


Abstract
We previously showed that ribosomes initiating translation of the B. subtilis hbs mRNA at a strong Shine–Dalgarno sequence block the 5′ exoribonuclease RNase J1 from degrading into the coding sequence. Here, we identify new and previously unsuspected features of this mRNA. First, we identify RNase Y as the endoribonuclease that cleaves the highly structured 5′-UTR to give access to RNase J1. Cleavage by RNase Y at this site is modulated by a 14-bp long-range interaction between the 5′- and 3-UTRs that partially overlaps the cleavage site. In addition to this maturation/degradation pathway, we discovered a new and ultimately more important RNase Y cleavage site in the very early coding sequence, masked by the initiating ribosome. Thus, two independent pathways compete with ribosomes to tightly link hbs mRNA stability to translation initiation; in one case the initiating ribosome competes directly with RNase J1 and in the other with RNase Y. This is in contrast to prevailing models in Escherichia coli where ribosome traffic over the ORF is the main source of protection from RNases. Indeed, a second RNase Y cleavage site later in the hbs ORF plays no role in its turnover, confirming that for this mRNA at least, initiation is key.
INTRODUCTION
To adapt to their environment, bacteria employ a variety of mechanisms to regulate gene expression at both transcriptional and post-transcriptional levels. Modification of mRNA stability is one strategy that allows rapid post-transcriptional changes in the levels of gene expression. Ribonucleases are the key actors in this process and around 35 RNases have thus far been identified in the two best-studied model bacteria Escherichia coli and Bacillus subtilis (1). Remarkably, only eight RNases are common to both organisms, highlighting the fact that these two bacteria have evolved with largely different arsenals with which to tackle the issue of mRNA decay. Even more remarkably, the essential enzymes are generally not conserved between these two bacteria; for example, in E. coli the main endoribonuclease involved in initiating mRNA decay is the essential enzyme RNase E (2), while this function is afforded by the non-homologous and non-essential enzyme RNase Y in B. subtilis (3–6). Their diverse RNases allow E. coli and B. subtilis to employ distinct strategies to modulate mRNA stability. In B. subtilis, mRNAs can be degraded by exoribonucleases from either their 3′ or 5′ extremities, catalysed by 3′-5′ exoribonucleases, mainly PNPase, or by the 5′-3′ exoribonuclease complex formed by RNases J1 and J2, respectively (7). Escherichia coli is limited to 3′-5′ exonucleolytic RNA degradation as a 5′-3′ exoribonuclease activity is not known to exist in this organism.
The decay of most bacterial mRNAs involves cooperation between endo- and exoribonucleolytic activities, because exoribonucleolytic degradation requires access to 5′ or 3′ extremities. The stem loop of the transcriptional terminator present at the 3′ end of most mRNAs protects them from 3′-5′ exoribonucleases, while 5′-proximal structures prevent RNA degradation by RNase J1 (8,9). Moreover, RNase J1 has been shown to require a 5′-monophosphate (or 5′ OH) extremity to efficiently degrade RNAs from the 5′ end (10). This can be achieved either by prior endoribonuclease cleavage (11) or by the action of an RNA pyrophosphohydrolase, e.g. RppH, that removes the protecting 5′ triphosphate group from primary transcripts (12).
In addition to secondary structures, non-coding RNAs, RNA binding proteins, and ribosomes have all been shown to impede mRNA decay from the 5′ end in B. subtilis (9,13,14). A ribosome bound to a strong Shine–Dalgarno (SD)-like sequence (even without an associated start codon) is sufficient to promote stability of many kilobases of downstream mRNA in this organism (13,15,16), while in E. coli only translating ribosomes can prevent mRNA degradation (17,18). In E. coli, ribosomes generally protect mRNA from degradation by masking endoribonucleolytic sites in the coding sequence. Decoupling transcription from translation of the lacZ transcript promotes instability, and degradation of the rpsO mRNA was shown to be mediated by endonucleolytic cleavage events that depend on translation (19,20). In B. subtilis, protection by a ribosome bound to an SD sequence is related to its ability to block the 5′-3′ progress of RNase J1 (10,21,22). These data raise the question of whether naked mRNA is actually a target of endoribonucleases in B. subtilis.
In this article, we focus on the model hbs mRNA, whose degradation has been shown to be highly sensitive to translation levels (21). The hbs gene codes for the essential histone-like protein that shares 58% identity with the HU-1 and HU-2 proteins of E. coli. It is highly expressed (∼50 000 molecules per cell), similar to the levels of ribosomal proteins (23). Two primary hbs transcripts possess long highly structured 5′ UTRs that protect the mRNA from direct 5′-3′ attack by RNase J1 (21,24). A third stable transcript was shown to be the result of mRNA maturation, consisting of endonucleolytic cleavage about 40 nucleotides (nts) upstream of the hbs start codon followed by 5′-3′ exonucleolytic degradation by RNase J1 until it bumps up against ribosomes initiating translation at the exceptionally strong hbs SD sequence (21). A small decrease in the strength of this SD/anti-SD interaction led to complete instability of the mature species, suggesting that the same pathway can be used to degrade the hbs mRNA at lower translation levels. Here, we identify RNase Y as the endoribonuclease that provides access to RNase J1 by cutting the hbs mRNA in the 5′-UTR. We also show that a long range interaction (LRI) between the 5′ and 3′ UTRs impacts that efficiency of this cleavage event. Lastly, our experiments reveal three additional RNase Y cleavages of the hbs mRNA both within the coding and non-coding portions of the transcript, which is remarkable for such a short (<600 nts) mRNA. Two cleavage sites within the coding sequence have remarkably different impacts on hbs mRNA stability as a function of translation, and suggest that for this mRNA at least, initiation of translation rather than ribosome coverage of the coding sequence per se is the key stability determinant.
MATERIALS AND METHODS
Strains and constructs
Strains and oligonucleotides used are given in Supplementary Tables S1 and S2, respectively.
The B. subtilis strains used in this study were derivatives of W168 (SSB1002; Supplementary Table S1). Two new strains lacking either RNase J1 or Y in which we could deplete the other enzyme, were constructed. Strain CCB637 was obtained by transforming the rnjA::kan mutation from strain CCB448 (4) into strain CCB338 txpA -10Δ Pxyl-rny rny::spc (25). Strain CCB760 was obtained by transforming the rny::spc mutation from strain CCB441 (4) into strain CCB034 (26). Overnight cultures of depletion strains grown in the presence of 1 mM IPTG (CCB760) or 2% xylose (CCB637) were washed twice in 2xYT medium and inoculated in fresh medium at an OD600 of 0.05, with or without IPTG (CCB760), or with either 2% xylose or 2% glucose (CCB637).
Most plasmids containing the mutant derivatives of hbsΔ were obtained using overlapping primer pairs CC1607/primer1 and primer2/CC572 and reamplified with CC1607/CC572 and then cloned between the AvaI/BamHI sites of plasmid pDG148 (27). The primers as well as the DNA templates used to construct these plasmids are listed in Supplementary Table S3. Plasmids pDG148hbsΔnoAUG and pDG148hbsΔnoSD were made by site directed mutagenesis. Note that all pDG148hbsΔ plasmids and mutant derivatives used here have an in-frame stop codon restored at codon 18 compared to those used in (21), to reconstitute the native hbs 3′-UTR and allow exploration of 3′-dependent pathways. All plasmids were verified by sequencing and then transformed into the wild-type B. subtilis strain SSB1002. Mutant constructs rny::spc, rnjA::spc or pnp::cm were subsequently introduced by transformation of strains expressing the mutant hbsΔ plasmid constructs with chromosomal DNA from strains CCB441, CCB434 or SSB1030, respectively. Successful transfer of chromosomal mutations was verified by PCR as described (4). Strains are listed in Supplementary Table S1. CCB1013 (pDG148-hbsΔ pnp::cm rny::spc) was obtained by transforming strain CCB755 (pDG148-hbsΔ pnp::cm) with chromosomal DNA from strain CCB455 (rny::spc pnp::kan) (4).
The hbsΔ::gfpSDwt fusion was obtained by three-fragment overlapping PCR using primer pairs CC1607/1702 and CC1705/572 with pDG148hbsΔ as a template, and CC1703/1704 with pCV0019 as a template (gift from Rut Carballido-Lopez). The overlapping fragment reamplified using primer pair CC1607/572 and was cloned between the AvaI/BamHI sites of plasmid pDG148 yielding pDG148hbsΔ::gfpSDwt. The pDG-148hbsΔ::gfpSDstrong mutant derivative was obtained by overlapping PCR (see Supplementary Table S3) and cloned between the AvaI/BamHI sites of plasmid pDG148. To introduce these fusions into the integrative vector pHM2 (28), the hbsΔ::gfpSDwt and hbsΔ::gfpSDstrong fusions were amplified from their pDG148 derivatives using the primer pair CC1607/572 and cloned between the EcoRI/BamHI sites of pHM2. The hbsΔ::gfpSDweak fusion was obtained by overlapping PCR using primer pairs CC840/1555 with pDG148hbsΔSDweak as DNA template, and CC1556/538 with pDG-148hbsΔ::gfpSDwt as a template. The overlapping hbsΔ::gfpSDweak fragment amplified using the nested primer pair CC1607/572 and was cloned between the EcoRI/BamHI sites of pHM2. These three plasmids were linearized with XbaI and transformed into SSB1002 (WT) for integration into the amyE locus.
RNA isolation and northern blots
RNA was isolated from mid-log phase B. subtilis cells growing 2xYT medium either by the glass beads/phenol method described in (29). Northern blots were performed as described previously (3).
GFP measurement
GFP fluourescence measurement was performed using the ClarioStar monochromator microplate reader (BMG-Labtech). The SSB1002, CCB822, CCB897 and CCB898 strains were grown in triplicate in a 96-well microplate (Falcon) in 200 μl of 2xYT medium at 37°C with agitation (200 rpm). Optical density was measured every 30 min for 10 h at 600 nm, with the following parameters for GFP measurement (excitation 470 nm, dichroic filter 491.2 and emission 520 nm).
Primer extension
Primer extension assays on total B. subtilis RNA have been described previously (26). Primer CC463 was used to map the 5′ ends of the ∼380 nt species and primer CC1924 was used for the ∼123 nt (C3-ter) degradation intermediate.
RESULTS
RNase Y cleaves within the hbs 5′ UTR to give access to RNase J1
The B. subtilis hbs gene is transcribed from two promoters, P3 and P1, about 210 and 180 nts, respectively, upstream of the hbs open reading frame (ORF). The 5′ untranslated region (UTR) is highly structured, with two major hairpins, hp1 and hp2, protecting the 5′ end of the shorter P1 transcript and an additional overlapping hairpin protecting the longer P3 transcript (Figure 1A) (21,24). For both primary transcripts, the nucleotides at the 5′ end are directly involved in base-pairing interactions rendering them resistant to immediate attack by the 5′-exoribonuclease RNase J1. Both transcripts end at a Rho-independent transcription terminator about 80 nts downstream of the hbs ORF, which protects them from 3′-exoribonucleases. We have previously shown that the P3 and P1 primary transcripts (570 and 540 nts, respectively) are efficiently cleaved by an unknown endoribonuclease at site (here called C2) between hp2 and the Shine–Dalgarno (SD) sequence. This gives rise to a tight cluster of mRNA degradation/maturation intermediates around 400 nts in length that accumulate in cells depleted for RNase J1 (21). The 5′ monophosphate extremities of these cleaved intermediates are highly sensitive to RNase J1, but the 5′-3′ exonucleolytic progression of this enzyme into the hbs coding sequence is blocked by a high rate of ribosome occupancy of the exceptionally strong SD sequence (10 canonical Watson-Crick and two G-U base-pairs with the 3′ end of 16S rRNA), leading to the accumulation of a highly stable mature 380-nt transcript in wild-type (wt) cells, here referred to as M.
Figure 1.

Endonucleolytic cleavage of the 5′-UTR by RNase Y provides access to the 5′-3′ exoribonuclease RNase J1 for hbs mRNA maturation. (A) Schematic of the B. subtilis hbs gene and transcripts. The P3 and P1 promoters and transcription terminator (ter) are indicated. C1–C6 refer to endonucleolytic cleavage sites mapped in the course of this and previous studies. Complementary sequences involved in a proposed long-range interaction (LRI) between the 5′ and 3′-UTRs are indicated. Transcripts are shown as wavy lines. Hp1 and hp2 refer to 5′ hairpin structures. Key endonucleolytic cleavage events are symbolized by scissors; the 5′-3′ exonuclease RNase J1 by a Pacman symbol. The positions of different features are given relative to the translation start site, designated as +1. (B) Northern blot of total RNA isolated from wild-type (wt), RNase Y (Δrny), RNase III (Δrnc) and RNase J1 (ΔrnjA) mutants at different times after rifampicin addition. Calculated half-lives of the major transcripts are given underneath the blot. Half-lives >30 min are referred to as stable. Average decay curves are shown in Supplementary Figure S1. (C) Northern blot of total RNA isolated from wt, ΔrnjA strains, Δrny and ΔrnjA Pxyl-rny and Δrny Pspac-rnjA depletion strains grown in the presence (+) or absence (–) of inducer (ind). For panels B and C, the migration positions of the P3 and P1 primary transcripts, the mature species (M) and the degradation intermediates cleaved at C1, C2, C3 and C4 are indicated. Blots were first probed with oligo CC287 (hbs), shown as a red line in panel A, and then reprobed with oligos CC1444 (tmRNA) or HP246 (5S rRNA) as a loading control. Number of biological replicates (n) = 2.
To identify the endoribonuclease that provides RNase J1 access to the 5′ UTR at C2, we analyzed the stability of hbs mRNAs in strains lacking the two main endoribonucleases known to be involved in mRNA turnover in B. subtilis, namely RNase Y and RNase III, at different times after rifampicin addition to block new transcription (Figure 1B). We also included a deletion mutant of the endo/exonuclease RNase J1 in this experiment, as our previous experiments had employed a more ambiguous depletion strain (21). As previously observed under RNase J1 depletion conditions, the deletion of the RNase J1 gene (ΔrnjA) led to an accumulation of three relatively stable degradation intermediates 485, ∼400 and ∼240 nts in size, while the 380-nt mature species (M) disappeared completely compared to wild-type (wt) (Figure 1B, compare lanes 19–24 to 1–6). The strong accumulation of the ∼400-nt species rules out RNase J1 as the endonuclease that cleaves at C2, a formal possibility that remained if depletion were incomplete in the earlier studies. The ∼240-nt species results from a number of tightly spaced endonuclease cleavages (C4) that occur within the hbs ORF, centred around position +122 relative to the start codon, while the 485-nt intermediate results from an endonucleolytic cleavage (C1) that occurs between the hp1 and hp2 (21). The endonuclease(s) responsible for the C1 and C4 cleavages is/are also unknown. Deletion of the gene encoding RNase III (Δrnc) had no effect on either the accumulation or half-life of any of the hbs transcripts compared to wt (Figure 1B, lanes 13–18), ruling out a role for this RNase in any of these three cleavages (C1, C2 or C4). On the other hand, deletion of the RNase Y gene (Δrny) led to an accumulation the P1 primary transcript and stabilization of both P3 and P1 (Figure 1B, lanes 7–12).
The stabilization of the primary transcripts in the absence of RNase Y suggested a possible role for this enzyme in hbs mRNA turnover and/or maturation. To test this hypothesis, we asked whether we could still detect the signature RNase J1-dependent degradation intermediates in cells lacking RNase Y. Since strains completely lacking both RNase Y and RNase J1 are inviable (4), we made double mutants lacking either RNase J1 or Y in which we could deplete the other enzyme (i.e.Δrny Pspac-rnjA and ΔrnjA Pxyl-rny). Northern blots of these strains grown in the presence or absence of inducer (IPTG or xylose) showed clearly that in the absence of RNase Y, depletion of RNase J1 no longer resulted in an accumulation of the ∼400-nt species (Figure 1C, lane 7 compared to lane 2 or 4). Similarly, depletion of RNase Y in cells completely lacking RNase J1 abolished the accumulation of the ∼400-nt species normally seen in a ΔrnjA strain (Figure 1C, lane 5 compared to lane 2 or 4). The 485 nt and ∼240-nt degradation intermediates were similarly lost in cells lacking RNase Y (Figure 1C, lane 7 compared to lane 2 or 4) or were strongly reduced in cells depleted for this enzyme (Figure 1C, lane 5 compared to lane 2 or 4). These experiments clearly show that RNase J1 access to the hbs mRNA is dependent on RNase Y cleavages at C1 and C2 in the 5′-UTR and C4 in the coding sequence. Endonucleolytic cleavage at C2 by RNase Y, followed by 5′-exoribonuclease degradation by RNase J1 appears to be the primary pathway for generating the mature 380-nt ribosome-protected hbs transcript, judging by the relative intensities of the C1 and C2 species in the absence of RNase J1.
The ∼380-nt RNAs in wt and Δrny strains are generated by different pathways
In light of these findings, the persistence of a ∼380-nt RNA in strains lacking RNase Y (e.g. Figure 1C, lane 3) was puzzling. We therefore asked if this species were identical in wt and Δrny strains, by mapping its 5′ end by primer extension assay (Figure 2A). In wt cells, the major 5′ end of the 380-nt mature species mapped to -21U, 8 nts upstream of the first G of the GGAGG SD sequence, as seen previously (21) (Figure 2). This band corresponds to the upstream border of the ribosome-protected M fragment, which we refer to as the ribosome heel-print. In cells lacking RNase J1, we detected the cluster of 5′ ends extending from positions -23 to -60 relative to the AUG (Figure 2, red arrows), centred around strong cleavages at positions -34/35 and -39/40 that account for most of the ∼400-nt signal seen on Northern blots (Figure 1B and C). Interestingly, most of these cleavages, collectively referred as C2, mapped to A-residues, suggesting that RNase Y has a preference for this nucleotide. In cells lacking or depleted for RNase Y, a new 5′ end was observed 4 nts upstream of the ribosome-protected 5′ end seen in wt cells (Figure 2, green arrow). Thus, the 380-nt ribosome-protected species observed in wt cells and the 384-nt RNA (M+4) that accumulates in Δrny strains correspond to distinct RNAs that are generated by different pathways. The ribonuclease responsible for this cleavage remains to be identified (see Discussion).
Figure 2.
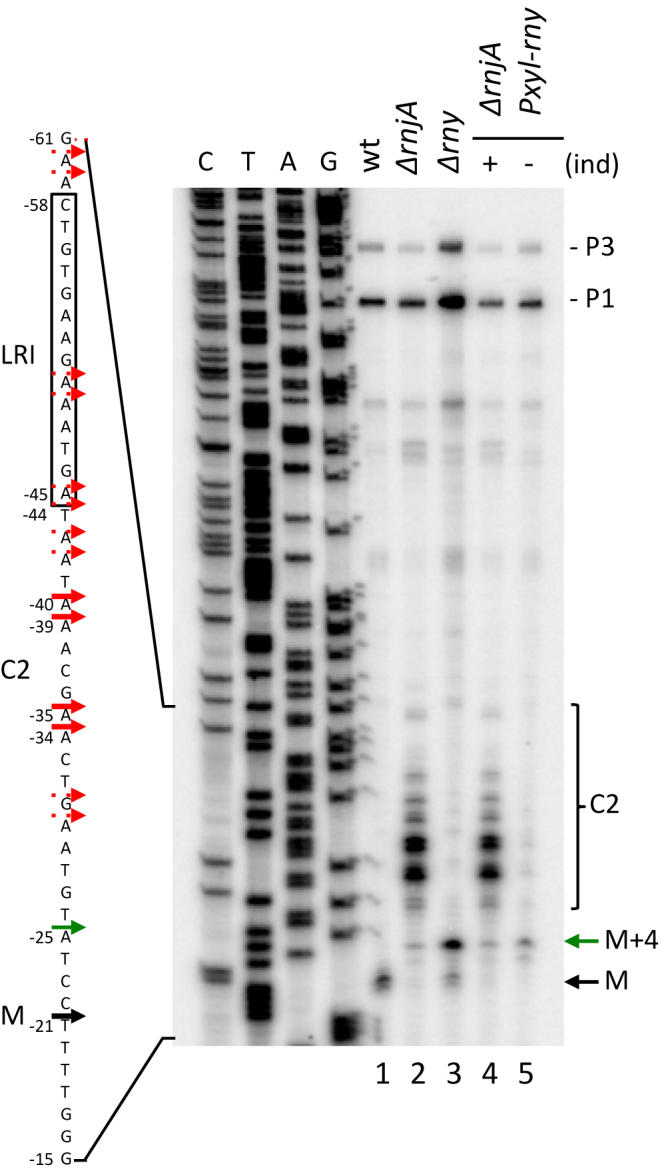
The 5′ end of the ∼380-nt species that accumulates in a Δrny strain is distinct from that of the mature species (M). Primer extension of total RNA isolated from wt, Δrny, ΔrnjA strains and the ΔrnjA Pxyl-rny depletion strain grown in the presence (+) and absence (–) of inducer (ind). C2 cleavage sites are shown as red arrows, the mature species (M) as a black arrow and the Δrny specific 5′ end (M+4) as green arrow (n = 2). The sequence is labeled as its complement to facilitate direct readout.
The strength of the SD sequence determines the accumulation level of the mature hbs mRNA but not its apparent stability
We previously showed that mutations that strengthened or weakened the SD sequence of hbsΔ were found to have a major impact on the steady state levels of the mature 380-nt RNA (21). To get further insight into the role of translation in determining the hbs degradation pathway, we examined the stabilities of the various hbs transcripts in mutants with stronger or weaker SD sequences, or where translation was completely abolished. To avoid potential pleiotropic effects of manipulating the essential hbs gene, these assays were performed on a plasmid-borne mini-hbs gene (hbsΔ) truncated after 17 amino-acids, but containing the native 5′ and 3′-UTRs (Figure 3A) (21). First, RNA stability assays of wt, Δrny and ΔrnjA cells expressing this plasmid confirmed that the maturation pathway of hbsΔ with a wt SD was very similar to the endogenous hbs mRNA (Supplementary Figure S2). In cells lacking RNase Y, we observed stabilization of the primary transcripts, in particular P1Δ. In the ΔrnjA strain, we saw a strong accumulation of a stable intermediate ∼20 nts greater in size than the mature species (MΔ), corresponding to the maturation intermediate cleaved at C2, in addition to the intermediate resulting from the cleavage between hp1 and hp2 at C1 (Supplementary Figure S2). The deletion in the hbs coding sequence abolished the internal cleavage site at C4; we thus did not see the corresponding species. However, the plasmid-borne overexpression allowed us to detect a new degradation intermediate resulting from cleavage early in the hbsΔ ORF that was stabilised in the absence of RNase J1. The mapping and the importance of this newly identified cleavage site (C3) will be discussed below.
Figure 3.

The efficiency of ribosome recruitment is the primary determinant for the level of mature hbsΔ mRNA. (A) Schematic of the B. subtilis hbsΔ construct. Labeling is as in Figure 1. The predicted interactions with the 3′ end of 16S rRNA for the hbsΔ SDwt, and SDstrong and SDweak mutant variants are indicated underneath the schematic. (B) Northern blot of total RNA isolated from strains carrying the hbsΔ SDwt, and hbsΔ SDstrong and hbsΔ SDweak mutant plasmids at different times after rifampicin addition (n = 2). Blots were first probed with oligo CC1997 (hbs), shown as a red line in panel A, and then reprobed with oligo HP246 (5S rRNA) as a loading control. Calculated half-lives of the major transcripts are given underneath the blot. Half-lives >30 min are referred to as stable. Average decay curves are shown in Supplementary Figure S1.
The SDstrong mutant was designed to contain 12 perfect Watson–Crick base-pairs with the 3′ end of 16S rRNA, while the SDweak mutant has only 7/12 base complementarity, but still retains the central GGAGG sequence key to translation initiation (Figure 3A) (21). The stabilities of the primary transcripts, P3Δ and P1Δ, and the ribosome-protected mature species MΔ were similar for the wt and SDstrong constructs (Figure 3B, lanes 1–6 and 13–18), with the MΔ species from the SDstrong variant accumulating to higher levels than wt, as seen previously (Figure 3B, compare zero time points in lanes 1 and 13) (21). In contrast, the primary transcripts were less stable and the mature species barely detectable (but nonetheless stable) with the SDweak construct (Figure 3B, lanes 7–12). Thus, although the levels of the ribosome-protected RNA (MΔ) vary between constructs in this experiment (Figure 3B, lanes 1, 7, 13), its stability does not. This suggests that the absolute levels of the mature species are determined by how quickly ribosomes are recruited to protect its 5′ extremity from ribonuclease attack, but that unprotected species are degraded almost instantaneously.
The exceptional strength of the wt hbs SD sequence and the SDstrong variant (ΔG = –17.8 and –18.5 kcal/mol, respectively) raised the question of whether these mRNAs are translated efficiently, i.e. can the ribosomes easily escape the SD or are they retained at the 5′ end of the coding sequence only protecting it from degradation? To answer this question, we made hbsΔ-gfp translational fusions integrated into the amyE site on the B. subtilis chromosome. This construct consisted of the two hbs promoters, the 5′ UTR, the first 17 codons of the hbsΔ coding sequence fused in frame to GFP, followed by the hbs 3′ UTR (Figure 4A). We observed that the hbsΔ-gfp primary transcripts are matured similarly to the hbsΔ mRNA to give rise to a stable mature mRNA (hbsΔ-gfp M) (Figure 4B). Thus, this region of hbs is able to confer maturation and stability on a heterologous mRNA. Moreover, analysis of the steady state mRNAs of the fusions with stronger or weaker SD sequences showed that they behaved similarly to the hbsΔ construct: the ribosome-protected transcript (M) from the hbsΔ-gfp SDstrong construct accumulated to higher levels than that with the wt SD (hbsΔ-gfp SDwt) and was undetectable with the hbsΔ-gfp SDweak construct (Figure 4C). Measurement of GFP activity showed that the translation levels of the hbsΔ-gfp SDweak fusion (ΔG = –11.3 kcal/mol) were ∼2-fold lower than that of SDwt, while translation of the hbsΔ-gfp SDstrong fusion was ∼2-fold higher, suggesting that ribosomes can indeed escape the strong SD sequence efficiently (Figure 4D).
Figure 4.

Ribosomes can efficiently escape the exceptionally strong SD sequence of the hbs mRNA. (A) Schematic of the B. subtilis hbsΔ-gfp construct. Labeling is as in Figure 1. (B) Northern blot of total RNA isolated from strains carrying the hbsΔ-gfp WT construct integrated into the chromosome in amyE at different times after rifampicin addition (n = 2). The blot was probed with oligos CC1708 (gfp; shown as a red line in panel A), CC287 (to reveal the native hbs mRNA) and then CC058 (16S rRNA) as a loading control. (C) Northern blot of total RNA isolated from strains carrying the hbsΔ-gfp SDwt, hbsΔ SDstrong and hbsΔ SDweak constructs (n = 2). The blot was probed with CC1708 (gfp) and then CC058 (16S rRNA) as a loading control. Ratios of total hbsΔ-gfp transcripts (P3+P1+M) to 16S rRNA are indicated under the blot. (D) GFP expression levels normalized to OD600 for the strains containing the hbsΔ-gfp SDwt, hbsΔ SDstrong and hbsΔ SDweak constructs (n = 3). Measurements were taken at OD600 = 1.
The mature hbs mRNA is fully protected from degradation even in the absence of ribosome flow over the internal RNase Y cleavage site
We had previously shown that simply the binding of the ribosome to the SD was sufficient to promote steady state accumulation of the processed hbsΔ mRNA using a construct in which the AUG start codon was mutated to AAA (hbsΔ noAUG; Figure 5A) (21). Here, we confirmed that although the mature hbsΔ fragment from the hbsΔ noAUG construct accumulated to slightly lower levels than the wt, it was still fully stable (Figure 5B; compare lanes 1–6 and 7–12). Thus, the mature hbsΔ mRNA is indeed protected from degradation with a single ribosome bound to the SD. The lower levels of the hbsΔ noAUG mature transcript compared to wt hbsΔ are likely explained by a lack of initiator tRNA contribution to initiation complex stability.
Figure 5.

Restoration of RNase Y cleavage site C4 within the hbs coding sequence does not destabilize the hbsΔ mRNA, even in the absence of ribosome traffic. (A) Schematics of the B. subtilis hbsΔ (above) and hbsΔ-C4 (below) mutant constructs. Labeling is as in Figure 1. Mutation of the AUG start codon to AAA in hbsΔ noAUG and hbsΔ-C4 noAUG is indicated with a downward pointing arrow. Mutation of the lysine codon 18 (AAA) to a stop codon (UAA) in hbsΔ-C4 UAA18 is indicated by a stop sign. (B) Northern blot of total RNA isolated from strains carrying the hbsΔ SDwt, and hbsΔ noAUG, hbsΔ-C4 noAUG and hbsΔ-C4 UAA18 mutant plasmids at different times after rifampicin addition (n = 2). The blot was probed with oligo CC429 (hbs), shown as a red line in panel A, and then HP246 (5S rRNA) as a loading control. Calculated half-lives of the major transcripts are given underneath the blot. Half-lives >30 min are referred to as stable. Average decay curves are shown in Supplementary Figure S1.
The generally accepted model in bacterial mRNA decay is that translating ribosomes stabilize mRNAs by masking endonuclolytic cleavage sites within their coding sequences. We therefore asked whether the hbs mRNA was an exception to this rule by probing whether translation itself, and not just the efficiency of ribosome binding to the SD, could have an impact the hbsΔ mRNA degradation. As noted above, the hbsΔ construct lacks the internal RNase Y cleavage site at C4 present in the endogenous hbs mRNA coding sequence. To determine whether this internal cleavage site contributed to the degradation of non-translated hbs mRNAs, we made two new hbsΔ constructs in which this site was restored. In one, the hbsΔ noAUG construct was extended by a further 33 codons (i.e. from 17 to 50 aa) to include the RNase Y cleavage site at C4 (hbsΔ-C4 noAUG; Figure 5A). In the other, we made a variant of hbsΔ-C4 with a wt start codon and a stop codon in position 18, creating hbsΔ-C4 UAA18 that terminates translation at the same location as in hbsΔ. Both hbsΔ-C4 UAA18 and hbsΔ-C4 noAUG constructs are predicted to allow unrestricted access to RNase Y at C4. We compared the mRNA stabilities of transcripts from these two constructs to their shorter hbsΔ variants by Northern blot at times after rifampicin addition to cells (Figure 5B). The results showed that the mature species (MΔ-C4) accumulated to relatively high levels and remained fully stable in both hbsΔ-C4 UAA18 and hbsΔ noAUG constructs (Figure 5B, lanes 7–12 and 13–18, respectively). Thus, the internal RNase Y cleavage site at C4 does not have a major impact on the degradation of the mature species, even when ribosomes no longer transit over the cleavage site.
Identification of a new RNase Y cleavage site under the initiating ribosome
The data described above indicate that the efficiency of ribosome recruitment to the SD sequence (and not ribosome transit) is the key determinant of the stability of the mature hbsΔ species. We next asked what would be the effect of completely abolishing ribosome access to the mRNA, by mutating the GGAGG sequence of the hbsΔ SD to CCCGG (hbsΔ noSD; Figure 6A). Although the mRNAs produced by this construct completely lacked the ribosome-protected species as anticipated (i.e. RNase J1 completely degrades the unprotected RNA following cleavage at C2), we were surprised to observe that the levels of both the P3 and P1 primary transcripts were significantly decreased compared to wt (Figure 6B, compare lane 7 to lane 1). We therefore considered possibility that the initiating ribosome covers yet another destabilizing cleavage site.
Figure 6.

Mutation of the Shine–Dalgarno (SD) sequence of the hbsΔ mRNA leads to additional RNase Y-dependent degradation. (A) Schematic of the B. subtilis hbsΔ noAUG and noSD mutant constructs. Labeling is as in Figure 1. The predicted interactions with the 3′ end of 16S rRNA for the hbsΔ noAUG and noSD mutant variants are indicated to the right of the schematic. (B) Northern blot of total RNA isolated from wt and Δrny strains carrying the hbsΔ SDwt, and hbsΔ noSD and hbsΔ noAUG mutant plasmids at different times after rifampicin addition (n = 2). The blot was probed with oligo CC1997 (hbs), shown as a red line in panel A, and then HP246 (5S rRNA) as a loading control. Calculated half-lives of the major transcripts are given underneath the blot. Average decay curves are shown in Supplementary Figure S3.
The role of RNase Y in the turnover of the hbsΔ noAUG and noSD mRNAs, was investigated by analysis of their stabilities in Δrny cells by northern blot. Like the SDwt hbsΔ construct, the P1 and P3 transcripts of the hbsΔ noAUG and noSD mRNAs were stabilized in the absence of RNase Y (Figure 6C). Thus, even when the hbsΔ mRNA is not translated, no other endoribonuclease besides RNase Y seems to make a significant contribution to its degradation. However, we noticed a much stronger accumulation of the primary transcripts (in particular P1) in the hbsΔ noSD construct in the Δrny strain compared to the other two constructs (Figure 6C, lanes 7–12, compared to 1–6 or 13–18). We therefore asked whether the same pathway was used for the degradation of hbsΔ noSD transcripts that are incapable of recruiting ribosomes. We performed primer extension assays in a ΔrnjA background to look for the characteristic cluster of 5′ ends resulting from RNase Y cleavages at C2 in the 5′ UTR in the context of the hbsΔ, hbsΔ noSD and hbsΔ noAUG constructs (Figure 7A). Although the intensity of signal was significantly reduced compared to hbsΔ SDwt and to hbsΔ noAUG, a cluster of 5′ ends was clearly visible with the hbsΔ noSD construct, indicating that RNase Y still cleaves at C2. More importantly, however, an intense new band whose 5′ end mapped to position +13 of the hbs coding sequence was observed primarily in the hbsΔ noSD construct (Figure 7A). The fact that the primary transcripts from the hbsΔ noSD construct accumulate strongly and are completely stable in the Δrny mutant suggests that RNase Y is also responsible for this new major cleavage event (C3). This strong cleavage site at C3 is masked by the initiating ribosome that protects as far as nt +16 of the hbs coding sequence in previous toe-print assays (21).
Figure 7.
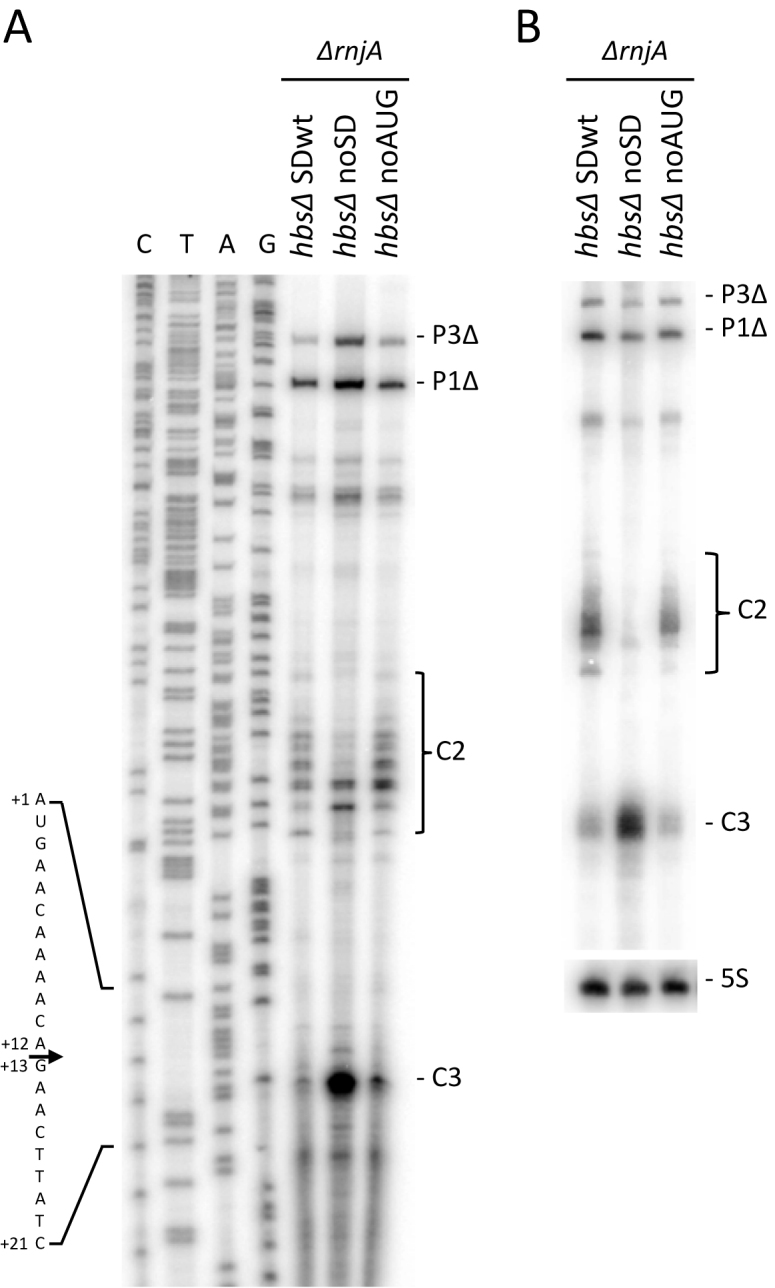
Strong cleavage of the hbsΔ mRNA at nt +12 by RNase Y in the absence of initiating ribosomes. (A) Primer extension of total RNA isolated from ΔrnjA strains carrying the hbsΔ SDwt, and hbsΔ noSD and hbsΔ noAUG mutant plasmids (n = 1), using primer CC1924 hybridising to just downstream of the start codon. C2 and C3 cleavage sites are shown, in addition to the 5′ ends corresponding to the P3Δ and P1Δ transcription start sites. The sequence is labeled as its complement to facilitate direct readout. (B) Northern blot of total RNA isolated from ΔrnjA strains carrying the hbsΔ SDwt, and hbsΔ noSD and hbsΔ noAUG mutant plasmids (n = 2). The blot was probed with oligo CC1924 (hbs) and then HP246 (5S rRNA) as a loading control.
The primer extension assay also suggested that cleavage at C3 may have a greater impact than C2 for the turnover of the untranslated hbsΔ noSD mRNA. We confirmed this by northern blot; in the ΔrnjA background, the intensity of the C3 degradation intermediate was significantly greater than that corresponding to C2 for hbsΔ noSD, whereas they had similar intensities for wt hbsΔ or hbsΔ noAUG, each of which are protected by ribosomes bound to the SD (Figure 7B). This experiment suggests that, of the two major pathways for degrading non-translated hbsΔ noSD mRNAs, i.e. cleavage at either C2 or C3 by RNase Y, followed in both cases by degradation by RNase J1, the new cleavage identified at C3 plays the more significant role.
The hbs mRNA is also cleaved in its 3′ UTR allowing 3′-5′ degradation by PNPase
The experiments performed thus far suggested that most of the degradation of the hbsΔ mRNA occurs from 5′ proximal cleavage events (C1, C2, C3). To determine whether the hbsΔ mRNA could also be degraded in the 3′ to 5′ direction from the 3′-UTR, we measured the accumulation and stability of degradation intermediates in a strain lacking PNPase activity, the major 3′-5′ exoribonuclease in B. subtilis (Figure 8). Using a probe (c) that overlaps the stop codon (–18 to +16 relative to UAA), four new degradation intermediates, approximately 275, 240, 110 and 75 nts in size, were detected in the Δpnp strain compared to wt (labeled in blue in Figure 8B, lane 2 compared to lane 1). A series of probes (a–f) were used to determine the 5′ and 3′ extremities of these four species (Supplementary Figure S4). Based on these data and their relative sizes and intensities, the ∼240 and ∼75-nt species are consistent with transcripts extending from the P1Δ promoter and the ribosome-protected 5′-end (MΔ) to just downstream of the hbsΔ stop codon, while the ∼275 and ∼110-nt intermediates have the same 5′ ends, but extend about 35 nts further into the 3′-UTR (Figure 8A). We refer to these new 3′ ends as C5 and C6, respectively. Half-life experiments show that the MΔ-C5 and MΔ-C6 species are highly stable in the Δpnp mutant (Figure 8C).
Figure 8.

Degradation of the hbsΔ mRNA can also be initiated within the 3′-UTR. (A) Schematic of the B. subtilis hbsΔ constructs showing positions of different oligonucleotide probes. Labeling is as in Figure 1. Transcripts are shown as wavy lines. Key cleavage events C2, C5, C6 are symbolized by scissors; PNPase by a brown Pacman symbol. (B) Northern blot of total RNA isolated from wt, Δpnp, Δpnp rny, ΔrnjA and Δrny strains carrying plasmids expressing the wt hbsΔ construct (n = 1). (C) Northern blot of total RNA isolated from wt and Δpnp strains carrying plasmids expressing the wt hbsΔ construct at different times after rifampicin addition (n = 1). Blots in panels B and C were probed with oligo CC1997 (hbs probe c) and then HP246 (5S rRNA) as a loading control. Degradation intermediates seen in the Δpnp strain are labeled in blue; those accumulating in the ΔrnjA strain are labeled in red.
To show these new 3′ ends were the result of endonucleolytic cleavages in the 3′-UTR, we performed experiments designed to detect the downstream cleavage fragments by hybridising total RNA isolated from a ΔrnjA strain with probes (e and f) overlapping the transcription terminator. With these probes, we detected four degradation intermediates about 175, 120, 70 and 45 nts in length (labeled in red in Supplementary Figure S4B). The two larger species correspond in size to species extending from the C2 and C3 cleavage sites to the transcription terminator, seen previously (e.g. Figure 7), while the two shorter species correspond well with the predicted sizes of the downstream fragments of endonucleolytic cleavage at C5 and C6, i.e. ∼80 and ∼45 nts, respectively. Thus, PNPase gains access to the hbsΔ 3′ UTR through two endonucleolytic cleavages, with C5 mapping to within a ∼10 window downstream of the stop codon and C6 mapping to around 45 nts from the 3′ end of the hbsΔ mRNA.
We next asked whether RNase Y was responsible for the cleavages in the 3′-UTR by probing RNA isolated from a Δpnp rny double mutant with probe (c) which detects all four degradation intermediates seen in the Δpnp single mutant. Although the ∼75 and ∼110-nt intermediates, corresponding to MΔ-C5 and MΔ-C6, essentially disappeared in the double mutant, the ∼275, ∼240-nt species, corresponding to P1Δ-C6 and P1Δ-C5, did not. These results clearly indicate that RNase Y is not the endonuclease that cleaves at C5 and C6. The absence of the MΔ-C5 and MΔ-C6 species in the Δpnp rny mutant is most likely explained by the lack of RNase Y cleavage at C2, required to produce the ribosome-protected 5′ end at MΔ. It should be stressed that the essentially full stability of the mature hbsΔ species in wt cells suggests that degradation initiated from C5 and C6 within the 3′ UTR, like that initiated from C4 within the coding sequence, are minor degradation pathways. Nonetheless, cleavage at these three sites may be important for the slow mechanism by which MΔ is eventually degraded.
Basepairing between the 3′ and 5′ UTR inhibits endonucleolytic cleavages in the hbs leader region
Based on in vitro structure probing data (24), we had previously proposed the existence of a long-range interaction (LRI) between the 5′ and 3′ UTRs of the hbs mRNA, through a 14 nt sequence lying just downstream of hp2 in the 5′ UTR that can base-pair with a fully complementary sequence (ΔG = –19 kcal/mol) just upstream of the transcription terminator in the 3′ UTR (Figure 9A). Since this complementarity partially overlaps the C2 cleavage site, we asked whether it could impact the 5′ degradation pathway in vivo. We mutated the LRI sequence in either the 3′ UTR (LRImut3’), the 5′UTR (LRImut5’) or a combination for both to restore complementarity (LRIcomp; ΔG = –17 kcal/mol) in the context of the plasmid-borne hbsΔ construct (Figure 9B). The hbsΔ LRImut5’ mRNA showed a strong accumulation of the mature transcript with a concomitant diminution in the stability of the primary transcripts (Figure 9B, compare lanes 13–18 with 1–6), consistent with the idea that disruption of the base-pairing with the 3′ LRI increases C2 cleavage efficiency in the 5′ UTR. We confirmed that we had not created a new cleavage site with the LRImut5’ mutation by examining the 5′ ends produced from this construct in a ΔrnjA strain. Indeed, the 5′ ends were qualitatively very similar but significantly more intense than the wt construct, consistent with increased cleavage efficiency at C2 (Figure 9C). The accumulation and stabilities of the primary and mature transcripts were restored to wt levels upon introduction of the compensatory mutation in the 3′ UTR (hbsΔ LRIcomp) (Figure 9B), confirming that this long-range interaction occurs in vivo and that it restricts the efficiency of the C2 cleavage.
Figure 9.

Impact of a long-range interaction (LRI) between 5′ and 3′ UTRs on hbsΔ mRNA cleavage and stability. (A) Secondary structure of the B. subtilis hbsΔ 5′-UTR and potential LRI identified in vitro (24). Hp1 and hp2 refer to 5′ hairpin structures. The transcription start sites from the P3 and P1 promoters are indicated. Mapped 5′ ends corresponding to C1 and C2 cleavage sites and the mature species (M) with the WT construct are indicated with red arrows. Strong cleavages seen in the LRImut3′ construct at –44/45 are shown with green arrows. (B) Northern blot of total RNA isolated from strains carrying the hbsΔ LRIwt, and hbsΔ LRImut3′, LRImut5′, and LRIcomp mutant plasmids at different times after rifampicin addition (n = 2). The blot was probed with oligos CC1997 (hbs) and then HP246 (5S rRNA) as a loading control. Calculated half-lives of the major transcripts are given underneath the blot. Half-lives >30 min are referred to as stable. Average decay curves are shown in Supplementary Figure S3. (C) Primer extension of total RNA isolated from ΔrnjA strains carrying the hbsΔ LRIwt, and hbsΔ LRImut3′, LRImut5′, and LRIcomp mutant plasmids, using primer CC463 (n = 1). Strongest C2 cleavage sites are shown as red arrows and the displacement of cleavage site in the strain carrying the LRI3′mut construct is shown as a green arrow. Additional 5′ ends corresponding to the P3Δ and P1Δ transcription start sites and the C1 cleavage site are indicated. The sequence is labeled as its complement to facilitate direct readout.
Mutation of the LRI in the 3′ UTR alone also accelerated the decay rate of the primary transcripts (Figure 9B, lanes 7–12). However, in this case, we did not observe a concomitant increase in the accumulation of the mature species MΔ, but rather a destabilization of this portion of the transcript, suggesting an acceleration of the 3′ pathway. In the primer extension assay, the two strongest cleavages in the 5′-UTR observed with the LRImut3’ construct in the ΔrnjA background were at positions -44/45, corresponding to the beginning of the overlap between C2 and the 5′ LRI sequence (see Figure 9A). In the wt construct, the strongest cleavages were at -34/35 and -39/40: thus the main cleavage events are shifted upstream in LRImut3’ (Figure 9A). Therefore, in addition to accelerating the 3′ pathway, the LRImut3′ mutation also qualitatively affects endonucleolytic cleavages in the 5′-UTR, further evidence that these two elements communicate.
The primer extension assay also revealed that, although the accumulation levels and decay rates of the primary and mature transcripts were restored in the compensatory mutant hbsΔ LRIcomp in Figure 9B, there are also some qualitative differences in the cleavage sites compared to wt (Figure 9C). Cleavage at C2 is quite weak in this mutant and is primarily restricted to -34/35, while cleavage at C1 is significantly increased. Although we do not fully understand the basis for these qualitative differences, the fact that combining the LRImut3’ and LRImut5’ mutations significantly alters the cleavage pattern seen with the LRImut5’ mutant alone further confirms that the 5′ and 3′ UTR sequences communicate to modulate hbsΔ degradation patterns.
DISCUSSION
In this paper, we have identified several distinct hbs mRNA maturation/degradation pathways, some of which are hypersensitive to the translational status of the mRNA. These pathways involve RNase Y cleavages at four sites (C1 to C4) in the 5′-UTR and hbs coding sequence, followed by the action of the 5′-3′ exoribonuclease RNase J1, and cleavages by unknown endonucleases at two sites (C5 and C6) in the 3′-UTR, followed by the action of the 3′-5′ exoribonuclease PNPase. The maturation pathway is initiated by RNase Y cleavages at C1 or C2 (primarily) in the hbs 5′ UTR. RNase J1 then attacks these newly exposed 5′ ends in 5′-3′ exonucleolytic mode until it encounters a ribosome bound to the exceptionally strong hbs SD sequence (21). In the absence of an initiating ribosome, this maturation pathway becomes a turnover pathway as RNase J1 continues degrading into the hbs coding sequence.
The two RNase Y cleavages sites identified within the hbs coding sequence (C3 and C4) can both potentially inactivate the mRNA, but have different functional relevance. C4, which is located in the middle of the hbs ORF plays a minor role, even when ribosomes are prevented from flowing over this cleavage site. This was particularly evident with the two hbsΔ constructs in which the C4 cleavage site was restored, but where ribosome traffic over the site was blocked either by mutating the start codon (hbsΔ-C4 noAUG) or terminating translation early (hbsΔ-C4 UAA18) in the open reading frame (Figure 5); the interruption of ribosome flow over C4 had no impact on the stability or the accumulation of the mature species (M) in either case.
The newly mapped C3 cleavage site after nt +12 of the hbsΔ coding sequence clearly has a more important functional role in mRNA decay. This was revealed by the construct lacking the SD sequence (hbsΔ noSD) in which no mature species was visible and the primary transcripts accumulated to significantly lower levels. We showed previously that ribosomes bound to the hbs SD sequence protect up to nt +16 in toe-print assays (21), thus shielding the hbs mRNA from RNase Y cleavage at C3 and degradation of the resulting downstream fragment by RNase J1. In the absence of an initiating ribosome, C3 is wide open to attack by RNase Y. It is remarkable that the hbs mRNA has two apparently redundant pathways to tightly couple a lack of translation initiation to mRNA decay: cleavage at either C2 or C3 by RNase Y, followed by degradation by RNase J1 in both instances. In one case the initiating ribosome competes directly with RNase J1, while in the other it competes directly with RNase Y. Our data (Figure 7B), would suggest that the newly discovered C3 site is more frequently used than C2 to begin the destruction of hbs transcripts lacking a ribosome bound to the SD, although we cannot rule out the possibility of sequential cleavages at C2 then C3 (or indeed C1, C2 and then C3) by RNase Y complicating this interpretation, as has been proposed for RNase E in E. coli (30,31). Indeed, sequential cleavages might explain why cleavage at C1 appears to be stronger than C2 in the LR1comp mutant (Figure 9C). In any case, these data suggest that, for the hbs mRNA at least, translation initiation is key for protection against mRNA decay and that ribosome traffic over the cleavage site in the middle of the coding sequence has little impact on mRNA stability. It remains to be seen how general this phenomenon may be in B. subtilis, but numerous other mRNAs have been described where ribosome binding to an SD or SD-like sequence can protect downstream untranslated mRNA (9,13,32–34). It should be noted that ribosomes bound to the SD sequence have also been shown to protect certain mRNAs from degradation in E. coli (35–37) even though this organism lacks an identified 5′-3′ exoribonuclease. However, the generally accepted model in E. coli is that ribosome flow over the coding sequence has the greatest impact on mRNA stability (38).
In the absence of RNase Y, a new 5′ extremity located only four nucleotides upstream of the matured transcript was identified (Figure 2). The (M+4) transcript has a similar stability to the mature transcript (M) suggesting that its 5′ extremity might also be protected by a ribosome. The hbs ribosome-binding site contains two overlapping potential SD sequences (GGAGGAGG; see Figure 3A). It is therefore possible that the 5′ extremity of the 383 nt RNA (M+4) is stabilized by sub-population of ribosomes bound to the upstream GGAGG sequence, predicted to be less optimal in terms of its global sequence context than the downstream site. When ribosomes occupy the upstream GGAGG, the C3 site at +12 would be predicted to be uncovered, which might account for the high sensitivity of the M+4 transcript to RNase Y. The identity of the ribonuclease that generates the 5′ end of the M+4 remains to be elucidated.
It is possible that the principle of ribosome binding site occupation blocking cleavage by RNase Y can be extended to other mRNAs in B. subtilis. We have previously noted that an unusually large proportion of mRNAs (>20%) in this organism have A-rich lysine codons (AAA or AAG) just downstream of their initiation codons (34). Since RNase Y has a preference for single-stranded A-rich sequences, it is possible that a portion of these are RNase Y sites protected by initiating ribosomes. Further experiments are required to uncover the full extent of this potential means to link mRNA decay to translation initiation levels in B. subtilis.
In E. coli, it has been shown that more than 6 nts base-pairing between the SD and anti-SD sequences can be detrimental for translation initiation, the additional stability making to more difficult for ribosomes to escape the ribosome binding site (39). This effect could be partially counteracted by adding AU-rich ribosomal protein S1 recognition sequences upstream. Our data suggest that B. subtilis ribosomes behave remarkably differently, having no difficulty escaping the ultra strong (über) SD sequence of the hbs mRNA, despite the lack of a ribosome-associated S1 protein in this organism (40). Indeed, the stronger the base-pairing, the better the yield of the hbsΔ-gfp fusion (Figure 4).
The two RNase Y-independent cleavages in the 3′ UTR (C5 and C6) appear to have little impact on hbs mRNA stability in the wt context. However, mutation of the 3′ LRI accelerated the degradation rate of the ribosome-protected species (Figure 9), suggesting that it is the strength of these cleavage sites that limits their impact. Indeed, we have recently shown that insertion of an efficient Rae1-dependent cleavage site in the hbsΔ 3′ UTR strongly reduces the half-life of the primary transcripts and completely destabilises the ribosome-protected species (34). Of the two natural cleavage sites in the hbs 3′ UTR, C5 appears to be significantly stronger, judging by the levels to which the corresponding degradation intermediates accumulate in Δpnp or ΔrnjA strains. Intriguingly, of the 6 total cleavage sites mapped in the hbs mRNA, two occur in each of the 5′ UTR, coding sequence and 3′ UTR elements, with a strong (C2, C3, C5) and weak (C1, C4, C6) cleavage site in each case. This is remarkable redundancy for such a short mRNA (<600 nts).
Cleavage at C2 by RNase Y leading to the production of the ribosome-protected species is influenced by the long-range interaction (LRI) between the 5′ and 3′ UTRs. Mutation of either the 5′ or 3′ UTR LRI sequence alone decreased the half-life of the hbs primary transcripts and a compensatory mutation restored these to wt levels. In the case of the 5′ LRI mutation, this was attributable to an opening of the duplex and an acceleration of the C2 cleavage, resulting in a strong accumulation of the ribosome-protected mature species (Figure 9B). The effect of the 3′ LRI mutation was more complex. First, we did not observe an acceleration of the C2 cleavage linked to accumulation of the mature species as we would have expected if we were simply opening the duplex in the 5′ UTR. Second, we observed a destabilisation of the mature species, which no longer has the 5′ LRI attached and therefore should not in theory be sensitive to duplex formation. These observations suggest that the nucleotide sequence introduced with the 3′ LRI mutation may have accelerated the 3′ pathway. Upon introduction of the compensatory mutation in the 5′ LRI, the fact that we return to wt transcript stabilities suggests this acceleration due to the 3′ LRI mutation is subordinate to duplex formation, at least as far as the primary transcripts are concerned. Although the model presented in Figure 9A suggests that the LRI between the 5′ and 3′ UTR occurs in cis, we cannot exclude the possibility that some of these interactions occur in trans, i.e. with other hbs mRNA molecules, particularly since this is a very highly expressed RNA. An interaction between a primary transcript with a compensatory 5′ LRI mutation and a mature transcript with a 3′ LRI mutation in trans could contribute to the re-stabilization of the mature species in the hbsΔ LRIcomp construct compared to hbsΔ LRImut3′. Interactions between the 5′ and 3′ UTRs that impact RNA degradation pathways have been seen previously in the icaR mRNA of Staphylococcus aureus and the hilD mRNA of Salmonella enterica (41,42).
The hbs LRI duplex is quite long (14 bp), but is mostly composed of A/U base-pairs with a relatively modest ΔG of -19 kcal/mol. We considered the possibility that an RNA chaperone such as Hfq might be involved in forming or stabilizing the duplex. However, neither the stability nor the accumulation of any of the hbs mRNAs were affected in a hfq deletion strain (data not shown). There is currently no clear function for Hfq of B. subtilis (43), a major player in sRNA regulation in E. coli (44). As the Hbs protein of B. subtilis and its ortholog the HU protein of E. coli have been described to have RNA binding activity in addition to their roles as a histone-like proteins (45,46), we also considered the possibility that Hbs might control its own synthesis by binding to the LRI duplex. However, we were unable to detect any interaction between Hbs and its own mRNA (data not shown). Thus the question of whether RNA binding proteins are involved in promoting or stabilising LRI duplex formation, and whether this is a regulatory event, remains open.
In summary, this paper shows how multiple complex and redundant RNA degradation pathways can exist for even very short RNAs, and how the coupling of translation to mRNA decay can be achieved in ways other than ribosome transit over cleavage sites within the coding sequence. Lastly, it reinforces previous observations that show how some of these processes can be modulated by long-range interactions between complementary elements on either side of the coding sequence, not just local changes in RNA structure as is more classically found in the literature.
Supplementary Material
ACKNOWLEDGEMENTS
We thank lab members and M. Springer for helpful discussion.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
CNRS [UPR 9073]; Université Paris VII-Denis Diderot; Agence Nationale de la Recherche [asSUPYCO, ARNr-QC to C.C. and BaRR to S.D.]. This work has been published under the framework of Equipex (Cacsice) and a LABEX programs (Dynamo) that benefit from a state funding managed by the French National Research Agency as part of the Investments for the Future program. Funding for open access charge: ANR grant ARNr-QC.
Conflict of interest statement. None declared.
REFERENCES
- 1. Condon C. Reference Module in Biomedical Sciences. 2014; Elsevier; doi:10.1016/B978-0-12-801238-3.02455-7. [Google Scholar]
- 2. Carpousis A.J., Luisi B.F., McDowall K.J.. Endonucleolytic initiation of mRNA decay in Escherichia coli. Prog. Mol. Biol. Transl. Sci. 2009; 85:91–135. [DOI] [PubMed] [Google Scholar]
- 3. Durand S., Gilet L., Bessieres P., Nicolas P., Condon C.. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 2012; 8:e1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Figaro S., Durand S., Gilet L., Cayet N., Sachse M., Condon C.. Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J. Bacteriol. 2013; 195:2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laalami S., Bessieres P., Rocca A., Zig L., Nicolas P., Putzer H.. Bacillus subtilis RNase Y activity in vivo analysed by tiling microarrays. PLoS One. 2013; 8:e54062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lehnik-Habrink M., Schaffer M., Mader U., Diethmaier C., Herzberg C., Stulke J.. RNA processing in Bacillus subtilis: identification of targets of the essential RNase Y. Mol. Microbiol. 2011; 81:1459–1473. [DOI] [PubMed] [Google Scholar]
- 7. Durand S., Tomasini A., Braun F., Condon C., Romby P.. sRNA and mRNA turnover in Gram-positive bacteria. FEMS Microbiol. Rev. 2015; 39:316–330. [DOI] [PubMed] [Google Scholar]
- 8. Condon C. What is the role of RNase J in mRNA turnover. RNA Biol. 2010; 7:316–321. [DOI] [PubMed] [Google Scholar]
- 9. Sharp J.S., Bechhofer D.H.. Effect of 5′-proximal elements on decay of a model mRNA in Bacillus subtilis. Mol. Microbiol. 2005; 57:484–495. [DOI] [PubMed] [Google Scholar]
- 10. Mathy N., Benard L., Pellegrini O., Daou R., Wen T., Condon C.. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007; 129:681–692. [DOI] [PubMed] [Google Scholar]
- 11. Yao S., Richards J., Belasco J.G., Bechhofer D.H.. Decay of a model mRNA in Bacillus subtilis by a combination of RNase J1 5′ exonuclease and RNase Y endonuclease activities. J. Bacteriol. 2011; 193:6384–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richards J., Liu Q., Pellegrini O., Celesnik H., Yao S., Bechhofer D.H., Condon C., Belasco J.G.. An RNA pyrophosphohydrolase triggers 5′-exonucleolytic degradation of mRNA in Bacillus subtilis. Mol. Cell. 2011; 43:940–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agaisse H., Lereclus D.. STAB-SD: a Shine–Dalgarno sequence in the 5′ untranslated region is a determinant of mRNA stability. Mol. Microbiol. 1996; 20:633–643. [DOI] [PubMed] [Google Scholar]
- 14. Glatz E., Nilsson R.-P., Rutberg L., Rutberg B.. A dual role for the Bacillus subtilis leader and the GlpP protein in the regulated expression of glpD: antitermination and control of mRNA stability. Mol. Microbiol. 1996; 19:319–328. [DOI] [PubMed] [Google Scholar]
- 15. Hue K.K., Cohen S.D., Bechhofer D.H.. A polypurine sequence that acts as a 5′ mRNA stabilizer in Bacillus subtilis. J. Bacteriol. 1995; 177:3465–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharp J.S., Bechhofer D.H.. Effect of translational signals on mRNA decay in Bacillus subtilis. J. Bacteriol. 2003; 185:5372–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joyce S.A., Dreyfus M.. In the absence of translation, RNase E can bypass 5′ mRNA stabilizers in Escherichia coli. J. Mol. Biol. 1998; 282:241–254. [DOI] [PubMed] [Google Scholar]
- 18. Komarova A.V., Tchufistova L.S., Dreyfus M., Boni I.V.. AU-rich sequences within 5′ untranslated leaders enhance translation and stabilize mRNA in Escherichia coli. J. Bacteriol. 2005; 187:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Braun F., Le Derout J., Regnier P.. Ribosomes inhibit an RNase E cleavage which induces the decay of the rpsO mRNA of Escherichia coli. EMBO J. 1998; 17:4790–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iost I., Dreyfus M.. The stability of Escherichia coli lacZ mRNA depends upon the simultaneity of its synthesis and translation. EMBO J. 1995; 14:3252–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daou-Chabo R., Mathy N., Benard L., Condon C.. Ribosomes initiating translation of the hbs mRNA protect it from 5′-to-3′ exoribonucleolytic degradation by RNase J1. Mol. Microbiol. 2009; 71:1538–1550. [DOI] [PubMed] [Google Scholar]
- 22. Yao S., Bechhofer D.H.. Processing and stability of inducibly expressed rpsO mRNA derivatives in Bacillus subtilis. J. Bacteriol. 2009; 191:5680–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ross M.A., Setlow P.. The Bacillus subtilis HBsu protein modifies the effects of alpha/beta-type, small acid-soluble spore proteins on DNA. J. Bacteriol. 2000; 182:1942–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daou-Chabo R., Condon C.. RNase J1 endonuclease activity as a probe of RNA secondary structure. RNA. 2009; 15:1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durand S., Gilet L., Condon C.. The essential function of B. subtilis RNase III is to silence foreign toxin genes. PLoS Genet. 2012; 8:e1003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Britton R.A., Wen T., Schaefer L., Pellegrini O., Uicker W.C., Mathy N., Tobin C., Daou R., Szyk J., Condon C.. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol. Microbiol. 2007; 63:127–138. [DOI] [PubMed] [Google Scholar]
- 27. Stragier P., Bonamy C., Karmazyn-Campelli C.. Processing of a sporulation sigma factor in Bacillus subtilis: how morphological structre could control gene expression. Cell. 1988; 52:697–704. [DOI] [PubMed] [Google Scholar]
- 28. Gendron N., Putzer H., Grunberg-Manago M.. Expression of both Bacillus subtilis threonyl-tRNA synthetase genes is autogenously regulated. J. Bacteriol. 1994; 176:486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bechhofer D.H., Oussenko I.A., Deikus G., Yao S., Mathy N., Condon C.. Analysis of mRNA decay in Bacillus subtilis. Methods Enzymol. 2008; 447:259–276. [DOI] [PubMed] [Google Scholar]
- 30. Durand S., Richard G., Bontems F., Uzan M.. Bacteriophage T4 polynucleotide kinase triggers degradation of mRNAs. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:7073–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 32. Bechhofer D.H., Dubnau D.. Induced mRNA stability in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hambraeus G., Karhumaa K., Rutberg B.. A 5′ stem-loop and ribosome binding but not translation are important for the stability of Bacillus subtilis aprE leader mRNA. Microbiology. 2002; 148:1795–1803. [DOI] [PubMed] [Google Scholar]
- 34. Leroy M., Piton J., Gilet L., Pellegrini O., Proux C., Coppee J.Y., Figaro S., Condon C.. Rae1/YacP, a new endoribonuclease involved in ribosome-dependent mRNA decay in Bacillus subtilis. EMBO J. 2017; 36:1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wagner L.A., Gesteland R.F., Dayhuff T.J., Weiss R.B.. An efficient Shine–Dalgarno sequence but not translation is necessary for lacZ mRNA stability in Escherichia coli. J. Bacteriol. 1994; 176:1683–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lodato P.B., Hsieh P.K., Belasco J.G., Kaper J.B.. The ribosome binding site of a mini-ORF protects a T3SS mRNA from degradation by RNase E. Mol. Microbiol. 2012; 86:1167–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richards J., Luciano D.J., Belasco J.G.. Influence of translation on RppH-dependent mRNA degradation in Escherichia coli. Mol. Microbiol. 2012; 86:1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dreyfus M. Killer and protective ribosomes. Prog. Mol. Biol. Transl. Sci. 2009; 85:423–466. [DOI] [PubMed] [Google Scholar]
- 39. Komarova A.V., Tchufistova L.S., Supina E.V., Boni I.V.. Protein S1 counteracts the inhibitory effect of the extended Shine–Dalgarno sequence on translation. RNA. 2002; 8:1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roberts M.W., Rabinowitz J.C.. The effect of Escherichia coli ribosomal protein S1 on the translational specificity of bacterial ribosomes. J. Biol. Chem. 1989; 264:2228–2235. [PubMed] [Google Scholar]
- 41. Ruiz de los Mozos I., Vergara-Irigaray M., Segura V., Villanueva M., Bitarte N., Saramago M., Domingues S., Arraiano C.M., Fechter P., Romby P. et al. . Base pairing interaction between 5′- and 3′-UTRs controls icaR mRNA translation in Staphylococcus aureus. PLoS Genet. 2013; 9:e1004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lopez-Garrido J., Puerta-Fernandez E., Casadesus J.. A eukaryotic-like 3′ untranslated region in Salmonella enterica hilD mRNA. Nucleic Acids Res. 2014; 42:5894–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rochat T., Delumeau O., Figueroa-Bossi N., Noirot P., Bossi L., Dervyn E., Bouloc P.. Tracking the elusive function of Bacillus subtilis Hfq. PLoS One. 2015; 10:e0124977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Updegrove T.B., Zhang A., Storz G.. Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 2016; 30:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakamura K., Yahagi S., Yamazaki T., Yamane K.. Bacillus subtilis histone-like protein, HBsu, is an integral component of a SRP-like particle that can bind the Alu domain of small cytoplasmic RNA. J. Biol. Chem. 1999; 274:13569–13576. [DOI] [PubMed] [Google Scholar]
- 46. Macvanin M., Edgar R., Cui F., Trostel A., Zhurkin V., Adhya S.. Noncoding RNAs binding to the nucleoid protein HU in Escherichia coli. J. Bacteriol. 2012; 194:6046–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.