

Abstract
Phylogenetic reconstruction from transposable elements (TEs) offers an additional perspective to study evolutionary processes. However, detecting phylogenetically informative TE insertions requires tedious experimental work, limiting the power of phylogenetic inference. Here, we analyzed the genomes of seven bear species using high-throughput sequencing data to detect thousands of TE insertions. The newly developed pipeline for TE detection called TeddyPi (TE detection and discovery for Phylogenetic Inference) identified 150,513 high-quality TE insertions in the genomes of ursine and tremarctine bears. By integrating different TE insertion callers and using a stringent filtering approach, the TeddyPi pipeline produced highly reliable TE insertion calls, which were confirmed by extensive in vitro validation experiments. Analysis of single nucleotide substitutions in the flanking regions of the TEs shows that these substitutions correlate with the phylogenetic signal from the TE insertions. Our phylogenomic analyses show that TEs are a major driver of genomic variation in bears and enabled phylogenetic reconstruction of a well-resolved species tree, despite strong signals for incomplete lineage sorting and introgression. The analyses show that the Asiatic black, sun, and sloth bear form a monophyletic clade, in which phylogenetic incongruence originates from incomplete lineage sorting. TeddyPi is open source and can be adapted to various TE and structural variation callers. The pipeline makes it possible to confidently extract thousands of TE insertions even from low-coverage genomes (∼10×) of nonmodel organisms. This opens new possibilities for biologists to study phylogenies and evolutionary processes as well as rates and patterns of (retro-)transposition and structural variation.
Keywords: retrotransposition, bears, Ursidae, phylogeny, evolution, transposable elements
Introduction
In an innovative study almost 20 years ago, rare genomic changes were used to confirm the close relationship between hippopotamus (Artiodactyla) and whales (Cetacea) (Shimamura et al. 1997; Nikaido et al. 1999). Transposable element (TE) insertions are a type of rare genomic changes that propagate in the genome via copy-and-paste (retrotransposons) or cut-and-paste (DNA transposons) mechanisms. Germline transposition events are passed on to descendants, making it possible to deduce their phylogenetic relationships (Shimamura et al. 1997; Nikaido et al. 1999). In contrast to nucleotide substitutions which are prone to homoplasy by parallelisms, convergence, and reversals, TE insertions are virtually homoplasy free. Parallel integration of TE insertions in the same loci in different species is highly improbable due to low-germline insertion rates and the presence of different active TE families (Ray et al. 2006). Finally, the exact removal of TE insertions is very rare and usually leaves a detectable genetic “scar” (van de Lagemaat et al. 2005). These features are very valuable for the understanding of deep or complex divergences, like the early radiation of mammals and birds (Churakov et al. 2009; Nishihara et al. 2009; Hallström and Janke 2010; Suh et al. 2015).
Detecting phylogenetically informative TE insertions was initially challenging because fully sequenced genomes were not available (Shimamura et al. 1997; Nikaido et al. 1999). Therefore, only experimental work could identify candidate TE loci of which often only a minor fraction were phylogenetically informative (Shimamura et al. 1997; Nikaido et al. 1999). Although, the increasing availability of genome assemblies and new methods have allowed computational identification of phylogenetically informative TE insertions, extending the taxon sampling for species without available genomes relied still on extensive experimental testing (Kriegs et al. 2006; Churakov et al. 2009). Alternative methods, not based on genome assemblies can only identify a limited number of informative TE insertions (Suh et al. 2012; Kuramoto et al. 2015). Finally, experimental enrichment protocols for TE insertions can identify thousands of informative loci, but require knowledge of the TE sequence and are biased toward loci with existing TE insertions (Platt et al. 2015). A recently developed bioinformatic approach to detect novel TE insertions uses the information from discordantly mapped paired-end short reads without requiring a de novo genome assembly for each species (Medvedev et al. 2009). Such “TE calling” methods have allowed biologists to study TE insertion dynamics and other structural variations (SV) on a population-scale (Hormozdiari et al. 2013; Sudmant et al. 2015). This approach has been successfully applied to the great apes and to mice (Nellåker et al. 2012; Hormozdiari et al. 2013) showing its potential for phylogenetic inference. However, as yet, no phylogenetic study has applied TE calling methods to nonmodel organisms, for which often only draft genome assemblies and low-coverage resequencing data are available.
A long-standing question in phylogenetics is determining the evolutionary history of bears (Ursidae), for which different scenarios have been reconstructed from analyses of mitochondrial, autosomal, and gonosomal DNA sequences. In particular, the six ursine species that include the polar (Ursus maritimus) and brown bear (Ursus arctos), share a complex evolutionary history due to their rapid radiation during the Pliocene (5–3.5 Million years ago (Ma)) (Kumar et al. 2017). The best studied examples are polar bears, which according to mitochondrial DNA (mtDNA) are nested within the brown bears (Yu et al. 2007). However, analyses of nuclear DNA showed that polar bears are genetically distinct and the sistergroup to brown bears (Hailer et al. 2012). The phylogeny of the American black bear (Ursus americanus) and the three South-East Asian bear species is less understood with deviating mtDNA and nuclear gene trees (Yu et al. 2007; Pagès et al. 2008; Kutschera et al. 2014). Phylogenomic analyses reconstructed the American black bear as the sister group to a monophyletic polar and brown bear lineage and show that the three South-East Asian bears form a clade with the Asiatic black bear (Ursus thibetanus) being the sister group to sun (Ursus malayanus) and sloth bear (Ursus ursinus) (Kutschera et al. 2014; Kumar et al. 2017).
The observed phylogenetic incongruence among bears can be caused by introgressive hybridization and incomplete lineage sorting (ILS) (Maddison 1997). As such, the analysis of genome-wide data is necessary to understand these complex processes (Delsuc et al. 2005). However, the lack of whole genome sequences inhibited efficient screening for phylogenetically informative TE insertion events until the polar bear genome sequence and genome data of all other bear species became available (Miller et al. 2012; Liu et al. 2014; Kumar et al. 2017). These new genome data have allowed us to detect TE insertions as additional independent phylogenomic markers to study the evolution of Ursidae. We developed the TeddyPi (TE detection and discovery for phylogenetic inference) pipeline to process data from TE and SV callers. TeddyPi pursues the idea of integrating different TE callers (Lin et al. 2015; Nelson et al. 2017) and extends it to routinely integrate TE insertion data sets from multiple samples to track integrations of TEs in orthologous loci and to create presence/absence tables for phylogenetic inference.
We applied TeddyPi to whole-genome sequencing data of all ursine bears and the monotypic subfamily Tremarctinae to extract phylogenetically informative markers that are independent from nucleotide substitution analyses. We aimed to study the evolutionary history of bears and test whether TE insertions identify the same signals of gene flow and ILS as in a previous nucleotide-based study (Kumar et al. 2017). This recently generated genome data of all ursine bears made it possible to observe nucleotide substitutions in the flanks around the TE insertions, that are mutationally saturated for deeper divergences. To validate the in silico TE calls made by TeddyPi, 151 loci were validated experimentally by PCR and Sanger sequencing. The TeddyPi pipeline extracted an extensive catalog of 150,513 TE insertions to reconstruct the first TE-derived species tree of bears and revealed varying rates of TE accumulation in their genomes.
Materials and Methods
The TeddyPi Pipeline
The TeddyPi pipeline is a modular framework to process TE and SV calls and to prepare data sets for phylogenetic inference. The application is written in Python and utilizes established code libraries for biological computing. Parameters and the filter pipeline are configured with comprehensively structured configuration files and allow the user to create tailor-made filtering pipelines for a variety of variant callers. The first module (teddypi.py) processes each sample genome individually and filters the output of the selected variant callers. Several filters and merge-functions are included in this module, and a flexible codebase allows implementation of new functions with little programming knowledge. In the same module, large deletions are transformed to reference-insertion calls on the basis of annotated TEs in the reference genome. It is also possible to make intersections or create nonredundant data sets of the input data in this step. In the second module (tpi_ortho.py), TE insertion data is combined across a set of samples (typically different taxa) to generate presence/absence matrices for reference insertions (Ref+) and nonreference insertions (Ref−) separately. Finally, the last module (tpi_unite.py), merges both matrices to a comprehensive presence/absence matrix that can be exported in tabular-text and NEXUS format. A schematic overview is shown in figure 1 and a flowchart of the pipeline in supplementary figure 1, Supplementary Material online. TeddyPi is open source and can be accessed on https://github.com/mobilegenome/teddypi, last accessed September 2017. Easy configuration and the modular architecture make it convenient to adapt TeddyPi to process data from a broad range of TE/SV callers or other integration pipelines such as SVMerge or McClintock (Wong et al. 2010; Nelson et al. 2017). TeddyPi can be applied to any group of organisms where accurate TE/SV calling is feasible.
Fig. 1.

—Schematic illustration of the TeddyPi pipeline. (1) Transposable Element (TE) and Structural Variation (SV) callers detect reference (Ref+, red) and nonreference (Ref−, blue) TE insertions from reads mapped to a reference genome. The boxed trees show a schematic phylogeny with the reference genome (Ref) and two other taxa (A and B). The TE insertion is shown by an arrow and indicates Ref+ and Ref− detection depending on which branch the TE inserted. (2) TE calls are filtered based on the polar bear genome annotation, call quality, and sequencing coverage across the genome. Different TE classes are collected separately. (3) Sets of TE calls (call sets) for each individual genome are merged to create a comprehensive presence/absence matrix (4) that is used for phylogenetic inference and (5) to select loci for in vitro validation.
Taxon Sampling and Genome Sequencing
Whole genome sequencing data generated with Illumina HiSeq technology from Kumar et al. (2017) and Miller et al. (2012) for six ursine bear species and the spectacled bear (Tremarctos ornatus) were obtained. For mapping, paired-end reads (100–125 base pairs (bp) long) were quality-trimmed with Trimmomatic (Bolger et al. 2014), mapped with BWA (Li and Durbin 2010), and duplicated reads were marked. In total, nine genomes with a mean coverage of 13.7× from seven species were analyzed (supplementary table 1, Supplementary Material online). In comparison to the giant panda (Ailuropoda melanoleuca) genome sequence (ailMel1), the polar bear genome sequence (Liu et al. 2014) has higher contiguity and contains potentially better-assembled repeats because it is based on longer sequencing reads. Therefore, the polar bear was the preferred choice for reference mapping.
Considerations for Nested Reference Genomes
Programs to detect TE insertions (in analogy to SNP callers named TE callers) depend on a pairwise comparison between the paired-end short reads of a sample and the reference genome the reads were mapped to. As most published TE callers can only detect nonreference (Ref−) TE insertions it is beneficial to have a reference genome that is phylogenetically placed as the outgroup to the taxa under study to detect insertions across the complete phylogeny (supplementary fig. 2, Supplementary Material online). If this is not possible, the use of only nonreference TE callers will lead to unresolved internodes and a skewed phylogenetic interpretation. For example, when the reference genome is nested inside the ingroup/tree, only TE insertions on the terminal branches are detectable and certain internodes cannot be resolved (supplementary fig. 2, Supplementary Material online). To overcome such a bias, reference (Ref+) TE insertions (i.e., those shared with the reference genome) need to be considered, too. Ref+ TE insertions can not be called directly, but are inferred from deletion calls made from the mapped short read data. These deletions resemble insertions in the reference genome sequence. From the reference genome, the sequence of the insertion can be extracted and screened for similarity to known TEs. TeddyPi inverts deletion calls that intersect with TE sequences in the reference genome to infer Ref+ TE insertions (supplementary fig. 3, Supplementary Material online).
Analysis of TEs in the Polar Bear Genome Sequence
Repetitive elements in the polar bear genome were identified using RepeatMasker in sensitive mode (-s) searching for carnivore-specific repeats (Repbase version 20140131). The script createRepeatLandscape.pl provided with RepeatMasker was used to calculate the repeat landscape. We explored the diversity of LINE1 copies in the polar bear genome to find active copies that can drive retrotransposition or inactive copies incapable of retrotransposition, for example, by the presence of premature stop codons in the open reading frame (ORF) 2 of the LINE1. The LINE1 ORF2 sequence was retrieved from a full-length LINE1 found on the polar bear Y chromosome (Bidon et al. 2015) and used as a BLAST query against the polar bear genome sequence (Altschul et al. 1990). Hits were filtered for full length, coding ORF2 copies and a maximum of three mismatches. Then, these sequences plus 7,000-bp flanking sequence on 5′ and 3′ ends were extracted from the polar bear genome sequence. Within these sequences, a BLAST search for a coding LINE1 ORF1 sequence was performed to find LINE1 copies containing two coding ORFs. As an additional proxy for LINE1 activity, we screened the polar bear and giant panda genome for the U6 snRNA (Accession No: M14486.1) using BLAST. According to Doucet et al. (2015), all hits with >97.5% identity, 26-bp alignment length and an E-value of < 10 were considered as full-length hits. Additionally, we annotated 146,268 gaps totaling to 38 mega base pairs (Mb) in the polar bear genome; the majority of these gaps (138,041) were >1 bp.
Detection of Nonreference (Ref−) TE Insertions
Reference mapped short reads were processed with RetroSeq (Keane et al. 2013) and Mobster (Thung et al. 2014) to identify insertions that are present in the corresponding genome while being absent in the reference genome. For RetroSeq, a minimum mapping quality of 30 and a TE mapping identity of 90% at 50% length were used. The upper coverage threshold was set to 2.5× of the samples’ sequencing depth. Mobster was run with default settings. A library of 593 carnivore specific TE sequences was retrieved from Repbase (Jurka et al. 2005), and used as consensus database for both programs. Mobster and RetroSeq used this database to identify reads that match the consensus TE sequence and thereby inferred the type of TE that has been integrated. In addition, RetroSeq identifies reads matching the RepeatMasker track of the reference genome. Using the TeddyPi pipeline, callsets (i.e., the sets of called TE variants) from RetroSeq and Mobster were filtered for calls falling within regions of undetermined bases (N) plus a window of 200 bp in the polar bear genome. Calls were also filtered, if they were supported by less than five reads or when located within 100 bp of annotated TEs of the same type in the polar bear genome. For stringency, both data sets were masked for regions that had a depth of coverage <33% or >250% of the mean sample coverage. Thereby, regions of ambiguously mapped reads or with insufficient coverage for TE calling were excluded. Only overlapping calls from both programs were further processed.
Detection of Reference Insertion (Ref+) TE Insertions
To detect TE insertions absent from at least one of the low-coverage bear genomes and present in polar bear reference genome (Ref+ insertions), Pindel (Ye et al. 2009), and Breakdancer (Chen et al. 2009) mined the genomes for deletions, that are indicative of insertions in the reference genome (Nellåker et al. 2012). Pindel uses split-read (SR) information to obtain breakpoint information at a single-nucleotide level resolution and was run with the following parameters –report_interchromosomal_events false, –anchor_quality 30, -w 40. Only deletions were considered for further processing. BreakDancer was run using a maximum variant size of 10 kilo bases (kb) and requiring at least five supporting reads to make a SV call. BreakDancer utilizes only discordant reads and does not utilize SRs for SV-calling. Therefore, start- and end-coordinates from the deletions were used. For each sample, book-ended (i.e., those directly after another) calls and overlapping calls were merged, filtered for N-regions (+200 bp flanking sequence) and tandem repeats (+50 bp) in the reference genome. All calls in regions with a depth of coverage <33% or >250% of the average were excluded. The calls from Pindel and Breakdancer were merged to a nonredundant set. The start/end coordinates or if available, the breakpoint of the deletion plus a window of ± 50 bp were used to detect intersections with annotated repeats in the polar bear reference genome. Deletion calls that matched duplicate RepeatMasker hits and appeared twice, were merged. When coordinates overlapped with more than one TE in the reference genome, and one was a recent SINE insertion (i.e., SINEC_Ame subfamily) while the other TE(s) were not known to be active within Carnivora, it was called as “SINE derived”. If coordinates overlapped with different types of annotated TEs, and more than one was potentially active, the event was recorded as “complex”. Predicted deletion loci of more than one sample were attributed to the same locus if both were intersecting with the reference TE and the distance was <100 bp. To obtain reference insertion (Ref+) calls, presence/absence information was inverted (supplementary fig. 2, Supplementary Material online).
Integration of Ref+ and Ref− Call Sets, Filtering, and Processing
To combine the insertion and deletion data sets, results were integrated across all species. This module of TeddyPi (tpi_ortho.py) loads the final call sets for all species, sorts these by position, and merges overlapping and book-ended calls if not done before. Then, BedTools window is called via pybedtools to create a presence/absence matrix (coded as 1 and 0, respectively) over all variants and taxa (variant × taxa) (Quinlan and Hall 2010; Dale et al. 2011).
Despite originating from the same insertion event, breakpoint estimates might differ slightly between taxa. Therefore, overlapping, book-ended, and events within 100 bp of each other were merged using BedTools. Presence/absence information from deletion calls was inverted (1 ↔ 0) to obtain reference insertions (Ref+) calls. The state of TE insertions in the reference genome was added with either 1 or 0 for Ref+ and Ref− events, respectively. Callsets for Ref+ and Ref− were saved as a tab-separated file and converted to a NEXUS character matrix using the python-nexus package (Greenhill S. unpublished).
Merging Ref+ and Ref− Callsets, and Correcting for Missing Data
Ref+ and Ref− data sets were merged in the tpi_unite.py module of TeddyPi and a final presence/absence matrix was created. A synthetic outgroup with state “0” for all loci was added. For the Ref− data set, loci were coded as missing data (“?” in the NEXUS matrix) for samples with an insufficient or excessive depth of coverage. The criteria were set for each sample individually to include only loci with coverage between 0.33× and 2.5× of the samples mean coverage.
Phylogenetic Inference from TE Insertion Calls
We processed SINE and LINE1 callsets separately and created Dollo parsimony trees in PAUP* (Swofford 2002) using the heuristic search with 500 replicates. Bootstrap support was calculated from 1,000 replicates. The trees were rooted using the synthetic outgroup. The number of SINE insertions for species-tree congruent and alternative topologies were obtained from the presence/absence matrices and analyzed using the KKSC test that conceptually transfers the D-statistics to TE insertion data (Durand et al. 2011; Kuritzin et al. 2016). The KKSC test evaluates the number of phylogenetically conflicting TE insertions among three taxa and uses binomial distribution to test for the probability of hybridization or ILS as cause of the observed insertion pattern.
Median networks for SINE insertions were calculated in SplitsTree 4 (Huson and Bryant 2006). Phylogenetic networks for Ref+ and Ref− data were calculated separately using all SINEs and LINE1s.
Estimating TE Insertion Rates
SINE and LINE insertion counts were extracted from the parsimony-tree branch lengths and were divided by the divergence times (in Myr) estimated previously (Kumar et al. 2017) to get estimates on the relative insertion rate. To estimate per-generation insertion rates, the generation time for the polar and brown bear was assumed to be 10 years (Tallmon et al. 2004; Cronin et al. 2009) and 6 years for the other bear species (Onorato et al. 2004; Kutschera et al. 2014).
Genomic Context of TE Insertions
The genomic context of the TE insertions was evaluated using the genome annotation from the polar bear genome (Liu et al. 2014). The TE insertion catalog was screened for overlaps with 3′- and 5′-UTRs, introns, exons, and intergenic regions.
Flanking Sequence Analysis of TE Insertion Loci
TE insertions and the substitutions in their flanking genomic regions are expected to share the same evolutionary history. We sought to explore the congruence in phylogenetic signal between TEs and flanking regions and to determine the range of the phylogenetic congruence (i.e., the spatial extent in bp) between them. To this end, consensus sequence alignments were created using substitution calls from Kumar et al. (2017). First, 10-kb sequence up- and downstream of the insertion site were extracted and the maximum likelihood (ML) phylogeny was inferred with RaxML (Stamatakis 2014) for each flank as well as for the concatenated sequence of both flanks. For automation and calling RAxML, the Dendropy package was utilized (Sukumaran and Holder 2010). To account for possibly misaligned reads around the insertion site, the first 500 bp on each side of the insertion site were excluded. The question was, whether the flanking sequence yields the same phylogenetic signal as the presence/absence pattern of the TE insertion. Therefore, we checked if the species carrying the TE insertion form a monophyletic group in the ML-trees using the ETE toolkit (Huerta-Cepas et al. 2016). Furthermore, to gain insight in the phylogenetic signal in the TE flanking region a sliding window approach was applied to the same 10-kb flanking regions using nonoverlapping 1-kb windows. For each window, sites were counted showing the same phylogenetic signal as the TE insertion and then divided by the number of segregating sites.
Experimental Validation Screening
From the in silico data set, loci were randomly selected for experimental verification. DNA samples from all ursine bears and the spectacled bear were included. For the Asian bear species and the spectacled bear, the same DNA samples were used for validation as for the Illumina genome sequencing. We selected loci containing TE insertions supporting different topologies (supplementary table 2, Supplementary Material online) including topologies in conflict with the species tree (e.g., presence in American black and Asiatic black bear or American black bear and sun bear).
For primer design, consensus sequence alignments that spanned 4-kb up- and downstream of the predicted TE insertion site were extracted from Kumar et al. (2017). PCR primers were generated with primer3 to be located ∼200 bp from the TE insertion site (Untergasser et al. 2012). Primers are listed in the supplementary data 1, Supplementary Material online. Each locus was amplified using 8 ng of DNA per species and Amplicon Taq (VWR) in a touchdown PCR. Banding patterns were examined using gel-electrophoresis agarose gels along with a DNA marker (ThermoFisher GeneRuler 1Kb). The fragment length of each PCR product was estimated and species that had the indication of a TE insertion were recorded. The PCR amplicons were Sanger-sequenced in both directions using the ABI 3730 DNA Analyzer. The type of the inserted sequence was determined by querying the sequence against Repbase (Jurka et al. 2005) (www.girinst.org; last accessed September 2017). For 13 markers, PCR products were sequenced of all or nearly all bear species to verify the phylogenetic information of the loci. The alignments were screened for the TE type, the orientation, target site duplications (TSDs), and the integrity of the flank. Two markers were specifically selected and sequenced to investigate the absence of a SINEC1_Ame in the polar bear (marker 40 and 122).
Experimentally confirmed insertion patterns were compared with the computationally predicted insertions at the same locus. We considered each matching insertion status (predicted: absence—PCR: absence/predicted: presence—PCR: presence) as correctly called. If the PCR product indicated presence of a TE insertion but no TE call was made, the locus was recorded as false negative (FN) and false positive (FP) for the opposite case. If a PCR reaction did not yield an amplicon for a locus, the locus was flagged as inconclusive.
Results
Transposable Elements in Ursine Bears
Our screening of the interspersed repeats in the polar bear reference genome identified 1,223,168 SINEs (8.4% of the genome), 978,888 LINEs (21.3%), 320,346 LTR retrotransposons (5.3%), as well as 340,447 DNA transposons (3.1%) (supplementary table 3, Supplementary Material online). In total, the polar bear genome comprised 38.1% interspersed repeats, similar to other carnivores like the giant panda, dog, or cat (Lindblad-Toh et al. 2005; Pontius et al. 2007; Li et al. 2010). The most abundant and recently active SINE-family in carnivore genomes is the lysine-tRNA derived SINEC (Walters-Conte et al. 2011). In Ursidae, SINEC1_AMe is the most frequent SINE subfamily in both the polar bear and giant panda genomes with 249,740 copies and 237,604 copies, respectively. SINEC1_Ame has a consensus length of 201 bp and was initially described from the giant panda genome (Li et al. 2010). SINEC elements are thought to be LINE1 propagated, and a screen for potentially active full-length LINE1s revealed 535 copies with two intact ORFs in the polar bear genome. The U6 snRNA that has been strongly associated with LINE1 activity in mammalian genomes (Doucet et al. 2015) was found in 67 copies in the polar bear genome sequence. Repeat landscapes of both the polar bear and giant panda genomes indicate the presence of low divergent and thus recently active SINEs (supplementary fig. 4, Supplementary Material online).
Detecting Ref− Insertions
In all analyzed samples, the programs RetroSeq (Keane et al. 2013) and Mobster (Thung et al. 2014) found 696,041 and 491,193 Ref− TE insertions, respectively (supplementary tables 4 and 5, Supplementary Material online). Despite the difference in numbers of raw calls, the number of SINEs and LINEs selected from the unfiltered data sets of RetroSeq and Mobster are very similar (∼300,000 SINEs, ∼135,000 LINEs). Still, data sets from both programs differed in susceptibility to the subsequent filtering pipeline, indicating differences in the overall call-quality (supplementary tables 4–7, Supplementary Material online). Thus, after filtering, 50% more SINEs were obtained from Mobster than from RetroSeq. For LINEs, 25% more calls from RetroSeq were retained by TeddyPi (supplementary table 8, Supplementary Material online). After merging data from RetroSeq and Mobster, the final Ref− insertion data set consisted of 84,462 SINEs and 7,734 LINEs (supplementary table 8, Supplementary Material online).
Detecting Ref+ Insertions
A different approach was necessary to identify Ref+ TE insertions due to nested position of the polar bear in the ursine species tree (supplementary fig. 2, Supplementary Material online). The two SV callers Pindel (Ye et al. 2009) and BreakDancer (Chen et al. 2009) identified 10,527,959 deletions in the nine bear genomes. Of these ∼10.5 million deletions (96.4%) were shorter than 100 bp and excluded from further processing. Length distributions of the deletion callsets showed distinct peaks of 200 bp and 6 kb, corresponding to full-length copies of SINEs and LINE1s, respectively (supplementary figs. 5 and 6, Supplementary Material online). After filtering, we retained 12,865 (Pindel) and 296,013 (BreakDancer) high-quality deletion calls that were between 100 bp and 10 kb long (supplementary tables 9 and 10, Supplementary Material online).
The majority (95%) of detected Pindel deletions were also identified by BreakDancer, suggesting a higher reliability at the expense of lower sensitivity in the program Pindel. The filtered data of both programs were merged into a nonredundant set of 295,434 deletion calls (supplementary table 11, Supplementary Material online). Of these, 270,689 (92%) matched TE annotations in the polar bear genome and hence were considered as Ref+ TE insertions. We detected 210,999 deletions that intersected SINE insertions in the polar bear genome. From 30,609 deletions matching LINE1 insertions, only a minor fraction (2.5%) was longer than 5 kb, the remaining copies were likely 5′-truncated (supplementary table 11, Supplementary Material online).
Phylogenetic networks generated from Ref+ and Ref− data sets, respectively, show that one type of detected insertions can only resolve one side of the phylogenetic tree (supplementary figs. 7 and 8, Supplementary Material online).
TE Insertion Rates in Ursine Bears
For both Ref+ and Ref− insertions, TeddyPi discovered on an average 10,000 and 20,000 TE insertions per genome, respectively (fig. 2a). The few TE insertions discovered in the two resequenced polar bears reflect the species’ low genetic diversity and are expected because the reference genome is of a conspecific individual. Compared with LINE1 insertions, novel SINE insertions were ∼6 times more frequent. TeddyPi identified 1.5 times more Ref+ than Ref− insertions in the bear genomes (fig. 2a). The highest number of TE insertions was found in the spectacled bear and the lowest number of TE insertions was identified in the two additional polar bear genomes (fig. 2a). For the other species, the numbers of identified TE insertion were homogeneous. As expected from their higher abundance, the genomic distance between SINE insertions was shorter than for LINEs (median distance: 10,010 and 73,240 bp, respectively) (fig. 2b). For the distance between SINEs, the upper bound was 330 kb. The upper bound of the LINE1 distances of >1 Mb indicates the presence of large genomic regions in which TeddyPi did not detect ursine-specific LINE1 insertions.
Fig. 2.

—Detection results for TE insertions calls and inferred TE insertion rates. (a) Counts of Ref − (left) and Ref + (right) TE calls per analyzed sample shown for long interspersed element (LINE) insertions (orange) and short interspersed element (SINE) insertions (blue). (b) Distance distribution of all detected TE insertions among all bears. Vertical dashed lines indicate median distances. (c) TE insertion rates as insertions per generation (ins/gen) for all ursine species were estimated for the terminal branches in a chronogram scaled to divergence times from Kumar et al. (2017).
The rate of TE mobilization is known to differ between lineages (Hormozdiari et al. 2013). Among bears, LINE1-mediated retrotransposition of LINEs and SINEs is ubiquitous, but insertion rates (i.e., the number of TE insertion per generation) were substantially higher in brown and polar bear (fig. 2c). With 0.12 SINE insertions per generation, the insertion rate in the brown bear genome was the highest. TE insertions into coding or regulatory regions disrupt reading frames or inhibit transcription, however beneficial and potentially adaptive TE insertions are known (Cordaux and Batzer 2009; Casacuberta and Gonzalez 2013; Hof et al. 2016). In bears, 97% of TE insertions integrated into noncoding regions and only a few are located in exons or potential regulatory regions (supplementary fig. 9, Supplementary Material online).
In Vitro Validation of the TE Prediction Accuracy
Predicting TE insertions from high-throughput sequencing data is challenging and prone to artifacts. We extracted 151 loci to perform validation assays using PCR and Sanger sequencing to assess the accuracy of the in silico predictions (supplementary data 1, Supplementary Material online). All Sanger-sequenced loci, for which the size of the PCR amplicon suggested a TE insertion, were validated as a SINEC1_Ame insertion. Furthermore, the target site duplications and breakpoints were identical among different taxa, thus indicating a single, unique integration event (supplementary fig. 10 and note 1, Supplementary Material online). The validation experiment showed that 90% of the Ref− TE calls were accurate and both, false positive (FPR) and false negative rates (FPR) were low (table 1). The results indicate that the Ref− callers are more likely to miss a true TE insertion than to return an artifact. Loci were randomly selected for PCR validation from the whole data set or predefined presence/absence patterns for phylogenetic hypotheses (supplementary table 2, Supplementary Material online). Irrespectively, of whether the hypothesis matched the species tree or is in conflict with it, 93% of the predictions were experimentally confirmed to be accurate (table 1, supplementary data 1, Supplementary Material online).
Table 1.
Summary of In Vitro TE Validation Experiments for Ref− and Ref+ Insertion Loci
| Type | Set | Informative Loci |
All Loci |
||||||
|---|---|---|---|---|---|---|---|---|---|
| N | TP | FP | FN | N | TP | FP | FN | ||
| Ref− | All Ref− | 80 | 0.90 | 0.04 | 0.06 | 111 | 0.87 | 0.07 | 0.06 |
| Hypothesis-driven | 48 | 0.93 | 0.03 | 0.04 | 71 | 0.88 | 0.05 | 0.07 | |
| Random | 32 | 0.82 | 0.05 | 0.06 | 40 | 0.80 | 0.13 | 0.07 | |
| Ref+ | All Ref+ | 31 | 0.74 | 0.23 | 0.04 | 40 | 0.70 | 0.26 | 0.03 |
| Pindel+Break Dancer | 17 | 0.76 | 0.23 | 0.02 | 20 | 0.71 | 0.28 | 0.01 | |
| Pindel | 8 | 0.79 | 0.14 | 0.07 | 10 | 0.70 | 0.24 | 0.06 | |
| BreakDancer | 6 | 0.67 | 0.31 | 0.02 | 10 | 0.71 | 0.27 | 0.14 | |
Note.—Results are shown for loci that were phylogenetically informative and all loci, that is, those lacking amplicons in more than one sample (All). The number of tested loci (N) and frequency of amplicon size differences that matched the computational prediction (true positives, TP), and false positively (FP) or false negatively (FN) predicted insertions are shown. For Ref− loci, random loci (Random), and loci predicted to support a specific phylogenetic hypothesis (Hypothesis-driven) were selected. For Ref+ markers, all loci were randomly selected.
In all 40 verified Ref+ TE insertion loci, an insertion was present in the polar bear genome, proving the reliability of our approach to select for Ref+ TE insertions. Prediction accuracy for Ref+ insertions in other species was 74% mainly attributed to a higher FPR than in Ref− insertions. A false positive Ref+ TE insertion call means that deletions were not recovered by SV callers, therefore Ref+ FPR should be considered as FNR.
For 111 loci, the PCR amplification yielded an unambiguous phylogenetic informative signal, that is, amplicon size differences with amplification success in all species. For 40 additional loci, one or more individual did not yield a PCR amplicon, and the locus was recorded as inconclusive. For all in vitro validated loci, we identified 17 loci with heterozygous SINE insertions (supplementary table 12, Supplementary Material online). In the brown bear, 17% of the amplified insertions were heterozygous. For the American black, Asiatic black, sun, sloth, and polar bear TE heterozygosity was 6% or less.
Interestingly, two SINE insertions (No. 40 and 122) were present in all ursine species except the polar bear. The flanks around the empty insertion site in the polar bear lack deletions and only the preintegration site was present compared with the other ursine bears. Other validated species-tree incongruent TE insertions (supplementary fig. 11, Supplementary Material online) support alternative tree topologies reflecting the mitochondrial phylogeny or previously identified gene-flow signals from individual gene trees (Yu et al. 2007; Kutschera et al. 2014; Kumar et al. 2017). For example, seven validated TE insertions were synapomorphic for American and Asiatic black bear and nine insertions were shared by Asiatic black bear and sloth bear.
Reconstructing the Phylogeny of Bears
The Ref+ and Ref− TE insertions were merged into a common data set comprised of 150,513 SINE and LINE1 insertions. From these, 71,444 (47.5%) of the TEs were phylogenetically informative and 70,356 (46.7%) were species-specific. We found 8,713 TE insertions being shared by all seven bear species. However, the numbers of shared TE insertions differ when applying maximum parsimony that accounts for missing data (fig. 3).
Fig. 3.

—Dollo parsimony tree of bears reconstructed from 132,039 SINE insertions. Parsimony inferred branch lengths indicate the number of SINE insertions on that branch. Most nodes received bootstrap support of 100% (not indicated). Bootstrap support <100% is shown in red. The rescaled consistency index is 0.567, indicating conflict in the data set.
We identified seven times more insertions of SINEs than LINEs (132,093 and 18,420, respectively) across the bear genomes. The phylogenetic analysis focused on SINE insertions because these are shorter than the mean insert size of the sequencing libraries and thus robustly recovered by TE and SV calling. A Dollo parsimony analysis of 132,093 SINE insertions resulted in a phylogenetic tree with 100% bootstrap support for all nodes, except for the node separating the two polar bear individuals (fig. 3). The tree clearly groups spectacled bears that belong to the family Tremarctinae, outside the ursine bears. Within Ursinae, the tree has two clades that consist of the polar, brown, and American black bear and the Asiatic black, sun and sloth bear, respectively. Sun and sloth bear form a sister group to the Asiatic black bear. Despite, having 100% bootstrap support and branches that are generally supported by several thousand independent SINE insertions, a rescaled consistency index of 0.567 indicated phylogenetic incongruence among the data.
To explore phylogenetic conflict, a network analysis of the same data revealed a tree-like network. Similarly to the Dollo parsimony tree, the network clearly separated the Asiatic black, sloth, and sun bear from the other three ursine bears by a long edge, that represented 3,305 SINE insertions (fig. 4). Still, strong conflict among the Asiatic black, sun, and sloth bear was indicated by an intertwined web, that also included common splits with the polar or brown bear. Polar and brown bear were grouped by an edge that represents 3,597 SINE insertions, but polar bears also shared 2,240 insertions with the American black bear.
Fig. 4.

—Median network from 132,093 SINE insertions. Parallel edges indicate shared splits between species. Major edges are colored, they separate the two major ursine clades (blue), or group together sun and sloth bear (purple), brown bear and polar bear (green) and American black and polar bear (yellow). Edge lengths indicate the number of shared SINE insertions as calculated by SplitsTree 4. For better readability the spectacled bear is not shown.
Phylogenetic conflict can be caused by hybridization or ancient polymorphisms that lead to allele sharing between nonsister group lineages and has been demonstrated for different ursine bears (Kutschera et al. 2014; Kumar et al. 2017). We stringently analyzed the phylogenetic conflict among Asiatic black, sun, and sloth bear using shared SINE insertions obtained from the presence/absence matrix without allowing for any missing data. The Asiatic black bear shares 278 SINE insertions with the sun bear and 265 SINE insertions with sloth bear. The monophyly of sun and sloth bear is supported by 168 SINE insertions. For these three taxa, statistical analyses using the KKSC test (Kuritzin et al. 2016) support the species-tree topology at high significance (bifurcation test, P = 2.325e-10) and reject hybridization between sun bear and the Asiatic black bear (hybridization test, P = 0.6060, supplementary table 13, Supplementary Material online). For the American and Asiatic black bear, 129 shared SINE insertions were recovered (fig. 5b), however the statistical significance of this result could not be assessed with existing methods. The monophyly of polar and brown bear is supported by 3,160 SINE insertions and the species-tree topology of polar, brown, and American black bear is significantly supported (tree test, P = 1.04e-159). The monophyly of all three species is supported by 2,178 SINE insertions (supplementary fig. 12, Supplementary Material online).
Fig. 5.
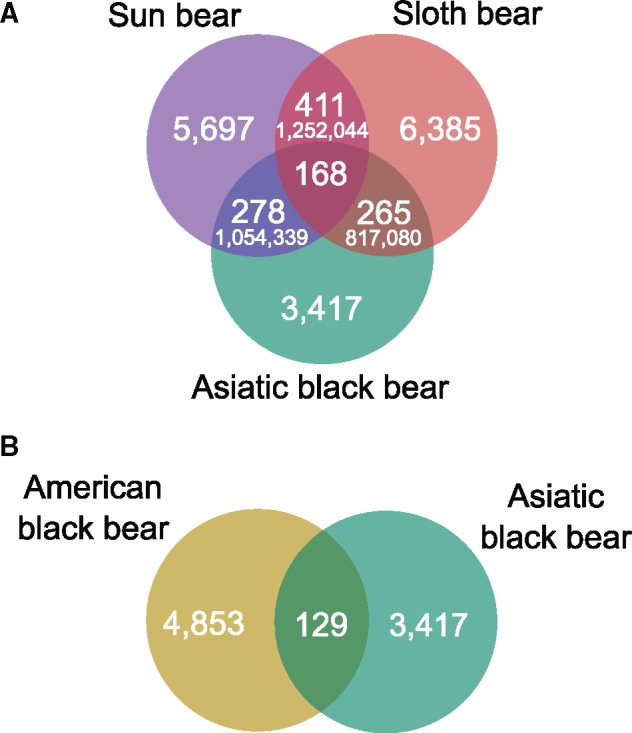
—Venn Diagrams depicting phylogenetic conflict among Asiatic black, sun, and sloth bear (a) and American black and Asiatic black bear (b). The amount of shared SINE insertions under Dollo parsimony are shown. The numbers in smaller font give the number of shared genome-wide nucleotide substitutions calculated using the D-statistics (Kumar et al. 2017).
Different Extent of Phylogenetic Signal in the Flanking Regions
Alignments of genomic sequences flanking phylogenetically informative TE insertion sites were analyzed for their phylogenetic signal as well as for congruence with the phylogenetic signal from the adjacent TE insertion. Up to 65% of the individual ML trees calculated from the flanking sequences were identical with the presence/absence pattern of the TE insertion (fig. 6). To investigate the spatial congruence between the TE insertion and its flanks in more detail, we measured the number of substitutions that reconstructed the same phylogeny as the TE insertion in 1-kb nonoverlapping windows extending up to 10 kb from the insertion site (fig. 6). TE supporting substitutions were elevated in the direct vicinity of the TE insertion site and then tapered off with distance from the insertion site. The frequency of supporting substitutions is highest at TE insertion sites that are congruent with the ursine species tree and lower for those with a conflicting signal. For example, among 215 orthologous TE insertions shared by all Asiatic bears, the average frequency of TE-supporting substitutions increased from 0.01 to 0.04 within the first 5 kb from both sides of the insertion site (fig. 6). For species-tree incongruent TE insertion loci, the elevation of TE-supporting substitutions was less pronounced and the stretch of spatial congruence was shorter. Substitution frequencies for phylogenies that are different to the TE insertion signal were generally not elevated toward the insertion site (supplementary fig. 13, Supplementary Material online). In cases of only a minor difference in the phylogenetic signal between substitutions and TE, substitution frequencies were increased (supplementary note 2, Supplementary Material online).
Fig. 6.

—Analysis of flanking sequences of TE insertions present in different groups of taxa. Left panel: Green branches in the phylogenetic tree indicate when the TEs integrated. Middle panel: Bar plots showing the frequency of ML-trees calculated from 10-kb flanking sequence on the 5′, 3′ end or a concatenation of both. Right panel: Frequency of substitutions that support the TE insertion signal in 1-kb windows around the insertion site. Frequencies are normalized by the number of segregating sites.
Discussion
Analyzing whole genome sequence data for TE insertions makes it possible to study the landscape of genetic variation at unprecedented extent and detail. However, it faces methodological challenges. Here, we developed the TeddyPi pipeline that integrates different available TE callers and applies stringent filtering to overcome limitations of TE calling. It produces an automated output of presence/absence tables of TE insertions that can be immediately used for phylogenetic analyses. The pipeline follows a “quality over quantity” approach to select highly reliable TE insertion loci. Recent phylogenomic studies suggest that genomes are often a mosaic of different genealogies caused by evolutionary processes such as introgressive hybridization or ILS (Mallet et al. 2016). To study such complex signals, sufficient character sampling is necessary. This can only be achieved by nucleotide-based genome analyses, or genome-wide and unbiased discovery of TE insertions (Kuritzin et al. 2016; Dodt WG, Gallus S, Matthew PJ, Nilsson MA, Unpublished data). TE insertion data provide an independent and robust molecular marker system to build phylogenies that are not based on sequence analysis (Shedlock et al. 2004).
SINE Insertions Recapitulate the Evolutionary History of Bears
Extensive phylogenetic discordance across loci has previously challenged the resolution of the bear phylogeny (Yu et al. 2007; Kutschera et al. 2014; Kumar et al. 2017). The TeddyPi pipeline extracted 132,093 SINE insertions from low-coverage data to build a reliable data set of phylogenetically informative TE markers to study the evolutionary history of bears. We reconstructed a well-supported phylogenetic species tree despite incongruent phylogenetic signals (figs. 3and 4). The three Asian bears form a clade that is consistent with coalescent analyses of genome sequence data (Kumar et al. 2017). However, this contrasts with previous studies, that placed the Asiatic black bear as sister group to the polar, brown, and American black bear clade or as sister group to the American black bear, respectively (Yu et al. 2007; Krause et al. 2008; Pagès et al. 2008). Despite significant bootstrap support for each node of the parsimony TE tree, the tree had a low-consistency index, indicating that many TE insertions conflict with the inferred phylogeny. Phylogenetic networks can depict such conflicting signals better than trees that force the data to a bifurcating model of evolution (Bapteste et al. 2013). The network analyses reveal that phylogenetic conflict among bears occurs mostly in the two main clades of the ursine subfamily (fig. 4). In particular, the Asiatic black, sun, and sloth bear that currently inhabit South-East Asia form a complex network. We explored this conflict further and found that the Asiatic black bear share almost identical numbers of orthologous SINE insertions with sun and sloth bear, respectively, thereby indicating ILS as the origin of the conflict (fig. 5 and supplementary table 13, Supplementary Material online). Despite reconstructing the same species tree, our detailed analyses contrast nucleotide-based analyses of millions of sites, that inferred ancestral hybridization as the main driver of phylogenetic conflict among these species (Kumar et al. 2017). To what extent hybridization occurred between bears and what caused the conflicting signal of single nucleotide substitutions and TE insertions remains to be further explored.
In previous mtDNA-based analyses, the Asiatic and American black bear have been placed as sister species (Yu et al. 2007; Krause et al. 2008). This is not supported by the majority of identified TE insertions. However, 129 SINE insertions are shared by American and Asiatic black bear (fig. 5). Therefore, the close relationship of the two black bears based on mtDNA analyses is likely a result of an ancient mitochondrial capture event and additional introgression of nuclear DNA carrying these TE insertions (Kutschera et al. 2014). An alternative scenario explaining the discordance between mtDNA and nuclear DNA phylogenies of American and Asiatic black bear involves nuclear swamping of the American black bear genome by brown bear alleles. In this scenario, the mitochondrial phylogeny reflects the true speciation history but was eventually obfuscated by introgression of brown bear DNA into the American black bear genome. This would produce a similar phylogenetic signal and artificially place the American black bear on the lineage leading to brown and polar bear (Kutschera et al. 2014). However, our network analysis and 99 SINE insertions shared by brown bear and American black bear give very little support for this hypothesis, suggesting that ancient hybridization between the two black bear species had a more pronounced effect on their genomes than nuclear swamping by brown bear DNA (fig. 3 and supplementary fig. 12, Supplementary Material online). If the phylogenetic conflict during the initial radiation of Ursinae was caused by ILS, approximately equal number of TE insertions supporting different evolutionary scenarios were expected. This is not evident from our analyses.
Differences in retrotransposition activity or demographic history can cause varying rates of TE insertions between lineages (Hormozdiari et al. 2013). Our insertion rate estimates were 0.022 SINE and 0.004 LINE1 insertions per genome per generation, which is half of the rate for humans (0.035 Alus and 0.008 LINE1s) (fig. 2c;Sudmant et al. 2015). Fixation of neutral or slightly deleterious TE insertions depends on genetic drift, which is stronger in small effective population sizes or on purifying selection, which is stronger in large populations (Charlesworth 2009; Gonzalez and Petrov 2012). The substantially higher insertion rates of TEs and the high heterozygosity rate in brown bear can thus be explained by the large population size that brown bears have maintained over long timespans (Miller et al. 2012). Polar bears exhibit a low heterozygosity of TE insertion, reflecting the species low genetic diversity as consequence of population bottlenecks (Hailer et al. 2012; Miller et al. 2012). In polar and brown bear, TE insertion rates are higher than in the other bears. The high TE insertion rate in polar and brown bears can also be explained by a retrotranspositional burst caused by hybridization (O’Neill et al. 1998; Dion-Côté et al. 2014). Bears are known to hybridize, and hybrids between polar and brown bears have been observed (Galbreath et al. 2008; Kelly et al. 2010). Additionally, a hybrid origin of polar bears has been proposed (Lan et al. 2016). Thus, consequent genetic introgression potentially leads to a burst of TE insertions in the species into which hybrids backcross and thus may explain the high TE insertion rate in brown and polar bears. This indicates, that there is no general genomic mechanism to suppress genomic insertions as was suggested by the absence of mitochondrial pseudogene insertions (Lammers et al. 2017).
The accompanying sequence-based analyses of the same data set enabled to examine the correlation of nucleotide substitutions and TEs for conflicting phylogenies (Kumar et al. 2017). Expectedly, TE insertions were several magnitudes less frequent than nucleotide substitutions. Yet, both analyses yielded the same phylogeny but differed in their interpretation of phylogenetic conflict (fig. 5). Huff et al. (2010) described that TE(s) have on an average older genealogies due to the rarity of TE insertion compared to the nucleotide mutation rate. Thus, TE loci have deeper coalescence times and a higher probability for ILS (Maddison 1997). Also, introgression of alleles carrying TE insertions might be less frequent because they can be deleterious due to genetic incomparability, that is, Dobzhansky–Muller incompatibilities (Dobzhansky 1941; Muller 1942). This highlights the need for nucleotide-based analyses in addition to genome wide analyses of TE insertions.
Quality over Quantity Approach for Phylogenetic Inference of TEs
Previous phylogenetic TE analyses relied on the availability of reference genomes which were often restricted to one species per order or family. For bears, draft genome assemblies of polar bear and giant panda are available (Li et al. 2010; Liu et al. 2014). Traditional in vitro approaches would have identified orthologous loci in both genomes, with one carrying a TE insertion that is experimentally tested for presence or absence in the other bears using PCR (Shedlock et al. 2004). Although the availability of two reference genomes is beneficial, unbiased identification of variable, that is, phylogenetically informative TEs across the complete taxon-sampling is not possible using this approach. Adding genomes from the entire ursine subfamily makes it possible to discover TE insertions free from sampling artifacts and to precisely extract phylogenetically informative markers. However, the nested position of the polar bear reference genome inside the species tree, the use of low-coverage genome data and misassembled regions in the reference genome were challenging for TE calling and required methodological refinements to increase prediction quality of TE insertions. These challenges were rarely discussed in other studies but are central when aiming for a large-scale identification of TE insertions from paired-end mapping data without introducing a sampling bias.
If the reference genome is nested inside the ingroup, as in the case of the polar bear inside Ursinae, a two-sided approach using Ref+ and Ref− insertions is necessary to yield support for all internodes in the resulting phylogenetic tree or network (supplementary fig. 2, Supplementary Material online). The polar bear genome sequence has a higher contiguity than that of the giant panda, a better assembly of repeats due to longer sequencing reads and it benefits from the low heterozygosity in polar bear. Compared with the polar bear reference genome, the giant panda genome is less suited to be used as a reference for mapping because of its high evolutionary distance to the other bear species, which diverged from the giant panda some 20 Ma. To solve this problem and to make TeddyPi more ubiquitously applicable, SV callers were integrated in the pipeline to deduce Ref+ insertions from deletions calls (Nellåker et al. 2012). Only few TE callers are specifically developed to detect Ref+ insertions. To our knowledge, only T-lex and T-lex2 (Fiston-Lavier et al. 2011, 2015) perform Ref+ insertion detection, but they are not compatible with the TeddyPi pipeline due to different file format requirements. Other programs, such as RetroSeq, Mobster and Jitterbug exclusively detect Ref− TE insertions (Keane et al. 2013; Thung et al. 2014; Hénaff et al. 2015). Depending on the mapping-signature utilized for SV-calling (split-reads, read-pairs, depth of coverage) detection results differed markedly between programs as exemplified by our results from Pindel and Breakdancer (supplementary tables 9 and 10, Supplementary Material online) and by results from other studies (Ewing 2015). Inconsistencies between different programs will affect the phylogenetic inference, which relies on precise presence/absence patterns of orthologous loci and make it necessary to integrate different SV callers as implemented in TeddyPi. Despite the general concordance of TE calls from Mobster and RetroSeq, only overlapping calls were used to increase the reliability of the calls. For TE calling, integration of multiple callers is recognized as an appropriate strategy to enhance the consistency of TE predictions (Lin et al. 2015; Nelson et al. 2017), and this functionality is implemented in TeddyPi for both, Ref+ and Ref− insertions. A true positive rate (TPR) of 93% for TE calls from the TeddyPi pipeline (table 1) is higher than the estimated sensitivity of RetroSeq for 10× whole genome sequencing data (Keane et al. 2013). The reliability of TeddyPi is equally good as estimates from Mobster analyses of high-quality human data (Thung et al. 2014). The false positive rate for Ref− TE calls is low (4%), but considerably higher for Ref+ insertions (23%). Thus, when possible, the use of a suitable outgroup genome to analyze only Ref− insertions for phylogenetic reconstruction is recommended.
Detecting TE insertions and SVs in resequenced whole genome data often have breakpoint inaccuracies within a margin of up to 50 bp (Ewing 2015). It is therefore not possible to distinguish between near or near-exact deletion or insertions. This can affect detecting ortholog events or analyzing genetic effects by intersection with coding sequences or regulatory regions (supplementary fig. 7, Supplementary Material online). Given the short length of regulatory sequences an overestimation of disrupting TE insertions can not be excluded. However, breakpoint inaccuracies are unlikely to have affected the detection of orthologous TE insertions because long near-exact indels occur at a very low level (van de Lagemaat et al. 2005). Therefore, they would have contributed only marginally to the observed phylogenetic conflict among bears.
Missing data and unplaced scaffolds are common in most genome assemblies, because of current technological limitations to sequence and assemble repetitive DNA. Thus, in genome sequences, sequence gaps are mostly caused by repetitive regions, such as TEs and satellite DNA. Long read sequencing technologies, such as PacBio or Nanopore, are expected to alleviate this problem considerably. The 2.3 Gb polar bear genome sequence was based on short read technology and based on an estimated genome size of 2.7 Gb for extant bears lacks 400 Mb of genomic information (Vinogradov 1998; Krishan et al. 2005; Liu et al. 2014). Another artifact from repetitive DNA in genome sequences are unassembled regions in the scaffolds (N-regions). TeddyPi utilized 38 Mb of N-regions in the polar bear genome as a proxy for poorly assembled regions, and all TE calls in their vicinity were excluded from the analyses. The removal of N-regions greatly increased the success rates in the experimental validation and show that this is a necessary step in TE calling, that previously has not been implemented. Another indicator of assembly quality and of the ability to confidently predict TEs is the mappability (or uniqueness) of short-reads to the reference genome. Mappability can be assessed by deviations of local coverage depth from the mean coverage. To account for poorly mapped regions, TE calls in regions of exceptionally low- and high coverage were coded as missing data. Another challenge to TE and SV calling comes from the random integration of TEs in the genome. Occasionally, TEs can randomly integrate into older TE sequences. If both TEs are of the same type, sequence reads will be ambiguously mapped to either the young or old TE. This increases the risk for false positive calls during TE calling. Therefore, TE calls located within annotated TEs of the same type were removed in the TeddyPi pipeline to increase the reliability of our phylogenetic markers.
Unlike for the human genome, a generally accepted standard or database of TE insertions does not exist for nonmodel organisms to compare our results to. Thus, detection sensitivity can only be estimated by experimental approaches. The validation experiments show that compared with standard TE callers, the rigorous approach of the TeddyPi pipeline substantially improves TE detection from nonmodel organism genomes that lack highly curated and well-annotated genome assemblies. For the polar bear genome sequence, every experimentally verified locus was confirmed for the presence of SINEC1_Ame, corroborating the assembly and RepeatMasker annotation for these loci. The presence of TSDs in all analyzed loci further strengthens the TeddyPi approach in identifying true, orthologous TE insertion events.
TE Insertions, Flanking Sequences, and Recombination Blocks in Ursine Bears
TE insertions share an evolutionary history with nucleotide substitutions occurring in their immediate genomic vicinity (Daly et al. 2001). If the TE insertion is neutral, the extent of linkage, that is, the size of a recombination block that carries the TE depends on the recombination rate and the demographic history of the genomic region (Ellegren and Galtier 2016). In great apes, phylogenetic congruence between the TE insertion and its flanking sequence was used to prove hemiplasy of the TE insertion (Hormozdiari et al. 2013), however nucleotide-homoplasy and uncertainties in tree-reconstruction of the specific regions can mislead such an analysis, especially for longer timescales (Suh et al. 2015). Ursine bears radiated ∼5 Ma, which left little time for flanking sequences to be saturated, allowing for nucleotide level comparisons. In bears, TE insertions and their flanking sequences share the same phylogenetic signal, but the extent of spatial congruence (i.e., linkage) is limited to a few kb and differs depending on the phylogenetic signal of the TE (fig. 6 and supplementary fig. 12, Supplementary Material online). The size of the recombination block, as evident from the extent of spatial congruence (fig. 6), gives a relative estimate of the time since the TE insertions. A lesser extent of spatial congruence around the species-tree incongruent TE insertions can be explained by an earlier TE integration and subsequent breakdown of the recombination blocks. TE insertions shared exclusively by American and Asiatic black bear have a narrow extent of spatial congruent substitutions, and thus are older than species-tree congruent TE insertions. If a locus originates from more recent introgression a wider extent of spatial congruence carrying the same phylogenetic signal is expected. The flanks of the orthologous TE insertions in the Asiatic bears share the same phylogenetic signal, and therefore show no homoplasy and suggest that ILS has contributed to the phylogenetic incongruence among these loci. For the Asiatic bears, we propose that ILS is the primary driver of phylogenetic incongruence causing high amounts of pairwise similarities (fig. 5a; Kutschera et al. 2014) and additionally, hybridization between Asiatic black and sun bear led to an excess of shared alleles between these species (fig. 5a; Kumar et al. 2017). Under the assumption that the current species tree of bears (fig. 3) reflects the speciation history, introgressive hybridization involving the American black bear must have occurred. However, in agreement with coalescent-based analyses (Kutschera et al. 2014), analyses of TE insertion patterns and their flanking regions (figs. 5and 6) indicate that the local geneologies are not yet sorted, thereby confounding introgression analyses. Although our sequence analyses of the TE flanking regions were restricted to one taxonomic group, it is evident that analyses of deeper divergences in any taxa will have shorter recombination blocks and thus fewer phylogenetic signatures. Thus, screening for flanking substitutions surrounding old TE insertions is likely to be uninformative due to the limited spatial congruence coupled with nucleotide saturation.
Conclusion
Twenty years after the successful introduction of TE insertions as phylogenetic markers, it is now possible to not only use a few but thousands of informative loci across the genome to reconstruct phylogenies of complete taxonomic groups. The TeddyPi pipeline allowed us to detect TE insertion in silico from nine bear genomes. Over 130,000 SINE insertions show that TE insertions are a major driver of genomic variation among ursine bears and reconstructed their phylogeny with virtually homoplasy-free evolutionary information. The TE phylogeny of bears confirm the presence of two distinct clades among Ursinae and significantly shows that Asiatic black, sun, and sloth bear form a monophyletic clade, despite a high degree of ILS. The conceptual framework of the integrated and stringent approach in TeddyPi allows an unbiased analysis of ancestry-informative TEs as a routine procedure in comparative genomic studies. Deciphering recent and complex speciation processes using TE insertions as well as nucleotide substitutions is subject to further analyses and important for our understanding of phylogenetics and speciation (Mallet et al. 2016).
Data Availability
The final TE data set, and primers for validation experiments are included as Supplementary Material online. TeddyPi is available at https://github.com/mobilegenome/teddypi, last accessed September 2017.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors thank Dije Tjwan Thung and Jayne Hehir-Kwa, The Radboud University Medical Center, for providing an unpublished version of Mobster, Thomas Keane, Wellcome Trust Sanger Institute, for advice in using RetroSeq, and Markus Pfenninger for helpful discussions. The authors are thankful to Kathinka Schulze and Clara Heumann-Kieser for performing validation experiments, Alison Eyres for English proof-reading, and five anonymous reviewers for helpful comments on earlier versions of this manuscript. Jón Baldur Hlíðberg (www.fauna.is) painted the bears in figure 3. The publication of this article was funded by the Open Access Fund of the Leibniz Association.
Author Contributions
F.L., M.A.N., and A.J. conceived and designed the study. F.L. developed TeddyPi and performed the computational analyses. S.G. and M.A.N. coordinated and performed experimental validation experiments. F.L. and M.A.N. wrote the manuscript with input from all co-authors. All authors read and approved the final manuscript.
Literature Cited
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Bapteste E, et al. 2013. Networks: expanding evolutionary thinking. Trends Genet. 29(8):439–441. [DOI] [PubMed] [Google Scholar]
- Bidon T, et al. 2015. Genome-wide search identifies 1.9 Mb from the Polar Bear Y chromosome for evolutionary analyses. Genome Biol Evol. 7(7):2010–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta E, Gonzalez J.. 2013. The impact of transposable elements in environmental adaptation. Mol Ecol. 22(6):1503–1517. [DOI] [PubMed] [Google Scholar]
- Charlesworth B. 2009. Effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 10(3):195–205. [DOI] [PubMed] [Google Scholar]
- Chen K, et al. 2009. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6(9):677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churakov G, et al. 2009. Mosaic retroposon insertion patterns in placental mammals. Genome Res. 19(5):868–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Batzer MA.. 2009. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 10(10):691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin MA, Amstrup SC, Talbot SL, Sage GK, Amstrup KS.. 2009. Genetic variation, relatedness, and effective population size of polar bears (Ursus maritimus) in the southern Beaufort Sea, Alaska. J Hered. 100(6):681–690. [DOI] [PubMed] [Google Scholar]
- Dale RK, Pedersen BS, Quinlan AR.. 2011. Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics 27(24):3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES.. 2001. High-resolution haplotype structure in the human genome. Nat Genet. 29(2):229–232. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Brinkmann H, Philippe H.. 2005. Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet. 6(5):361–375. [DOI] [PubMed] [Google Scholar]
- Dion-Côté A-M, Renaut S, Normandeau E, Bernatchez L.. 2014. RNA-seq reveals transcriptomic shock involving transposable elements reactivation in hybrids of young lake whitefish species. Mol Biol Evol. 31(5):1188–1199. [DOI] [PubMed] [Google Scholar]
- Dobzhansky T. 1941. Genetics and the origin of species. 2nd ed West Sussex: Columbia University Press. [Google Scholar]
- Doucet AJ, Droc G, Siol O, Audoux J, Gilbert N.. 2015. U6 snRNA pseudogenes: markers of retrotransposition dynamics in mammals. Mol Biol Evol. 32(7):1815–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EY, Patterson N, Reich D, Slatkin M.. 2011. Testing for ancient admixture between closely related populations. Mol Biol Evol. 28(8):2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Galtier N.. 2016. Determinants of genetic diversity. Nat Rev Genet. 17(7):422–433. [DOI] [PubMed] [Google Scholar]
- Ewing AD. 2015. Transposable element detection from whole genome sequence data. Mob DNA 6:24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiston-Lavier A-S, Barron MG, Petrov DA, Gonzalez J.. 2015. T-lex2: genotyping, frequency estimation and re-annotation of transposable elements using single or pooled next-generation sequencing data. Nucleic Acids Res. 43(4):e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiston-Lavier A-S, Carrigan M, Petrov DA, González J.. 2011. T-lex: a program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 39(6):e36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbreath GJ, Hunt M, Clements T, Waits LP.. 2008. An apparent hybrid wild bear from Cambodia. Ursus 19(1):85–86. [Google Scholar]
- Gonzalez J, Petrov DA.. 2012. Evolution of genome content: population dynamics of transposable elements in flies and humans In: Anisimova M, editor. Evolutionary genomics: statistical and computationla methods. Vol. 855 New York: Springer-Humana, p. 361–383. [DOI] [PubMed] [Google Scholar]
- Hailer F, et al. 2012. Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science 336(6079):344–347. [DOI] [PubMed] [Google Scholar]
- Hallström BM, Janke A.. 2010. Mammalian evolution may not be strictly bifurcating. Mol Biol Evol. 27(12):2804–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hénaff E, Zapata L, Casacuberta JM, Ossowski S.. 2015. Jitterbug: somatic and germline transposon insertion detection at single-nucleotide resolution. BMC Genomics 16:768.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof AEV, et al. 2016. The industrial melanism mutation in British peppered moths is a transposable element. Nature 534(7605):102–105. [DOI] [PubMed] [Google Scholar]
- Hormozdiari F, et al. 2013. Rates and patterns of great ape retrotransposition. Proc Natl Acad Sci. 110(33):13457–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Serra F, Bork P.. 2016. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol Biol Evol. 33(6):1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff CD, Xing J, Rogers AR, Witherspoon D, Jorde LB.. 2010. Mobile elements reveal small population size in the ancient ancestors of Homo sapiens. Proc Natl Acad Sci U S A. 107(5):2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Bryant D.. 2006. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 23(2):254–267. [DOI] [PubMed] [Google Scholar]
- Jurka J, et al. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110(1–4):462–467. [DOI] [PubMed] [Google Scholar]
- Keane TM, Wong K, Adams DJ.. 2013. RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics 29(3):389–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly BP, Whiteley A, Tallmon D.. 2010. The Arctic melting pot. Nature 468(7326):891.. [DOI] [PubMed] [Google Scholar]
- Krause J, et al. 2008. Mitochondrial genomes reveal an explosive radiation of extinct and extant bears near the Miocene-Pliocene boundary. BMC Evol Biol. 8:220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegs JO, et al. 2006. Retroposed elements as archives for the evolutionary history of placental mammals. PLoS Biol. 4(4):537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishan A, et al. 2005. DNA index, genome size, and electronic nuclear volume of vertebrates from the Miami Metro Zoo. Cytometry A 65A(1):26–34. [DOI] [PubMed] [Google Scholar]
- Kumar V, et al. 2017. The evolutionary history of bears is characterized by gene flow across species. Sci Rep. 7:46487.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto T, Nishihara H, Watanabe M, Okada N.. 2015. Determining the position of storks on the phylogenetic tree of waterbirds by retroposon-insertion analysis. Genome Biol Evol. 7(12):3180.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuritzin A, Kischka T, Schmitz J, Churakov G.. 2016. Incomplete lineage sorting and hybridization statistics for large-scale retroposon insertion data. PLoS Comput Biol. 12(3):e1004812.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutschera VE, et al. 2014. Bears in a forest of gene trees: phylogenetic inference is complicated by incomplete lineage sorting and gene flow. Mol Biol Evol. 31(8):2004–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers F, Janke A, Rücklé C, Zizka V, Nilsson MA.. 2017. Screening for the ancient polar bear mitochondrial genome reveals low integration of mitochondrial pseudogenes (numts) in bears. Mitochondrial DNA B 2:251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T, et al. 2016. Genome-wide evidence for a hybrid origin of modern polar bears. BioRxiv. doi.org/10.1101/047498.
- Li H, Durbin R.. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, et al. 2010. The sequence and de novo assembly of the giant panda genome. Nature 463(7279):311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Smit S, Bonnema G, Sanchez-Perez G, de Ridder D.. 2015. Making the difference: integrating structural variation detection tools. Brief Bioinform. 16(5):852–864. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438(7069):803–819. [DOI] [PubMed] [Google Scholar]
- Liu S, et al. 2014. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157(4):785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison WP. 1997. Gene trees in species trees. Syst Biol. 46(3):523–536. [Google Scholar]
- Mallet J, Besansky N, Hahn MW.. 2016. How reticulated are species? BioEssays 38(2):140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev P, Stanciu M, Brudno M.. 2009. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods 6:13–20. [DOI] [PubMed] [Google Scholar]
- Miller W, et al. 2012. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc Natl Acad Sci. 109:E2382–E2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1942. Isolating mechanisms, evolution and temperature. Biol Symp. 6:71–125. [Google Scholar]
- Nellåker C, et al. 2012. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 13(6):R45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MG, Linheiro RS, Bergman CM.. 2017. McClintock: an integrated pipeline for detecting transposable element insertions in whole genome shotgun sequencing data. G3 Genes Genomes Genet. 7:2763–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido M, Rooney AP, Okada N.. 1999. Phylogenetic relationships among cetartiodactyls based on insertions of short and long interpersed elements: hippopotamuses are the closest extant relatives of whales. Proc Natl Acad Sci. 96(18):10261–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Maruyama S, Okada N.. 2009. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc Natl Acad Sci. 106(13):5235–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill RJ, O’Neill MJ, Graves JA.. 1998. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature 393:68–72. [DOI] [PubMed] [Google Scholar]
- Onorato DP, Hellgren EC, Van Den Bussche RA, Doan Crider DL.. 2004. Phylogeographic patterns within a metapopulation of black bears (Ursus americanus) in the American southwest. J Mammal. 85(1):140–147. [Google Scholar]
- Pagès M, et al. 2008. Combined analysis of fourteen nuclear genes refines the Ursidae phylogeny. Mol Phylogenet Evol. 47(1):73–83. [DOI] [PubMed] [Google Scholar]
- Platt RN, et al. 2015. Targeted capture of phylogenetically informative ves SINE insertions in genus Myotis. Genome Biol Evol. 7(6):1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontius JU, et al. 2007. Initial sequence and comparative analysis of the cat genome. Genome Res. 17(11):1675–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DA, Xing J, Salem A-H, Batzer MA.. 2006. SINEs of a nearly perfect character. Syst Biol. 55(6):928–935. [DOI] [PubMed] [Google Scholar]
- Shedlock AM, Takahashi K, Okada N.. 2004. SINEs of speciation: tracking lineages with retroposons. Trends Ecol Evol. 19(10):545–553. [DOI] [PubMed] [Google Scholar]
- Shimamura M, et al. 1997. Molecular evidence from retroposons that whales form a clade within even-toed ungulates. Nature 388(6643):666–670. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526(7571):75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh A, Kriegs JO, Donnellan S, Brosius J, Schmitz J.. 2012. A universal method for the study of CR1 retroposons in nonmodel bird genomes. Mol Biol Evol. 29(10):2899–2903. [DOI] [PubMed] [Google Scholar]
- Suh A, Smeds L, Ellegren H.. 2015. The dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol. 13(8):e1002224.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran J, Holder MT.. 2010. DendroPy: a Python library for phylogenetic computing. Bioinformatics 26(12):1569–1571. [DOI] [PubMed] [Google Scholar]
- Swofford D. 2002. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland, Massachusetts: Sinauer Associates.
- Tallmon DA, Bellemain E, Taberlet P, Swenson JE.. 2004. Genetic monitoring of Scandinavian brown bear effective population size and immigration.DeWoody, editor. J Wildl Manage. 68:960–965. [Google Scholar]
- Thung DT, et al. 2014. Mobster: accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 15(10):488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, et al. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40(15):e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Lagemaat LN, Gagnier L, Medstrand P, Mager DL.. 2005. Genomic deletions and precise removal of transposable elements mediated by short identical DNA segments in primates. Genome Res. 15(9):1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov AE. 1998. Genome size and GC-percent in vertebrates as determined by flow cytometry: the triangular relationship. Cytometry 31(2):100–109. [DOI] [PubMed] [Google Scholar]
- Walters-Conte KB, Johnson DLE, Allard MW, Pecon-Slattery J.. 2011. Carnivore-specific SINEs (Can-SINEs): distribution, evolution, and genomic impact. J Hered. 102(Suppl 1):S2–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Keane TM, Stalker J, Adams DJ.. 2010. Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol. 11(12):R128.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z.. 2009. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25(21):2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Li Y-W, Ryder OA, Zhang Y-P.. 2007. Analysis of complete mitochondrial genome sequences increases phylogenetic resolution of bears (Ursidae), a mammalian family that experienced rapid speciation. BMC Evol Biol. 7:198.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The final TE data set, and primers for validation experiments are included as Supplementary Material online. TeddyPi is available at https://github.com/mobilegenome/teddypi, last accessed September 2017.