


Abstract
Single-molecule force spectroscopy provides insight into how proteins bind to and move along DNA. Such studies often embed a single-stranded (ss) DNA region within a longer double-stranded (ds) DNA molecule. Yet, producing these substrates remains laborious and inefficient, particularly when using the traditional three-way hybridization. Here, we developed a force-activated substrate that yields an internal 1000 nucleotide (nt) ssDNA region when pulled partially into the overstretching transition (∼65 pN) by engineering a 50%-GC segment to have no adjacent GC base pairs. Once the template was made, these substrates were efficiently prepared by polymerase chain reaction amplification followed by site-specific nicking. We also generated a more complex structure used in high-resolution helicase studies, a DNA hairpin adjacent to 33 nt of ssDNA. The temporally defined generation of individual hairpin substrates in the presence of RecQ helicase and saturating adenine triphosphate let us deduce that RecQ binds to ssDNA via a near diffusion-limited reaction. More broadly, these substrates enable the precise initiation of an important class of protein–DNA interactions.
INTRODUCTION
Single-molecule force spectroscopy (SMFS) allows the dynamics and energetics of individual protein–nucleic-acid complexes to be studied, revealing important mechanistic details (1–4). Studies probing enzymes have typically relied upon double-stranded DNA (dsDNA) as a substrate and are often initiated by adding in a critical component (e.g. nucleotide triphosphates) (5). However, a wide array of proteins, including motor proteins such as translocases and helicases, may not bind directly to dsDNA, but instead bind to single-stranded DNA (ssDNA) and/or at ssDNA/dsDNA junctions. Unfortunately, inefficiencies in preparing substrates containing sections of ssDNA within longer dsDNA molecules hamper their widespread application, particularly for optical-trapping-based assays that require micrometer-scale dsDNA.
An important innovation in high-resolution SMFS helicase studies was the development of the hairpin-unwinding assay (6). In this assay, a long dsDNA molecule containing a short hairpin and a small single-stranded nucleic-acid region is stretched (Figure 1A). The single-stranded region is typically slightly larger than the helicase’s footprint, so that the helicase binds within a few nucleotides (nt) of the hairpin. Unwinding activity is detected via length changes in the taut DNA substrate. For every 1 base pair (bp) of the hairpin unwound, 2 nt of ssDNA are put under tension. Compared to directly measuring helicase motion along dsDNA (1 bp = 0.34 nm), the hairpin-unwinding assay provides a 3-fold mechanical amplification since the contour length of the taut DNA increases by 1.12 nm (2 × 0.56 nm/nt) (7,8). Although initially applied to an RNA helicase (6) and the ribosome (9), such assays have been extended to studies of DNA helicases using both optical traps (10,11) and magnetic tweezers (12,13). Unfortunately, the standard protocol for making these substrates involves a three-way hybridization (Figure 1B, Left) (6), typically a laborious and inefficient process in our hands and others (14,15). Such assays are additionally complicated by the relatively low throughput of high-resolution optical traps (16) and by fluid flows necessary to initiate unwinding [e.g. introducing adenine triphosphate (ATP)].
Figure 1.
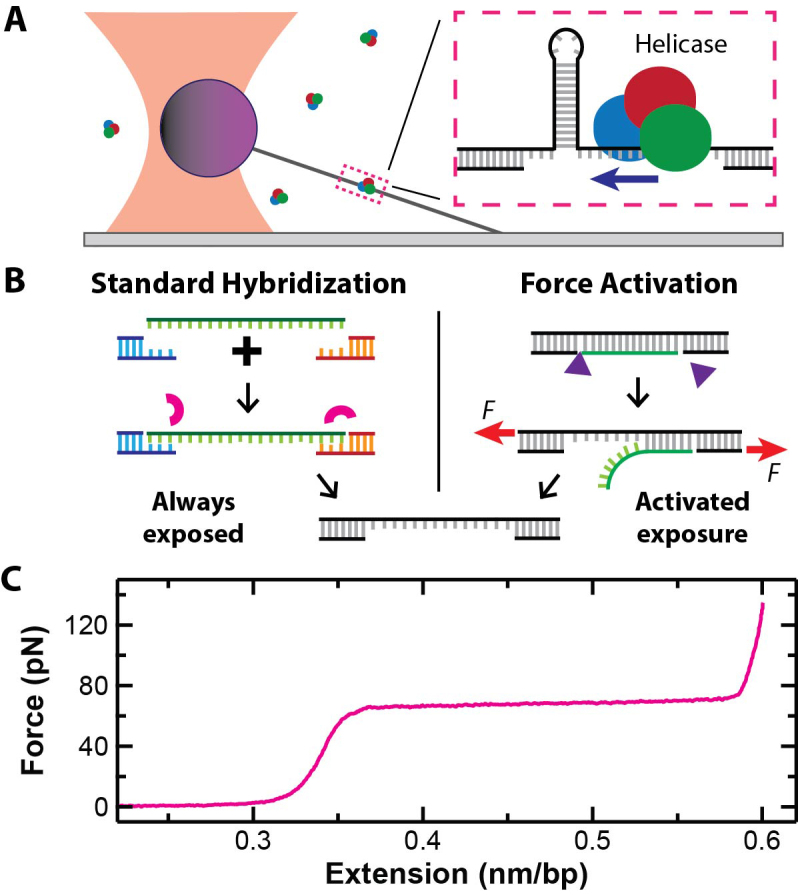
(A) A cartoon of a single-molecule hairpin-unwinding assay using a surface-coupled optical trap. Inset: a DNA hairpin is unwound by a helicase that initially binds to the small segment of ssDNA adjacent to the hairpin. (B) Left: the standard method to create ss-dsDNA substrates for such assays is via a three-way hybridization. The small arcs represent ligase molecules. Right: a force-activated substrate contains a pair of enzymatically induced, site-specific nicks located on the same strand. Small triangles represent nicking enzymes. Stretching this dsDNA molecule partway into the overstretching transition induces dissociation. (C) A force-extension curve for a 2-μm-long dsDNA molecule shows the overstretching transition at 65 pN as a function of the normalized extension (nm/bp).
An alternative means to generate ssDNA is to mechanically denature dsDNA using force. In the first experiment to demonstrate purely force-induced dissociation (i.e. no added chemical denaturants), an atomic force microscope was used to rapidly stretch the DNA (v = 0.15–3 μm/s) (17). The resulting dissociation of a full ssDNA strand occurred at a high force (120–250 pN depending on v) relative to DNA’s overstretching transition (65 pN) (7,18). As background, DNA overstretching is characterized by a 70% increase in the extension of dsDNA at 65 pN (Figure 1C) and often occurs by force-induced ssDNA dissociation starting from free ends or nicks, commonly referred to as ‘peeling’ (19–21). However, previous experiments that pulled dsDNA into and even just past the overstretching transition did not lead to its dissociation into two complementary strands (7,8,18–23) [except when the DNA was ≤30 bp long (24)]. Strand stabilization during and after overstretching is speculated to arise from islands of repeated GC base pairs (25) that suppress local DNA melting and peeling (19–21). This conjecture explains the much higher forces required to induce strand dissociation in the rapid pulling assay (17,25) and more recent results where sustained force distinctly above the overstretching transition was needed for dissociation (75 pN for ≥5 s) (23). This latter protocol, we note, did generate the controlled release of large segments of ssDNA defined by a single site-specific nick. Despite this success (23), several important issues remain in such substrates: (i) high sustained forces can lead to tether failure, (ii) force-activation efficiency decreases under ionic conditions that stabilize base-pair formation (e.g. 150 mM NaCl) and (iii) more sophisticated DNA structures, such as the one needed for the hairpin-unwinding assay, have not been realized.
To overcome these limitations, we developed two force-activated DNA substrates. In the first substrate (Figure 1B, Right), a pair of enzymatically introduced nicks were separated by 1 kbp. The 50%-GC sequence between these nicks had no adjacent GC base pairs and, as a result, dissociated when we stretched the construct partway into the overstretching transition. Importantly, the construct contained no ssDNA until activated, providing user-controlled initiation of protein–ssDNA interaction at the single-molecule level. We designed our second substrate to leverage this capability to study helicases using the hairpin-unwinding assay. We then sequentially activated individual molecules of this hairpin-unwinding substrate to measure the on rate (kon) for a DNA helicase binding to ssDNA, traditionally a difficult parameter to deduce in a SMFS assay (13).
MATERIALS AND METHODS
Sequence design of the 50%-GC DNA substrate
The 50%-GC substrate consisted of a 1002 bp long section where each AT bp is adjacent to a GC bp. The exact sequence was designed using custom software. The code produced the sequence in one hundred ten-bp segments by randomly selecting bases to insert into each AT or GC spot, and the full sequence was run through an autocorrelation routine to ensure no large sequence complements were present that would yield stable secondary structure formation. For this proof-of-concept demonstration, we chose a 1000-bp segment so that we balanced the cost of synthesis with investigating a sufficiently long DNA segment that would be expected to exhibit canonical high-force overstretching characteristics (17). After this 1000-bp sequence was defined, we introduced recognition sites for the nicking enzymes (Nt.BspQI and Nb.BsmI), the latter adding 2 nt to the 50%-GC sequence. To stabilize the remaining dsDNA, we added in GC-clamp sequences next to the site-specific nicking site. We also added traditional endonuclease recognition sites (PflmI and BbsI) so that this designed sequence could be ligated into a larger construct. This full sequence was ordered from Genscript (Piscataway, NJ, USA) (given in Supplementary Table S1).
Embedding the 50%-GC DNA substrate into a larger construct
To facilitate optical trapping, a larger final construct consisting of a 1667 bp ‘left handle’ and a 3670 bp ‘right handle’ was assembled from sections of the M13mp18 plasmid. Briefly, we individually polymerase chain reaction (PCR) amplified and purified the left handle, right handle and 50%-GC segments. The PCR products of the left handle and the 50%-GC sequence were then cut at their PflmI endonuclease sites, combined together at an equimolar ratio and ligated. The purified ligated product and the right handle were then cut with BbsI and ligated together. We then cloned this fully ligated construct into a bacterial plasmid. The plasmid was transformed into competent Escherichia coli, grown up, and then isolated. The isolated plasmids were verified by sequencing. A full description of the details of substrate preparation, including all materials and conditions used, is available in the Supplementary Data.
Importantly, the above process only had to be performed once. Once the full construct was in a plasmid, it was efficiently produced for single-molecule application via a single PCR followed by site-specific nicking, without the need for any ligation. Specifically, for our optical-trapping application, we used primers that were 5′-labeled with biotin and dibenzocyclooctyne (DBCO). DBCO is a copper-free click chemistry reagent that reacts with an azide moiety. The resulting purified and 5′-labeled PCR product was then nicked sequentially with the restriction endonucleases Nt.BspQI and Nb.BsmI.
Assembly of hairpin-unwinding DNA construct
We used a 20-bp tetraloop hairpin for the unwinding construct, slightly modifying the sequence of a previously published hairpin (20R50–4T) (26). Additionally, our 33-nt helicase landing site was based on a previously published assay; oligo #3 in ref (27). Encoding the hairpin into the substrate was a technical challenge, since a 20-bp hairpin within a PCR primer was undesirable (Supplementary Figure S1A). We solved this problem by encoding each half of the hairpin stem, as well as the loop region, into separate PCR primers. Each primer also had one nick site for the endonuclease Nb.BbvCI and one primer contained the sequence for the helicase landing site. Critically, the restriction site for the endonuclease BstXI was encoded within the loop region of each primer (Supplementary Figure S1B). Each primer was then used to create a separate PCR product, DNA molecules we referred to as the ‘left’ and ‘right’ halves. The BstXI sites on both molecules were then cut, yielding complementary, non-palindromic 4-nt ssDNA overhangs. The left and right halves were then annealed and ligated (Supplementary Figure S1C). The resulting final construct was inserted into a plasmid for subsequent use as a PCR template. We expect that the formation of a stable intrastrand hairpin within the template strand during PCR extension (∼70°C) will limit the length of the hairpin stem that can be produced by this strategy without additional post-PCR enzymatic steps.
To improve the strength of the coupling between the DNA and the optically trapped, streptavidin-coated beads, our forward primer for the single-molecule substrate contained 4 biotins spaced every 9–10 bp. The reverse primer contained a 5′-DBCO. The resulting purified and 5′-labelled PCR product was then nicked with Nb.BbvCI. A full description of the details of substrate preparation, including all materials and conditions used, is available in the Supplementary Data with primer sequences listed in Supplementary Table S2.
Assembly of force-activated single-molecule assay
For our optical-trapping assay, we used streptavidin-coated polystyrene beads and epoxy-stabilized flow cells. Streptavidin was coupled to 750-nm-diameter polystyrene beads (Invitrogen/Molecular Probes, C3724) using an established coupling protocol (28). We assembled the flow cells from a coverslip, a slide and double-sided sticky tape, similar to prior work (28), for a final channel volume of ∼15 μl. However, instead of relying upon non-specific adsorption of proteins to clean glass, we functionalized the surface with silane-PEG-azide using a recently developed protocol (29). This allowed us to covalently couple the DBCO-labeled DNA to the surface via a copper-free click chemistry reaction. Specifically, stock 5′-labeled DNA was diluted to 100 pM in dilution buffer [20 mM HEPES (pH 7.5), 100 mM KCl, 1 mM EDTA]. After pre-rinsing each flow cell with 200 μl of dilution buffer, we then flowed 15 μl of dilute DNA into each flow cell and let them incubate in a humidity chamber for 3 h at room temperature. The flow cells were then washed with 200 μl of phosphate-buffered saline (pH 7.4) and 200 μl of force-extension working buffer [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.4% Tween-20]. Next, we flowed 15 μl of 2.5 pM streptavidin-coated polystyrene beads in working buffer into each cell and incubated them for 1 h at room temperature. Finally, the cells were washed with 400 μl of working buffer and sealed using nail polish.
Optical-trapping assay
The flow cells were then mounted on a previously described optical-trapping microscope (30). To probe individual DNA molecules, we visually located bead–DNA complexes exhibiting tethered particle motion. Such beads were then trapped and positioned ∼400 nm above the surface. We then laterally aligned the DNA’s anchor point to the cover slip with the optical trap's vertical axis by performing a 2D-centering routine via a set of force-extension curves along each coordinate axis (i.e. x-y axes) (28). We then repeated this process of setting the bead height and centering the anchor point one additional time. The resulting force-extension curves were also used to screen for single-molecule attachment by selecting for tethers that exhibited the correct contour length and persistence length. We note that the 2D stretching geometry inherent in our surface-coupled assay was incorporated using the standard geometric correction (31). To test for force activation, we set the trap to a high stiffness (ktrap = 0.7 pN/nm), moved the coverslip a pre-set distance at 2 μm/s, and immediately reversed that motion while digitizing bead position data at 120 kHz for the records shown in Figures 2 and 3. For Figure 4, we started with the dsDNA molecule pre-tensioned to 8 pN and then moved the stage at 5 μm/s to decrease the time associated with force-activation. For presentation, force-extension curves were averaged to 500 Hz. All experiments were performed at 25°C.
Figure 2.

Forced dissociation of a 1002-nt DNA segment at ∼65 pN. (A) Substrate schematic showing the 1002-bp 50% GC segment, nick sites, GC clamps and 5′-end labels. Nt.BspQI and Nb.BsmI are enzymes that introduce site-specific nicks. (B) A set of three sequential force-extension curves at v = 2 μm/s shows hysteresis in the stretching cycle but no permanent ssDNA strand dissociation when stretching the DNA molecule only 30% of the way through the overstretching transition (black). The stage motion was reversed immediately to relax the DNA to zero force (cyan), and then stretched again (magenta). (C) A similar set of three force-extension curves but showing force-activaton via ssDNA dissociation when the construct was pulled 50% of the way through the overstretching transition. Color coding is the same as in B. The thick gray curve is a representative full overstretching curve.
Figure 3.

Force activation of a substrate for high-resolution studies of a DNA helicase. (A) Schematic of the substrate with a more detailed view of the region around the strand to be displaced. Enzymatically induced nick sites (Nb.BbvCl), GC clamps and 5′-end labels shown. (B) A set of three force-extension curves at v = 2 μm/s from a stretch-relax-stretch cycle shows no hysteresis when the maximum force was kept just below the overstretching transition (black, cyan and magenta, respectively). (C) A similar set of three force-extension curves but showing force-activation when the construct was pulled 7% of the way into the overstretching transition. Inset: a magnified view of the force-extension curves shows hairpin folding (cyan) and unfolding (magenta) after activation. As expected, these curves span the initial force-extension curve of the fully dsDNA construct (black).
Figure 4.

Force-activation enables efficient determination of kon for a helicase via a single-molecule assay. (A) An extension-versus-time trace prior to and immediately after force activation of the hairpin-unwinding substrate in the presence of RecQΔH and saturating ATP. After a time t1, unwinding of the newly created hairpin led to an increase in extension when measured under constant load (F = 8 pN). Note, the decrease in extension after activation matched the expected value (10 nm). Activation occurred in ∼0.5 s (orange bar). (B) Normalized histogram of t1 at two different enzyme concentrations well below the KD of RecQΔH for ssDNA. An exponential fit to the histograms yielded the average time, denoted τ1. All the data were acquired in a single day [N = 70 at 200 pM and N = 67 at 100 pM].
Force-extension analysis of force-activated constructs
To accurately analyze force-extension curves at relatively high force (F > 10 pN), we used an extensible worm-like-chain (eWLC) model (31,32). Our goal was to determine the amount of ssDNA generated after force activation, not whether a single DNA molecule was being stretched (which was determined during the aforementioned 2D-centering routine). For this process, we fit the total extension as a sum of dsDNA and ssDNA extensions, using the Odijk eWLC for dsDNA (32) and the extensible freely-jointed-chain for ssDNA (7). We achieved repeatable results by fixing the dsDNA contour length to the known number of base pairs in the construct (using 0.338 nm/bp) and fitting for the ssDNA contour length in both the initial and post-activation curves. The difference between the fitted ssDNA contour lengths yields the number of ssDNA nt created, while also removing any absolute offsets due to variability in bead size and linker length. In this process, we fixed the persistence length (p) and stretch modulus (K) for ss and dsDNA (ssDNA: P = 0.75 nm and K = 800 pN; dsDNA: P = 45 nm and K = 1200 pN). For the hairpin construct, we also took into account the hairpin's finite width of 2 nm. The reported uncertainties throughout this work are the standard deviations in the fitted parameters as calculated by our fitting routine (WaveMetrics, Igor Pro 6) and propagated as required.
RecQΔH assay and analysis
RecQΔH was generously supplied by Yeonee Seol and Keir Neuman using a previously detailed purification protocol (13). This 8.5 μM stock RecQΔH was stored at −80°C in 30 mM Tris–HCl (pH 7.5), 200 mM NaCl, 1 mM β-mercaptoethanol and 10% glycerol. For daily use, an aliquot was thawed on ice and then sequentially diluted 10-fold per dilution to 850 pM using a dilution buffer [30 mM Tris–HCl (pH 7.5), 100 mM NaCl, 10% glycerol]. To improve precision, we prepared the 100 pM and 200 pM RecQΔH concentrations from the same individual 850 pM dilution. The final assay had either 100 pM or 200 pM RecQΔH in helicase buffer [30 mM Tris–HCl (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 0.4% Tween-20, 1 mM dithiothreitol (DTT) and 1 mM ATP]. We prepared the flow cells and anchored the fully duplex hairpin-unwinding DNA, as described above. We then incubated the flow cells with ∼75 μg/ml bovine serum albumin (Sigma Aldrich, A3059–10G) for 30 min, washed with 200 μl of helicase buffer and sealed the flow cells with nail polish.
To perform the single-molecule assay, we mounted the flow cell on the optical-trapping microscope and visually located a tethered bead–DNA complex. As described above, each individual bead-DNA tether underwent a 2D-centering routine that aligned the DNA anchor point with the optical trap and verified an individual DNA molecule was being stretched. Next, we stretched the DNA to 8 pN using ktrap = 0.2 pN/nm. A software-based force clamp modulated ktrap to keep the force at 8 pN while data were collected at 120 kHz and averaged to 50 Hz. At a user-determined time, the substrate activation process was started: (i) the force clamp was deactivated, (ii) ktrap was set to 0.4 pN/nm, (iii) the stage moved 500 nm at 5 μm/s to stretch the DNA and (iv) the stage was immediately moved back its starting location at the same speed. After this activation, we reset ktrap to 0.2 pN/nm and re-engaged the force clamp. As expected, force-activation of the hairpin-unwinding substrate led to the expected extension change (10 nm) (see Figure 4A). We collected the data for Figure 4 from two slides at each concentration, with a total of 67 records collected at 100 pM and 70 at 200 pM. All experiments were performed at 25°C. Our calculation of kon and comparison to previous findings is given in the Supplementary Data.
RESULTS AND DISCUSSION
Rapid dissociation of a 1000-bp 50%-GC substrate at 65 pN
We hypothesized that efficient strand dissociation at 65 pN could be promoted by engineering out GC-islands. To do so, we developed a 1002-bp target sequence that had a 50%-GC content but no adjacent GC base pairs (i.e. strictly alternates between GC and AT base pairs). Dissociation of this target sequence was facilitated by a pair of site-specific nicks that were enzymatically induced (via Nt.BspQI and Nb.BsmI) as shown in Figure 2A. Peeling within the larger DNA construct [6449 bp (≈2.2 μm)] was partially suppressed by introducing GC clamps (see the ‘Materials and Methods’ section and Supplementary Data for details). This micrometer-scale DNA construct was compatible with our surface-coupled optical-trapping assay (Figure 1A). Similar to earlier work (33), we used biotin and DBCO functional groups to improve the mechanical robustness of our assay. In particular, the click-chemistry-based covalent linkage to the coverslip provided enhanced mechanical strength relative to the digoxigenin–anti-digoxigenin bond traditionally used in DNA-based SMFS assays (1–4).
To demonstrate force-activation, we first anchored our 50%-GC construct to an azide-functionalized coverslip (29) and then attached it to a streptavidin-coated polystyrene bead. We next used a back-and-forth stretching protocol where the coverslip was moved relative to a stationary optical trap. Our protocol started with the DNA anchor point co-aligned with the optical trap. From this state, we rapidly executed a stretch-relax-stretch cycle by moving the coverslip a predetermined distance at v = 2 μm/s with no delay between changes in direction. By varying the predetermined distance, we controlled how far into the overstretching transition the DNA was extended (Figure 2B and C). As expected, the initial force-extension curve (black) for F < 40 pN was well fit by an extensible worm-like chain model (31) for dsDNA with contour length equal to that of the full construct (Supplementary Figure S2). This agreement shows that the nicked DNA was fully double stranded prior to force activation.
By pulling our engineered DNA construct halfway through the overstretching transition, we achieved essentially instantaneous dissociation of a 1002-nt ssDNA segment. Notably, rapid force-activation of this substrate required pulling the construct a minimum distance into the overstretching transition. In particular, for small (≤30%) excursions into the overstretching transition, we observed the canonical hysteresis between the stretching and relaxation force-extension curves for such a dynamic assay (7). This hysteresis was caused by peeling of ssDNA from free ends or nicks, but such peeling intermediates quickly reformed fully duplex DNA once the force was sufficiently reduced (Figure 2B, cyan) (19,22). On the other hand, when the construct was stretched 50% into the overstretching transition (Figure 2C), hysteresis was observed between the initial and subsequent force-extension curves coupled with a permanent increase in the contour length of the activated substrate. To determine the amount of ssDNA generated, we compared worm-like chain fits for the initial and second stretching curves (black and magenta, respectively). This analysis yielded 996 ± 9 nt of ssDNA (best fit ± SD), in quantitative agreement with the 1002 nt ssDNA expected from our construct (Supplementary Figure S2). Importantly, every DNA construct that completed the more extensive stretching cycle was activated (N = 22).
The observed maximum distance into the overstretching transition of 30% before permanent strand displacement suggests that peeling from free ends was concurrent with the force-induced dissociation of the target strand at v = 2 μm/s. This interpretation is based upon previous work, which showed peeling from free ends or nicks is required for hysteresis while hysteresis is not observed in a topologically closed but rotationally free substrates (22). To be quantitative, this 30% excursion is 2-fold larger than that needed to overstretch just the 1002-bp nicked region. Hence, we expect that there is peeling from the free ends of our construct as well as the site-specific nicks. Such peeling had no adverse effects in this construct, but extra GC clamps could be added to suppress it, if needed.
Beyond achieving efficient generation of ssDNA, these substrates also proved the conjecture that GC-islands lead to strand stabilization during and after overstretching (25). Unlike extensive previous studies over the last 20 years that used natural sequences containing GC islands (7,8,18–23), our substrate had exactly a 50%-GC content with no adjacent GC base pairs. Upon stretching, this engineered substrate still exhibited the standard overstretching transition at 65 pN, but we now observed dissociation of a large strand (1 kbp) halfway through the overstretching transition. Overall, this result, in conjunction with aforementioned 20 years of previous studies on natural DNA sequences (7,8,18–23), shows that GC islands are critical to strand stabilization. Moreover, we expect that strand dissociation within the 65-pN plateau generalizes to arbitrary lengths of 50%-GC segments above a minimum threshold of ∼20–30 bp (24), though maximum distance into the overstretching transition before permanent stand displacement should vary.
Force-activation of a hairpin-unwinding DNA substrate
Having established our ability to rapidly dissociate such large segments of ssDNA, we next focused on triggering the formation of the more complicated structure needed for the hairpin-unwinding assay. To do so, we designed a substrate for a 3′-to-5′ DNA helicase that consisted of a 33-nt binding site adjacent to a 20-bp hairpin capped by a 4-nt loop. The sequence encoding this structure was embedded in an 83-bp region flanked by two site-specific nicking sites within a larger construct with a total size of 5529 bp (≈1.8 μm) (Figure 3A). The 20-bp hairpin had a 50%-GC content and the 33-nt section had a 58%-GC content. We engineered GC clamps into the shorter dsDNA handle to suppress unwanted fraying. To increase the strength of the DNA attachment to the streptavidin-coated bead, we replaced the single 5′-biotin with 4-biotin labels spaced approximately every helical turn within one PCR primer. Encoding the hairpin into the substrate was a technical challenge, since a 20-bp hairpin within a PCR primer was undesirable. We solved this problem by initially dividing each strand of the hairpin stem into two separate DNA molecules (see ‘Materials and Methods’ section and Supplementary Figure S1). The combined molecule was then cloned into a plasmid for subsequent use as a PCR template.
Since the nicked strand contained only 83 nt, we first wanted to verify that stretching into the overstretching transition was still required for activation. Thus, we repeated the rapid stretch-relax-stretch protocol with a peak force of 55 pN, a high force just below the overstretching transition (Figure 3B). No hysteresis was observed. Hence, no peeling occurred. Next, we repeated the stretch-relax-stretch protocol but stretched the construct just into the overstretching transition (7%). In this case, hysteresis coupled with a contour length increase in the force-extension curves was observed (Figure 3C). Importantly, folding and unfolding transitions consistent with hairpin formation and rupture were now observed at lower forces (Figure 3C, Inset). As expected at v = 2 μm/s, there was now hysteresis in the force-extension curves at low forces due to the non-equilibrium folding and unfolding of the DNA hairpin, similar to pioneering studies of RNA hairpins (34). We note that substrate activation was successful in 100% of attempts (N > 300) at two relatively high ionic concentrations that stabilize base pair formation (150 mM NaCl or 50 mM NaCl + 5 mM MgCl2). To verify that the proper structure was formed, we analyzed segments of the second stretching curve prior to and after hairpin rupture using worm-like chain fits (Supplementary Figure S3). This analysis yielded 45 ± 1 nt (best fit ± SD), in agreement with the expected 44 nt introduced by hairpin rupture. Hence, we generated a valid substrate for the hairpin-unwinding assay via force activation.
Single-molecule determination of kon for RecQ DNA helicase
To demonstrate how such force-activated substrates can enhance studies of helicases, we measured the on-rate (kon) for a DNA helicase binding to ssDNA, traditionally a difficult parameter to determine in a single-molecule assay (13). This difficulty arises, in part, from the time associated with buffer exchange and flow-induced mechanical perturbations that degrade high-resolution single-molecule assays. In our assay, we started with a pure dsDNA construct—our inactive hairpin substrate—stretched to 8 pN and premixed with the helicase and saturating ATP [i.e. 1 mM ATP versus KM = 16 μM (13)]. We then rapidly activated the substrate (v = 5 μm/s) and measured the time (t1) between activation and the start of unwinding (Figure 4A). As expected, in all records, unwinding was only observed after activation. This time t1 is the sum of the time it takes the helicase to bind to the newly created 33-nt ssDNA (ton) and the time it takes the helicase to translocate along ssDNA to the dsDNA hairpin and begin unwinding (ttrans). By performing the experiment at saturating ATP but a low enzyme concentration, ttrans was made small relative to t1 (∼0.3 versus ∼10 s, where the maximum expected ttrans ≈ 19/70 nt/s) (35,36). Hence, our measurement of t1 yields ton, which in turn yields kon ( = 1/(τon [E]) where τon = <ton>.
We used E. coli RecQ DNA helicase in this initial study, since RecQ is well characterized and can bind to an internal segment of ssDNA prior to unwinding dsDNA (37). As background, RecQ family helicases unwind the complex DNA structures that form as intermediates in many processes used to maintain the genome, and mutations in human RecQ helicases are associated with a number of diseases (e.g. Bloom’s and Werner’s syndromes) (37). Structurally, RecQ is a multidomain protein with a core catalytic activity composed of a pair of RecA-like domains stabilized with a zinc-binding domain. This catalytic activity is tuned by two auxiliary domains, a winged-helix domain and a helicase-and-RNaseD–C terminal (HRDC) domain. To focus on RecQ’s helicase activity, we used a truncated version, RecQΔH, which lacks its HRDC domain (38). In comparison to wild type RecQ, RecQΔH shows no change in processivity nor mechanochemical coupling (39), but removal of the HRDC domain increases ATPase activity by a factor of 2, and weakens binding to ssDNA by a factor of 2–5 (i.e. increased KD) (36).
To determine kon, we measured t1 at two enzyme concentrations that were much lower than RecQΔH’s KD for ssDNA (90 nM) (36). In this limit, t1 (and thus ton) should scale inversely with enzyme concentration. Specifically, we measured 70 individual molecules at 200 pM and 67 at 100 pM, all in a single day. Histograms of t1 were well fit by a single exponential and yielded average values of τ1 = 13.7 ± 0.7 s and 26.3 ± 1.8 s (best fit ± SD) for 200 and 100 pM, respectively (Figure 4B). Importantly, our results agreed with the expectation that t1 at 100 pM should be twice t1 at 200 pM. The dependence of t1 on concentration coupled with the exponential distribution in t1 at fixed concentration shows that the dsDNA hairpin was unwound by a RecQΔH monomer rather than a dimer, an ongoing question for RecQ (40,41) and other helicases (42). Further analysis—under the assumption that t1 ≈ ton—yielded kon = 3.7 ± 0.6 × 108 M−1s−1 (best fit ± SD), where the reported uncertainty includes both the uncertainty in the fit as well as enzyme concentration. Our value for kon approximately equals the ‘diffusion limit,’ the maximum rate at which a molecule undergoing a random walk in 3D can collide with its target (43).
Our single-molecule determination of kon for RecQΔH is in close agreement with the one prior measurement from an ensemble stopped-flow assay (35). In that study, a 54-nt segment of ssDNA was used in contrast to the 33-nt segment in the present work. Our value for kon is ∼1.5-fold larger than the previous determination (see the Supplementary Data for calculation). Potential causes for this difference—albeit a relatively small one for a kinetic parameter—include the taut conformation of our ssDNA and the fact that it is flanked by long dsDNA handles, which could facilitate the diffusional search process (43).
One way our force-activated hairpin-unwinding assay can be improved is by decreasing the activation time (currently 0.5 s). The fundamental limit for this time is set by the round trip time of the stretch-relax cycle. This limit varies with stretching velocity and distance traveled by the stage motion. For the present construct held at 8 pN, the round trip distance was 1000 nm. Shorter constructs would decrease this time and are also advantageous since high-resolution assays benefit from stiffer linkers. In addition, future work is needed to fully investigate the velocity dependence of force activation, as we found that a small percentage (30%) of the 50%-GC substrates failed to activate when pulled halfway into the overstretching transition at 20 μm/s, whereas all of the substrates activated at 2 μm/s.
CONCLUSION
By quickly stretching DNA molecules partway into the overstretching transition, we induced dissociation of a 1-kbp segment of ssDNA from doubly nicked dsDNA. This success was enabled by engineering the target DNA sequence to avoid repeated GC base pairs (while introducing repeated GC base pairs to stabilize specific parts of the construct). Importantly, after such engineering, these substrates were efficiently produced by PCR and site-specific nicking. In addition to dissociating large segments of ssDNA, we temporally controlled the generation of a more complicated DNA structure useful in single-molecule assays, so that initial binding times could be precisely measured. Specifically, in the presence of a helicase and saturating ATP, we force-activated a DNA hairpin flanked by 33 nt of ssDNA. We highlighted the efficiency of this assay by determining kon for a helicase from data acquired within a single day. More broadly, this assay illustrates the means by which force-activation can improve the throughput of future high-resolution, single-molecule studies of protein–DNA interactions in both dual-beam (44) and active-stage-stabilization (30) optical-trapping assays. Beyond improvements in such protein-DNA studies, the present work also provides a means for determining the minimum number of repeated GC base pairs needed for interstrand stabilization after overstretching, which remains an open question in the mechanics of DNA overstretching despite more than two decades of research (3,7,18).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Yeonee Seol and Keir Neuman for discussions and generously providing RecQΔH, Ty Miller and Guy Margalit for preparation of azide-functionalized surfaces, and George Emanuel for designing the 50%-GC sequence. Mention of commercial products is for information only; it does not imply NIST’s recommendation or endorsement. T.T.P. is a staff member of NIST’s quantum physics division.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Health and the University of Colorado, Molecular Biophysics Training Grant [T32 GM-065103 to S.R.O.]; National Science Foundation Graduate Research Fellowship [DGE 1144083 to S.R.O.]; National Science Foundation [DBI-1353987 to T.T.P., PHY-1734006]; National Institute of Standards and Technology (NIST). Funding for open access charge: NIST.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bustamante C., Bryant Z., Smith S.B.. Ten years of tension: single-molecule DNA mechanics. Nature. 2003; 421:423–427. [DOI] [PubMed] [Google Scholar]
- 2. Chemla Y.R. Revealing the base pair stepping dynamics of nucleic acid motor proteins with optical traps. Phys. Chem. Chem. Phys. 2010; 12:3080–3095. [DOI] [PubMed] [Google Scholar]
- 3. Chaurasiya K.R., Paramanathan T., McCauley M.J., Williams M.C.. Biophysical characterization of DNA binding from single molecule force measurements. Phys. Life Rev. 2010; 7:299–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou J., Schweikhard V., Block S.M.. Single-molecule studies of RNAPII elongation. Biochim. Biophys. Acta. 2013; 1829:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yin H., Wang M.D., Svoboda K., Landick R., Block S.M., Gelles J.. Transcription against an applied force. Science. 1995; 270:1653–1657. [DOI] [PubMed] [Google Scholar]
- 6. Dumont S., Cheng W., Serebrov V., Beran R.K., Tinoco I.J., Pyle A.M., Bustamante C.. RNA translocation and unwinding mechanism of HCV NS3 helicase and its coordination by ATP. Nature. 2006; 439:105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith S.B., Cui Y., Bustamante C.. Overstretching of B-DNA: the elastic response of individual double-stranded and single stranded DNA molecules. Science. 1996; 271:795–799. [DOI] [PubMed] [Google Scholar]
- 8. McCauley M.J., Williams M.C.. Mechanisms of DNA binding determined in optical tweezers experiments. Biopolymers. 2007; 85:154–168. [DOI] [PubMed] [Google Scholar]
- 9. Wen J.D., Lancaster L., Hodges C., Zeri A.C., Yoshimura S.H., Noller H.F., Bustamante C., Tinoco I.. Following translation by single ribosomes one codon at a time. Nature. 2008; 452:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morin J.A., Cao F.J., Lazaro J.M., Arias-Gonzalez J.R., Valpuesta J.M., Carrascosa J.L., Salas M., Ibarra B.. Active DNA unwinding dynamics during processive DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8115–8120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qi Z., Pugh R.A., Spies M., Chemla Y.R.. Sequence-dependent base pair stepping dynamics in XPD helicase unwinding. Elife. 2013; 2:e00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ribeck N., Kaplan D.L., Bruck I., Saleh O.A.. DnaB helicase activity is modulated by DNA geometry and force. Biophys. J. 2010; 99:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seol Y., Strub M.P., Neuman K.C.. Single molecule measurements of DNA helicase activity with magnetic tweezers and t-test based step-finding analysis. Methods. 2016; 105:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paik D.H., Roskens V.A., Perkins T.T.. Torsionally constrained DNA for single-molecule assays: an efficient, ligation-free method. Nucleic Acids. Res. 2013; 41:e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lipfert J., Koster D.A., Vilfan I.D., Hage S., Dekker N.H.. Single-molecule magnetic tweezers studies of type IB topoisomerases. Methods Mol. Biol. 2009; 582:71–89. [DOI] [PubMed] [Google Scholar]
- 16. Perkins T.T. Angstrom-precision optical traps and applications. Ann. Rev. Biophys. 2014; 43:279–302. [DOI] [PubMed] [Google Scholar]
- 17. Rief M., Clausen-Schaumann H., Gaub H.E.. Sequence-dependent mechanics of single DNA molecules. Nat. Struct. Biol. 1999; 6:346–349. [DOI] [PubMed] [Google Scholar]
- 18. Cluzel P., Lebrun A., Heller C., Lavery R., Viovy J.L., Chatenay D., Caron F.. DNA: An extensible molecule. Science. 1996; 271:792–794. [DOI] [PubMed] [Google Scholar]
- 19. van Mameren J., Gross P., Farge G., Hooijman P., Modesti M., Falkenberg M., Wuite G.J., Peterman E.J.. Unraveling the structure of DNA during overstretching by using multicolor, single-molecule fluorescence imaging. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18231–18236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X., Chen H., Le S., Rouzina I., Doyle P.S., Yan J.. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching by single-molecule calorimetry. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. King G.A., Gross P., Bockelmann U., Modesti M., Wuite G.J., Peterman E.J.. Revealing the competition between peeled ssDNA, melting bubbles, and S-DNA during DNA overstretching using fluorescence microscopy. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:3859–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paik D.H., Perkins T.T.. Overstretching DNA at 65 pN does not require peeling from free ends or nicks. J. Am. Chem. Soc. 2011; 133:3219–3221. [DOI] [PubMed] [Google Scholar]
- 23. Candelli A., Hoekstra T.P., Farge G., Gross P., Peterman E.J., Wuite G.J.. A toolbox for generating single-stranded DNA in optical tweezers experiments. Biopolymers. 2013; 99:611–620. [DOI] [PubMed] [Google Scholar]
- 24. Morfill J., Kuhner F., Blank K., Lugmaier R.A., Sedlmair J., Gaub H.E.. B-S transition in short oligonucleotides. Biophys. J. 2007; 93:2400–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Clausen-Schaumann H., Rief M., Tolksdorf C., Gaub H.E.. Mechanical stability of single DNA molecules. Biophys. J. 2000; 78:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woodside M.T., Behnke-Parks W.M., Larizadeh K., Travers K., Herschlag D., Block S.M.. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:6190–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harmon F.G., Kowalczykowski S.C.. RecQ helicase, in concert with RecA and SSB proteins, initiates and disrupts DNA recombination. Genes Dev. 1998; 12:1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perkins T.T., Dalal R.V., Mitsis P.G., Block S.M.. Sequence-dependent pausing of single lambda exonuclease molecules. Science. 2003; 301:1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walder R., LeBlanc M.-A., Van Patten W.J., Edwards D.T., Greenbert J.A., Adhikari A., Okoniewski S.R., Sullan R.M.A., Rabuka D., Sousa M.C. et al. . Rapid characterization of a mechanically labile α-helical protein enabled by efficient site-specific bioconjugation. J. Am. Chem. Soc. 2017; 139:9867–9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carter A.R., Seol Y., Perkins T.T.. Precision surface-coupled optical-trapping assays with 1 base-pair resolution. Biophys. J. 2009; 96:2926–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang M.D., Yin H., Landick R., Gelles J., Block S.M.. Stretching DNA with optical tweezers. Biophys. J. 1997; 72:1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Odijk T. Stiff chains and filaments under tension. Macromolecules. 1995; 28:7016–7018. [Google Scholar]
- 33. Eeftens J.M., van der Torre J., Burnham D.R., Dekker C.. Copper-free click chemistry for attachment of biomolecules in magnetic tweezers. BMC Biophys. 2015; 8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liphardt J., Onoa B., Smith S.B., Tinoco I.J., Bustamante C.. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001; 292:733–737. [DOI] [PubMed] [Google Scholar]
- 35. Kocsis Z.S., Sarlos K., Harami G.M., Martina M., Kovacs M.. A nucleotide-dependent and HRDC domain-dependent structural transition in DNA-bound RecQ helicase. J. Biol. Chem. 2014; 289:5938–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harami G.M., Nagy N.T., Martina M., Neuman K.C., Kovacs M.. The HRDC domain of E. coli RecQ helicase controls single-stranded DNA translocation and double-stranded DNA unwinding rates without affecting mechanoenzymatic coupling. Sci. Rep. 2015; 5:11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bernstein K.A., Gangloff S., Rothstein R.. The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 2010; 44:393–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bernstein D.A., Zittel M.C., Keck J.L.. High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 2003; 22:4910–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harami G.M., Seol Y., In J., Ferencziova V., Martina M., Gyimesi M., Sarlos K., Kovacs Z.J., Nagy N.T., Sun Y. et al. . Shuttling along DNA and directed processing of D-loops by RecQ helicase support quality control of homologous recombination. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E466–E475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rad B., Forget A.L., Baskin R.J., Kowalczykowski S.C.. Single-molecule visualization of RecQ helicase reveals DNA melting, nucleation, and assembly are required for processive DNA unwinding. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6852–E6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang X.D., Dou S.X., Xie P., Hu J.S., Wang P.Y., Xi X.G.. Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 2006; 281:12655–12663. [DOI] [PubMed] [Google Scholar]
- 42. Comstock M.J., Whitley K.D., Jia H., Sokoloski J., Lohman T.M., Ha T., Chemla Y.R.. Direct observation of structure-function relationship in a nucleic acid-processing enzyme. Science. 2015; 348:352–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. von Hippel P.H., Berg O.G.. Facilitated target location in biological systems. J. Biol. Chem. 1989; 264:675–678. [PubMed] [Google Scholar]
- 44. Shaevitz J.W., Abbondanzieri E.A., Landick R., Block S.M.. Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature. 2003; 426:684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.