into immunodeficient mice (Figures 2 and 3) and for quantitation of stem/progenitor sizes in primary human samples (Figure 1). For FACS purification, additional antibodies (anti-CD2, anti-CD4, anti-CD8, anti-CD235a–glycophorin) against lymphoid (to avoid graft-versus-host disease) and erythroid cells (to reduce ineffective cell number injected) but not myeloid cells (anti-CD14 and anti-CD16) were used. For Figure 1, cells were stained with lineage cocktail–fluorescein isothiocyanate (CD3 [MϕP9], 14 [3G8], 16 [NCAM16.2], 19 [SJ25C1], 20 [SK7], 56 [L27]), and CD34-PerCP (8G12), CD38-V450 (HIT2), CD45RA-APC H7 (HI100), CD90-PE Cy7 (5E10), and CD123-APC (7G3). All antibodies were from BD (Oxford, UK). In Figures 2 and 3, the following antibodies were used: lineage cocktail: anti-CD2 (RPA-2.10), CD3 (HIT3a), CD4 (RPA-T4), CD8 (RPA-T8), CD19 (HIB19), CD20 (2H7), and GPA (GA-R2) (all from e-Bioscience, Hatfield, UK). Following this, cells were stained with QDOT605-conjugated goat F(ab′) anti-mouse immunoglobulin G (H+L; Invitrogen, Paisley, UK). Cells were then washed and subsequently stained with fluorescein isothiocyanate–conjugated anti-CD38 (HIT2; e-Bioscience), phycoerythrin-conjugated anti-CD45RA (HI100; e-Bioscience), PerCP-conjugated anti-CD34 (581; BioLegend, London, UK), PECy7-conjugated anti-CD123 (6H6; e-Bioscience), and biotin-conjugated anti-CD90 (5E10; e-Bioscience). Finally, cells were stained with a streptavidin-conjugated APC-eFluor 780 (e-Bioscience). All samples were double sorted where cell numbers permitted (95% of sorts).
Figure 2.

Dynamic changes in immunophenotypic compartments in the progression from CP to BP-CML. (A) Bar graphs of mean sizes of indicated populations (x-axis) as a percentage of bone marrow Lin−CD34+ population (y-axis). Error bar corresponds to standard error of the mean: *P < .05; **P < .01; ***P < .001. (B) Tabular representation of the data in panel A.
Figure 3.

Functional LSC activity in myeloid BP-CML. (A) Purities of immunophenotypic populations (expressed as percentage), after FACS sorting, used in primary xenotransplantation from 5 patients (COL091, COL091R, CML371, CML002, and HER002). (B) Number of mice with human cell engraftment above engraftment threshold (defined as 0.1% human CD45+CD33+CD19− cells) out of the total number of mice injected. Myeloid engraftment or absence of engraftment is indicated. ND, nondetected. (C) Primary engraftment (4 patients: COL091, COL091R, CML371, and CML002, x-axis) in 1 to 6 mice from the indicated population. Black and red dots show the engraftment level in individual mice. y-axis: mean percentage human (h)CD45+CD33+CD19− cell engraftment/total live MNC. x-axis: injected cell fraction. Red line: engraftment threshold. (D) Number of human cells (y-axis) injected in primary mice from indicated population from 4 patients: COL091, CML371, COL091R, and CML002 (x-axis) in 1 to 6 mice. Black dots: engrafted mice. Red dots: mice that failed to engraft above the engraftment level of 0.1%.
Figure 1.

Expansion of LMPP-, GMP-, MPP-, and CMP-like populations in myeloid BP-CML. Representative FACS plots of CD34+ enriched (A) normal bone marrow; (B) CP-CML; (C) AP-CML; myeloid BP-CML with (D) MPP-like and CMP-like populations; or (E) LMPP-like and GMP-like populations. Numbers of samples studied are shown on the right. Markers studied are shown below plots. Numbers in gates are the mean of all samples within the group expressed as a percentage of Lin−CD34+ cells.
Fluorescence in situ hybridization (FISH) analysis
FACS-sorted cells were incubated at 37°C for 15 minutes in a hypotonic solution (0.075 M KCl). Cells were then centrifuged at 1500 rpm for 5 minutes and resuspended in fixative (3:1 methanol:acetic acid) added in a dropwise manner while continuously vortexing. Cells were incubated at room temperature for 5 minutes and centrifuged at 12 000 rpm for 2 minutes. The cells were washed twice in fixative (12 000 rpm for 2 minutes) before resuspension in 1 mL fresh fixative. Fixed cell suspension (3 µL) was dropped onto a glass slide and air-dried, and cell density was checked using a phase contrast microscope. Probe mixes were prepared according to manufacturer’s instructions and 2 μL was added and covered with a coverslip sealed with rubber solution. The slide was placed in a hybridization chamber, heated to 75°C for 5 minutes and then 37°C overnight. Coverslips were removed, and slides were washed in a 0.4× saline sodium citrate/3% Nonidet P-40 wash buffer at 72°C for 2 minutes and then a 2× saline sodium citrate/1% Nonidet P-40 wash buffer at room temperature for 2 minutes. 4′,6-Diamidino-2-phenylindole mounting medium (Vector Laboratories, Peterborough, UK) was applied to the slide; a coverslip was attached, and the slide was analyzed using a Zeiss Axio Imager Z2 and Cytovision software from Leica Biosystems. For multiprobe studies, probes to BCR-ABL fusion and deletions of p53 and iso(17)q were custom designed and manufactured (Empire Genomics) with BCR fluorescently labeled in green, ABL in Texas red, TP53 in gold, and MPO (iso17q) in aqua. All probes were used following the manufacturer’s instructions. Standard BCR-ABL pattern is R1G1F2. We also observed 2 patterns of atypical BCR-ABL profiles: R1G1F3 and R1G1F1.
Results
Immunophenotypic analysis of CML progression from CP to myeloid BP
We first compared the size of different immunophenotypic hemopoietic stem and progenitor (HSPC) compartments: HSC (Lin−CD34+CD38−CD90+CD45RA−), MPP (Lin−CD34+CD38−CD90−CD45RA−), LMPP (Lin−CD34+CD38−CD90−CD45RA+), CMP (Lin−CD34+CD38+CD45RA−CD123+), GMP (Lin−CD34+CD38+CD45RA+CD123+), and megakaryocyte-erythroid progenitor (MEP; Lin−CD34+CD38+CD45RA−CD123−) in normal, CP-, accelerated phase (AP)-, and myeloid BP-CML (Figure 1A-E). When compared with normal, CP-, and AP-CML (Figure 1A-C, respectively), in myeloid BP-CML, there are 2 distinct immunophenotypic patterns: a dominant MPP-like/CMP-like phenotype (Figure 1D) and a dominant LMPP-like/GMP-like phenotype (Figure 1E). Consequently, there are striking differences in the sizes of cellular subcompartments within the Lin−CD34+ population between normal, CP-, AP-, and myeloid BP-CML (Figure 2A-B). LMPP-like cells were <0.5% of Lin−CD34+ in normal, CP-, and AP-CML, but significantly increased with progression to LMPP-like/GMP-like BP-CML (18.1%; P < .01). GMP-like cells decreased as disease progressed from normal and CP- to AP-CML (P < .05), but significantly increased on progression to LMPP-like/GMP-like BP-CML (P < .05). The MEP fraction was significantly expanded in CP- and AP-CML (P < .01), but significantly decreased on progression to BP-CML (P < .001) (Figure 2A-B). In contrast, the size of the immunophenotypic HSC compartment remained relatively constant despite disease progression. In myeloid BP samples, with what we have termed an MPP-like/CMP-like profile, the overall percentage of CMP-like and MPP-like cells was not significantly increased.
In summary, we demonstrate dynamic changes in size of the different immunophenotypic HSPC-like compartments with disease progression in CML and heterogeneity of HSPC-like populations in myeloid BP-CML. As a next step, we proceeded to functionally characterize the different HSPC-like compartments in myeloid BP-CML.
LSC function in myeloid BP-CML
To identify which cell compartments contain leukemia-propagating function, we purified HSPC-like populations from 5 BP-CML patient samples (COL091, COL091R, CML371, CML002, and HER002) (Figure 3A). For patient COL091, 2 samples were tested: the initial myeloid BP diagnostic sample and a follow-up sample at relapse (COL091R), 9 months after therapy with palliative oral 6-mercaptopurine chemotherapy. Samples were tested for engraftment in primary immunodeficient murine recipients (Figure 3B). Human myeloid-only leukemic engraftment was detected in 4 of 5 samples (Figure 3C) by immunophenotype and FISH analysis for leukemia-associated mutations (see “Analysis of clonal evolution in HSPC populations in BP-CML”). HER002 failed to engraft despite injection of up to 2 × 105 cells (data not shown).
Two engrafting samples had large MPP-like/CMP-like compartments (COL091, CML371). In samples with expanded MPP-like/CMP-like populations, HSC-like, MPP-like, CMP-like, and MEP-like populations reproducibly engrafted. The GMP-like population variably engrafted, and the LMPP-like population was too small to purify.
Two engrafting samples had expansion of LMPP-like/GMP-like compartments (COL091R and CML002). In both samples, all HSPC-like populations that could be purified engrafted in primary recipient mice (Figure 3C). Because varying numbers of cells were available and thus injected from each immunophenotypic compartment, we considered whether failure to engraft primary recipients could be explained by injection of low cell numbers. In all 4 samples, there were examples of engrafting populations (black dots) that had been injected at lower cell numbers compared with nonengrafting populations (red dots) (Figure 3D). Failure to engraft may have occurred either because (i) LSC frequency was low in nonengrafting populations and cell numbers did not permit establishing LSC frequencies by limiting dilution analysis; (ii) cell populations were too small to purify (eg, MEP-like compartment in COL091); (iii) we had insufficient cell numbers to inject into several mice (eg, GMP-like compartment in CML371).
For 2 samples (COL091 and CML002), we purified cell populations from primary engrafted subpopulations (Figure 4A) and analyzed engraftment in secondary recipient mice (Figure 4B). There were 2 aims of the experiment. First, we wanted to establish if patient HSPC populations could serially engraft leukemia. For COL091, HSC-like, CMP-like, and GMP-like populations serially propagated leukemia (ie, had LSC function). For sample CML002, both the LMPP- and the GMP-like populations from the patient serially engrafted. Collectively, these data demonstrate multiple LSC populations in myeloid BP-CML.
Figure 4.

Hierarchical relationships of normal HSPCs are not maintained in BP-CML. (A) Percentage purities of population, after FACS sorting, injected for secondary transplant (Tx) from 2 patients (COL091, CML002). (B) Secondary engraftment of cells from 2 patients (COL091 and CML002). x-axis: bottom: patient sample populations injected into primary mice; top: population from primary engrafted mouse injected into secondary recipient. y-axis: mean percentage human (h)CD45+CD33+CD19− cell engraftment/total live MNC. Red line: engraftment threshold. Each dot represents 1 injected mouse: black dots: engrafted mice; red dots: mice that failed to engraft.
Second, we asked if the leukemic HSPC populations were organized in a hierarchical manner akin to the hierarchy seen in normal hemopoiesis. In COL091, the HSC-like population from the patient generated an HSC-like population and downstream progenitor populations (Figure 4A-B), but only the HSC-like and CMP-like populations from the primary mice engrafted secondary mice. Furthermore, when the CMP-like population from the patient was injected into primary mice, it generated an MPP-like population in addition to CMP-like and GMP-like progenitor-like populations. Both MPP-like and CMP-like populations from primary mice were able to engraft secondary recipients. However, although GMP-like cells from the patient-generated CMP-like and GMP-like populations, only the GMP-like cells from primary mice were able to engraft secondary mice. From patient CML002, both LMPP-like and GMP-like cells generated both LMPP-like and GMP-like populations in primary mice that could be purified and that were transplantable in secondary recipients. Based on these limited immunophenotypic analyses of engrafting populations, one interpretation of the data is that the leukemia is not hierarchically organized. An alternative interpretation is that the cell surface markers used to immunophenotype normal HSPC populations do not reliably separate out hierarchically organized populations in BP-CML.
Analysis of clonal evolution in HSPC populations in BP-CML
Clonal evolution, often associated with acquisition of additional cytogenetic abnormalities (ACAs) beyond t(9:22), is a marker of disease progression to AP- and BP-CML. Thus, we wanted to understand in which cell compartments ACAs were present in BP-CML by identifying clonal structures in myeloid BP-CML patients. Furthermore, given the multiple LSC populations in BP-CML, we asked if there was a correlation between LSC function and cytogenetic heterogeneity. Finally, we asked how accurately clonal structure in patients was captured in the experimental immunodeficient mouse model.
Karyotypic analysis of patient cells identified abnormalities in addition to t(9:22) in 4 patient samples (COL091, COL091R, CML371, and CML002) (supplemental Table 1). In 3/4 cases (COL91, COL091R, and CML002), we detected the ACAs by multicolor FISH at a level of >5%. Thus, we examined clonal structure in these cases in all available HSPC-like cell subpopulations from patient samples (Figure 5A-Ci-iii) and engrafted mice (Figure 6). For some subpopulations and in some engrafted mice, insufficient cells were available for analysis.
Figure 5.

Clonal structures of stem/progenitor populations in myeloid BP-CML. (A-C) Data from patients CML002, COL091, and COL091R. Clone identities denoted by circles or bars. (i) Immunophenotypic HSPC populations in patient sample. y-axis: proportion of population as percentage of Lin−CD34+ cells. (ii) Clonal composition of purified patient populations is based on FISH analysis. x-axis: HSPC population. y-axis: frequency of clones per population. (iii) Clonal hierarchies inferred from FISH data. (D) FISH images: ABL (red); BCR (green); BCR-ABL fusion (F); p53/17p13 (gold); MPO/17q22 (aqua). Atypical BCR-ABL is defined by 1 or 3 BCR-ABL fusions. 17p13 loss is defined by p53 loss. Isochrosome 17q is defined by 3 MPO signals. Numbers of signals detected are indicated.
Figure 6.

Analysis of clonal structures in NSG mice after transplantation of stem/progenitor populations. Frequency and type of leukemic clones in individual engrafted mice. Injected populations indicated. Results from 1° (A, B, C) and 2° transplantation (A and B) are shown. BM, bone marrow; NA, cells unavailable for FISH analysis.
Taking data from all 3 patient samples and engrafted mice together, the following points emerge. First, in the 3 patients, the ACAs involved heterozygous loss of chromosome 17p (detected with a TP53 probe), and in 2/3 cases, the ACAs also involved heterozygous loss of isochromosome 17q, consistent with previous data showing chromosome 17 aberrations are common in the clonal evolution of CML.15 Second, in all 3 cases, ACAs are detected in multiple immunophenotypic compartments regardless of which immunophenotypic compartments are expanded (Figure 5A-Ci-iii). Third, in all 3 cases, there were differences in clonal composition of cells in the patient and in the mice. For example, in the patient sample COL091, all 3 clones in patient cells had a standard BCR-ABL FISH signal (standard BCR-ABL, standard BCR-ABL plus 17p13 loss, and standard BCR-ABL plus 17p13 loss with isochromosome 17q) (Figure 5B). In contrast, in secondary transplanted mice, clones with an aberrant BCR-ABL signal were detected (Figure 6B). In 1 of these mice, the aberrant BCR-ABL clones comprised 90% of engrafted cells. In the relapsed sample COL091R, there was evidence of clones in the transplanted mice not detected in the patient sample (Figure 6C).
Discussion
BP-CML is a serious unmet clinical need with poor outcomes and 5-year survival rates <20% with the only aggressive, curative option, conventional chemotherapy followed by allogeneic stem cell transplant. Thus, there is an urgent need to improve our understanding of the biology of BP-CML to identify novel improved therapeutic strategies. As a step toward this goal, we have fully characterized the leukemia stem and progenitor cell populations in myeloid BP-CML.
We demonstrate in myeloid BP-CML the sizes of the immunophenotypic stem/progenitor compartments are heterogeneous and often show expanded progenitor populations. Serial transplantation indicates that LSCs can reside in any of the immunophenotypically defined HSPC populations. Concordantly, we show that ACAs are also present in the multiple immunophenotypic HSPC-like populations with LSC potential.
Our data are in contrast to previous reports that suggested LSCs in myeloid BP-CML were present only in a transformed GMP population.14,16 However, these studies did not systematically assess stem cell function in all human immunophenotypic HSPC compartments in immunodeficient mice. Considering the limited number of samples we have assessed, further studies will be required to validate how representative our data are of myeloid BP-CML.
Separately, data presented here are consistent with and extend our prior studies, which demonstrated expanded progenitor compartments in de novo AML and established the presence of functional LSCs in GMP-like and LMPP-like populations11 by now showing functional LSCs present in MPP-like/CMP-like and HSC-like compartments. However, an important caveat in the interpretation of the data is that cell surface marker expression used to define current normal HSPCs compartments may be altered by transformation leading to aberrant marker expression. This caveat prevents an unequivocal determination of whether the varied immunophenotypically defined stem/progenitor populations are truly different. Future studies of global RNA profiling of myeloid BP-CML LSCs will address this question as we have done previously in AML.11,12 However, it is noteworthy that in our previous study,11 immunophenotypic primary human LMPP-like and GMP-like LSC populations were most closely related to normal LMPP and GMP, not just on the basis of cell surface markers but also on the basis of the whole transcriptomes.
The inability to relate the leukemic cell populations back to normal HSPC beyond immunophenotype also does not allow us to determine in which compartment ACAs are first acquired; rather a more limited conclusion can be made that they are present in all leukemic stem/progenitor compartments.
In all 3 samples, we demonstrate that clonal structures in the patient are not faithfully recapitulated in the mouse (striking examples, Figure 6Biii and Figure 6Ciii). These discrepancies may arise because of clonal selection in the mouse of minor clones from the patient, not detected by FISH (100 cells were analyzed by FISH), suggesting the selection pressures for clonal expansion in the patient and the mouse differ. Alternatively, it is possible that clonal evolution occurs in the mouse. We cannot distinguish between these possibilities. Regardless of which explanation is correct, the data would support the hypothesis that the widely used NSG immunodeficient mouse model does not accurately model clonal competition and stem cell function. This mirrors other recent data wherein engrafting potential did not match clonal composition in the patient.12,17
In summary, our data conclusively show that functional LSCs reside in multiple immunophenotypically distinct HSPC populations in myeloid BP-CML. Moreover, FISH analysis demonstrates clonal evolution in all HSPC compartments, including the HSC-like populations. Future studies will focus on identifying deregulated pathways (in addition to WNT/β-catenin) in myeloid BP-CML, which may be amenable to therapy in combination with TKIs to reduce resistance and improve patient outcomes.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients who kindly donated samples, the staff in the MDSBio study, Glasgow and Liverpool for processing samples, and the UK National Cancer Research Network CML Subgroup. The authors are grateful to Avril Morris for assistance with access to fluorescence microscopy.
D.K. and P.V. were supported by Cancer Research UK (CRUK) Program grant CRUK: C7893/A12796; P.V. acknowledges funding from Medical Research Council (MRC) Disease Team Award I (G1000729) and Disease Team Award II (MR/L008963/1), the MRC Molecular Haematology Unit Award (MC_UU_12009/11), and the Oxford Partnership Comprehensive Biomedical Research Centre (National Institute for Health Research Biomedical Research Centre Funding scheme). M.C. acknowledges funding from Scottish Funding Council (SCD/04) and Bloodwise (11017). This study was supported by the Glasgow Experimental Cancer Medicine Centre, which is funded by CRUK and by the Chief Scientist's Office (Scotland). The Howat Foundation and Kay Kendall Leukaemia Fund support the FACS facility at the Paul O’Gorman Leukaemia Research Centre.
Authorship
Contributions: D.K., R.K., N.G., H.M., and M.C. performed and designed experiments and analyzed data; M.H., L.R., R.E.C., and M.C. provided patient samples; R.E.C. and P.V. designed experiments and analyzed data. All authors contributed to writing the manuscript.
Conflict-of-interest disclosure: M.C. has received research funding from Bristol-Myers Squibb and honoraria from Bristol-Myers Squibb, Novartis, Pfizer, and Ariad. R.E.C. has received research funding and honoraria from Novartis, BMS, and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Paresh Vyas, MRC Molecular Haematology Unit and Centre for Haematology, Weatherall Institute of Molecular Medicine and Cancer and Haematology Unit, Churchill Hospital, University of Oxford and Oxford University Hospitals NHS Trust, Oxford OX3 9DU, United Kingdom; e-mail: paresh.vyas@imm.ox.ac.uk; and Mhairi Copland, Paul O'Gorman Leukaemia Research Centre, College of Medical, Veterinary, & Life Sciences, Institute of Cancer Sciences, University of Glasgow, Gartnavel General Hospital, 1053 Great Western Rd, Glasgow G12 0ZD, United Kingdom; e-mail: mhairi.copland@glasgow.ac.uk.
References
- 1.Jabbour EJ, Cortes JE, Kantarjian HM. Tyrosine kinase inhibition: a therapeutic target for the management of chronic-phase chronic myeloid leukemia. Expert Rev Anticancer Ther. 2013;13(12):1433-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737-747. [DOI] [PubMed] [Google Scholar]
- 3.Soverini S, Colarossi S, Gnani A, et al. ; GIMEMA Working Party on Chronic Myeloid Leukemia. Contribution of ABL kinase domain mutations to imatinib resistance in different subsets of Philadelphia-positive patients: by the GIMEMA Working Party on Chronic Myeloid Leukemia. Clin Cancer Res. 2006;12(24):7374-7379. [DOI] [PubMed] [Google Scholar]
- 4.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shlush LI, Zandi S, Mitchell A, et al. ; HALT Pan-Leukemia Gene Panel Consortium. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia [published correction appears in Nature. 2014;508(7496):420 (Note: Yousif, Fouad [added])]. Nature. 2014;506(7488): 328-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woll PS, Kjällquist U, Chowdhury O, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo [published correction appears in Cancer Cell. 2014;25(6):861 and Cancer Cell. 2015;27(4): 603,-605]. Cancer Cell. 2014;25(6): 794-808. [DOI] [PubMed] [Google Scholar]
- 8.Busque L, Patel JP, Figueroa ME, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goardon N, Marchi E, Atzberger A, et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138-152. [DOI] [PubMed] [Google Scholar]
- 12.Quek L, Otto GW, Garnett C, et al. Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med 2016;213(8):1513-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. 1999;94(6):2056-2064. [PubMed] [Google Scholar]
- 14.Jamieson CH, Ailles LE, Dylla SJ, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351(7):657-667. [DOI] [PubMed] [Google Scholar]
- 15.Verma D, Kantarjian H, Shan J, et al. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer. 2010;116(11):2673-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abrahamsson AE, Geron I, Gotlib J, et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci USA. 2009;106(10):3925-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klco JM, Spencer DH, Miller CA, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.