

Abstract
Edaravone (Radicava): a novel neuroprotective agent for the treatment of amyotrophic lateral sclerosis
INTRODUCTION
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a fatal degenerative disease that affects the motor neurons connecting the brain and spinal cord, leading to eventual paralysis and death. Approximately 5,600 individuals are diagnosed with ALS annually in the United States, and as many as 30,000 Americans are currently affected. Although rare, ALS is the most common motor neuron disease; it affects people of all races and ethnicities, but has a higher prevalence among Caucasians.1
In patients with ALS, the brain loses the ability to control muscle movement when the neurons controlling mobility begin to die, resulting in complete paralysis in its latter stages. Early symptoms of the disease include muscle twitching, cramping, stiffness, or weakness, and eventually slurred speech and difficulty chewing or swallowing. Psychological and cognitive difficulties are also seen in patients with ALS, including involuntary laughing or crying, depression, impaired executive functions, and maladaptive social behavior. Advanced stages of the disease feature symptoms such as muscle atrophy, spasticity, cramps, and weakness, all of which progressively worsen. The average life expectancy of a person with ALS is two to five years from time of diagnosis, with death resulting from respiratory failure (e.g., aspiration pneumonia) and medical conditions related to immobility. About half of patients with ALS live at least three years or more after diagnosis; 20% live five years or more; and up to 10% survive for more than 10 years.1–4
ALS was first described in 1869 by the French neurologist Jean-Martin Charcot. The disease gained wide recognition in the United States after the baseball player Lou Gehrig announced his ALS diagnosis in 1939. The disorder causes “amyotrophy”—the atrophy of muscle fibers—and “lateral sclerosis”—the changes seen in the lateral columns of the spinal cord when upper motor neuron axons in these areas degenerate and are replaced by fibrous astrocytes. Although the cause of ALS is unknown, about 5% of patients have a family history of the disease. Studies conducted in twins show a genetic contribution with a heritability of about 61%.2,5–14
Although there is no cure for ALS, available treatments can extend the length of quality of life in most patients. As the mainstay of ALS therapy, the American Academy of Neurology recommends adaptive treatments directed at the clinical manifestations of the disease, which include enteral nutrition via percutaneous endoscopic gastrostomy to stabilize body weight in patients with impaired oral intake, noninvasive ventilation to treat respiratory insufficiency to prolong survival and slow the decline of forced vital capacity (FVC), and mechanical insufflation/exsufflation to clear secretions in patients with reduced peak cough flow, particularly during an acute respiratory infection. The first drug approved by the Food and Drug Administration (FDA) for the treatment of ALS was riluzole, which should be offered to all patients with ALS to slow disease progression.15–17 In May 2017, the FDA approved edaravone (Radicava, Mitsubishi Tanabe Pharma America), a novel neuroprotective agent indicated to slow the advance of ALS.
DESCRIPTION
Edaravone, a member of the substituted 2-pyrazolin-5-one class, has the chemical name 3-methyl-1-phenyl-2-pyrazolin-5-one. It is a white crystalline powder with a melting point of 129.7° C and is freely soluble in acetic acid, methanol, or ethanol, as well as slightly soluble in water or diethyl ether. The molecular formula of edaravone is C10H10N2O (Figure 1), and the molecular weight is 174.20. Edaravone is available as a clear, colorless liquid provided as a sterile injection solution supplied for intravenous (IV) infusion in a polypropylene bag containing 30 mg edaravone in 100 mL of isotonic solution. Inactive ingredients include L-cysteine hydrochloride hydrate, sodium bisulfite, sodium chloride for isotonicity, and phosphoric acid and sodium hydroxide to adjust the solution to a pH of 4.17
Figure 1.
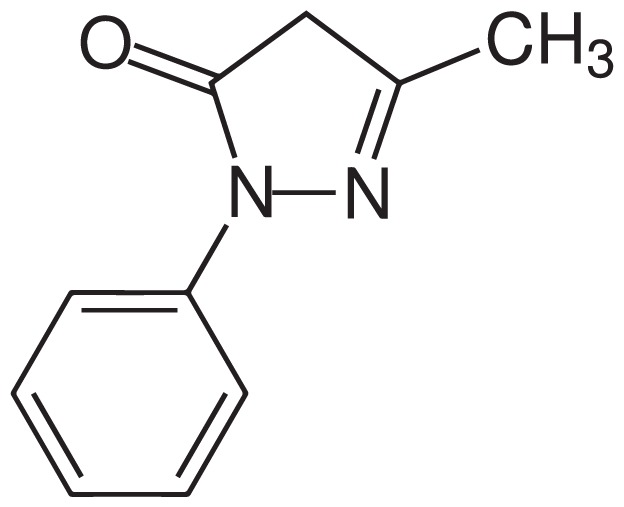
Chemical Structure of Edaravone17
MECHANISM OF ACTION
Although the exact mechanism of action of edaravone in the treatment of ALS is unknown, its therapeutic effect may be due to its known antioxidant properties; oxidative stress is a part of the process that kills neurons in patients with ALS.17,18
PHARMACOKINETICS
The maximum plasma concentration (Cmax) of edaravone was reached at the end of IV administration during clinical trials. Studies also showed a trend of a greater than dose-proportional increase in area under the concentration-time curve (AUC) and Cmax of edaravone, but no plasma drug accumulation was observed with multiple dose administration. The drug is 92% protein bound, primarily to albumin, and in the range of 0.1 to 50 micromol/L will yield no concentration dependence. The mean terminal elimination half-lives of edaravone and its metabolites are 4.5–6.0 hours and 2.0–2.8 hours, respectively. The drug is metabolized to pharmacologically inactive sulfate and glucuronide conjugates by multiple uridine diphosphate glucuronosyltransferase (UGT) isoforms in the liver and kidney, particularly during glucuronide conjugation. Edaravone is mainly detected as the sulfate conjugate in human plasma formed by sulfotransferases. It was excreted mainly in the urine as its glucuronide conjugate form after 70% to 90% of the dose was administered during the studies. Approximately 5% to 10% and 1% or less of the drug was recovered in the urine as sulfate conjugate and unchanged, respectively.17
The pharmacokinetics of edaravone were not affected by age in the geriatric population during clinical trials, and no significant differences in Cmax and AUC were observed between Japanese and Caucasian subjects. There are no pharmacokinetic data available from studies in patients with renal or hepatic impairment.17
PIVOTAL CLINICAL TRIALS
A double-blind, parallel-group, placebo-controlled, phase 3 trial was conducted to establish the efficacy and safety of edaravone for the treatment of ALS. This 36-week confirmatory trial consisted of a 12-week preobservation period followed by a 24-week treatment period. Eligible patients (age range, 20–75 years) in the studies met the revised Airlie House diagnostic criteria defined as “definite ALS,” “probable ALS,” or “probable laboratory-supported ALS.” At baseline, patients had to be able to feed themselves and require no assistance in activities of daily living; start the trial within three years of the onset of ALS; and have an FVC of at least 70%. Patients with dyspnea and deteriorating respiratory function were excluded. Patients were randomized to receive either placebo (n = 104) or edaravone (n = 102) 60 mg IV infusion once daily for 14 days followed by a 14-day observation period (initial treatment cycle). Patients then continued to receive edaravone 60 mg or placebo for 10 of 14 days in cycles 2 to 6, each followed by a 14-day observation period. Primary efficacy was evaluated based on the change in revised ALS Functional Rating Scale (ALSFRS–R) scores. Changes in FVC, grip strength (left/right mean), pinch strength (left/right mean), modified Norris scale score, ALS Assessment Questionnaire–40, and time to death or a specified state of disease progression (incapable of independent ambulation, loss of function in upper limbs, tracheotomy, artificial respirator with intubation, or tube feeding) were used to evaluate secondary endpoints.
The changes in ALSFRS–R scores at week 24 were −6.35 ± 0.84 in the placebo group (n = 99) and −5.70 ± 0.85 in the edaravone group (n = 100). The difference of 0.65 ± 0.78 (P = 0.411) was not statistically significant. The only statistically significant outcome from the secondary measures was the pinch strength of edaravone-treated patients (−0.83 ± 0.15) compared with placebo-treated patients (1.03 ± 0.15), a difference of 0.20 ± 0.14 (P = 0.165). There were no significant differences in the safety profile between the edaravone and placebo groups, but the trial did not show efficacy of edaravone to delay the progression of ALS.19
However, a six-month, randomized, placebo-controlled, double-blind study conducted in Japanese patients with ALS demonstrated the efficacy of edaravone for the treatment of ALS. The patients were randomized to receive placebo (n = 68) or edaravone (n = 69) 60 mg via IV infusion over 60 minutes based on the scheduled protocol, which included an initial treatment cycle with daily dosing for 14 days, followed by a 14-day drug-free period (cycle 1) and subsequent treatment cycles with daily dosing for 10 of 14 days, followed by 14-day drug-free periods (cycles 2–6). Change in the ALSFRS–R total scores from baseline to week 24 was the primary efficacy endpoint. The decline in ALSFRS–R scores from baseline was significantly less in the edaravone group (−5.01 ± 0.64) compared with the placebo group (−7.50 ± 0.66) (P = 0.0013).17
WARNINGS AND PRECAUTIONS
Edaravone is contraindicated in patients with a history of hypersensitivity to edaravone or any of the other ingredients in the product. Based on post-marketing reports, patients receiving edaravone should be monitored carefully for hypersensitivity reactions, such as redness, wheals, and erythema multi-forme, as well as for signs and symptoms of anaphylaxis, which include urticaria, decreased blood pressure, and dyspnea. If hypersensitivity reactions occur, discontinue edaravone, and manage and monitor patients until the condition resolves. Because edaravone contains sodium bisulfite, monitor patients for sulfite-related allergic reactions, including anaphylactic symptoms. Overall prevalence of sulfite sensitivity among the general population is unknown, but it is more common in patients with asthma.17
Adverse Events
In U.S. clinical trials and in post-marketing data from outside the U.S., the most serious adverse effects reported with edaravone treatment included hypersensitivity and sulfite allergic reactions, including anaphylactic symptoms. Bruising or contusions, gait disturbance, headache, dermatitis, and eczema were the most common adverse reactions observed in 10% or greater of edaravone-treated patients during the studies. The adverse reactions that occurred in 2% or more of edaravone-treated patients and at least 2% more frequently than in the placebo group appear in Table 1.17
Table 1.
Adverse Events Occurring in ≥ 2% of Edaravone-Treated Patients and ≥ 2% More Frequently Than in Placebo Patients*17
| Adverse Event | Edaravone (n = 184) % | Placebo (n = 184) % |
|---|---|---|
| Contusion | 15 | 9 |
| Gait disturbance | 13 | 9 |
| Headache | 10 | 6 |
| Dermatitis | 8 | 5 |
| Eczema | 7 | 4 |
| Respiratory failure, respiratory disorder, hypoxia | 6 | 4 |
| Glycosuria | 4 | 2 |
| Tinea infection | 4 | 2 |
Data from pooled, placebo-controlled trials.
Patients that are receiving edaravone are advised to notify health care providers immediately or go to the nearest emergency room if experiencing hives, swelling of the lips, tongue, or face, fainting, difficulty breathing, wheezing, trouble swallowing, dizziness, itching, or an asthma attack.17
Use in Specific Populations17
Pregnancy and Lactation
No human data are available regarding edaravone-induced fetal developmental risk in pregnant women. At therapeutic doses, edaravone was associated with developmental effects, such as increased mortality, decreased growth, delayed sexual development, and altered behavior, in animal studies. The excretion of edaravone in human milk, its effects on the breastfed infant, or the effects on milk production are unknown.
Geriatric and Pediatric Use
No differences in the safety and efficacy of edaravone were observed in geriatric patients with ALS compared with the adult population younger than age 65 years, based on three placebo-controlled trials; however, older patients may exhibit greater sensitivity to the drug. Edaravone has not been evaluated in patients younger than the age of 18 years.
Renal and Hepatic Impairment
The pharmacokinetics of edaravone in patients with renal or hepatic impairment have not been studied. Dosage adjustment is not required in patients with renal impairment or mild or moderate hepatic impairment.
Drug–Drug Interactions
In vitro studies show that therapeutic doses of edaravone and its metabolites are not inhibited by cytochrome P450 (CYP) enzymes, UGT isoforms, or major transporters in humans. In addition, the active drug and metabolites of edaravone at clinical doses do not induce CYP1A2, CYP2B6, or CYP3A4.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Edaravone has not been evaluated for its carcinogenic potential, but demonstrated negative mutagenesis in vitro and in vivo.
DOSAGE AND ADMINISTRATION17
The recommended dose of edaravone is 60 mg administered via 60-minute IV infusion once daily for 14 days as the initial treatment cycle, followed by a 14-day drug-free period. Subsequent treatment cycles consist of once-daily dosing for 10 of 14 days, each followed by a 14-day drug-free period.
Edaravone 60 mg is administered from two consecutive single-dose polypropylene bags each containing 30 mg of edaravone in 100 mL of clear, colorless aqueous solution. The infusion rate is approximately 1 mg or 3.33 mL per minute. The manufacturer recommends that no other medications should be injected or mixed with edaravone in the infusion bag.
P&T COMMITTEE CONSIDERATIONS
Edaravone is worthy of formulary consideration for patients with ALS. One 60-minute 60-mg infusion (supplied as two 30-mg/100-mL infusion bags) has an average wholesale price of $1,303.20 It may be a viable alternative after the failure of riluzole therapy, the first and only oral treatment for ALS. Riluzole delays the onset of ventilator dependence or tracheostomy in select patients and may increase survival by approximately two to three months. In the Japanese trial of edaravone, the drug significantly slowed the decline in ALSFRS–R scores compared with placebo. There are no significant safety warnings with edaravone, and clinical trials demonstrated no significant differences between the placebo and edaravone groups with respect to death, serious adverse effects, or treatment discontinuation. Edaravone requires IV infusion on a regular dosing schedule, which may be limited by the availability of infusion centers and may pose travel challenges for patients with this progressive, degenerative disease.
CONCLUSION
The pathophysiology of ALS remains poorly understood. Riluzole, the first FDA-approved treatment for the disease, provides limited benefit to patients. More recently, edaravone, a potent pyrazolone free radical scavenger and antioxidant, was approved for the treatment of ALS and is theorized to decrease the effects of oxidative stress in ALS. The safety of edaravone in patients with severe renal impairment, end-stage renal disease, or moderate-to-severe hepatic impairment remains to be studied. Further clinical studies with longer durations are warranted to better establish the efficacy of edaravone for the treatment of ALS.
Footnotes
Disclosure: The author reports no commercial or financial interests in regard to this article.
REFERENCES
- 1.Association ALS. Quick facts about ALS and the ALS Association. [Accessed September 15, 2017]. Available at: www.alsa.org/news/media/quick-facts.html.
- 2.Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7(11):639–649. doi: 10.1038/nrneurol.2011.153. [DOI] [PubMed] [Google Scholar]
- 3.Walling AD. Amyotrophic lateral sclerosis: Lou Gehrig’s disease. Am Fam Physician. 1999;59(6):1489–1496. [PubMed] [Google Scholar]
- 4.Kimura F, Fujimura C, Ishida S, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology. 2006;66(2):265–267. doi: 10.1212/01.wnl.0000194316.91908.8a. [DOI] [PubMed] [Google Scholar]
- 5.King SJ, Duke MM, O’Connor BA. Living with amyotrophic lateral sclerosis/motor neurone disease (ALS/MND): decision-making about “ongoing change and adaptation”. J Clin Nurs. 2009;18(5):745–754. doi: 10.1111/j.1365-2702.2008.02671.x. [DOI] [PubMed] [Google Scholar]
- 6.Eisen A. Amyotrophic lateral sclerosis: A 40-year personal perspective. J Clin Neurosci. 2009;16(4):505–512. doi: 10.1016/j.jocn.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 7.Phukan J, Hardiman O. The management of amyotrophic lateral sclerosis. J Neurol. 2009;256(2):176–186. doi: 10.1007/s00415-009-0142-9. [DOI] [PubMed] [Google Scholar]
- 8.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks BR. Managing amyotrophic lateral sclerosis: slowing disease progression and improving patient quality of life. Ann Neurol. 2009;65(suppl 1):S17–S23. doi: 10.1002/ana.21544. [DOI] [PubMed] [Google Scholar]
- 10.Al-Chalabi A, Fang F, Hanby MF, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry. 2010;81(12):1324–1326. doi: 10.1136/jnnp.2010.207464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hudson AJ. The motor neuron diseases and related disorders. Clinical Neurology. 1996;4:11–14. [Google Scholar]
- 12.Kanouchi T, Ohkubo T, Yokota T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? J Neurol Neurosurg Psychiatry. 2012;83(7):739–745. doi: 10.1136/jnnp-2011-301826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218–1226. doi: 10.1212/WNL.0b013e3181bc0141. Erratum in: Neurology 2010;74(9):781; Neurology 2009;73(24)2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227–1233. doi: 10.1212/WNL.0b013e3181bc01a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radicava (edaravone) prescribing information. Jersey City, New Jersey: MT Pharma America, Inc; May, 2017. [Google Scholar]
- 18.Brooks BR, Miller RG, Swash M, Munsat TL World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 19.Abe K, Itoyama Y, Sobue G, et al. Confirmatory double-blind, parallel-group, placebo controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7–8):610–617. doi: 10.3109/21678421.2014.959024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Red Book Online. Ann Arbor, Michigan: Truven Health Analytics; [Accessed November 14, 2017]. [Google Scholar]