

Key Points
We identify genes prognostic of disease relapse in patients allografted for AML.
Mutational profiles often change at relapse postallograft, which may have implications for the design of posttransplant interventions.
Abstract
Disease relapse is the major cause of treatment failure after allogeneic stem cell transplantation (allo-SCT) in acute myeloid leukemia (AML). To identify AML-associated genes prognostic of AML relapse post–allo-SCT, we resequenced 35 genes in 113 adults at diagnosis, 49 of whom relapsed. Two hundred sixty-two mutations were detected in 102/113 (90%) patients. An increased risk of relapse was observed in patients with mutations in WT1 (P = .018), DNMT3A (P = .045), FLT3 ITD (P = .071), and TP53 (P = .06), whereas mutations in IDH1 were associated with a reduced risk of disease relapse (P = .018). In 29 patients, we additionally compared mutational profiles in bone marrow at diagnosis and relapse to study changes in clonal structure at relapse. In 13/29 patients, mutational profiles altered at relapse. In 9 patients, mutations present at relapse were not detected at diagnosis. In 15 patients, additional available pre–allo-SCT samples demonstrated that mutations identified posttransplant but not at diagnosis were detectable immediately prior to transplant in 2 of 15 patients. Taken together, these observations, if confirmed in larger studies, have the potential to inform the design of novel strategies to reduce posttransplant relapse highlighting the potential importance of post–allo-SCT interventions with a broad antitumor specificity in contrast to targeted therapies based on mutational profile at diagnosis.
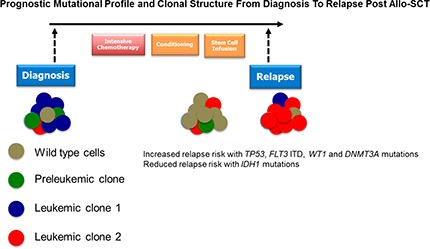
Introduction
Allogeneic stem cell transplantation (allo-SCT) is an increasingly important treatment in adult acute myeloid leukemia (AML). Although allo-SCT delivers potent antileukemic therapy through both dose intensification and the genesis of a potent graft-versus-leukemia (GVL) effect, disease relapse occurs in 30% to 70% of patients and represents the major cause of treatment failure.1,2 Novel treatment strategies to reduce disease relapse are therefore urgently required. A number of disease- and transplant-specific factors such as presentation karyotype, disease status at transplant, and intensity of conditioning regimen are known to predict relapse risk (RR).3-5 Although mutations in both FLT3 and NPM1 are prognostic of posttransplant outcome, there has been only limited analysis of the prognostic impact of mutations in other AML-associated genes after allo-SCT.6 Similarly, there have been no studies of changes in clonal structure in longitudinal samples from diagnosis, from pretransplant, and at relapse. This has hampered the rational development of novel treatment strategies to reduce post–allo-SCT relapse.
Next-generation sequencing (NGS) has identified almost all common recurrent exonic genetic mutations in AML. Importantly, NGS identified gene mutations prognostic of clinical outcome after intensive chemotherapy (IC), as well as identifying molecular targets for targeted therapy.7-11 These studies provided insight into AML clonal structure at both disease presentation and relapse after IC.12,13 Taken together, such studies refined risk stratification in IC-treated patients. Given the distinct kinetic and curative mechanisms in allo-SCT, compared with IC, it is plausible that different disease-related AML-specific mutations may be prognostic of relapse post–allo-SCT.
Posttransplant administration of either cellular or pharmacological therapy represents a key approach to reduce disease relapse after allo-SCT. Such therapies can augment GVL (eg, donor lymphocyte infusions [DLI]14 or checkpoint inhibitors15), or manipulate disease relapse kinetics (eg, FLT3 inhibitors or Azacitidine16-19). Critically, rational deployment of posttransplant interventions would be aided by greater understanding of actionable AML mutations in recurrent disease postallograft, which is currently limited.
To begin to address these deficits, we have characterized genes prognostic of outcome after allo-SCT and studied clonal structures at diagnosis and disease relapse.
Patients and methods
These studies were approved by the local institutional review board under protocol 06/Q1606/110 (MDSBio).
Patients and transplant characteristics
One hundred thirteen patients allografted for AML at 2 major United Kingdom transplant centers were studied (demographics summarized in Table 1). All patients provided consent for data collection. Presentation karyotype was available in 112 patients and was classified according to Medical Research Council (MRC) criteria.20 One hundred five patients were in remission at the time of transplant (complete remission 1 [CR1], n = 78; CR2, n = 27). Seventy-five patients were transplanted using a reduced intensity conditioning regimen according to European Society for Blood and Marrow Transplantation criteria, and 38 received a myeloablative conditioning regimen. Forty-nine patients had disease recurrence with >5% blasts morphologically posttransplant.
Table 1.
Characteristics of patient cohort
| Characteristic | Number | Percentage |
|---|---|---|
| Age, y | ||
| Age range | 16-70 | |
| Median age | 49 | |
| Sex | ||
| Male | 58 | 51 |
| Female | 55 | 49 |
| Cytogenetic risk stratification | ||
| Favorable risk | 5 | 4 |
| Intermediate risk | 85 | 75 |
| Adverse risk | 22 | 20 |
| Unknown | 1 | 1 |
| Disease status at transplant | ||
| CR1 | 78 | 69 |
| CR2 | 27 | 24 |
| Not in CR | 8 | 7 |
| Relapse posttransplant | 49 | 43 |
| Transplant conditioning | ||
| Reduced intensity | 75 | 66 |
| Myeloablative | 38 | 34 |
| Conditioning regimen | ||
| Flu/Mel/Campath | 58 | 51 |
| Cy/TBI | 29 | 26 |
| Flu/Mel | 9 | 8 |
| Flu/Cy/TBI | 5 | 4 |
| Bu/Cy | 5 | 4 |
| FLAMSA/TBI/ATG | 3 | 3 |
| FLAMSA/Bu/ATG | 1 | 1 |
| TBI/Flu/Cy/ATG | 1 | 1 |
| Cy/TBI/Campath | 1 | 1 |
| Bu/Flu/Thio/ATG | 1 | 1 |
| Donor source | ||
| Sibling | 49 | 43 |
| Unrelated | 61 | 54 |
| Cord | 3 | 3 |
| CMV recipient/donor | ||
| Negative/Negative | 44 | 39 |
| Positive/Positive | 33 | 29 |
| Positive/Negative | 25 | 22 |
| Negative/Positive | 11 | 10 |
| Equivocal/Negative | 1 | 1 |
| Status at last contact | ||
| Alive | 61 | 54 |
| Dead | 52 | 46 |
ATG, antithymocyte globulin; Bu, busulfan; CMV, cytomegalovirus; Cy, cyclophosphamide; FLAMSA, fludarabine, cytarabine, amsacrine, cyclosphosphamide; Flu, fludarabine; Mel, melphalan; Thio, thiotepa; TBI, total body irradiation.
NGS targeted resequencing
Targeted resequencing was performed on 35 genes frequently mutated in AML and myeloid malignancies on genomic DNA (gDNA) from bone marrow aspirate (BMA) samples obtained at presentation in all 113 patients. A similar analysis was performed on BMA samples from 29 patients who relapsed postallograft. In 15 of these patients, additional samples collected immediately prior to transplant were subjected to a similar analysis allowing comparative analysis of genetic mutations at diagnosis, pretransplant, and relapse. Multiplex polymerase chain reaction (PCR) was performed with 373 amplicons on a Fluidigm Access Array (supplemental Table 1A). Amplicons covered areas with high frequency of AML gene mutations (hotspots). If there were no hotspots previously reported in COSMIC (http://cancer.sanger.ac.uk/cosmic), the entire exon was studied. The exons covered in each of the 35 genes are listed in supplemental Table 1B. Sequencing was performed using Illumina MiSeq (300-bp paired-end). Greater than 1.1 × 105 bases were covered in paired-end sequencing per sample, with an average read depth of 1339 reads (range 0-3308) per amplicon (supplemental Table 1A). Technical replicates were performed with 2 amplicon libraries per sample in independent experiments.
Bioinformatics
Sequencing quality was assessed using FASTQC (Samtools) and aligned using a Burroughs-Wheeler Aligner algorithm in Stampy.21 A Phred score of 30 was set as a minimum quality threshold for variant calling. We used 3 different variant callers: GATK,22 VARSCAN,23 and Pindel.24 As germ line DNA was not available for analysis, we implemented criteria to optimize calling of disease-associated mutations and to exclude likely germ line single-nucleotide polymorphisms or technical artifacts. Inclusion criteria for variant calling and filtering were as follows: (i) nonsynonymous mutations; (ii) mutations in protein coding regions; (iii) concordance between technical replicates; (iv) concordance between GATK and VARSCAN; (v) previous documentation as a somatic mutation in hematopoietic samples in COSMIC; (vi) novel truncating variants (nonsense, deleterious missense/indels, variants affecting splicing) not found in human variation databases (ESP6500, 1000 genomes, and dbSNP) with a frequency of >0.0014 (0.14%); (vii) novel variants not previously reported but occurring in the same codon as a previously documented somatic variant; (viii) novel variants not previously reported but occurring within 3 codons of a previously documented somatic variant. Putative variants were filtered using the following parameters: minimum coverage 100 reads; minimum variant frequency 0.05; minimum read depth of variant 5; and P value <.05. Additional exclusion criteria for variants were the following: (i) variants predicted to result in a silent amino acid change; (ii) known polymorphisms present in human variation databases at a population frequency of >0.0014 (0.14% reflecting the population incidence of myeloid disease) except for when the variant is a known somatic variant in COSMIC; (iii) variants not previously noted in hematopoietic disease (documented in <1 hematopoietic samples in COSMIC); (iv) 1-bp indels present adjacent to regions of >4 homopolymer bases. Putative variants were further validated by visualization using the Integrated Genome Viewer. Recurrent somatic variants previously reported in hematological disease in COSMIC but not in myelodysplastic syndromes or AML were validated by Sanger sequencing.
Identification of FLT3 internal tandem duplication (ITD) by capillary electrophoresis and NGS
We identified the FLT3 ITD mutation by semiquantitative PCR of gDNA and/or complementary DNA and fluorescent capillary electrophoresis. Estimates of FLT3 ITD variant allele frequency (VAF) were made by area-under-peak analysis. We detected FLT3 ITD by NGS on 60% (26/43) of patients known to be FLT3 ITD-positive by capillary electrophoresis (supplemental Table 1C), using primers that were designed to cover exons 13-15 in the FLT3 gene. We used Pindel in addition to VARSCAN and GATK to improve the detection rate. We then interrogated the NGS reads for the presence of the ITD using a sequence specific for that patient’s ITD. Using this method, we estimated the threshold of detection to be ∼0.1% (or 1:1000).
Droplet digital PCR
Our ability to detect mutations with low VAFs by NGS was, on occasion, hampered by high false positive VAFs (range from <0.1% to 1.8%; assessed using wild-type control gDNA). In cases where this background was >0.1%, droplet digital PCR (ddPCR) improved sensitivity of detection by 10- to 100-fold (up to 0.002% in our study). ddPCR was also discriminatory for NGS variant calls where the NGS VAF was close to (within twofold) the background threshold.
We used ddPCR to look for mutations detected at relapse but apparently absent or ambiguous at diagnosis and/or CR in 5 patients using the BIORAD platform.10 Sequences of primers (Thermo Fisher Scientific, Hemel Hempstead, United Kingdom) and dual-labeled hydrolysis probes (3′ fluorescent reporter: hexachloro-fluorescein–labled probe complementary to wild-type sequence, 6-FAM–labeled probe complementary to mutant sequence; and a 5′ quencher: BHQ-1; Eurofins Genomics, Ebersberg, Germany) are listed in supplemental Table 1D. gDNA (40-60 ng/well) or whole-genome amplification gDNA (2 μL/well of a 1:20 whole-genome amplification product, RepliG; Qiagen, Manchester, United Kingdom) was subjected to ddPCR, and fluorescence of droplets was analyzed using a 2-color detector (FAM: wild-type/hexachloro-fluorescein: mutant). Wild-type gDNA was spiked with a mutant control sample of known VAF to give a predicted VAF of 0.1% as a detection control. Detection controls for all individual variants were successfully scored mutant with an average VAF of 0.074% (range 0.044% to 0.102%), whereas all pure wild-type controls did not show any mutant droplets.
Cytogenetic analysis
Cytogenetic analysis of patient samples was performed at local hospitals by chromosomal banding analysis (maximum 20 metaphases) and fluorescent in situ hybridization. Karyotypes were classified using published MRC AML group criteria as described by Grimwade et al.20
Statistical methodology
Statistical analyses were performed in Stata 14.1. Univariate and multivariate Cox proportional hazards models were used for the following outcomes: relapse-free survival (RFS) and overall survival (OS). RR was analyzed using a competing risks model (Fine and Gray)25 considering death as a competing risk. Survival analysis was started at date of transplant.
Multivariate analysis included adverse cytogenetics, age, donor source, transplant conditioning (myeloablative/reduced intensity), disease status at time of transplant, and cytomegalovirus status of recipient and donor. A gene mutation was only included in the univariate and multivariate analyses if at least 5 patients had a mutation in that gene. Each gene mutation was assessed individually with covariables described above. Given the small numbers of patients with each mutation, P values <.1 are considered significant in this exploratory analysis of potential prognostic factors. Forest plots were created in R and contain all gene mutation hazard ratios for outcome.
Results
Mutation profile of patient cohort at diagnosis
Our patient cohort consisted of AML patients wherein allo-SCT was offered on the basis of age, performance status, cytogenetic and molecular risk, and response to induction chemotherapy. We detected 262 mutations in 102/113 (90%) patients (Figure 1A; supplemental Table 2; supplemental Table 3A). The frequency of mutations in the studied genes is shown in Figure 1B. The mean number of mutations per patient in our cohort is 2.3 (median = 3) compared with a mean of 3.4 and 5.2 mutations in the 2 other large published cohorts of 200 and 1540 unselected adults with AML7,11 (Figure 1C). However, as we used a more limited panel of amplicons, the breadth of coverage of mutations is less. Therefore, we compared our data with data from these 2 cohorts curated to mirror the same breadth of genotyping as we had performed (Figure 1D). This cohort of allo-SCT patients is significantly enriched for the presence of FLT3 ITD, KIT, TET2, RUNX1, and SRSF2 mutations and depleted for NPM1 mutations and point mutations in FLT3 (Figure 1D).
Figure 1.

Mutation profile of patient cohort at diagnosis. (A) Heat map detailing mutation distribution among patient cohort with individual cytogenetic risk stratification and relapse/nonrelapse status post–allo-SCT. (B) Frequency of mutations in the cohort. (C) The number of mutations detected per patient in our cohort. (D) Comparison of our cohort with 2 published cohorts of de novo AML patients from the Cancer Genome Atlas (TCGA)7 and Papaemmanuil et al11 (Papa). TCGA and Papa datasets were curated to exclude mutations that would not have been covered by our NGS panel (TCGA-restricted and Papa-restricted). Fisher’s exact test was used to compare frequency of mutations in the indicated genes between our cohort and the restricted TCGA and Papa cohorts. Genes and frequencies highlighted in blue are those where P < .05.
Impact of diagnostic mutational profile on transplant outcome
Forty-nine patients relapsed after allogeneic transplantation. In multivariate analysis mutations in WT1 (P = .018), DNMT3A (P = .045), FLT3 ITD (P = .071), and TP53 (P = .06) were associated with an increased RR (Figure 2A). The presence of a TP53 mutation was associated with decreased RFS (P = .025) and OS (P = .007) (Figure 2B-C). IDH1 mutation was associated with a reduction in RR (P = .018) and improved RFS (P = .024) and OS (P = .032) (Figure 2A-C). There was no correlation between the number of mutations detected per patient and outcome (data not shown).
Figure 2.

Forest plots of RR, OS, and RFS (multivariate analysis) in our patient cohort. (A) RR per gene mutation in all patients. (B) OS per gene mutation in all patients. (C) RFS per gene mutation in all patients. *P < .10. CI, confidence interval; HR, hazard ratio.
Genetic characterization of disease relapse posttransplant
In order to further characterize the molecular basis of disease relapse after allo-SCT, we next performed mutational analysis in 29 patients wherein paired diagnostic and relapse samples were available (Figure 3A; supplemental Table 3B). Changes in genetic profile (acquisition or loss of genetic or karyotypic abnormalities) were documented at disease relapse in 23/29 (79%) patients. This suggests that therapy (either IC or allo-SCT) selects changes in clonal structure in most patients.
Figure 3.

Patients with or without genetic changes between diagnosis and relapse. (A) One hundred thirteen patients in our study were divided into those who relapsed after allo-SCT and those who did not. Patients were further classified according to those where changes to the profile of genetic aberrations, either in karyotype or in genetic mutations between diagnosis and relapse. n, number of patients. (B) Graph showing 13 patients who had changes (gains and losses) of the number of genetic mutations between diagnosis and relapse.
In 13/23 patients, a distinct genetic mutational profile was documented at relapse, 6 of whom demonstrated concurrent cytogenetic evolution. Genetic mutations, either not detected at diagnosis or present at VAF of <2% (in patients 56 and 112), were subsequently detected at VAF frequencies of at least 5% in 10/13 patients. In these 13 patients, 7 patients had gains only; 3 had losses only, and 3 had gains and losses (Figure 3B). On average, 1.3 additional mutations were detected at relapse compared with diagnosis. Mutations either acquired de novo, or selected for, at relapse included the following: TET2 (n = 4), NRAS, WT1, ETV6, RUNX1, DNMT3A, TP53 (n = 2), NPM1, IDH1, FLT3 ITD, and PHF6.
We detected change in karyotype alone in 10 patients and, consistent with previous reports, a gain of 4.6 karyotypic abnormalities per patient was observed.26 Of note, 10/23 (43%) patients with paired karyotyping analysis acquired complex karyotypic abnormalities (>3) between diagnosis and relapse (supplemental Tables 4-6).
Genetic characterization of disease at relapse in comparison with pretransplant characteristics
Next, we asked if genetic mutations detected at posttransplant relapse were detectable prior to allo-SCTafter IC. If mutations were detectable, then pretransplant sampling could be of value in predicting mutational composition at relapse. Furthermore, mutations detected prior to transplant and at relapse, but not at diagnosis, are likely selected by IC.
In 14/29 relapsed patients (11 in CR and 3 not in CR: patients 61, 62, and 84) BMA samples taken immediately prior to transplant were tested. In another patient (65), we tested a pretransplant sample taken 1 month prior to the patient achieving CR pre–allo-SCT, when the patient had active disease (∼50% blasts). NGS and ddPCR afforded a detection threshold between ∼1:300 (0.35%, range 0.01%-4.34%) and 1:30 000 (0.003%, range 0.002%-0.013%), respectively. We detected mutations pre–allo-SCT in 9/15 patients (patients 60, 65: supplemental Table 4; patients 35, 84, 92: supplemental Table 5; patients 1, 61, 62, 64: supplemental Table 7; examples shown in Figure 4A-C) with a wide range of VAFs (0.06%-85.13%). These mutations were previously implicated in preleukemic and leukemic hemopoiesis (ASXL1, DNMT3A, and TET2, IDH2 and NPM1, PHF6, PTPN11, SRSF2, RUNX1, TP53, and WT127-33). Taken together, this demonstrates a substantial clonal burden pre–allo-SCT.
Figure 4.

Graphs showing changes to genetic mutation profile variant at diagnosis, post-IC/pre–allo-SCT, and relapse posttransplant in AML patients. The key shown in (A) applies to (A-C). (A) Patients (61 and 62) where genetic mutations are present at diagnosis were detected at CR and recurred at relapse. (B) Patients (35 and 68) who acquired new mutations at relapse that were not detected at diagnosis or CR. (C) Patients (65 and 92) who acquired new mutations at relapse that were detected pre–allo-SCT. (D) The changes to clonal structure in patients 65 and 92 from diagnosis (Dx) (blue shading), pre–allo-SCT (green shading), and relapse (pink shading) inferred from changes of VAF. Two possible examples of clonal structure of 65 and the most likely structure for 92 are depicted. Letters represent mutations (shown in key) that cooccur in the same clone/ subclone. Red X, elimination of subclone.
In 9 patients where mutations were seen pre–allo-SCT, 3 patterns of changes were seen. First, in 4 patients, the same genes were mutated at diagnosis, pre–allo-SCT and relapse (patients 1, 61, 62, 64, supplemental Table 7; examples in Figure 4A), indicating failure of IC to eradicate clones that subsequently drive relapse post–allo-SCT. Patients 61 and 62 had persistent disease pretransplant, with ∼35% and ∼50% blasts, respectively, and additional cytogenetic anomalies were detected pre–allo-SCT. In patient 61, clones with t(3;5) and t(5;16) are not present at diagnosis, present pretransplant, and are lost at relapse. In contrast, mutations in NPM1 and WT1, and the FLT3 ITD, are present at changing VAFs at all time points. One interpretation is that the cytogenetic abnormalities t(3;5) and t(5;16) may have been acquired after the mutations. If true, clones with these karyotypic abnormalities are likely selected by IC but eliminated post–allo-SCT (Figure 4C).
Second, in 3 patients, mutations detected at relapse were not present at diagnosis or immediately prior to allo-SCT (patients 25, 35, and 68; supplemental Tables 4-6; examples in Figure 4B), indicative of clones selected post–allo-SCT. Again there was heterogeneity in the patterns of mutational change. In patient 35, SRSF2 and IDH2 mutations (VAF 41% to 44% at diagnosis and CR) were likely clonal heterozygous mutations that persisted from diagnosis to CR. This is consistent with these mutations being in preleukemic clones (Figure 4B). At relapse, the VAF of these mutations dipped to 21%, but mutations in NRAS Q61K and DNMT3A G706W were detected at a VAF of 18% to 21%, consistent with these being markers of a leukemic relapse clone.
In patient 68, the data are most consistent with an NPM1 mutant leukemic clone that was cytoreduced by IC such that at CR it was not detected (Figure 4B). At relapse, the NPM1 mutant clone reemerged and 2 TET2 mutations were detected for the first time. However, as there was ∼15% residual donor cells in the relapse sample (inferred from cytogenetic analysis), we cannot exclude the possibility that the TET2 mutations were donor derived. Both TET2 mutations have previously been reported in myelodysplastic syndrome.34
Finally, in 2 patients (65 and 92), complex and distinct changes in clonal structures occurred from diagnosis to pre–allo-SCT (presumably reflecting selection by IC or possibly de novo mutation), and from pre–allo-SCT to relapse (Figure 4C; summarized in Figure 4D and supplemental Tables 4 and 5).
At diagnosis, 1 interpretation of the data for patient 65 is that there is at least 1 stem clone with at least 3 clonal heterozygous mutations (RUNX1 D198G, SRSF2 P95A, and TET2 V1718L: VAF 48% to 56%). In addition, there is a subclonal RUNX1 L175P mutation (VAF 7%). In the intermediate sample taken after the second course of IC when there was persistent disease (∼55% blasts), there is loss of a subclone with RUNX1 L175P mutation, but emergence of a subclone with PHF6 C85X (VAF 25%), which if heterozygous, may be present in all blasts. There is a difference in the VAF between TET2 (51%) on the 1 hand and SRSF2 and RUNX1 D198G (34% to 39%) on the other hand. It is unclear if a difference in VAF between TET2 and SRSF2 and RUNX1 is due to technical error or if TET2 is present in >1 clone. At relapse, post–allo-SCT, a TET2 Q1084P mutation (VAF 27%), is detected for the first time, suggesting ongoing clonal selection, although it remains possible that this variant could be donor derived. Both TET2 Q1084P and trisomy 13 mark the relapse clone.
This complex, shifting clonal landscape is also seen in patient 92. At diagnosis, the following mutations are detected: CSF1R G747R, TET2 H949R (VAF 46% to 47%), and SRSF2 P95L (VAF 72%). As SRSF2 mutations are rarely homozygous, it is possible that the VAF of >50% in SRSF2 is consistent with an additional copy of mutated SRSF2. The patient also had an IDH1 R132C mutation (VAF 41%). At CR CSF1R and TET2, VAFs are unchanged, but the SRSF2 VAF falls to 44%, concurrent with a loss of the IDH1 mutant clone. At CR, a subclonal TP53 R136H mutation (VAF 4%) is detected. At relapse, the mutant TP53 R136H clone expands to likely become clonal (VAF 49%) with a further likely clonal TP53 P48fs mutation (VAF 50%), which is most likely on the other TP53 allele. Accompanying this, at relapse, 5 patient male clones are detected, 4 of which have karyotypic abnormalities (none of which are complex) together with some residual female donor cells. In both cases, analysis of sequential samples provides some insight into potential changes in clonal structure through therapy. In these 2 cases and in other previous cases, definitive description of clonal structures requires single-cell genotyping approaches.
Taken together, these sequential mutational snapshots from diagnosis to pre–allo-SCT and relapse, on limited numbers of patients, are highly informative. They show the considerable heterogeneity in evolving clonal landscapes with selection from diagnosis with IC and further selection postallograft.
Discussion
The development of novel therapeutic strategies to reduce relapse in patients allografted for AML is critically dependent on characterization of the biology of disease recurrence after allo-SCT. We have identified genes prognostic of disease relapse after allo-SCT. In addition, our data demonstrate evolution of clonal structure at relapse. Both observations have implications for clinical practice. We performed mutation profiling using targeted NGS for recurrent mutations in 35 genes in myeloid malignancy in 113 patients receiving an allo-SCT, 49 of whom relapsed. In contrast to previous studies of patients treated with IC, this transplant cohort was enriched for FLT3 ITD, KIT, TET2, RUNX1, and SRSF2 mutations and underrepresented for NPM1 mutations. Although mutations in TET2, RUNX1, SRSF2 are typically associated with myelodysplastic syndrome and secondary AML, many patients with a clinical history of de novo AML have mutations in these genes, and other genes that typically were first identified in chronic myeloid malignancies.11 Importantly, mutational profiling in secondary AML, therapy AML and de novo AML, improves the classification of AML ontogeny.35 Taken together, these data demonstrate the potential utility of detailed genetic characterization to better characterize AML allo-SCT patient cohorts.
At diagnosis, mutations in WT1, FLT3, DNMT3A, and TP53 are associated with an increased risk of relapse posttransplant, and IDH1 mutations associated with a reduced RR. Both FLT3 ITD and TP53 mutations have previously been correlated with an increased risk of relapse after allo-SCT.5,6,36 Furthermore, a similar study (112 patients and resequencing of 26 genes) has recently reported that mutations in WT1 correlate with an increased RR post–allo-SCT.6 It is not known if the immunogenicity of WT1 mutant clones is altered, but our studies support further evaluation of WT1 mutations on tumor alloreactivity. Mutations in DNMT3A and FLT3 ITD were correlated with increased RR but not decreased survival. These 2 genes are frequently mutated in the same patients (7/11 DNMT3A-mutated relapsed patients also had FLT3 ITD) but are largely mutually exclusive with TP53 and WT1 mutations, where patients do have poorer survival. Taken together, this raises the possibility that DNMT3A/FLT3 ITD patients are more responsive to salvage therapies. Previous studies of IDH1 mutations on outcome after IC have reported either no impact on or an increased rate of disease recurrence.37-39 Therefore, our data demonstrating a reduced risk of relapse after allo-SCT in patients with an IDH1 mutation raise the possibility that the genetic factors determining outcome after IC may differ from those determining relapse after allo-SCT.
Caution should be exercised in the interpretation of our data given our small cohort size. Nevertheless, our data clearly support further larger studies, which should ideally be prospective. A further limitation of our study is that, compared with more extensive genotyping methods (eg, whole exome/ genome sequencing), we may have failed to identify mutations in less frequently mutated AML-associated genes, and critically, variants in genes that regulate immune response. These variants will likely shed important insights into both GVL and graft-versus-host disease and are likely to be missed by studies focusing on patients treated only with IC. In this context, it is of interest that IDH1 mutations modulate DNA methylation status in leukemic blasts40 and potentially their ability to be recognized by the donor alloimmune response. Such an argument may equally apply to other variants that regulate the epigenome.
Our serial characterization of the mutational landscape, albeit in only 15 patients, from diagnosis, to pre–allo-SCT, and at relapse provides a window on the remarkable heterogeneity of clonal dynamics during therapy. Despite this heterogeneity, some principles may be emerging, which need to be validated through larger studies. First, AML-associated mutations present at diagnosis were commonly detected prior to allo-SCT (in 9/15 patients who relapsed). These patients subsequently relapsed with reexpansion of preexisting clones (4/9 patients), or with evidence of subclonal selection rather than through development of an unrelated disease-driving clone (5/9 patients). It is difficult to definitively prove that emergence of new clones is due to subclonal selection or acquisition of new mutations from therapy, as it is usually impossible to rule out selection of minor clones present below the threshold of detection. However, much of current data supports clonal selection.41 Second, detection of subclonal change in the BMA immediately prior to transplant suggests that in some patients genetic drivers of disease relapse are selected by pretransplant chemotherapy, which may play an important role in determining RR posttransplant. It also highlights the potential importance of genetic analysis immediately prior to transplant, rather than just at diagnosis, in terms of determining RR. Third, in a proportion of patients, mutations documented at relapse postallograft were not detectable at presentation or immediately prior to transplant. Thus, distinct posttransplant selection pressures may operate in such patients. These data also confirm that care must be exercised in choice of genetic markers to detect minimal residual disease (MRD)10 to monitor outcome posttransplant. Flow MRD, especially quantitation of leukemic stem cell compartments42 post–allo-SCT, is feasible and may be a more stable MRD technique in this setting.43 Fourth, in 10/23 patients whose genetic profile changed from diagnosis to relapse did so by acquiring a complex karyotype at relapse, consistent with previous data.26,44 Frequent occurrence of complex karyotype cells at relapse raises the hypothesis that these cells have a clonal advantage. Thus, genes that either drive chromosomal instability or allow a cell to tolerate chromosomal instability are likely to be important in therapy resistance. Interestingly, not all 10 of these patients had detectable mutations in TP53, suggesting other genes may play a role in acquisition or tolerance of a complex karyotype.
Our demonstration of differences in cytogenetic abnormalities and gene mutations at relapse compared with diagnosis in 23/29 patients has ramifications for the considerable ongoing work to develop posttransplant strategies to reduce relapse. These strategies include either administration of cellular therapies (eg, DLI) to directly augment GVL or targeted drugs (eg, FLT3 inhibitors) to slow disease growth posttransplant to provide a longer time period for GVL to develop. Detection of genetic and cytogenetic changes at relapse but not diagnosis suggests posttransplant interventions with a broader antileukemic activity, such as DLI, or checkpoint inhibitors that are indeterminate of AML genetic and cytogenetic changes may be more broadly useful. Furthermore, our data caution against the choice of targeted therapies for administration posttransplant based solely on the diagnostic mutational profile.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients who kindly donated samples, the staff in the National Cancer Research Network Bio study, and the Birmingham sample banks.
L.Q., M.M., A.K., and P.V. were supported by MRC Molecular Haematology Unit Award (MC_UU_12009/11), MRC Disease Team Award (G1000729 and MR/L008963/1), the Oxford Partnership Comprehensive Biomedical Research Centre (National Institute for Health Research Biomedical Research Centre Funding scheme). P.F., M.G., and C.C. were supported by the Birmingham Cancer Research UK Experimental Cancer Medicine Centre.
Authorship
Contribution: L.Q., P.F., P.V., and C.C. designed the study and led the experimental work; I.A. and K.W. led the statistical analysis; M.M., A.K., C.G., A.T., and C.W. contributed to the experimental work; S.J., K.P., and M.G. led the cytogenetic analysis; R.D., M.R., A.P., P.V., and C.C. contributed patient data; and A.B., C.W., and J.W. performed data analysis. All authors critically reviewed the manuscript and had the final responsibility for the decision to submit.
Conflicts-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Paresh Vyas, MRC Molecular Haematology Unit, Weatherall Institute of Molecular Medicine, University of Oxford, Oxford OX3 9DU, United Kingdom; e-mail: paresh.vyas@imm.ox.ac.uk; and Charles Craddock, Centre for Clinical Haematology, Queen Elizabeth Hospital, Edgbaston, Birmingham B15 2TH, United Kingdom; e-mail: charles.craddock@uhb.nhs.uk.
References
- 1.Appelbaum FR. Optimising the conditioning regimen for acute myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22(4):543-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bejanyan N, Weisdorf DJ, Logan BR, et al. . Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant. 2015;21(3):454-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter RB, Sandmaier BM, Storer BE, et al. . Number of courses of induction therapy independently predicts outcome after allogeneic transplantation for acute myeloid leukemia in first morphological remission. Biol Blood Marrow Transplant. 2015;21(2):373-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craddock C, Nagra S, Peniket A, et al. . Factors predicting long-term survival after T-cell depleted reduced intensity allogeneic stem cell transplantation for acute myeloid leukemia. Haematologica. 2010;95(6):989-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmid C, Labopin M, Socié G, et al. ; Acute Leukemia Working Party of the European Group of Blood and Bone Marrow Transplantation. Outcome of patients with distinct molecular genotypes and cytogenetically normal AML after allogeneic transplantation. Blood. 2015;126(17):2062-2069. [DOI] [PubMed] [Google Scholar]
- 6.Luskin MR, Carroll M, Lieberman D, et al. . Clinical utility of next-generation sequencing for oncogenic mutations in patients with acute myeloid leukemia undergoing allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22(11):1961-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel JP, Gönen M, Figueroa ME, et al. . Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivey A, Hills RK, Simpson MA, et al. ; UK National Cancer Research Institute AML Working Group. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374(5):422-433. [DOI] [PubMed] [Google Scholar]
- 11.Papaemmanuil E, Gerstung M, Bullinger L, et al. . Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding L, Ley TJ, Larson DE, et al. . Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krönke J, Bullinger L, Teleanu V, et al. . Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100-108. [DOI] [PubMed] [Google Scholar]
- 14.Schmid C, Labopin M, Nagler A, et al. ; EBMT Acute Leukemia Working Party. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: a retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. J Clin Oncol. 2007;25(31):4938-4945. [DOI] [PubMed] [Google Scholar]
- 15.Davids MS, Kim HT, Bachireddy P, et al. ; Leukemia and Lymphoma Society Blood Cancer Research Partnership. Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med. 2016;375(2):143-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschan-Plessl A, Halter JP, Heim D, Medinger M, Passweg JR, Gerull S. Synergistic effect of sorafenib and cGvHD in patients with high-risk FLT3-ITD+AML allows long-term disease control after allogeneic transplantation. Ann Hematol. 2015;94(11):1899-1905. [DOI] [PubMed] [Google Scholar]
- 17.Craddock C, Jilani N, Siddique S, et al. . Tolerability and clinical activity of post-transplantation azacitidine in patients allografted for acute myeloid leukemia treated on the RICAZA Trial. Biol Blood Marrow Transplant. 2016;22(2):385-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platzbecker U, Wermke M, Radke J, et al. . Azacitidine for treatment of imminent relapse in MDS or AML patients after allogeneic HSCT: results of the RELAZA trial. Leukemia. 2012;26(3):381-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Metzelder SK, Schroeder T, Finck A, et al. . High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26(11):2353-2359. [DOI] [PubMed] [Google Scholar]
- 20.Grimwade D, Hills RK, Moorman AV, et al. ; National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354-365. [DOI] [PubMed] [Google Scholar]
- 21.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21(6):936-939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKenna A, Hanna M, Banks E, et al. . The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koboldt DC, Chen K, Wylie T, et al. . VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spencer DH, Abel HJ, Lockwood CM, et al. . Detection of FLT3 internal tandem duplication in targeted, short-read-length, next-generation sequencing data. J Mol Diagn. 2013;15(1):81-93. [DOI] [PubMed] [Google Scholar]
- 25.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94(446):496-509. [Google Scholar]
- 26.Bacher U, Haferlach T, Alpermann T, et al. . Comparison of cytogenetic clonal evolution patterns following allogeneic hematopoietic transplantation versus conventional treatment in patients at relapse of AML. Biol Blood Marrow Transplant. 2010;16(12):1649-1657. [DOI] [PubMed] [Google Scholar]
- 27.Jan M, Snyder TM, Corces-Zimmerman MR, et al. . Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149):149ra118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal S, Fontanillas P, Flannick J, et al. . Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genovese G, Kähler AK, Handsaker RE, et al. . Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477-2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klco JM, Spencer DH, Miller CA, et al. . Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379-392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shlush LI, Zandi S, Mitchell A, et al. ; HALT Pan-Leukemia Gene Panel Consortium. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia [published correction appears in Nature. 2014;508(7496):420 (Yousif, Fouad [added]]. Nature. 2014;506(7488): 328-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quek L, Otto GW, Garnett C, et al. Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. Journal of Experimental Medicine 2016;Jul 4. pii: jem.20151775. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 34.Ramos F, Robledo C, Izquierdo-García FM, et al. ; Spanish Group for Myelodysplastic Syndromes (GESMD). Bone marrow fibrosis in myelodysplastic syndromes: a prospective evaluation including mutational analysis. Oncotarget. 2016;7(21):30492-30503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindsley RC, Mar BG, Mazzola E, et al. . Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahn JS, Kim HJ, Kim YK, et al. . Transplant outcomes of the triple-negative NPM1/FLT3-ITD/CEBPA mutation subgroup are equivalent to those of the favourable ELN risk group, but significantly better than the intermediate-I risk group after allogeneic transplant in normal-karyotype AML. Ann Hematol. 2016;95(4):625-635. [DOI] [PubMed] [Google Scholar]
- 37.DiNardo CD, Ravandi F, Agresta S, et al. . Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamaguchi S, Iwanaga E, Tokunaga K, et al. . IDH1 and IDH2 mutations confer an adverse effect in patients with acute myeloid leukemia lacking the NPM1 mutation. Eur J Haematol. 2014;92(6):471-477. [DOI] [PubMed] [Google Scholar]
- 39.Ravandi F, Patel K, Luthra R, et al. . Prognostic significance of alterations in IDH enzyme isoforms in patients with AML treated with high-dose cytarabine and idarubicin. Cancer. 2012;118(10):2665-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Figueroa ME, Abdel-Wahab O, Lu C, et al. . Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong TN, Ramsingh G, Young AL, et al. . Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craddock C, Quek L, Goardon N, et al. . Azacitidine fails to eradicate leukemic stem/progenitor cell populations in patients with acute myeloid leukemia and myelodysplasia. Leukemia. 2013;27(5):1028-1036. [DOI] [PubMed] [Google Scholar]
- 43.Bradbury C, Houlton AE, Akiki S, et al. . Prognostic value of monitoring a candidate immunophenotypic leukaemic stem/progenitor cell population in patients allografted for acute myeloid leukaemia. Leukemia. 2015;29(4):988-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmidt-Hieber M, Blau IW, Richter G, et al. . Cytogenetic studies in acute leukemia patients relapsing after allogeneic stem cell transplantation. Cancer Genet Cytogenet. 2010;198(2):135-143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.