


Abstract
The quantitative specificity of the STAT1 transcription factor was determined by measuring the relative affinity to hundreds of variants of the consensus binding site including variations in the length of the site. The known consensus sequence is observed to have the highest affinity, with all variants decreasing binding affinity considerably. There is very little loss of binding affinity when the CpG within the consensus binding site is methylated. Additionally, the specificity of mutant proteins, with variants of amino acids that interact with the DNA, was determined and nearly all of them are observed to lose specificity across the entire binding site. The change of Asn at position 460 to His, which corresponds to the natural amino acid at the homologous position in STAT6, does not change the specificity nor does it change the length preference to match that of STAT6. These results provide the first quantitative analysis of changes in binding affinity for the STAT1 protein, and several variants of it, to hundreds of different binding sites including different spacer lengths, and the effect of CpG methylation.
INTRODUCTION
The STAT family of TFs play important roles in the differentiation of immune cell types as well as in their responses to various stimuli (1,2). Mutations in several members of STAT family of proteins have been shown to play critical roles in autoimmune disorders as well as primary immunodeficiency syndromes (3–7). Uzel et al. described patients with IPEX-like phenotypes from five different STAT1 mutations, all of which resulted in increased and prolonged phosphorylation of STAT1 in response to IFN-ϒ, IL-6 and IL-21 (7). Other examples of monogenic cause involve gain of function (GOF) mutations in STAT3 and STAT1, resulting in autoimmunity (8–10). Soltesz et al. reported gain of function of STAT1 signaling resulting from either hyper-phosphorylation or impaired dephosphorylation, which is the consequence of mutations in the coiled-coil and DNA binding domains (DBD) of the protein (10). Alternatively, stronger binding of the mutant DBD of STAT1 to the substrate DNAs may also play a role in GOF eventually leading to autoimmunity (10).
Overexpression of STAT3 (11) and STAT5 (12) were found to be linked with tumorigenesis. Furthermore, in cells, cytokine independence and expression of anti-apoptotic proteins were observed due to consecutive phosphorylation and DNA binding activities of STAT5 with mutations in DNA binding domain (13). Presence of tetrameric STAT5, which could result from mutation in transactivation domain, was also strongly correlated with tumorigenicity in mice and humans (14,15). Under chemical induction, mice lacking STAT1 develop tumors faster than wild-type individuals (16). In many instances, STAT1, STAT3 and STAT5 were identified to be either constitutively activated in certain types of tumors or required for the phenotype of oncogenic cell lines (17), with STAT1 and STAT3 sometimes playing opposing roles (18).
The activation of STAT proteins is achieved by a single consensus tyrosine (Tyr) phosphorylation, which has been shown to regulate the partitioning of STAT1 protein between different dimer conformations (19,20). Although STAT proteins are known to regulate different genes, they all have a high affinity towards the gamma activator sequence (GAS) element. By using a selection experiment, Horvath et al. showed that the sequence for optimal DNA binding with STAT1 is TTCC(C/G)GGAA, which has a core GAS element with the half palindromes (underlined), ‘TTC’ and ‘GAA’, separated by three nucleotides (21). It has also been demonstrated that STAT6 has a preference for binding to a GAS sequence in which the half palindromes are separated by four nucleotides (N4 sites), whereas STAT1 and STAT5 prefer half palindromes separated by three nucleotides (N3 sites) (22,23). Structures of protein–DNA complexes have been determined by X-ray crystallography for STAT1, STAT3 and STAT6 (23–25). Surprisingly, there is only one amino acid that makes direct hydrogen bond interactions with the DNA bases, to both the C and adjacent T in each half palindrome. That amino acid is an asparagine in STAT1 and STAT3 (N460 in STAT1 and N466 in STAT3) and a histidine in STAT6 (H415). In STAT6, H415 has been shown to be critical for the preference of N4 sites over N3 sites, and the mutant H415N reverses that, so the preference is for N3 sites (23). There is an additional lysine residue (K336 in STAT1, K340 in STAT2 and K284 in STAT6) that makes water-mediated hydrogen bonds to the bases in the spacer region of the GAS sequence (23–25). There are two additional residues (S459 and Q463 in STAT1, S465 and Q469 in STAT3 and V414 and Q418 in STAT6) that form a hydrophobic pocket around the methyl-groups of the two Ts in the half-palindrome and presumably contribute to binding specificity (23–25).
Our interest is in determining the quantitative specificity of STAT1 and identifying changes in specificity that may occur with mutations in the specificity determining residues, K336, S459, N460 and Q463. Although many mutations have been observed in STAT proteins, only one is known to occur in those amino acids, Q463H (26,27). A recent study performed alanine-scanning mutagenesis across the entire coiled-coil and DNA-binding domains (28). They found that substitution of alanine for any of the specificity determining residues, as well as the variant Q463H, were inactive in driving expression of a reporter gene from a GAS sequence (28). But that result does not rule out the possibility that those mutant proteins might have high affinity for alternative binding sites. We took advantage of an earlier observation that introduction of two cysteine residues in the C-terminal SH2 domain of STAT3 creates a variant that dimerizes without phosphorylation and is active both in vitro and in vivo (11). We used Spec-seq, which provides high-resolution measurements of relative binding affinity to hundreds of binding-site variants in parallel (29–31), to determine the specificity of wild-type STAT1, including sensitivity to CpG methylation and variation in the length of the spacer region, and to several variant proteins with alterations in the specificity-determining residues.
MATERIALS AND METHODS
Determination of relative-affinity using Spec-Seq
Protein−DNA interaction is measured by the dissociation constant, KD, of the binding equilibrium. KD is defined as the reciprocal of the association constant, KA¸ which is the ratio of the equilibrium concentrations of reactants and the DNA−protein complex:
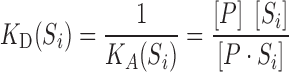 |
(1) |
In a competitive environment, the ratio of the concentrations of the bound and unbound species determines the relative affinity of the competing DNA binding sites (29–31):
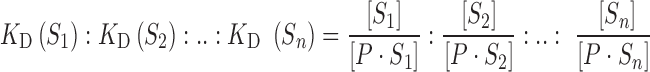 |
(2) |
In a binding reaction, involving TF and a library of DNAs, the concentration of bound or unbound species are directly proportional to the number of individual DNA molecules in bound or unbound fractions, respectively. Therefore, the relative affinity is measured by the ratio of numbers of the individual sites in each fraction:
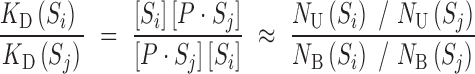 |
(3) |
where NB and NU are the numbers of a species (S) in bound or unbound fractions, respectively. Current high-throughput sequencing technologies allow parallel measurement of NB and NU for thousands of DNAs. In-gel separation of protein−DNA complex (bound) from unbound DNAs (unbound) allowed us to measure relative ratio of each species in these fractions (see Scheme 1, Supplemental materials). The natural logarithm of these ratios is the relative free energies of binding in units of kT (k = Boltzmann's constant and T = temperature used in experiments).
Protein expression
Based on an early report (32), STAT1 (Figure 1A) was expressed in a truncated form without the N-terminal domain (1–131 amino acids), which is responsible for the tetramerization of STAT proteins. Truncated STAT1 (amino acids 132–713) was expressed in Escherichia coli cells under T7 promoter inducible by IPTG. A 24-nucleotide (TGGTCTCACCCGCAGTTCGAAAAA) sequence was attached at the 3′-end of the constructs for encoding an additional eight amino-acid (WSHPQFEK) long peptide (Strep-tag) for purification. Active dimers of STAT1 were obtained by incorporating additional mutations in the Src homology 2 (SH2) domain. Bromberg et al. previously demonstrated the use of a STAT3 construct with two mutations, replacing native amino acids (A661 and N663) by cysteine, which led to the nuclear localization of STAT3 (11). The modified STAT3 dimerized by forming disulphide bridges in the Src-domain and demonstrated DNA binding in vitro and in vivo. The tSTATcc construct was made with amino acid replacements A656C and N658C (which are homologous to A661 and N663 of STAT3) (Figure 1B).
Figure 1.

Synthesis of tSTATcc. (A) Domains of STAT1. STAT1 has two isoforms. STAT1α is a 750 amino acid long protein, whereas STAT1β is 850 amino acid long. tSTATcc was a truncated variant of STAT1α (132–713 amino acid, 63 KDa) with additional mutations replacing A656 and N658 by cysteines. (B) The sequence alignment of the SH2 domains of STAT1, STAT3 and STAT4, showing where the cysteines are substituted. (C) Polyacrylamide gel visualization of tSTATcc. The dimer band appears in the native gel and disappears when run in a denaturing gel in presence of DTT.
In vivo protein expression was done by adding IPTG to E. coli BL-21 (DE-3) cell culture, containing DHFR-control vector (NEB PUREexpress), with cDNA of tSTATcc, at OD600 = 0.6. The cultures were incubated for six hours at 37°C before lysing the cells by sonication. The separation of proteins from the cellular debris was performed by centrifugation at 15 000 rpm. The supernatant was filtered and loaded directly on a Strep-Tactin column. The protein was eluted in buffer containing 100 mM Tris–HCl, 150 mM NaCl, 2.5 mM desthiobiotin and 1 mM EDTA. Dimers of tSTATcc were visualized in a 10% Tris-glycine SDS-PAGE, in absence of reducing agents such as, β-mercaptoethanol or DTT (Figure 1C). The monomeric tSTATcc was visualized (63 kDa) in a denaturing 10% Tris-glycine SDS-PAGE gel after incubating the protein sample in 100 mM DTT (Figure 1C).
Protein concentration was measured by using the equation: C = (1.55*A280) – (0.76*A260), where C is the concentration of the protein in mg/ml, A280 is the absorbance of protein samples at 280 nm and A260 is the absorbance at 260 nm. The protein concentration obtained by this method was comparable with BCA protein quantification assay results.
The mutant proteins were synthesized by employing site directed mutagenesis at specific positions of DNA binding domain (DBD) of STAT1. Eight PCR (polymerase chain reaction) site-directed mutagenesis reactions were carried out using Q5 Mutagenesis kit (NEB). The forward and reverse primers for each PCR mutagenesis are listed in the Supplementary materials (Supplementary Figure S1). NEB base changer web-tool was used to design the primers to replace the native amino acids K336, S459, N460 or Q463 of tSTATcc construct by alanine, arginine or histidine. PCR reactions were performed in 25 μl reaction mixtures, following the procedure described in the kit manual. Each reaction mixture contained 25 ng tSTATcc template, encoded in a DHFR-control vector (NEB), 125 ng of forward and reverse primers, 10 nmol of dNTPs, 2.5 units of DNA polymerase in 35 mM Tris–HCl (pH 8.0) containing 12 mM potassium acetate, 5mM DTT, 0.05% Triton X-100 and 0.05 mM EDTA. The products were ligated with Q5 ligase master mix and transformed in DH5α E. coli cells. Purified plasmids were verified by sequencing.
Library design and preparation of dsDNA substrate
The binding models for STAT1 and the mutant constructs were generated using the consensus (GAS) sequence and libraries based on GAS sequences. The libraries (Figure 2A) were designed with randomized (no more than four positions at a time) GAS sequences. The synthesis of double strand (dsDNA) libraries were initiated by mixing 100 pmol single-strand (ss) degenerate template oligos (Figure 2A) with 125 pmol FAM-labeled reverse complement primer, F1 (Supplementary Figure S2). A 10-s denaturation at 90°C followed by a 10-min annealing and extension at 52°C in presence of Taq polymerase afforded duplex DNAs. The DNA libraries were purified by PCR purification columns (QIAGEN), after the digestion of excess ssDNA by incubating the mixture in presence of exonuclease (NEB Exo I) for 30 min at 37°C.
Figure 2.

(A) DNA libraries. Designed based on GAS-consensus sequence. The randomised region was flanked by two primer-binding regions (P1 and P2) for amplification and indexing, followed by sequencing. (B) Binding energy logo for tSTATcc, involving the single-mutant of the preferred sequences in the Sp-3nu and Pa-CCG libraries. All energies are in units of kT. (C) Comparison between the relative binding energies of Sp-3nu and Pa-CCG libraries from two separate experiments. (D) Ratio of the binding energies of sequences in Sp-3nu (blue) and Pa-CCG (green) library that either have at least one CG dinucleotide (circles) or do not have any CG dinucleotide (triangles). Y-axis represent libraries treated with CpG methyl transferase whereas, X-axis represent untreated libraries.
Four additional libraries with 5′ mononucleotide barcodes (Supplemental Figure S3) were designed by either randomizing the palindrome or spacer region of the consensus GAS sequences and treated with or without CpG methyltransferase (M.SssI) following vendor's (NEB) protocol. Briefly, 1 μg duplex DNA libraries were incubated with 160 μM S-adenosylmethionine (SAM) and 4 unit of CpG methyltransferase for 1 h at 37 °C. The libraries were further purified by QIAGEN PCR purification columns. To ensure efficient enzymatic methylation both the methylated and unmethylated versions of the Pa-CCG libraries (PA-treated and PA-untreated in Supplementary Figure S3) were treated with restriction enzyme HpaII that cuts at CCGG (which every sequence contains) only if it is unmethylated. Supplemental Figure S4 shows that PA-untreated is nearly completely cut whereas PA-treated is completely resistant to cutting.
EMSA and Sample preparation for sequencing
The protein−DNA binding reactions were done in 1× NEB CutSmart buffer (50 mM KOAc, 20 mM Tris-OAc, 10 mM Mg(OAc)2, 100 μg/ml BSA, pH 7.9) supplemented with 10% glycerol. Fifty nmol of the FAM-labeled DNA libraries were incubated at 4°C for 30 min with varying concentrations of wild type protein, tSTATcc, or the mutants in 15 μl reaction volume. The reaction mixtures were run (4°C) in 10% Tris-glycine PAGE gel at 200 V for 1 h. The FAM-labeled DNA fragments in the bound (slow migrating band) and unbound (fast migrating bands) were visualized by a BioRad imager with a 520 nM bandpass filter (Supplemental materials, Scheme 1, Figure S5). The visible bands were excised from the gels and DNAs were extracted and purified using acrylamide extraction buffer (100 mM NH4OAc, 10 mM Mg(OAc)2, 0.1% SDS) and QIAGEN gel purification columns, respectively. The DNAs were amplified and barcoded by indexed Illumina-primers. The combination of either P1 or PE1-LT and P2 (Supplementary Figure S2) were used for the amplification of Sp-3nu, Pa-CCG and Sp-4nu (Figure 2A) or sequences with specific variable spacers (Supplementary Figure S6). Similarly, PE1 and PE2 were used for the methylation sensitivity assay. The sequencing results from Illumina 1 × 75 Myseq runs were filtered and sorted based on conserved regions and barcodes. For each library, the ratio of individual sequence in bound and unbound reads was calculated as a measurement of relative binding affinity (Equation (3)) compared to the consensus sequence.
RESULTS AND DISCUSSION
Synthesis of tSTATcc as a model protein
Although STAT proteins were recognized as important signal transducing proteins and transcription factors for decades, a thorough binding model for STAT has been unavailable. The development of Spec-seq (30,31) allows us to accurately measure relative affinities of TFs for thousands of sites in parallel. Here, we synthesized a truncated version of STAT1, which was used to generate a binding model for the protein. Most of the DNA binding studies with STAT1 (Figure 1A) was done in the past with a truncated STAT1α (∼60 kDa), which was phosphorylated by purified kinases to obtain active dimers (33). Another approach involved incorporation of mutations, which resulted in the replacement of native amino acids (A661 and N663) by cysteines in the SH2 domain of STAT3 (Figure 1B). The cysteine mutation allowed STAT3 to form active dimers via disulphide bonds, which ultimately led to nuclear localization and DNA binding (11). In this study, we took a similar approach by replacing two amino acids of STAT1, A656 and N658 by cysteines. The resulting protein, tSTATcc, showed dimer formation when visualized in a gel without the presence of reducing agents such as DTT or β-mercaptoethanol (Figure 1C, left panel). The dimers gradually disappeared with the addition of DTT as viewed in Figure 1C, right panel. The protein was purified from E. coli BL21 (DE3) cells and displayed complex formation with duplex DNA containing a GAS sequence (TTCCCGGAA) (Supplementary Figure S5). The construct, tSTATcc was used in combination of several DNA libraries for generating binding models for STAT1. Additionally, the construct was used as a model for the study of the effect of DNA binding for proteins with mutated amino acids in the DBD.
Library design and relative binding affinity
STAT1 is known to have strong affinity for GAS sequence. The crystal structures of both STAT1 (25) and STAT3 (24) reveal that these proteins form a symmetric complex with the palindromic 9-bp GAS site. We generated two libraries by dividing the GAS sequence in a central 3-bp spacer and two 3-bp flanking regions. We refer to these half-palindromic regions as L for left-half site (TTC) and R for right-half site (GAA). The spacer region (CCG) was referred to as S. The library Sp-3nu (Figure 2A) was synthesized with a degenerate S, whereas Pa-CCG had a randomized L site (Figure 2A). These two libraries added to tSTATcc and relative binding affinity of all 128 sites were determined using Spec-seq (Supplemental materials, Scheme 1). Figure 2B describes the relative binding specifies of Sp-3nu and Pa-CCG in form of energy logo (34,35). The energy logos show the change in free energy of binding for the single-mutant variants of the strongest binding sites, CCG, for the spacer, and TTC, for the L site. Most variant binding sites had an energy difference greater than 1 kT between the single mutants. The energy weight matrices (ePWMs) resulted from Spec-seq libraries, Sp-3nu and Pa-CCG, were used to generate ePWMs for the whole 6-bp (palindrome and spacer) sequences (Supplementary Figure S7). Since the L-site and the R-site of the 9-bp sequence are palindromic, we assumed that the PWM for the R-site (Pa-CCG) will be reverse complementary to the L-site. The reproducibility of the Spec-seq method was determined by comparing the relative binding energies of Sp-3nu and Pa-CCG libraries from two different experiments. Figure 2C shows the ratio of the binding energies of 64 Sp-3nu (red circles) and 64 Pa-CCG (blue triangles) sites from two separate experiments involving tSTATcc. The data were fitted to a straight line (r2 > 0.8). The mean differences between 64 SP-3nu and 64 Pa-CCG sites in two experiments were 0.22 kT and 0.13 kT, respectively, which is within the range of expected noise in our data (31).
Effect of DNA-methylation on STAT1 binding
CpG methylation is a major epigenetic factor in gene expression. CpG methylation in the promoter region of Interferon Regulatory Factor 8 (IRF8), which contains a GAS sequence and is regulated by STAT1, reduced activation of the gene in colon carcinoma cell line (36). STAT1 was shown to bind the promoter of IRF8 irrespective of methylation status, but we were prompted to assess the general sensitivity of STAT1 binding to CpG methylation. Libraries Sp-treated and PA-treated were subjected to CpG methyltransferase to generate methylated DNAs (Supplementary Figures S3 and S4). These libraries along with the unmethylated variants, Sp-untreated and PA-untreated, showed unaltered specificity of STAT1 binding. Even though all of the sequences of PA-treated library can be methylated due to the presence of at least one CG dinucleotide, only 34 out of the 64 sequences in the Sp-treated library can be methylated. The binding energies of these sequences were derived from Spec-seq experiment using an equimolar mixture of the internally barcoded methylated and unmethylated libraries (Supplementary Figure S3, underlined nucleotide). Figure 2D shows the ratio of CpG methyltransferase treated (Y axis) and untreated (X axis) 30 sequences that didn’t have any occurrence of a CG dinucleotide (blue triangles) in the spacer region. Additionally, the rest of the library (34 in number) that had at least one CG dinucleotide (Blue circles) was also compared. Clearly methylation on these sequences did not alter tSTATcc binding. Similarly, the comparison of treated and untreated palindrome libraries, reveals unaltered STAT1 binding (Figure 2D). Despite having a CG in the spacer, these sequences were further separated into two groups based on whether or not they have an additional CG in the palindrome region. The sequences with an additional CG in the palindrome are represented by green circles in Figure 2D, whereas sequences that don’t have a CG dinucleotide in the palindrome region are represented by green triangles. These results clearly show that methylation of GAS sequences does not alter STAT1 binding affinity. Therefore, the lack of activation of the IRF8 promoter when methylated indicates involvement of other methylation sensitive transcription factors.
Binding model for tSTATcc-N460H and comparison with wild type
The crystal structures of STAT1 and STAT3 suggest that only a single amino acid, N460 of STAT1 and N463 of STAT3, directly interacts with the bases of the palindromes (L and R) via hydrogen bonding (24,25). In STAT6 the homologous amino acid is H415 (23). STAT6 still favors the GAS sequence but with a preference for N4 sites over N3. It was recently shown that the STAT6 mutant H415N prefers the N3 spacer (23). We synthesised a mutant (tSTATcc-N460H) by replacing the native amino acid of tSTATcc by histidine and studied the DNA binding specificities of the 128 sites from two DNA libraries (Sp-3nu and Pa-CCG). The mutant tSTATcc-N460H has specificity very similar to the wild type tSTATcc (Figure 3A) but with slightly higher specificity for the palindromic part (TTC) and slightly less for the spacer (Figure 3B). We also studied the preference of N460H mutation in tSTATcc for N3 and N4 sites using a randomized 4 bp library, (SP-4nu, Figure 2A) and using a smaller library of specific N3 and N4 sites (Supplemental Figure S6) including those tested previously (23). We find that STAT1 N460H has the same preference for N3 sites over N4 sites as the wt protein (Figure 3C and Table 1). This shows that other parts of the protein, besides just the residue at 460, are required to allow high affinity binding to the longer spacer. This result is similar to that observed in a comparison of the Lac Repressor with the PurR protein (37). While the Lac Repressor (LacI) can bind with similar affinity to sites with spacers of 2–4 bp, PurR highly prefers just 2 bp spacers. To convert PurR to LacI-like specificity, where it could also bind with high affinity to sites with longer spacers, required mutations in both the DNA-interacting residues and those in a nearby loop region connecting two parts of the DNA-binding domain. Presumably additional changes in STAT1 could allow it to prefer N4 sites, as in STAT6.
Figure 3.

(A) Binding energy logo for tSTATcc-N460H, involving the single-mutant of the preferred sequences in the Sp-3nu and Pa-CCG libraries. (B) Evaluation of the binding energies of tSTATcc or tSTATcc-460H. Libraries with variable 3-base spacer (Sp-3nu) and 3-base palindrome (Pa-CCG) were used for the comparison. (C) Binding energies of tSTATcc or tSTATcc-460H was compared using Sp-4nu (Figure 2A) library. Note that energy = 0 is for the consensus GAS sequence which is not included in the sequences shown in this plot but was included in the mixture of sequences included in the binding reaction.
Table 1. Relative binding energies for the members of variable-spacer library.
| Normalized ΔΔG binding (kT)s | ||
|---|---|---|
| Sequence | tSTATcc | tSTATcc-460H |
| 1C_GAS: TTTTTTCGGGAAAA | 1.6 | 1.8 |
| 2C_GAS: TTTTTCCGGGAAAA | 0 | 0 |
| 3C_GAS: TTTCCCGGGAAAA | 1.6 | 1.6 |
| 0T_GAS: TTTTTTTAGGAAAA | 1.5 | 1.8 |
| 1T_GAS: TTTTTCTAGGAAAA | 1.7 | 1.7 |
| 2T_GAS: TTTTCCTAGGAAAA | 1.9 | 1.7 |
Effect of mutations of DNA-binding amino acids on binding specificity
We also investigated the effect of mutations of the DNA-binding amino acids by synthesizing mutant tSTATcc proteins. Figure 4 shows the amino acids of STAT1 interacting with the DNA, based on the crystal structures of STAT1 and STAT3 (24,25), that are thought to primarily determine the specificity of the proteins. Beside N460 that makes direct hydrogen bonds to the C and adjacent T of the palindrome, three additional amino acids are thought to contribute to DNA binding. K336 interacts with the spacer sequence via hydrogen bonding through a water molecule and S459 and Q463 form a hydrophobic pocket for the methyl group of the T in the first base of GAS sequence. Recently all four of those amino acids were replaced by alanine and shown to be inactive in driving a reporter gene via a GAS sequence (28). We wondered if replacing those amino acids by alanine, or with the large charged amino acid arginine, might alter the sequence preference of STAT1. Therefore, we produced seven additional variants of the tSTATcc protein: K336A, S459A, N460A, Q463A, S459R, N460R and Q463R. These seven mutant proteins were used in combination with libraries, Sp-3nu and Pa-CCG (Figure 2), to measure relative affinities using the Spec-seq method. Surprisingly, each of those mutations renders the protein nearly non-specific (Figure 5). The preferred sequences (which are not usually the GAS sequence) generally have energy differences of less than 0.2kT from alternative sequences, which is within the variance of our measurements (Figure 2C). That is, even though we can plot energy logos in Figure 5, comparing the energy scale of the logos of the wt protein and the N460H variant (also shown in Figure 5), indicates that the differences are not significantly different from experimental noise. We also include in Figure 5 the ‘information content’ for each protein, calculated from the energy matrices (Supplemental Figure S7) for the 9-long binding sites. While the wt protein and the N460H variant have 2.68 and 3.43 bits, respectively, the other variant proteins all have <0.23 bits, confirming that they are all essentially non-specific in their binding affinity.
Figure 4.

Interaction between the GAS sequence and major-groove binding amino acids in the DNA binding region of STAT1. The protein binds as a dimer and only one of the two monomers is shown here. The other monomer interacts with the DNA in the same way but in the opposite orientation of this sequence. The nucleotide sequence is shown in the box and the bases are color-coded for emphasizing the variation of molecular interaction with the functional groups of amino acids. Blue circles represent hydrogen-bond donors, whereas green and yellow circles represent hydrogen bond acceptors and methyl group on T, respectively. Additionally, the black circles represent water molecule mediated H-bonding and the hydrogen bonding is shown by the arrows.
Figure 5.
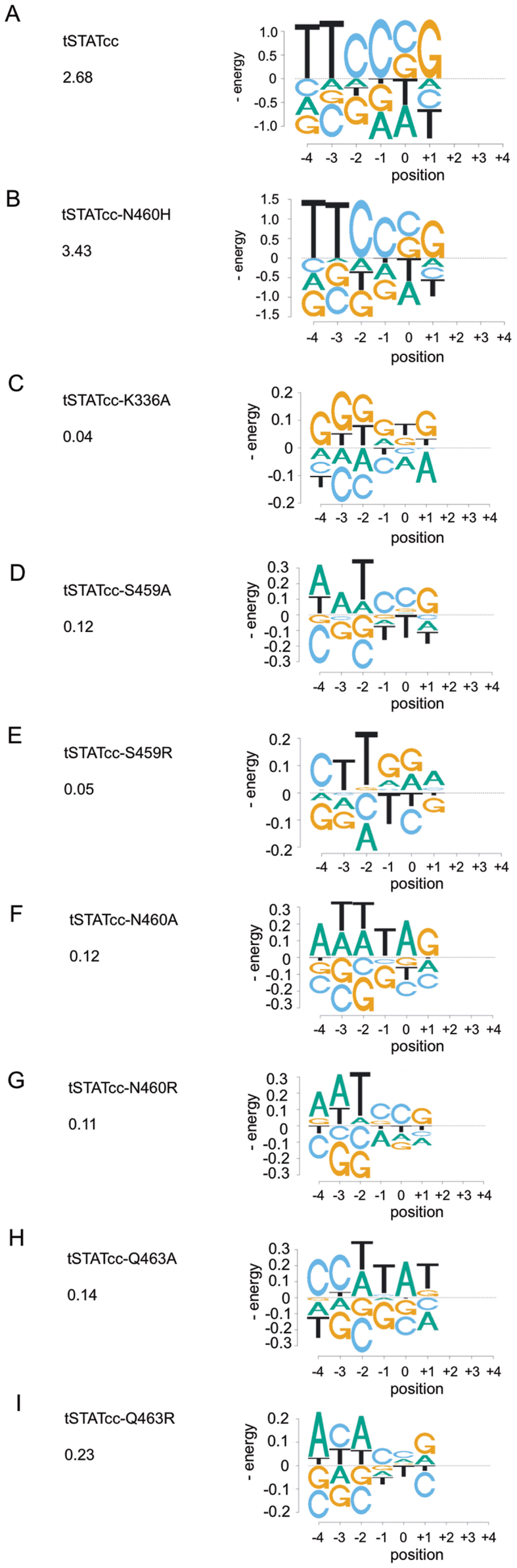
Binding energy logos involving tSTATcc and the mutants of STAt1. (A) tSTATcc. (B) tSTATcc-N460H. (C) tSTATcc-K366A. (D) tSTATcc-S459A. (E) tSTATcc-S459R. (F) tSTATcc-N460A. (G) tSTATcc-N460R. (H) tSTATcc-Q463A. (I) tSTATcc-Q463R. Column 1 indicates the specific mutation in the STAT1 construct and the information content for the entire binding site (which is calculated a 2x IC of position 1–3 plus IC of positions 4–6). Column 2 represents binding energy logos generated by considering single nucleotide mismatches from the preferred sequence.
CONCLUSION
We have generated a binding model for STAT1 based on a construct which successfully allowed the active dimerization of the protein, in vitro, without phosphorylation. STAT1 shows a strong preference for the consensus GAS sequence, TTCCCGGAA, but is insensitive to methylation of the CpG within that sequence. One mutant protein, N460H, which corresponds to the change in a DNA-binding residue that occurs in STAT5 and STAT6 proteins, has almost no effect on the specificity or on the spacing preference between the half-sites. We also tested seven additional mutations, at three additional positions in the DNA-binding domain. Surprisingly, instead of altering the specificity of STAT1, they all eliminate specificity almost entirely. Although only N460 makes direct hydrogen bonds to the bases in the major groove, the other amino acids that interact with the DNA via water-mediated hydrogen bonds and hydrophobic contacts, are critical to the specificity of STAT1. This is consistent with prior alanine-scanning results that showed loss of activation of a promoter containing a GAS sequence (28), but further shows not just altered specificity, but essentially complete loss of specificity. We speculate that the substitutions we made to those amino acids, both to alanine and arginine, may interfere with proper folding of the DNA-binding domain or they may disrupt a very tight interface between the protein and DNA that is required for high specificity binding.
ACCESSION NUMBERS
Raw reads for all of the experiments can be found in the NCBI short read archive under accession GSE95526.
Supplementary Material
Footnotes
Present address: Gary D. Stormo, Department of Genetics and Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, MO 63108-8510, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
ACKNOWLEDGEMENTS
We thank the members of the Stormo laboratory for their comments and suggestions and also the anonymous reviewers who made valuable suggestions that improved the quality of the presentation.
FUNDING
National Institutes of Health [HG000249]. Funding for open access charge: National Institutes of Health [HG000249].
Conflict of interest statement. None declared.
REFERENCES
- 1. Kiu H., Nicholson S.E.. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012; 30:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Shea J.J., Plenge R.. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012; 36:542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dupuis S., Jouanguy E., Al-Hajjar S., Fieschi C., Al-Mohsen I.Z., Al-Jumaah S., Yang K., Chapgier A., Eidenschenk C., Eid P. et al. Impaired response to interferon-[alpha]/[beta] and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003; 33:388–391. [DOI] [PubMed] [Google Scholar]
- 4. Holland S.M., DeLeo F.R., Elloumi H.Z., Hsu A.P., Uzel G., Brodsky N., Freeman A.F., Demidowich A., Davis J., Turner M.L. et al. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007; 357:1608–1619. [DOI] [PubMed] [Google Scholar]
- 5. Liu L., Okada S., Kong X.-F., Kreins A.Y., Cypowyj S., Abhyankar A., Toubiana J., Itan Y., Audry M., Nitschke P. et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 2011; 208:1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Minegishi Y., Saito M., Tsuchiya S., Tsuge I., Takada H., Hara T., Kawamura N., Ariga T., Pasic S., Stojkovic O. et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007; 448:1058–1062. [DOI] [PubMed] [Google Scholar]
- 7. Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E. et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation–polyendocrinopathy–enteropathy–X-linked–like syndrome. J. Allergy Clin. Immunol. 2013; 131:1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flanagan S.E., Haapaniemi E., Russell M.A., Caswell R., Allen H.L., De Franco E., McDonald T.J., Rajala H., Ramelius A., Barton J. et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014; 46:812–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haapaniemi E.M., Kaustio M., Rajala H.L.M., van Adrichem A.J., Kainulainen L., Glumoff V., Doffinger R., Kuusanmäki H., Heiskanen-Kosma T., Trotta L. et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood. 2015; 125:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soltész B., Tóth B., Shabashova N., Bondarenko A., Okada S., Cypowyj S., Abhyankar A., Csorba G., Taskó S., Sarkadi A.K. et al. New and recurrent gain-of-function STAT1 mutations in patients with chronic mucocutaneous candidiasis from Eastern and Central Europe. J. Med. Genet. 2013; 50:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bromberg J.F., Wrzeszczynska M.H., Devgan G., Zhao Y., Pestell R.G., Albanese C., Darnell J.E. Jr. Stat3 as an Oncogene. Cell. 1999; 98:295–303. [DOI] [PubMed] [Google Scholar]
- 12. Constantinescu S.N., Girardot M., Pecquet C.. Mining for JAK–STAT mutations in cancer. Trends Biochem. Sci. 2008; 33:122–131. [DOI] [PubMed] [Google Scholar]
- 13. Nosaka T., Kawashima T., Misawa K., Ikuta K., Mui A.L.F., Kitamura T.. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 1999; 18:4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. John S., Vinkemeier U., Soldaini E., Darnell J.E., Leonard W.J.. The significance of tetramerization in promoter recruitment by Stat5. Mol. Cell. Biol. 1999; 19:1910–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moriggl R., Sexl V., Kenner L., Duntsch C., Stangl K., Gingras S., Hoffmeyer A., Bauer A., Piekorz R., Wang D. et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005; 7:87–99. [DOI] [PubMed] [Google Scholar]
- 16. Kaplan D.H., Shankaran V., Dighe A.S., Stockert E., Aguet M., Old L.J., Schreiber R.D.. Demonstration of an interferon γ-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. 1998; 95:7556–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bromberg J. Stat proteins and oncogenesis. J. Clin. Invest. 2002; 109:1139–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Avalle L., Pensa S., Regis G., Novelli F., Poli V.. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAKSTAT. 2012; 1:65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bonham A.J., Wenta N., Osslund L.M., Prussin I.I.A.J., Vinkemeier U., Reich N.O.. STAT1:DNA sequence-dependent binding modulation by phosphorylation, protein:protein interactions and small-molecule inhibition. Nucleic Acids Res. 2013; 41:754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wenta N., Strauss H., Meyer S., Vinkemeier U.. Tyrosine phosphorylation regulates the partitioning of STAT1 between different dimer conformations. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:9238–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horvath C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000; 25:496–502. [DOI] [PubMed] [Google Scholar]
- 22. Ehret G.B., Reichenbach P., Schindler U., Horvath C.M., Fritz S., Nabholz M., Bucher P.. DNA binding specificity of different STAT proteins: comparison of in vitro specificity with natural target sites. J. Biol. Chem. 2000. [DOI] [PubMed] [Google Scholar]
- 23. Li J., Rodriguez J.P., Niu F., Pu M., Wang J., Hung L.W., Shao Q., Zhu Y., Ding W., Liu Y. et al. Structural basis for DNA recognition by STAT6. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:13015–13020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker S., Groner B., Muller C.W.. Three-dimensional structure of the Stat3[beta] homodimer bound to DNA. Nature. 1998; 394:145–151. [DOI] [PubMed] [Google Scholar]
- 25. Chen X., Vinkemeier U., Zhao Y., Jeruzalmi D., Darnell J.E. Jr, Kuriyan J.. Crystal Structure of a Tyrosine Phosphorylated STAT-1 Dimer Bound to DNA. Cell. 1998; 93:827–839. [DOI] [PubMed] [Google Scholar]
- 26. Chapgier A., Boisson-Dupuis S., Jouanguy E., Vogt G., Feinberg J., Prochnicka-Chalufour A., Casrouge A., Yang K., Soudais C., Fieschi C. et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006; 2:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boisson-Dupuis S., Kong X.F., Okada S., Cypowyj S., Puel A., Abel L., Casanova J.L.. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012; 24:364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kagawa R., Fujiki R., Tsumura M., Sakata S., Nishimura S., Itan Y., Kong X.F., Kato Z., Ohnishi H., Hirata O. et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J. Allergy Clin. Immunol. 2016; doi:10.1016/j.jaci.2016.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sasse S.K., Zuo Z., Kadiyala V., Zhang L., Pufall M.A., Jain M.K., Phang T.L., Stormo G.D., Gerber A.N.. Response element composition governs correlations between binding site affinity and transcription in glucocorticoid receptor feed-forward loops. J. Biol. Chem. 2015; 290:19756–19769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stormo G.D., Zuo Z., Chang Y.K.. Spec-seq: determining protein–DNA-binding specificity by sequencing. Brief. Funct. Genomics. 2015; 14:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuo Z., Stormo G.D.. High-resolution specificity from DNA sequencing highlights alternative modes of lac repressor binding. Genetics. 2014; 198:1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vinkemeier U., Cohen S.L., Moarefi I., Chait B.T., Kuriyan J., Darnell J.E.. DNA binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J. 1996; 15:5616–5626. [PMC free article] [PubMed] [Google Scholar]
- 33. Choudhury G.G., Ghosh-Choudhury N., Abboud H.E.. Association and direct activation of signal transducer and activator of transcription1alpha by platelet-derived growth factor receptor. J. Clin. Invest. 1998; 101:2751–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Foat B.C., Morozov A.V., Bussemaker H.J.. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006; 22:e141–e149. [DOI] [PubMed] [Google Scholar]
- 35. Stormo G.D. Modeling the specificity of protein–DNA interactions. Quant. Biol. 2013; 1:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McGough J.M., Yang D., Huang S., Georgi D., Hewitt S.M., Röcken C., Tänzer M., Ebert M.P.A., Liu K.. DNA methylation represses IFN-γ–induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol. Cancer Res. 2008; 6:1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zuo Z., Chang Y., Stormo G.D.. A quantitative understanding of lac repressor's binding specificity and flexibility. Quant. Biol. 2015; 3:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.