

Abstract
Candida albicans is an important opportunistic fungal pathogen capable of causing both mucosal and disseminated disease. Infections are often treated with fluconazole, a front-line antifungal drug that targets the biosynthesis of ergosterol, a major component of the fungal cell membrane. Resistance to fluconazole can arise through a variety of mechanisms, including gain-of-function mutations, loss of heterozygosity events and aneuploidy. The clinical isolate P60002 was found to be highly resistant to azole-class drugs, yet lacked mutations or chromosomal rearrangements known to be associated with azole resistance. Transcription profiling suggested that increased expression of two putative drug efflux pumps, CDR11 and QDR1, might confer azole resistance. However, ectopic expression of the P60002 alleles of these genes in a drug-susceptible strain did not increase fluconazole resistance. We next examined whether the presence of three copies of chromosome 4 (Chr4) or chromosome 6 (Chr6) contributed to azole resistance in P60002. We established that Chr4 trisomy contributes significantly to fluconazole resistance, whereas Chr6 trisomy has no discernible effect on resistance. In contrast, a Chr4 trisomy did not increase fluconazole resistance when present in the standard SC5314 strain background. These results establish a link between Chr4 trisomy and elevated fluconazole resistance, and demonstrate the impact of genetic background on drug resistance phenotypes in C. albicans.
Keywords: Candida, drug resistance, aneuploidy, fluconazole, efflux pumps
Abbreviations
ABC, ATP-binding cassette; MDR, multidrug resistance; MFS, major facilitator superfamily; qRT-PCR, quantitative reverse-transcription PCR; PDR, pleiotropic drug resistance; UTR, untranslated region.
Introduction
Mechanisms for generating resistance to stress are critical for the survival of microbial species. The fungus Candida albicans resides as a benign commensal in most humans, but overgrowth of the natural host niche is capable of producing both superficial and life-threatening infections [1, 2]. Symptomatic infections are especially prevalent in immunocompromised patients and those undergoing organ transplant or chemotherapy, and during heavy steroid or antibiotic use [3, 4]. Four classes of antifungal drugs are generally used in the preventative care and treatment of C. albicans infections: azoles, polyenes, allylamines and echinocandins [5]. The triazole drug fluconazole is frequently used in the clinic due to its low cost, efficacy, lack of toxicity and ease of administration [6]. Consequently, administration of prophylactic fluconazole in cases of immune suppression and in neonates has become widespread [7]. Fluconazole acts to block activity of lanosterol 14α-demethylase (the product of the ERG11 gene) leading to ergosterol depletion and accumulation of toxic sterol byproducts in the cell membrane [8]. These changes cause reduced membrane fluidity and increased membrane leakage, and ultimately inhibit fungal cell growth and division [9, 10].
Reports of resistance to fluconazole have increased due to more frequent use of immunosuppression and antifungal prophylaxis [11, 12]. The fungistatic nature of fluconazole may also facilitate the emergence of resistance during prolonged treatment. Common mechanisms of fluconazole resistance fall into three categories: (1) genetic alterations to the Erg11 drug target, (2) compensatory changes in ergosterol biosynthesis and (3) reduced effective drug concentrations inside the cell. More specifically, point mutations in ERG11 have been identified that reduce or abolish the binding capacity of fluconazole to its target protein [13, 14]. Alternatively, increased expression of the ERG11 gene can occur due to increased gene dosage or due to trans-acting mutations, thereby reducing drug efficacy [15, 16]. For example, a zinc cluster transcription factor, Upc2, activates ERG11 gene expression and hyperactive alleles of Upc2 have been identified in fluconazole-resistant isolates [17, 18]. An alternative metabolic mechanism of resistance occurs when loss-of-function mutations in ERG3 prevent the accumulated 14α-methyl sterol from being converted into toxic 3,6-diol derivatives [19]. Finally, increased activity of drug transporters can deplete the intracellular accumulation of fluconazole to promote resistance. For example, the ABC transporters encoded by Cdr1 and Cdr2, as well as the major facilitator superfamily (MFS) drug efflux pump encoded by Mdr1, can reduce fluconazole concentrations by active efflux [20, 21]. Hyperactive alleles of the Mrr1, Mrr2, or Tac1 transcription factors also increase drug efflux through upregulation of CDR1 and CDR2 genes [22, 23].
Fluconazole resistance often arises through multiple mechanisms and even single mutations can impact resistance through pleiotropic effects on multiple gene classes. For example, hyperactive Upc2 can directly upregulate expression of both ERG3 and ERG11, as well as the drug efflux pumps CDR1 and MDR1 [24]. In the clinic, drug resistance often develops through the progressive accumulation of multiple independent mutations that cause incremental increases in resistance. Each mutation confers additive resistance that results in full protection when present in combination [25–27].
Large-scale genomic changes such as chromosomal rearrangements and aneuploidy can also drive the emergence of fluconazole resistance. In particular, formation of an isochromosome comprising the two left arms of Chr5 (i5L) confers high levels of resistance to azole drugs in C. albicans [15]. The left arm of Chr5 encodes both the fluconazole target Erg11 and the transcription factor Tac1. Elevated expression of these genes due to increased copy number in strains carrying i5L mediates increased drug resistance [28]. Changes in the complement of chromosomes have also been associated with drug resistance in other fungal pathogens [29]. In general, the emergence of aneuploid forms provides a way for cells to rapidly generate genotypic and phenotypic diversity without permanently committing to the mutant genotype [30–32]. This rapid but imperfect mechanism of adaptation can subsequently be replaced by more refined adaptive changes that have a lower fitness cost [33].
In some cases, clinical infections with C. albicans persist even when azole drugs are used at concentrations well above the minimum inhibitory concentration (MIC) [34]. In these examples, cell subpopulations continue to grow above their MIC in a phenomenon known as tolerance or heteroresistance [35]. Robust production of β-glucan and extracellular DNA during biofilm formation promotes tolerance to multiple antifungal agents in C. albicans [36]. Defects in intrinsic cellular function such as the calcineurin pathway, Hsp90, and membrane trafficking through endosomes can also contribute to azole tolerance [37–39]. Mutations leading to elevated azole tolerance may commonly precede progression to full drug resistance [40].
The prevalence of fluconazole resistance in clinical C. albicans isolates can vary significantly, with reports of resistance in 2–25 % of isolates [41–43]. While sequencing can identify causative mutations in known resistance loci, many resistant isolates lack any established signatures of drug resistance [44]. In particular, it is possible that aneuploid forms other than i5L contribute to azole resistance in C. albicans. Multiple chromosomal aneuploidies have been observed in fluconazole-resistant isolates in addition to i5L, but a causal relationship between many of these structural changes and resistance has not been established [15, 45–47]. Chromosomal changes may be a direct consequence of fluconazole actively inducing the formation of aneuploid cells through disruption of the cell cycle [48]. Extensive karyotypic variation is commonly observed in C. albicans isolates [49–51] and reveals that cells can adopt a range of ploidy states from haploid to tetraploid, as well as multiple aneuploid variants [47, 52, 53]. It is therefore an important question to determine which chromosomal aneuploidies are simply a consequence of drug-induced genomic instability, and which provide a selective advantage in the presence of the drug.
Here, we investigated drug resistance in clinical isolate P60002, which lacks any established signatures of fluconazole resistance [54]. Two putative efflux pumps, CDR11 and QDR1, exhibited significantly elevated transcript levels in P60002 compared to fluconazole-susceptible strains. However, ectopic expression of the corresponding P60002 alleles in a susceptible isolate did not significantly alter fluconazole resistance. Instead, we demonstrated that drug resistance in P60002 is due, at least in part, to the presence of a Chr4 trisomy. Loss of the trisomic chromosome significantly reduced fluconazole resistance, although the euploid derivative still exhibited clinically significant levels of resistance. In contrast, derivatives of SC5314 that carried a Chr4 trisomy did not show elevated fluconazole resistance. Our results therefore establish that the presence of a trisomic Chr4 contributes to fluconazole resistance in P60002, but does so in a strain-background-dependent manner.
Methods
Passaging of C. albicans strain P60002
Ninety-six individual colonies of strain P60002 were cultured in liquid medium by serial passaging for 18 days. Cells were cultured in 900 µl yeast extract, peptone and dextrose (YPD) medium at 30 °C on a shaking platform. Every 24 h, 15 µl was transferred to 885 µl of fresh YPD medium.
Strain construction
The strains are listed in Table S1 (available in the online Supplementary Material). Transformations were performed using lithium acetate as previously described [55]. To integrate CDR11 and QDR1 alleles from P60002 into SC5314 (including 5′ UTR, promoter, coding sequence and 3′ UTR), both genes were cloned by PCR from P60002 genomic DNA into pSFS2a [56] using NotI and SacII restriction sites using the primers listed in Table S2. These plasmids were linearized using NotI for CDR11 or SacI for QDR1 prior to transformation. Integration at the endogenous CDR11 or QDR1 loci was confirmed by PCR using the primers listed in Table S2. Tests for multiple integration events were performed using primers flanking the restriction site used for linearization, but they failed to produce a band, suggesting that single copies of the plasmids integrated into each strain.
Fluconazole disk diffusion assay
Cells for each strain (including different karyotype configurations) were cultured overnight in YPD. Optical density measurements were used to dilute the cultures to 0.04 OD ml−1 (800 000 cells ml−1) and 70 µl was plated onto solid YPD agar. Inoculated plates were left for 1 h to dry and a single 25 µg fluconazole disc (Liofilchem, TE, Italy) was placed in the centre of the plate. Cells were allowed to grow for 48 h at 30 °C and images were taken using a Nikon SX40HS Powershot (Nikon, Tokyo, Japan). Drug resistance was quantified using the diskImageR program, which allows the analysis of drug-response parameters [57]. The ‘resistance score’ was calculated as: ((max score−RAD20)/max score)+(0.5*(1-(max score−RAD20)/max score)*FoG80), where RAD20 corresponds to the point where a 20 % reduction in growth occurs, and FoG80 corresponds to the fraction of growth achieved within the zone where there is 80 % growth inhibition [57]. Combining the RAD20 and FoG80 terms provided a measurement of overall drug resistance.
Broth microdilution assay
Susceptibility to fluconazole using the broth microdilution assay utilized previously published protocols by the National Committee for Clinical Laboratory Standards [58]. Briefly, serial dilutions of fluconazole (from 256 to 0.25 µg ml−1) were prepared in RPMI 1640 broth (Life Technologies, Carlsbad, CA, USA) and transferred to 96-well plates, with wells without drug serving as controls. Cells from saturated cultures grown in liquid YPD medium were diluted to an initial OD of 0.03 ml–1 in each well and allowed to grow for 24 h. Each strain was tested in biological triplicates with technical duplicates.
Quantitative reverse-transcription PCR (qRT-PCR)
RNA was prepared using the MasterPure yeast RNA extraction kit (Epicentre, Madison, WI, USA) and cDNA was synthesized using Superscript IV reverse transcriptase with a d(T)18 oligonucleotide primer (Thermo Fischer, Waltham, MA, USA). Transcript abundance was quantified by real-time PCR (qRT-PCR) with SYBR green incorporation using a Lightcycler 96 (Roche, Mannheim, Germany). Absolute quantification using the second derivative maximum value was used to calculate the threshold cycle (ΔCT) for each gene using ACT1 as a control. The primers are listed in Table S2. All qRT-PCR results represent the average abundance of at least three independent experiments per strain.
ddRAD-Seq analysis of chromosomal copy number
DNA was isolated from 56 passaged P60002 isolates using a MasterPure yeast DNA purification kit (Epicentre, Madison, WI, USA) according to the manufacturer’s instructions. The resulting DNA was suspended in 35 µl of TE (Tris, pH 8.0, EDTA) and stored at 4 °C for library preparation. Isolate DNA was prepared for double-digest restriction-site-associated DNA sequencing using the protocol described in [59]. Briefly, DNA from each sample was digested with BamHI and NdeI restriction enzymes to produce non-complementary overhangs. P1 and P2 adapters that were complementary to each of the overhangs were ligated to the DNA fragments. The P1 adapters encoded unique five-basepair (bp) barcodes and the P2 adapters corresponded to unique six bp barcodes. Adapter-ligated pools of eight samples each were gel-excised between 350–450 bp to limit the DNA library to fragments within that size range. These resulting pools were subsequently purified and amplified using universal Illumina P5 and P7 amplification primers. The prepared libraries were sequenced at the Missouri Genome Sequencing Facility.
The read quality of the sequenced libraries was assessed using FastQC [60]. Reads were demultiplexed using scripts available from the Quantitative Insights into Microbial Ecology (QIIME) consortium. Reads from each sample were individually aligned to the P60002 reference genome [54] using Bowtie2 [61]. Read counts and chromosomal copy numbers were inferred from previously developed custom scripts [62] that were adapted to C. albicans. Seven of the 56 samples were removed from analysis due to low read counts or aberrant rDNA overrepresentation and 49 isolates were used for further analysis.
Growth assays
P60002 isolates that were trisomic for Chr4 and Chr6 (CAY7401, CAY704), trisomic for Chr4 (CAY7403, CAY7404, CAY7405), or disomic for both chromosomes (CAY7417, CAY7418, CAY7420) were grown at 30 °C in liquid YPD or synthetic complete media containing 2% dextrose (SCD) medium overnight. The cultures were then diluted 1 : 200 into fresh YPD or SCD medium. Optical density was measured every 15 min for 48 h with a plate reader (Tecan, Mannedorf, Switzerland) and the polynomial measurement of the curve used to derive relative doubling times.
Rhodamine 123 accumulation assay
P60002 isolates that were trisomic for Chr4 and Chr6 (CAY7401, CAY704), trisomic for Chr4 (CAY7403, CAY7404, CAY7405), or disomic for both chromosomes (CAY7417, CAY7418, CAY7420) were grown in YPD medium to mid-logarithmic phase. We pelleted 107 cells and washed them twice with phosphate-buffered saline (PBS). The cells were then resuspended in PBS supplemented with 10 mM glucose and grown at 37 °C in the presence of 13 µM rhodamine 123 (Rh123) for 30 min. Cells were washed twice with PBS and OD and the fluorescence was measured using a Qubit 3.0 fluorimeter (ThermoFisher Scientific, Waltham, MA, USA). The dynamic range was assayed by either not adding Rh123 during the incubation phase or not washing the cells in PBS following incubation. This produced a range from 110 to 900 000 relative fluorescent units. Twelve biological replicates were performed for each strain.
Results
Previous characterization of a set of 21 sequenced C. albicans isolates identified two strains, P94015 and P60002, to be resistant to fluconazole (MIC >256 µg ml−1) [54]. P94015 encoded known resistance mutations in both ERG11, encoding the molecular target of fluconazole, and TAC1, encoding the major transcription factor controlling expression of the drug efflux pump genes CDR1 and CDR2 [54]. However, interrogation of the P60002 genome sequence did not reveal any mutations associated with drug resistance, yet this isolate was highly resistant to azoles compared to other C. albicans strains, including the standard laboratory strain, SC5314 (Fig. 1). The majority of C. albicans isolates are diploid [54], yet the sequenced isolate of P60002 was trisomic for two chromosomes, Chr4 and Chr6, which we investigated further, as outlined below.
Fig. 1.
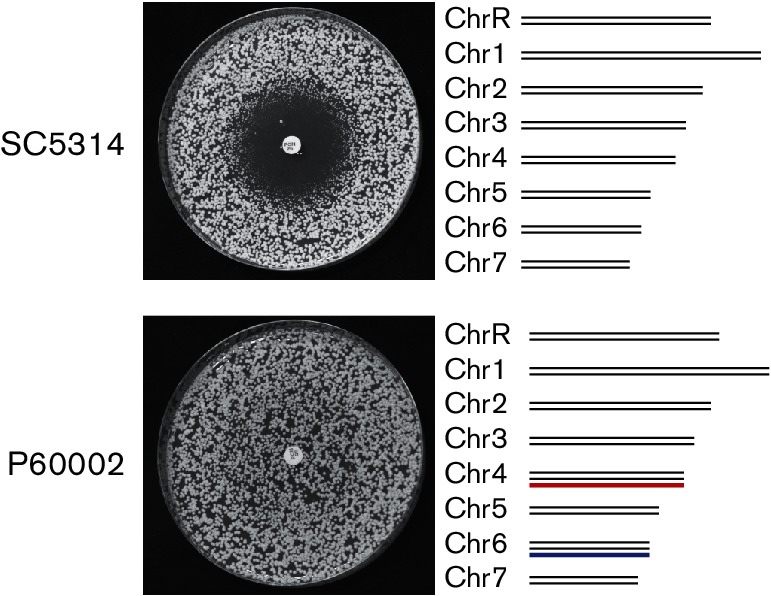
Fluconazole resistance in clinical isolate P60002. C. albicans strains SC5314 and P60002 were plated onto YPD medium and allowed to grow in the presence of a 25 µg fluconazole-containing disc. Both strains are diploid, but whereas SC5314 is euploid, P60002 is trisomic for Chr4 (red) and Chr6 (blue).
Increased expression of drug transporters in P60002
To identify gene expression changes that may contribute to fluconazole resistance, RNA-Seq was previously used to compare transcript abundance between P60002 and SC5314 [54]. Analysis of these data revealed expression differences in three genes with known or putative roles in fluconazole resistance. Specifically, P60002 expressed two putative drug transporters, CDR11 and QDR1, at a significantly higher level than SC5314 (Fig. 2a), and also overexpressed UPC2, encoding a major transcriptional regulator of ergosterol synthesis. CDR11, located on Chr3, belongs to the pleiotropic drug resistance (PDR) subfamily of ATP-binding cassette (ABC) transporters, whereas QDR1 on ChrR encodes a major facilitator superfamily drug efflux pump. We note that elevated expression of UPC2 (located on Chr1) did not lead to a corresponding increase in expression of its target genes in the ergosterol biosynthetic pathway (Fig. 2a), and so the role of this gene was not further investigated.
Fig. 2.

Analysis of the role of CDR11 and QDR1, encoding putative drug efflux pumps, in fluconazole resistance. (a) RNA-Seq expression of select SC5314 and P60002 transcripts. P60002 overexpressed three genes implicated in fluconazole resistance relative to SC5314: UPC2, CDR11 and QDR1. (b) Ectopic expression of the P60002 alleles of CDR11 and QDR1 in SC5314 increased transcript levels to a similar extent to those present in P60002. (c) Analysis of drug resistance in SC5314 strains ectopically expressing P60002 alleles of CDR11 or QDR1. Cells were plated onto YPD and allowed to grow in the presence of a 25 µg fluconazole disc. Plates were photographed after 2 days. (d) Quantitative analysis of fluconazole resistance in SC5314, P60002 or SC5314-derived strains expressing P60002 alleles of CDR11 and QDR1.
To determine if the P60002 alleles of CDR11 or QDR1 contributed to increased fluconazole resistance, allelic swaps of these loci from P60002 to the azole-susceptible SC5314 strain were performed. Multiple SNPs differentiate the P60002 and SC5314 alleles of these two genes; 48 and 8 SNPs exist in CDR11 (1 per 94 nt) and QDR1 (1 per 191 nt), respectively (Tables S3, S4). Each gene, including the predicted promoter and untranslated regions (UTRs), was cloned from P60002 and transformed into SC5314 to integrate at the endogenous locus. Importantly, expression of the P60002 alleles in SC5314 increased transcript abundance to similar levels to those of the endogenous genes in P60002 (Fig. 2b). We suggest that elevated expression of the two genes in the recipient strains is due to polymorphisms that distinguish the gene promoters in P60002 and SC5314 (see Tables S5, S6). Thus, introduction of the P60002 allele of CDR11 into SC5314 increased expression of this gene 46-fold, while introduction of the P60002 allele of QDR1 increased total QDR1 expression in SC5314 10-fold (Fig. 2b). Introduction of the P60002 CDR11 allele reduced the zone of drug clearance by a small but not significant amount (P=0.21, Fig. S1). However, neither of the engineered SC5314 strains showed increased fluconazole resistance (CDR11, P=0.48; QDR1, P=0.96, Fig. 2c, d).
Chromosome 4 trisomy contributes to increased drug resistance in P60002
The P60002 isolate is trisomic for two chromosomes, Chr4 and Chr6 (Fig. 1), and we tested whether the presence of either of the supernumerary chromosomes contributes to elevated fluconazole resistance. P60002 was serially passaged in YPD medium for 18 days to induce spontaneous loss of one or both of the supernumerary chromosomes (Fig. 3a). Forty-nine isolates [including multiple day 0 (D0) and day 18 (D18) time points] were genotyped by double-digest restriction-site-associated DNA sequencing (ddRAD-Seq). Surprisingly, the majority (20/26) of sequenced isolates from the D0 samples were euploid (Fig. 3b), indicating that the original P60002 isolate consisted of cells in a mixture of different ploidy states. In fact, of the 15 isolates sequenced at both D0 and D18 points, 10 were euploid prior to passaging. Two passaged isolates were initially trisomic for both Chr4 and Chr6; one retained the trisomic chromosomes during passaging, while the other lost both supernumerary chromosomes. All three isolates that contained a single trisomic chromosome prior to passaging (two for Chr4 and one for Chr6) became disomic for these chromosomes by day 18. Together, these experiments identified P60002 populations with different copy numbers for Chr4 and Ch6, and established that isolates trended towards euploidy during in vitro passaging.
Fig. 3.

Chr4 trisomy contributes to increased fluconazole resistance in P60002. (a) Single colonies of P60002 were serially passaged for 18 days to induce loss of Chr4 and/or Chr6 trisomies. Forty-nine isolates were genotyped by ddRAD-Seq to assess chromosome copy number. (b) Schematic showing P60002-derived isolates that were disomic or trisomic for Chr4 and/or Chr6 combined for both D0 and D18 time points. (c) Analysis of fluconazole resistance in P60002-derived isolates that were disomic or trisomic for Chr4 and Chr6. The dashed circle indicates an area where cells grew more slowly when lacking the Chr4 trisomy. (d) Analysis of fluconazole resistance in SC5314 or in an aneuploid SC5314 derivative that harbours a Chr4 trisomy. (e) Fluconazole resistance in SC5314 and P60002 strains that were disomic or are trisomic for Chr4 and/or Chr6. Drug resistance for SC5314 isolates that were trisomic for Chr4 was collected from six independent aneuploid strains. (f) Optical density of broth microdilution assays across a range of fluconazole concentrations for P60002- and SC5314-derived strains that were disomic or trisomic for Chr4 or Chr6 (values determined after 24 h). Each strain–ploidy combination is represented by three biological replicates. (g) Doubling times for different karyotypic forms of P60002 when grown in liquid YPD or SCD medium. (h) Measurement of rhodamine 123 accumulation in P60002 or derivatives to assay MDR efflux pump activity. All plate images in the figure were taken after 2 days.
To assess the potential contributions of trisomic chromosomes to drug resistance in P60002, isolates encoding different disomic/trisomic combinations of Chr4 and Chr6 were grown in the presence of fluconazole. As expected based on our previous analysis [54], P60002 isolates that were trisomic for both chromosomes grew up uniformly in the presence of the fluconazole diffusion disk, demonstrating resistance even to high levels of the drug (Fig. 3c). In contrast, the standard laboratory strain SC5314 exhibited a large zone of clearance around the disk, which was indicative of drug susceptibility (Fig. 3d). Euploid derivatives of P60002 that had lost both Chr4 and Chr6 trisomies displayed intermediate levels of resistance, in which colonies still formed close to the fluconazole disk but with a reduced size (Fig. 3c). These growth characteristics are indicative of drug tolerance, reflecting a reduced ability of the drug to inhibit C. albicans growth [57]. Importantly, P60002 isolates that were trisomic for Chr4 exhibited similar fluconazole resistance levels to isolates that were trisomic for both Chr4 and Chr6 (P=1), whereas those that were trisomic for Chr6 showed resistance phenotypes that were similar to those of euploid P60002 cells (compared to trisomic Chr4 and Chr6: trisomic Chr6; P=0.016, disomic; P=0.015, Fig. 3c, e). These results implicate loci present on Chr4 as making key contributions to fluconazole resistance in P60002. We also compared the growth of euploid and aneuploid versions of P60002 in a fluconazole broth microdilution assay. Using this assay we found that karyotypic variants of P60002 displayed no noticeable growth differences across a range of fluconazole concentrations. This reflects how different resistance phenotypes are observed between C. albicans isolates grown on solid media versus liquid media (Fig. 3f) [57, 63–65].
The presence of supernumerary chromosomes in C. albicans isolates is often associated with reduced fitness relative to euploid isolates [66–68]. To determine the impact of supernumerary chromosomes on doubling times in P60002, the growth rates of aneuploid and euploid strains were measured in YPD and SCD media. Interestingly, strains carrying the Chr4 trisomy had shorter doubling times than strains that were euploid or strains that were trisomic for both Chr4 and Chr6 when cultured in YPD medium (Fig. 3g). In contrast, no significant differences in doubling times existed when strains were grown in SCD. These results indicate that the presence of a supernumerary chromosome does not always lead to a significant growth defect, at least when comparing doubling times during exponential growth under replete nutrient conditions.
To determine if Chr4 trisomy also increases fluconazole resistance in other C. albicans strain backgrounds, we took advantage of parasexual derivatives of SC5314 that are trisomic for this chromosome [68]. We utilized five derivatives of SC5314 that have a Chr4 trisomy and examined their growth in the presence of fluconazole diffusion disks. Notably, SC5314 strains that were trisomic for Chr4 did not exhibit elevated resistance relative to the euploid control (P=0.56, Figs 3d–f and S2). However, one strain that was trisomic for both ChrR and Chr4 did show increased fluconazole resistance relative to the control (P=0.02, Fig. S2). Thus, while gene dosage of loci on Chr4 plays a significant role in promoting fluconazole resistance in P60002, this effect is dependent on strain background and is not typically observed in the standard laboratory strain of C. albicans.
Chr4 trisomy does not alter MDR efflux pump activity
To investigate if efflux pump activity provides elevated fluconazole resistance in P60002 isolates that are trisomic for Chr4, we performed Rh123 accumulation assays in strains with different combinations of Chr4 and Chr6. Removal of Rh123 from the cell requires activity of the multidrug-resistance (MDR) class of efflux pumps and correlates with azole resistance in C. albicans [69, 70]. Decreased Rh123 retention suggests hyperactive drug efflux that could also provide elevated drug resistance. All karyotypic variants of P60002 accumulated similar levels of Rh123 (Fig. 3h), suggesting that the expulsion of azoles by MDR efflux pumps does not contribute to elevated fluconazole resistance in P60002 harbouring a trisomic Chr4.
Discussion
In this study, we investigated the genetic basis of fluconazole resistance in C. albicans isolate P60002. This isolate was of significant interest as it lacked any polymorphisms or gene expression patterns known to be associated with fluconazole resistance. Furthermore, we showed that increased expression of two genes, CDR11 and QDR1, encoding putative drug efflux pumps had no effect on fluconazole resistance when transferred into the laboratory strain, SC5314. Instead, we established that a Chr4 trisomy makes a significant contribution to azole resistance in P60002, further extending the link between karyotypic change and drug resistance in fungal species.
Previous studies have demonstrated that many fluconazole-resistant isolates of C. albicans are aneuploid [15, 32, 45, 71, 72], although the precise relationship between specific aneuploid forms and drug resistance remains unclear. Fluconazole exposure was recently shown to induce aneuploid formation in C. albicans due to cell cycle aberrations [48], and a subset of these chromosomal alterations are known to provide fitness advantages in the presence of the drug. The most direct link between aneuploidy and drug resistance involves the formation of i5L aneuploids, which are commonly encountered in the clinic and increase fluconazole resistance through elevated ERG11 and TAC1 expression [15, 28]. More recent evidence suggests that trisomy of ChrR also contributes to increased azole resistance in C. albicans [46], and we similarly observed elevated fluconazole resistance in an SC5314-derived strain that was trisomic for both ChrR and Chr4 (Fig. S1). However, it is likely that many of the karyotypic changes observed in clinical isolates do not contribute directly to drug resistance. In addition, some aneuploid forms could represent a transitory state before cells achieve a more stable or less costly mechanism of resistance [45]. For example, studies in Saccharomyces cerevisiae have shown that supernumerary chromosomes can promote rapid adaptation to stressful environments, but more refined solutions then evolve with lower fitness costs than aneuploidy [33].
Fluconazole resistance appears to be multifactorial in isolate P60002, with Chr4 trisomy being one factor contributing to overall drug resistance. This isolate was recovered from the bloodstream of a patient and belongs to clade SA [54], but no other clinical information is available about its history. The genome-sequenced P60002 isolate was trisomic for both Chr4 and Chr6, and upon passaging in the laboratory both of the trisomic chromosomes were often lost, indicating that the supernumerary chromosomes had limited stability. This could be due to a fitness cost associated with aneuploidy, as an imbalanced karyotype has generally been associated with slower proliferation rates in a variety of species [73–75]. Notably, however, P60002 isolates that were trisomic for Chr4 grew marginally faster than euploid derivatives, at least when meassuring exponential growth in rich culture conditions. This is consistent with previous suggestions that C. albicans is well suited to tolerating aneuploidy [67], and with experiments in S. cerevisiae where some aneuploids were found to grow faster than isogenic euploids under a subset of conditions [15, 75, 76]. We also note that an increase in Chr4 copy number occurred in C. albicans isolate T118 when serially passaged in the presence of fluconazole, although the change in copy number was not shown to contribute to increased drug resistance [67].
Loss of the Chr4 trisomy reduced, but did not eliminate, the resistance profile observed in P60002. Thus, P60002 strains disomic for Chr4 were still highly tolerant to high concentrations of azoles, with extensive growth up to the fluconazole disk. This suggests that the resistance phenotype observed in the aneuploid P60002 strain is due to mechanisms that promote both tolerance and resistance to fluconazole. Derivatives that are disomic for Chr4 are no longer resistant but still exhibit considerable tolerance to the drug. Loss-of-function mutations for genes associated with C. albicans tolerance are not evident in P60002 [35, 38, 54], suggesting that additional mechanisms contribute to the underlying tolerance phenotype of euploid P60002 cells to fluconazole.
Analysis of fluconazole resistance in C. albicans has often focused on ERG11 polymorphisms, copy number variation in key transcriptional regulators, or gene expression levels of drug efflux pumps. P60002 displayed elevated expression of two members of the ABC and MFS drug efflux transporter gene families, CDR11 and QDR1, and ectopic expression of the P60002 alleles in SC5314 led to increased expression of these genes in this strain background. Elevated expression of the ectopic alleles in SC5314 is likely due to promoter polymorphisms that increase transcription, or to coding polymorphisms that alter RNA stability. Despite elevated expression of CDR11 and QDR1, drug resistance was not enhanced in the fluconazole-susceptible SC5314 strain. This is consistent with a recent study that found no difference in azole susceptibility in QDR1-null mutants of SC5314 [77]. It is therefore apparent that as yet unknown mechanisms, in addition to the contribution of Chr4 trisomy, promote drug resistance in P60002. By extension, our results emphasize the difficulty of making genotype-to-phenotype connections, even in cases where strong candidate genes are predicted to underlie drug resistance.
Acquisition of drug resistance often occurs by incremental increases in tolerance to the drug. For example, previous studies established that activating mutations in UPC2 and TAC1 make additive contributions to drug resistance [25]. Resistance in P60002 also appears to be due to the combinatorial effects of multiple genetic alterations. The presence of the Chr4 trisomy confers an increase in fluconazole resistance, but other genetic changes must also contribute to overall drug resistance, as loss of this trisomy generated P60002 derivatives that still exhibited tolerance to the drug. Loss of the Chr4 trisomy occurs in the absence of selective pressure, demonstrating that it is readily dispensable in the absence of drug.
We also examined whether Chr4 trisomy provides increased fluconazole resistance to C. albicans isolates from other strain backgrounds. Along with P60002, two additional clinical isolates in the same collection were found to be trisomic for Chr4 (isolates 12C and P78042), but neither of these isolates exhibited elevated drug resistance [54]. To further address the relationship between Chr4 trisomy and drug resistance, we examined multiple derivatives of SC5314 that were trisomic for Chr4 [68]. None of the SC5314 strains that were trisomic for only Chr4 showed elevated fluconazole resistance, however, indicating that the influence of Chr4 trisomy is dependent on strain background. These findings establish that certain genetic changes have universal effects on drug resistance independent of strain background, but others vary considerably between isolates, and are consistent with observations in other systems where genetic background has been shown to have a major effect on mutant phenotypes [78, 79]. Future investigations will therefore need to carefully consider the role of genetic interactions in the emergence of drug resistance to generate a comprehensive understanding of this important problem.
Funding information
This work was supported by National Institutes of Health grants AI081704 and AI122011 (to R. J. B.), a PATH award from the Burroughs Wellcome Fund (to R. J. B.) and a NIH Research Supplement to Promote Diversity in Health-Related Research AI081704-S1 (to M. Z. A.).
Acknowledgements
We would like to thank Christina Cuomo and Diego Martinez for assistance in analysis of RNA sequencing data and Matthew Hirakawa and Iuliana Ene for feedback on the manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Supplementary Data
References
- 1.Beck-Sague C, Banerjee S, Jarvis WR. Infectious diseases and mortality among US nursing home residents. Am J Public Health. 1993;83:1739–1742. doi: 10.2105/AJPH.83.12.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, et al. Hidden killers: human fungal infections. Sci Transl Med. 2012;4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 4.Calderone RA, Fonzi WA. Virulence factors of Candida albicans. Trends Microbiol. 2001;9:327–335. doi: 10.1016/S0966-842X(01)02094-7. [DOI] [PubMed] [Google Scholar]
- 5.Sanglard D, Odds FC. Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis. 2002;2:73–85. doi: 10.1016/S1473-3099(02)00181-0. [DOI] [PubMed] [Google Scholar]
- 6.Liu W, Tan J, Sun J, Xu Z, Li M, et al. Invasive candidiasis in intensive care units in China: in vitro antifungal susceptibility in the China-SCAN study. J Antimicrob Chemother. 2014;69:162–167. doi: 10.1093/jac/dkt330. [DOI] [PubMed] [Google Scholar]
- 7.Manzoni P, Farina D, Leonessa M, Priolo C, Gomirato G. Use of prophylactic fluconazole in a neonatal intensive care unit: efficacy is similar to that described in adult high-risk surgical patients. Crit Care. 2006;10:402. doi: 10.1186/cc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geber A, Hitchcock CA, Swartz JE, Pullen FS, Marsden KE, et al. Deletion of the Candida glabrata ERG3 and ERG11 genes: effect on cell viability, cell growth, sterol composition, and antifungal susceptibility. Antimicrob Agents Chemother. 1995;39:2708–2717. doi: 10.1128/AAC.39.12.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abe F, Usui K, Hiraki T. Fluconazole modulates membrane rigidity, heterogeneity, and water penetration into the plasma membrane in Saccharomyces cerevisiae. Biochemistry. 2009;48:8494–8504. doi: 10.1021/bi900578y. [DOI] [PubMed] [Google Scholar]
- 10.Mishra NN, Prasad T, Sharma N, Gupta DK. Membrane fluidity and lipid composition of fluconazole resistant and susceptible strains of Candida albicans isolated from diabetic patients. Braz J Microbiol. 2008;39:219–225. doi: 10.1590/S1517-83822008000200004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, et al. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 12.Marchaim D, Lemanek L, Bheemreddy S, Kaye KS, Sobel JD. Fluconazole-resistant Candida albicans vulvovaginitis. Obstet Gynecol. 2012;120:1407–1414. doi: 10.1097/AOG.0b013e31827307b2. [DOI] [PubMed] [Google Scholar]
- 13.Vanden Bossche H, Marichal P, Willemsens G, Bellens D, Gorrens J, et al. Saperconazole: a selective inhibitor of the cytochrome P-450-dependent ergosterol synthesis in Candida albicans, Aspergillus fumigatus and Trichophyton mentagrophytes. Mycoses. 1990;33:335–352. doi: 10.1111/myc.1990.33.7-8.335. [DOI] [PubMed] [Google Scholar]
- 14.Kelly SL, Lamb DC, Loeffler J, Einsele H, Kelly DE. The G464S amino acid substitution in Candida albicans sterol 14α-demethylase causes fluconazole resistance in the clinic through reduced affinity. Biochem Biophys Res Commun. 1999;262:174–179. doi: 10.1006/bbrc.1999.1136. [DOI] [PubMed] [Google Scholar]
- 15.Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribeiro MA, Paula CR. Up-regulation of ERG11 gene among fluconazole-resistant Candida albicans generated in vitro: is there any clinical implication? Diagn Microbiol Infect Dis. 2007;57:71–75. doi: 10.1016/j.diagmicrobio.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 17.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhäuser J, et al. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell. 2008;7:1180–1190. doi: 10.1128/EC.00103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.MacPherson S, Akache B, Weber S, de Deken X, Raymond M, et al. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother. 2005;49:1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly SL, Lamb DC, Corran AJ, Baldwin BC, Kelly DE. Mode of action and resistance to azole antifungals associated with the formation of 14α-methylergosta-8,24(28)-dien-3β,6α-diol. Biochem Biophys Res Commun. 1995;207:910–915. doi: 10.1006/bbrc.1995.1272. [DOI] [PubMed] [Google Scholar]
- 20.Dunkel N, Blass J, Rogers PD, Morschhäuser J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol. 2008;69:827–840. doi: 10.1111/j.1365-2958.2008.06309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coste A, Turner V, Ischer F, Morschhäuser J, Forche A, et al. A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics. 2006;172:2139–2156. doi: 10.1534/genetics.105.054767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2 . Eukaryot Cell. 2004;3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Liu JY, Shi C, Li WJ, Zhao Y, et al. Mutations in transcription factor Mrr2p contribute to fluconazole resistance in clinical isolates of Candida albicans. Int J Antimicrob Agents. 2015;46:552–559. doi: 10.1016/j.ijantimicag.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Heilmann CJ, Schneider S, Barker KS, Rogers PD, Morschhäuser J. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob Agents Chemother. 2010;54:353–359. doi: 10.1128/AAC.01102-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasse C, Dunkel N, Schäfer T, Schneider S, Dierolf F, et al. The stepwise acquisition of fluconazole resistance mutations causes a gradual loss of fitness in Candida albicans. Mol Microbiol. 2012;86:539–556. doi: 10.1111/j.1365-2958.2012.08210.x. [DOI] [PubMed] [Google Scholar]
- 26.Jensen RH, Astvad KM, Silva LV, Sanglard D, Jørgensen R, et al. Stepwise emergence of azole, echinocandin and amphotericin B multidrug resistance in vivo in Candida albicans orchestrated by multiple genetic alterations. J Antimicrob Chemother. 2015;70:2551–2555. doi: 10.1093/jac/dkv140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White TC. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob Agents Chemother. 1997;41:1482–1487. doi: 10.1128/aac.41.7.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selmecki A, Gerami-Nejad M, Paulson C, Forche A, Berman J. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol. 2008;68:624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 29.Kwon-Chung KJ, Chang YC. Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog. 2012;8:e1003022. doi: 10.1371/journal.ppat.1003022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep. 2012;13:515–527. doi: 10.1038/embor.2012.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen G, Rubinstein B, Li R. Whole chromosome aneuploidy: big mutations drive adaptation by phenotypic leap. Bioessays. 2012;34:893–900. doi: 10.1002/bies.201200069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selmecki A, Forche A, Berman J. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot Cell. 2010;9:991–1008. doi: 10.1128/EC.00060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yona AH, Manor YS, Herbst RH, Romano GH, Mitchell A, et al. Chromosomal duplication is a transient evolutionary solution to stress. Proc Natl Acad Sci USA. 2012;109:21010–21015. doi: 10.1073/pnas.1211150109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallè F, Sanguinetti M, Colella G, Di Onofrio V, Torelli R, et al. Oral candidosis: characterization of a sample of recurrent infections and study of resistance determinants. New Microbiol. 2011;34:379–389. [PubMed] [Google Scholar]
- 35.Cohen NR, Lobritz MA, Collins JJ. Microbial persistence and the road to drug resistance. Cell Host Microbe. 2013;13:632–642. doi: 10.1016/j.chom.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taff HT, Mitchell KF, Edward JA, Andes DR. Mechanisms of Candida biofilm drug resistance. Future Microbiol. 2013;8:1325–1337. doi: 10.2217/fmb.13.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luna-Tapia A, Tournu H, Peters TL, Palmer GE. Endosomal trafficking defects can induce calcium-dependent azole tolerance in Candida albicans. Antimicrob Agents Chemother. 2016;60:7170–7177. doi: 10.1128/AAC.01034-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanglard D, Ischer F, Marchetti O, Entenza J, Bille J. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Mol Microbiol. 2003;48:959–976. doi: 10.1046/j.1365-2958.2003.03495.x. [DOI] [PubMed] [Google Scholar]
- 39.Hill JA, Ammar R, Torti D, Nislow C, Cowen LE. Genetic and genomic architecture of the evolution of resistance to antifungal drug combinations. PLoS Genet. 2013;9:e1003390. doi: 10.1371/journal.pgen.1003390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamb DC, Corran A, Baldwin BC, Kwon-Chung J, Kelly SL. Resistant P45051A1 activity in azole antifungal tolerant Cryptococcus neoformans from AIDS patients. FEBS Lett. 1995;368:326–330. doi: 10.1016/0014-5793(95)00684-2. [DOI] [PubMed] [Google Scholar]
- 41.Rodloff C, Koch D, Schaumann R. Epidemiology and antifungal resistance in invasive candidiasis. Eur J Med Res. 2011;16:187–195. doi: 10.1186/2047-783X-16-4-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neves-Junior A, Cartágenes-Pinto AC, Rocha DA, de Sá LF, Junqueira ML, et al. Prevalence and fluconazole susceptibility profile of Candida spp. clinical isolates in a Brazilian tertiary hospital in Minas Gerais, Brazil. An Acad Bras Cienc. 2015;87:1349–1359. doi: 10.1590/0001-3765201520140717. [DOI] [PubMed] [Google Scholar]
- 43.Pfaller MA, Jones RN, Messer SA, Edmond MB, Wenzel RP. National surveillance of nosocomial blood stream infection due to Candida albicans: frequency of occurrence and antifungal susceptibility in the SCOPE Program. Diagn Microbiol Infect Dis. 1998;31:327–332. doi: 10.1016/S0732-8893(97)00240-X. [DOI] [PubMed] [Google Scholar]
- 44.Perea S, López-Ribot JL, Kirkpatrick WR, McAtee RK, Santillán RA, et al. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2001;45:2676–2684. doi: 10.1128/AAC.45.10.2676-2684.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, et al. The evolution of drug resistance in clinical isolates of Candida albicans. Elife. 2015;4:e00662. doi: 10.7554/eLife.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li X, Yang F, Li D, Zhou M, Wang X, et al. Trisomy of chromosome R confers resistance to triazoles in Candida albicans. Med Mycol. 2015;53:302–309. doi: 10.1093/mmy/myv002. [DOI] [PubMed] [Google Scholar]
- 47.Perepnikhatka V, Fischer FJ, Niimi M, Baker RA, Cannon RD, et al. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J Bacteriol. 1999;181:4041–4049. doi: 10.1128/jb.181.13.4041-4049.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrison BD, Hashemi J, Bibi M, Pulver R, Bavli D, et al. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol. 2014;12:e1001815. doi: 10.1371/journal.pbio.1001815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rustchenko-Bulgac EP. Variations of Candida albicans electrophoretic karyotypes. J Bacteriol. 1991;173:6586–6596. doi: 10.1128/jb.173.20.6586-6596.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iwaguchi S, Homma M, Tanaka K. Variation in the electrophoretic karyotype analysed by the assignment of DNA probes in Candida albicans. J Gen Microbiol. 1990;136:2433–2442. doi: 10.1099/00221287-136-12-2433. [DOI] [PubMed] [Google Scholar]
- 51.Pfaller MA, Rhine-Chalberg J, Redding SW, Smith J, Farinacci G, et al. Variations in fluconazole susceptibility and electrophoretic karyotype among oral isolates of Candida albicans from patients with AIDS and oral candidiasis. J Clin Microbiol. 1994;32:59–64. doi: 10.1128/jcm.32.1.59-64.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickman MA, Zeng G, Forche A, Hirakawa MP, Abbey D, et al. The 'obligate diploid' Candida albicans forms mating-competent haploids. Nature. 2013;494:55–59. doi: 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selmecki A, Bergmann S, Berman J. Comparative genome hybridization reveals widespread aneuploidy in Candida albicans laboratory strains. Mol Microbiol. 2005;55:1553–1565. doi: 10.1111/j.1365-2958.2005.04492.x. [DOI] [PubMed] [Google Scholar]
- 54.Hirakawa MP, Martinez DA, Sakthikumar S, Anderson MZ, Berlin A, et al. Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 2015;25:413–425. doi: 10.1101/gr.174623.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burrack LS, Applen SE, Berman J. The requirement for the Dam1 complex is dependent upon the number of kinetochore proteins and microtubules. Curr Biol. 2011;21:889–896. doi: 10.1016/j.cub.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reuss O, Vik A, Kolter R, Morschhäuser J. The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene. 2004;341:119–127. doi: 10.1016/j.gene.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 57.Gerstein AC, Rosenberg A, Hecht I, Berman J. diskImageR: quantification of resistance and tolerance to antimicrobial drugs using disk diffusion assays. Microbiology. 2016;162 doi: 10.1099/mic.0.000295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clinical and Laboratory Standards Institute . Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard. CLSI Document M27-A. Wayne, PA: 1997. [Google Scholar]
- 59.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. www.bioinformatics.babraham.ac.uk/projects/fastqc
- 61.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan Z, Hays M, Cromie GA, Jeffery EW, Scott AC, et al. Aneuploidy underlies a multicellular phenotypic switch. Proc Natl Acad Sci USA. 2013;110:12367–12372. doi: 10.1073/pnas.1301047110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sewell DL, Pfaller MA, Barry AL. Comparison of broth macrodilution, broth microdilution, and E test antifungal susceptibility tests for fluconazole. J Clin Microbiol. 1994;32:2099–2102. doi: 10.1128/jcm.32.9.2099-2102.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koga-Ito CY, Lyon JP, Resende MA. Comparison between E-test and CLSI broth microdilution method for antifungal susceptibility testing of Candida albicans oral isolates. Rev Inst Med Trop Sao Paulo. 2008;50:7–10. doi: 10.1590/S0036-46652008000100002. [DOI] [PubMed] [Google Scholar]
- 65.Zomorodian K, Bandegani A, Mirhendi H, Pakshir K, Alinejhad N, et al. In vitro susceptibility and trailing growth effect of clinical isolates of Candida species to azole drugs. Jundishapur J Microbiol. 2016;9:e28666. doi: 10.5812/jjm.28666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Magee BB, Magee PT. WO-2, a stable aneuploid derivative of Candida albicans strain WO-1, can switch from white to opaque and form hyphae. Microbiology. 1997;143:289–295. doi: 10.1099/00221287-143-2-289. [DOI] [PubMed] [Google Scholar]
- 67.Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 2009;5:e1000705. doi: 10.1371/journal.pgen.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forche A, Alby K, Schaefer D, Johnson AD, Berman J, et al. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol. 2008;6:e110. doi: 10.1371/journal.pbio.0060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clark FS, Parkinson T, Hitchcock CA, Gow NA. Correlation between rhodamine 123 accumulation and azole sensitivity in Candida species: possible role for drug efflux in drug resistance. Antimicrob Agents Chemother. 1996;40:419–425. doi: 10.1128/aac.40.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Neyfakh AA, Bidnenko VE, Chen LB. Efflux-mediated multidrug resistance in Bacillus subtilis: similarities and dissimilarities with the mammalian system. Proc Natl Acad Sci USA. 1991;88:4781–4785. doi: 10.1073/pnas.88.11.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerstein AC, Berman J. Shift and adapt: the costs and benefits of karyotype variations. Curr Opin Microbiol. 2015;26:130–136. doi: 10.1016/j.mib.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bennett RJ, Forche A, Berman J. Rapid mechanisms for generating genome diversity: whole ploidy shifts, aneuploidy, and loss of heterozygosity. Cold Spring Harb Perspect Med. 2014;4:a019604. doi: 10.1101/cshperspect.a019604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sheltzer JM, Amon A. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends Genet. 2011;27:446–453. doi: 10.1016/j.tig.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siegel JJ, Amon A. New insights into the troubles of aneuploidy. Annu Rev Cell Dev Biol. 2012;28:189–214. doi: 10.1146/annurev-cellbio-101011-155807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, et al. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, et al. Identification of aneuploidy-tolerating mutations. Cell. 2010;143:71–83. doi: 10.1016/j.cell.2010.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shah AH, Singh A, Dhamgaye S, Chauhan N, Vandeputte P, et al. Novel role of a family of major facilitator transporters in biofilm development and virulence of Candida albicans. Biochem J. 2014;460:223–235. doi: 10.1042/BJ20140010. [DOI] [PubMed] [Google Scholar]
- 78.Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, et al. Genotype to phenotype: a complex problem. Science. 2010;328:469. doi: 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vu V, Verster AJ, Schertzberg M, Chuluunbaatar T, Spensley M, et al. Natural variation in gene expression modulates the severity of mutant phenotypes. Cell. 2015;162:391–402. doi: 10.1016/j.cell.2015.06.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.