


Abstract
RNA-binding proteins (RBPs) play an important role in RNA post-transcriptional regulation and recognize target RNAs via sequence-structure motifs. The extent to which RNA structure influences protein binding in the presence or absence of a sequence motif is still poorly understood. Existing RNA motif finders either take the structure of the RNA only partially into account, or employ models which are not directly interpretable as sequence-structure motifs. We developed ssHMM, an RNA motif finder based on a hidden Markov model (HMM) and Gibbs sampling which fully captures the relationship between RNA sequence and secondary structure preference of a given RBP. Compared to previous methods which output separate logos for sequence and structure, it directly produces a combined sequence-structure motif when trained on a large set of sequences. ssHMM’s model is visualized intuitively as a graph and facilitates biological interpretation. ssHMM can be used to find novel bona fide sequence-structure motifs of uncharacterized RBPs, such as the one presented here for the YY1 protein. ssHMM reaches a high motif recovery rate on synthetic data, it recovers known RBP motifs from CLIP-Seq data, and scales linearly on the input size, being considerably faster than MEMERIS and RNAcontext on large datasets while being on par with GraphProt. It is freely available on Github and as a Docker image.
INTRODUCTION
RNA-binding proteins (RBPs), a class of proteins able to bind RNA molecules, play a vital role in processes such as RNA localization, RNA editing, RNA stability and splicing (1). In human cells, hundreds of RBPs have been discovered but the detailed functions of only a few have been explored so far (1,2). RBPs are known to recognize RNA molecules by their nucleotide sequence as well as their three-dimensional structure. Moreover, it has been found that many RBPs prefer binding to RNAs in specific structural contexts (Figure 1) (3,4). To characterize the function of an RBP, it is crucial to first identify its interaction partners, i.e. the regulated gene transcripts. In most cases, the RNA targets of an RBP share at least one common local sequence or structure preference—a so-called motif, which fits into the binding pocket of the protein and thus facilitates the recognition of the RNA by the protein.
Figure 1.

Visual representation of an RNA structure. The five structural contexts are represented by five colors: stems (red), exterior loops (light green), hairpin loops (purple), internal loops (blue), and multiloops (dark green). Figure adapted from (3).
Several approaches for motif finding, i.e. computationally extracting an unknown sequence motif from a set of target sequences, have been developed for transcription factor (TF) binding sites in DNA sequences. They can be categorized into four major classes (5): (i) enumerative algorithms count the occurrences of exact k-mers in a sequence set to find over-represented words (6–8); (ii) algorithms based on expectation maximization (EM) simultaneously optimize a position weight matrix (PWM) description of a sequence motif (9) and probabilities of motif starts in the associated sequences. A popular implementation of the EM algorithm is the MEME software (10); (iii) Algorithms based on probabilistic optimization, such as Gibbs sampling, iteratively sample from the conditional distribution of one motif start at a time (11); (iv) affinity-based (motif) models parametrize and fit a function representing the binding affinity of a protein (e.g. a TF) for a set of words (12–14).
Compared to experimental methods for detecting TF binding sites on DNA, high-throughput protocols for protein-RNA interactions are relatively new. Among them, in-vitro evolutionary methods, such as SELEX (15) and RNAcompete (16), identify high-affinity RNA ligands within pools of randomly or specifically selected sequences. Alternatively, various crosslinking and immunoprecipitation (CLIP) methods have been introduced (17–19), which rely on covalent crosslinking of an RBP to its RNA target in living cells, followed by isolation of RBP-RNA fragments and deep sequencing. The RNA sequences (reads) produced by CLIP-Seq protocols can be mapped back to the genome, and peak calling tools, such as Piranha (20) or PARalyzer (21), can be used to identify high-fidelity RBP binding sites from the read levels.
Although much work has been done in the area of DNA motif finding, few approaches have been developed for RNA motifs (Table 1). One of the reasons is that the binding of RNA depends not only on the RNA’s nucleotide sequence but also on its 3D structure. Consequently, motif finders that work well for finding sequence motifs in linear DNA cannot easily be applied to RNA, but have to be extended to take the RNA secondary structure into account. This is a difficult task due to the noisy nature of computational RNA secondary structure prediction, the fuzzy patterns of sequence-structure motifs, the large set of input sequences where the motif could possibly be contained, and the potentially large number of false positives among the RBP binding sites called from CLIP-Seq experiments (22).
Table 1. A selection of RNA motif finding algorithms.
| Motif finder | Operating principle | Structure incorporated? | Reference |
|---|---|---|---|
| Oligo-Analysis | Enumeration | No | van Helden et al. (23) |
| cERMIT | Enumeration | No | Georgiev et al. (8) |
| AptaTRACE | Enumeration | Sequence and structure modeled separately | Dao et al. (24) |
| - | Suffix trie | No | Brāzma et al. (25) |
| MEME | EM | No | Bailey et al. (26) |
| MatrixREDUCE | Least-squares fit | No | Foat et al. (27) |
| MotifSampler | Gibbs sampling | No | Thijs et al. (28) |
| BioProspector | Gibbs sampling | No | Liu et al. (29) |
| GibbsST | Gibbs sampling | No | Shida et al. (30) |
| MEMERIS | EM | Single-strandedness guides sequence motif finding | Hiller et al. (31) |
| Zagros | EM | Sequence and pairedness modeled together | Bahrami-Samani et al. (32) |
| RNAcontext | Limited-memory BFGS | Sequence and structure modeled separately | Kazan et al. (4) |
| GraphProt | Graph/SVM | Sequence and structure modeled as hypergraph | Maticzka et al. (33) |
The third column indicates whether the motif finder incorporates RNA secondary structure in any way.
Existing RNA motif finders either address only part of the problem or employ machine learning models which are harder to interpret in terms of sequence-structure preferences (Table 1). The first tool to incorporate RNA secondary structure into motif prediction was MEMERIS (31), an extension of the MEME EM algorithm. It uses single-strandedness information as a prior to guide motif finding to single-stranded regions based on the assumption that most RBPs prefer to bind in single-stranded regions. However, recent studies have shown that several RBPs bind to stem-like regions, rather than to single-stranded loops (3). Therefore, the main limitation of MEMERIS is that it misses binding motifs for proteins with stem-loop preference. Moreover, it does not take into account the full spectrum of RNA structures.
Another extension to the MEME software is Zagros introduced by Bahrami-Samani et al. (32). It accounts for secondary structure (paired or unpaired only) and crosslinking modifications in the EM framework.
Several methods exist to incorporate the full secondary structure into RNA motif finding. In 2010, Kazan et al. introduced RNAcontext, an affinity-based model which learns both sequence and structure preferences of an RBP, considering several structural contexts (4). RNAcontext learns the RBP binding affinity and optimizes the model’s parameters from both sequence k-mers and predicted structure profiles from input sequences. Affinity values are obtained by experimental assays such as RNAcompete, or can be set to discrete classes, e.g. bound and unbound from CLIP-Seq experiments. RNAcontext outperformed MEMERIS in classifying bound versus unbound RNA sequences on a selection of nine proteins (4). GraphProt by Maticzka et al. uses graph kernel-based support vector machines (SVM) trained on a large number of features from a hypergraph to learn sequence and structure preferences of RBPs (33) from high-throughput data. GraphProt produces a motif visualization in the form of separate sequence and structure logos. A sequence logo is a graphical representation of the preferences of an RBP for the four nucleotides at each binding site position (e.g. 1 to 6). A structural logo, as introduced by Maticzka et al., is a graphical representation of the preferences of an RBP for 5 different RNA structure types (e.g. loops, stems, multiloop, external loops, internal loops) at each base position in the binding site (33). Feature interpretation from the hypergraph SVM model is not straightforward and the two logos are indirectly generated from the top-scoring k-mer nucleotide sequences and structure profiles. Alternatively, the trained model can be used to predict novel binding sites in the same organism. Like RNAcontext, GraphProt requires both a positive and a negative input dataset for training. In a binary classification setting it was shown to be superior to RNAcontext for a large set of RBPs (33).
While RNAcontext and GraphProt are designed to accurately distinguish bound from unbound sites, a tool for de novo identification of sequence-structure motifs from RBP-bound sequences is lacking. In this paper, we propose ssHMM (sequence-structure hidden Markov model), a novel tool to identify de novo sequence-structure motifs in a set of RNA sequences bound by a certain RBP. Our method, trained on CLIP-Seq experimental data, and based on hidden Markov models to represent both sequence and structure preferences has several advantages compared to previous approaches: (i) It identifies a combined sequence-structure motif which characterizes the unique features of the RBP binding site rather than outputting two separate logos for sequence and secondary structure; (ii) It models a spectrum of five different structural contexts (stem and four different single-stranded loop contexts) instead of defining a general propensity for single-stranded regions; (iii) In contrast to discriminative approaches which focus on finding the optimal separation between positives (RBP sites) and negatives (background sequences), it is designed with the purpose of producing an interpretable motif model which can be intuitively visualized and easily understood.
We demonstrate the ability of our tool to recover RBP sequence-structure motifs from both synthetic and real CLIP-Seq data, including novel motifs such as a sequence-structure preference for the YY1 protein. Our analysis revealed also that the structure preference of an RBP weakens with increasing strength of the sequence motif.
In addition, ssHMM proved to be considerably faster than both MEMERIS and RNAcontext on large datasets, and therefore suitable for NGS data applications. ssHMM is freely available for download on Github (github.molgen.mpg.de/heller/ssHMM) and the Docker hub. It is easy to use and can be applied to characterize novel motifs in any set of input RNA sequences.
MATERIALS AND METHODS
Here, we present ssHMM, a de novo motif discovery tool which combines hidden Markov models (HMMs) with Gibbs sampling to learn the joint sequence and structure binding preferences of an RBP. The states of the model represent five different structural contexts of RNAs: stem, hairpin loop, multiloop, internal loop, and exterior loop (Figure 1), while its emissions represent the four RNA nucleotides. The rationale behind this topology is that RBPs might recognize their RNA targets by both their nucleotide sequence and their structure. ssHMM is trained on high-throughput RNA-binding protein data from CLIP-Seq experiments or any other experimental protocol yielding large numbers of RNA sequences. After training, the resulting model can be visualized as an intuitive graph logo (Figure 2). Aside from describing the model, its training and its visualization, we also explain how the synthetic and biological datasets were generated and how they were used to evaluate our tool.
Figure 2.

Overview of the motif finder workflow. (1) A CLIP-Seq experiment yields nucleotide sequences of RNAs (in uppercase) bound by a specific RBP. The genomic region surrounding the binding site was added in lowercase to benefit the next step. (2) The most likely structures of each nucleotide sequence are computed by a structure prediction tool (RNAshapes and RNAstructure). (3) The ssHMM is trained with the nucleotide and structure sequences. (4) The final model is visualized as a graph logo.
The ssHMM sequence-structure model
The sequence-structure hidden Markov model (ssHMM) constitutes the core of the motif finder presented here. It is trained on two types of ‘sequences’: the actual RNA nucleotide sequences corresponding to the RBP binding sites as detected by means of high-throughput experiments and their corresponding RNA structures (Figure 2). Abstracting from the predicted base pairing, the latter are encoded as sequences of symbols representing different structural contexts. ssHMM models the RNA-protein binding site as a set of symbol-emitting states. The symbols are the four nucleotides A, C, G and U. Each combination of binding site position P ∈ {1..n} and structural context C ∈ {E, I, S, H, M} is represented by exactly one state (Figure 3). The states and transition probabilities of the HMM represent the different RNA structures and the transitions between them, respectively. The emission probabilities, on the other hand, represent the RNA nucleotides and the probabilities of them being observed in a specific structural context. The motif length n of the binding site needs to be chosen by the user prior to training. It is recommended to train ssHMM with different biologically meaningful motif lengths for an RBP binding site (e.g. from 4 to 12) and to inspect the resulting average information content per position. For best results, a good compromise between motif length and information content needs to be found. An empirical rule for that is described in Section 1.2.1, Additional File 1.
Figure 3.

Topology of the ssHMM. Each combination of binding site position (columns) and structural context (rows) is represented by one state. The structural contexts are E(xterior), I(nternal or bulge), S(tem), H(airpin) and M(ultiloop). Note that every transition in the model proceeds immediately to the next binding site position and that exactly n nucleotides are emitted by the HMM from Start to End.
Model training
In order to train the ssHMM, the following two sets of variables need to be estimated:
Motif start positions
The RBP binding motif is typically much shorter than the input RNA sequences and it is unknown where it is located in each of the long RNA sequences. This information, however, is needed because the HMM models only the short motif. For an RNA sequence k of length l and a motif length n, the zero-based motif start position is defined as 0 ≤ ik ≤ l − n. During the training, ik is estimated for every RNA sequence and is sufficient to determine both the motif start sk = ik and end ek = ik + n − 1.
Best structure
RNAs can often fold into multiple thermodynamically stable structures. This is why most structure prediction tools compute several highly probable secondary structure conformations for each RNA nucleotide sequence. We employed the tools RNAstructure and RNAshapes (35,36) to predict RNA structure states. RNAstructure predicts the lowest free energy structure as well as a number of suboptimal structures for a given RNA sequence. RNAshapes, in contrast, builds upon the concept of abstract shapes (i.e. classes of structures with similar features) to avoid predicting many highly similar and redundant structures. The user can choose which tool to use for structure prediction in ssHMM. Besides RNAstructure and RNAshapes, any tool producing a set of highly probable structures in the required structure format for each sequence can be used. Regardless of the tool, ssHMM chooses one structure from this set for every RNA sequence during its training. It is selected in an iterative fashion based on its fit to the ssHMM model (see the following section).
Gibbs sampling procedure
To estimate these two sets of unknowns, we use a Gibbs sampling approach. Thus, we both train the ssHMM and estimate the two unknown variables for each sequence at the same time.
At the outset of the Gibbs sampling procedure, the unknown variables have to be initialized, for instance by choosing random values (see Supplementary Figure S1 in Additional File 1). Then, an iterative optimization process begins that alternates between re-estimating the ssHMM and the unknown variables. In each of the following iterations, one RNA nucleotide sequence together with its corresponding RNA structure sequences is left out. An iteration consists of two estimation steps:
-
The ssHMM is re-estimated using all sequences except the left-out one. The current best structure and motif start positions can be used to retrieve both the nucleotide motif occurrence and the structure motif occurrence from each sequence. The nucleotide motif occurrence denotes a series of HMM emissions (an emission sequence) while the structure motif occurrence denotes a series of HMM states (a path). Using all emission sequences and state paths, it is possible to calculate a maximum likelihood estimate for the model parameters. To estimate the transition probabilities δi → j, the number of transitions ti, j between each pair of states i and j in the state paths can be counted. The maximum likelihood estimate of the transition probability between two states is then
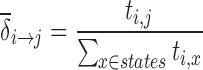
(1) To estimate the emission probabilities λk: b, the number of emissions ek(b) of symbol b ∈ A, C, G, U from state k can be counted in the emission sequences, and the maximum likelihood estimate of the emission probability from a state is then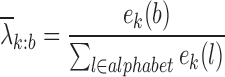
(2) The initial probabilities πk can be estimated by counting how often each state appears as the first state s1 in all state paths.
-
The motif start position and best structure of the left-out sequence is re-estimated given the ssHMM (i.e. given δ, λ, and π from point 1). For every possible combination of the two, we can calculate the conditional probability
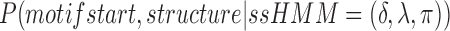
(3) Motif start and best structure unambiguously define an emission sequence and a state path. We compute the joint probability of the emission sequence and the state path which is equivalent to the conditional probability. The new motif start position and best structure for the left-out sequence is drawn randomly according to the distribution of the conditional probabilities.
The execution is terminated when the increase in joined sequence-structure likelihood compared to the last three iterations drops below a user-defined threshold (see Section 1.2.4 in Additional File 1).
Gibbs sampling initialization
Gibbs samplers run the danger of becoming trapped in local optima depending on their initial values. Therefore, it is advisable to choose these values carefully. In our approach, two initial values need to be determined at the outset for every sequence: one structure from the set of highly probable structures and one motif start position. From the structures, we initially always choose the one with the highest probability as determined by the structure prediction tool (i.e. the highest structure prediction score). For choosing the initial motif start positions, we implemented two different approaches:
Random Initial motif start positions are drawn randomly. Depending on the drawn values, Gibbs sampling may yield very different results;
Baum-Welch Initial motif start positions are determined using a sequence-only HMM. With the Baum-Welch algorithm, the sequence-only HMM is trained on the sequences to learn the strongest sequence motif. Afterwards, the Viterbi algorithm is used to locate that sequence motif in each sequence, and the starting index of the sequence motif is taken as the initial motif start position.
The Baum-Welch algorithm is subject to the local maxima problem as well, i.e. different initializations yield different motifs. Therefore, we run the Baum-Welch procedure on 100 randomly initialized sequence-only HMMs and choose the HMM with the highest likelihood. The Baum-Welch initialization approach yielded substantially better results than random initialization (see Supplementary Figures S3 and S4, Additional File 1). Therefore, ssHMM uses by default the Baum-Welch initialization.
ssHMM visualization
The trained HMM can be visualized as a graph in which each HMM state is represented by one node. Similar to a sequence logo, the nodes of the graph visualize the emission preferences of the corresponding HMM state with stacks of colored nucleotide letters. These stacks indicate which bases are prevalent at each binding site position in each structural context. The transition probabilities between the HMM states are visualized as arrows. The thicker an arrow between two states, the more likely is a transition between the two. Arrows corresponding to transition probabilities lower than 5% are not displayed to increase clarity. For more information on the output of ssHMM, see Section 1.3 in Additional File 1.
Dataset generation
For the evaluation of our motif finder, we collected two kinds of sequence datasets: randomly generated synthetic datasets and biological datasets derived from CLIP-Seq experiments on >20 different proteins. We evaluated the performance of ssHMM on both types of datasets and compared it with three approaches for the detection of RNA sequence-structure patterns: MEMERIS (version 1.0), RNAcontext, and GraphProt (version 1.1.1) (4,31,33). All evaluation data can be found on Github at github.molgen.mpg.de/heller/ssHMM_data.
Synthetic datasets
Synthetic sequences are generated specifically to contain a certain implanted sequence motif. We followed the protocol devised by Bahrami-Samani et al. (32) to generate datasets that contain sequence motifs of length 6, but adapted it to our purposes. We generated 24 such datasets with four different properties (Supplementary Table S2 in Additional File 1): (i) average information content per position (1.0 / 0.5), (ii) background sequence type (uniformly random / 3′UTR) and (iii) whether a certain fraction of motifs (10%/50%/100%) was implanted into (iv) a hairpin or stem context.
Each of the 24 datasets is comprised of 100 sequence sets. A sequence set consists of 2000 RNA sequences with their corresponding shapes. For each sequence set, one random position probability matrix (PPM) of length 6 with the given average per-position information content was created and stored for later evaluation.
From this PPM, motif occurrences were drawn for each of the 2000 sequences in the sequence set and implanted into background sequences of length 50 at random locations. Depending on the dataset, the background sequences were either generated by drawing from a uniform distribution over the four nucleotides (datasets *.A to *.F) or randomly sampled from the set of human three prime untranslated regions (3′UTRs, datasets *.G to *.M). Because 3′UTRs are known to contain many RBP binding sites and confounding signals, the 3′UTR datasets reflect real data particularly closely.
In order to assess the influence of the structural context on sequence motif recovery, it was ensured for each dataset that a minimum fraction of the 2000 motifs, 10%, 50% or 100%, was implanted into a hairpin loop or stem, respectively. In order to deliberately implant a motif in a hairpin loop, we reverse complemented a 10 nt sequence from one side of the motif and copied it to the other side. This drives the formation of variable size hairpin loops around the motifs. To confirm that the implanted motif really falls into an hairpin, we ran the structure prediction tool on the resulting sequence and retained it only if the entire motif was predicted to be in a hairpin. To deliberately implant a motif in a stem, we followed a simpler approach: relying on chance, the implantation was repeated with new background sequences until the structure prediction tool predicted the motif to be in a stem context.
CLIP-Seq datasets
We retrieved 25 different CLIP-Seq datasets for 27 different RBPs from various sources (Supplementary Table S7 in Additional File 1). Most datasets were downloaded from the doRiNA (version 2.0), a database of manually curated RBP binding site data (37). With the exception of three mouse datasets, all experiments were conducted in human HEK293 and HeLa cells. From the 25 datasets, 17 were generated with PAR-CLIP, 7 with HITS-CLIP, and 1 with iCLIP. These correspond to already processed binding site sequences provided as genomic coordinate files in Browser Extensible Data (BED) file format. The median lengths of such sequences lay between 22 bp (FXR2) and 164 bp (YY1). Lengths distributions for five selected proteins are shown in Supplementary Figure S7 (Additional File 1).
For testing all tools in discriminative mode positive as well as negative (i.e. bound and unbound) sequences are required. For each of the 25 proteins, the positive set consisted of the CLIP-Seq sequences for that protein. The corresponding negative set was formed by a subset of the CLIP-Seq sequences from all other 24 datasets and was of the same size as the positive set. In addition, each negative set was created in such a way that the length distribution of the selected sequences was as similar as possible to the positive dataset.
For both sets, secondary structures were predicted with RNAshapes (version 2.1.6), with command line options -o 1 (choosing output type 1) and -r (calculates structure probabilities) and RNAstructure with default parameters. Binding sites were elongated by 20 bases on each side prior to structure prediction. The dotbracket output of RNAshapes was converted to a string of structural context symbols using the forgi (version 0.2) Python library. The string encodes the predicted structural context of each nucleotide in the input sequence with the corresponding symbol: E for exterior loop, I for internal loop, S for stem, H for hairpin loop, and M for multiloop.
Although elongated binding sites were used for secondary structure prediction (see Section 3.2 in Additional File 1), motif finding by ssHMM is restricted to the experimentally determined binding sites. This is to ensure that retrieved motifs are correct, as unpaired bases (external context) which are predicted at the sequence ends are a mere artifact of the cutpoint sensitivity of the structure prediction tool, and should not be part of the retrieved motif.
Evaluation on synthetic datasets
The sequence motif recovery performance of the motif finders on synthetic datasets was evaluated using Tomtom (version 4.11.1), the motif comparison tool of the MEME software suite. After training on the synthetic sequences, all motif finders produce a sequence logo that can be expressed in terms of a PPM. This PPM contains the probability of every base at every position and we call it the recovered PPM. The PPM that was used to generate the synthetic sequences is called the original PPM. We compiled a target motif database of all 100 original PPMs of a dataset and compared each of the 100 recovered PPMs individually against this target motif database. We report the q-value of the highest ranking match between a recovered motif and the corresponding original motif. A q-value threshold of 0.05 was used to compute the fraction of recovered motifs for each dataset and tool.
We also analyzed how accurately ssHMM recovers the structure preference from the synthetic datasets with hairpin motif. In the nine datasets H.A to H.I, either 10%, 50% or 100% of the motifs were implanted into a hairpin context. Together with the remaining 90%, 50% or 0% of motifs which can lie in a hairpin by chance, the estimated hairpin fractions of the datasets are 28%, 60% and 100%, respectively. From the models trained on the different datasets, we extracted the recovered hairpin preference. This is defined as the transition probability between the start state and the H1 state in a trained ssHMM. Finally, we analyzed how well the recovered hairpin preferences reflect the estimated hairpin fractions.
Evaluation on CLIP-Seq datasets
To evaluate the performance of ssHMM on real CLIP-Seq datasets, we performed several tests: (i) we checked that the trained motif finder can generally distinguish between real binding sites (positives) and background sites (negatives); (2) we compared ssHMM and the other tools in a classification setting; (3) we analyzed the information content of the recovered motifs and (iv) carried out a qualitative analysis by assessing the resemblance of the recovered motifs to RBP motifs known from the literature.
Fisher’s exact test
In the first analysis, we used Fisher’s exact test to confirm that our motif finder is able to distinguish real binding sites from background sites. For this, the protein datasets were split into training and test data. Motif models for all proteins were trained on the training data portions and were then used to classify the sequences from the test portions. The optimal cutoff between positives and negatives was determined by optimizing on the P-values obtained by Fisher’s exact test. Finally, the P-values of the optimal cutoffs were adjusted using Benjamini & Hochberg correction (38).
Classification analysis
We compared ssHMM to MEMERIS, RNAcontext and GraphProt on a classification task. We executed MEMERIS in three settings, with pi=0 (strong prior directs motif finding to single-stranded regions), pi=100 (effectively finds sequence-only motif), and the in-between setting of pi=1. Initially, each protein dataset was split into a training and a test set. Then, both ssHMM and MEMERIS were trained on the positives of each training set to retrieve a motif model. A background model was obtained by training each tool on a random subsample from the mixture of all CLIP-Seq sequences. For each dataset, the log likelihood ratios of the motif model versus the background model were used as final scores to classify the test sequences. From the scores, the area under the Precision-Recall curve (AUCPR) was computed.
RNAcontext and GraphProt were trained on positives and negatives from the training set. Subsequently, separate test sets were used to produce Precision-Recall curves (see Additional File 4). We also evaluated whether sampling over all predicted secondary structures offers a benefit for ssHMM in comparison to using always the optimal structure.
Information content
We measured the ability of ssHMM to retrieve informative motifs given a set of binding site sequences by computing the information content (IC) of the retrieved motif model. Three variants of the motif’s IC on three different alphabets A were computed: IC of the sequence-only motif with A = {A, C, G, U}, IC of the structure-only motif with A = {E, I, S, H, M}, and IC of a combined sequence-structure motif with A = {A, C, G, U} × {E, I, S, H, M}. We further distinguished two ways of computing the information content (Section 3.7 in Additional File 1): Firstly, and similarly to GraphProt, the information content can be obtained from the top-scoring 1000 sequences. Alternatively, the information content can be directly computed from the trained model.
RESULTS
ssHMM learns interpretable sequence-structure motifs
ssHMM produces graph models of RBP binding affinity that connect the sequence and structure part of an RBP binding site in a natural and intuitive way. The underlying hidden Markov model is able to estimate the sequence and structure binding specificities of a protein simultaneously: while the nodes of the graph express preferences for individual nucleotides in different structural contexts, the arrows represent more or less likely transitions between structural contexts (Supplementary Figure S2, Additional File 1).
Compared to RNAcontext and GraphProt models, which only allow extracting separate sequence and structure logos from the classified sequences, the output of ssHMM can be directly interpreted as the most likely sequence-structure motif from the RBP binding data, giving for the first time detailed insights into the interdependency between sequence and structure preference. Therefore, unlike existing methods, ssHMM can model the occurrences of different sequence motifs in different structural contexts, as in the case of DGCR8 and YY1 (discussed in the next paragraph). This also helps to elucidate whether a specific structural context is required or not for a certain sequence motif. In addition, ssHMM incorporates five structural contexts into the motif model, unlike MEMERIS which distinguishes only double-stranded and unstructured regions. This gives a more precise description of the preferred structural contexts of an RBP, when, for instance, the RBP has a specific preference for multiloop, rather than single-stranded regions in general.
ssHMM recovers both new and validated motifs from CLIP-Seq data
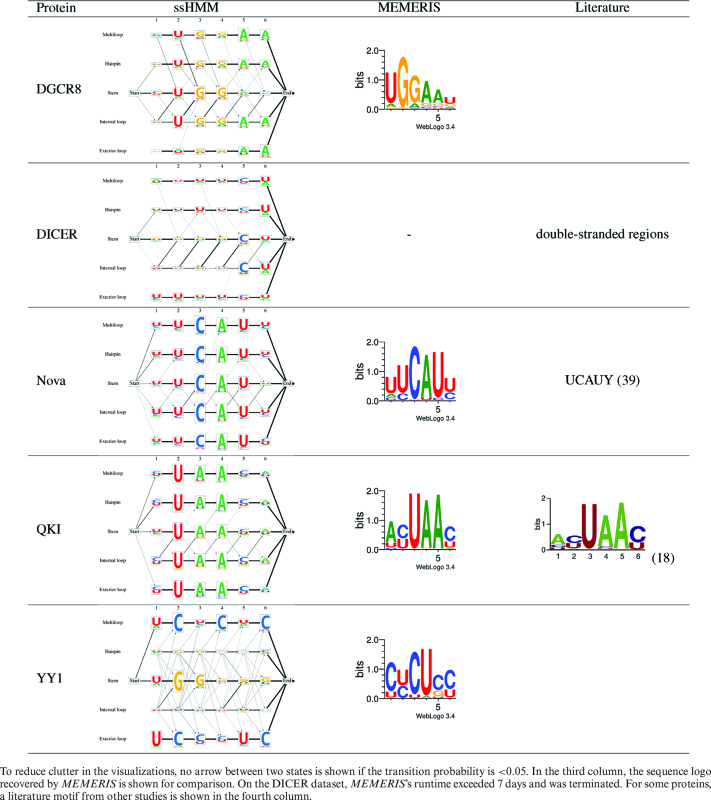
We applied ssHMM to biological datasets derived from 25 CLIP-Seq experiment datasets. Table 2 shows the output of our motif finder for five selected CLIP-Seq datasets. For three of the proteins, Nova, QKI and DICER, sequence and/or structure motifs have been previously characterized which confirm that ssHMM recovers correct motifs from biological data.
Table 2. Visualization of trained ssHMMs for five selected CLIP-Seq datasets.
|
Nova
Nova is an RBP exclusively expressed in neurons within the central nervous system and is involved in RNA alternative splicing and RNA metabolism. ssHMM recovers a clear UCAU sequence motif and U-rich flanking positions. This agrees with the motifs from MEMERIS and findings from other studies (3,39) which identified unstructured and U-rich regions as preferred binding environment for NOVA. The visualized ssHMM displays a considerable preference for the single-stranded multiloop and hairpin loop contexts.
QKI
The Quaking homolog (QKI) is an RBP regulating pre-mRNA splicing, mRNA export, mRNA stability, and protein translation (40). The ssHMM visualization for this protein shows a strong UAA sequence motif across all structural contexts. This is in accordance with the motifs retrieved by MEMERIS, RNAcontext, GraphProt and other previous studies (18). Our motif finder also found that QKI prefers to bind hairpin loops.
DICER
The protein DICER is a key player in the microRNA biogenesis pathway. It is specifically involved in the processing of microRNA precursors (pre-miRNAs) into double-stranded RNA fragments which then give rise to the ∼21-nt-long mature microRNAs. It is known that DICER binds double-stranded RNA structures and that this structural context determines its specificity, while nucleotide sequence does not play a role in determining DICER efficiency (41). ssHMM confirms previous observations about DICER’s binding preference for the stem context, the lack of a strong sequence binding motif and the preference for C nucleotides in the penultimate terminal stem position (41).
For DGCR8 and YY1, no RNA binding preference has been previously described.
DGCR8
The RBP DGCR8, together with Drosha, forms the so-called Microprocessor complex involved in microRNA biogenesis. It is responsible for recognizing and releasing pre-miRNA hairpins from large primary microRNA (pri-miRNA) transcripts. The two proteins in the complex have distinct and complementary tasks: DGCR8 recognizes and binds primary miRNAs while Drosha cuts them and thus converts them to pre-miRNAs (42).
The sequence-structure preference of DGCR8 is as yet not very well known. Previous studies have found that DGCR8 binds both double-stranded and single-stranded transcripts with similar affinity (43). ssHMM applied to DGCR8 PAR-CLIP data recovered a UGGAA sequence motif, identically retrieved by MEMERIS and, in more fuzzy form, by GraphProt. When comparing the sequence motifs from the different structural contexts based on ssHMM we can observe that, while the hairpin and internal loop contexts display the full UGGAA sequence motif, the stem context only exhibits the shortened motif UGG. ssHMM also reflects a strong preference for the stem context which is in accordance with the fact that DGCR8 contains two double-stranded RNA-binding domains (44). Our results are also in line with the findings from two previous studies (45,46) which identified a highly-conserved UG dinucleotide motif in a stem context at RNA positions –14 and –13 from the Drosha cleavage sites to be involved in enhanced pri-miRNA processing. Neither study provides, however, any reason for the molecular mechanisms behind the function of the UG motif. From our ssHMM analysis, we can suggest that the UG dinucleotide might be contributing to the specificity of DGCR8 binding in a double-stranded structural context.
YY1 is an interesting example of how ssHMM can be used to model the multi-motif sequence-structure preference of an important binding factor. It is therefore described in detail in the next paragraph.
For the full list of results from all datasets, including the literature motifs, motifs recovered by MEMERIS, RNAcontext and GraphProt, and their corresponding information contents, see Additional File 2. For all 25 protein datasets, the sequence-structure motifs recovered by ssHMM were robust to the choice of the particular structure prediction tool (RNAshapes or RNAstructure, see Additional File 3) and to the elongation span of the sequences subject to structure prediction (see Section 3.2, Additional File 1).
ssHMM recovers a dual motif for RNA-binding TF YY1
Recently, TFs that bind both DNA and RNA have gained considerable attention thanks to their prominent role in RNA-mediated transcriptional regulation of gene expression (34). YY1 is a multi-function transcription factor involved in many regulatory processes. Recent evidence suggested that YY1 interacts with several RNAs, including, but not limited to the lncRNA Xist. Xist is involved in X-chromosome gene inactivation, where YY1 might have a role in tethering Xist to chromatin (47). While the DNA binding motif of YY1 is known, its RNA specificity has been poorly investigated.
We analyzed a YY1 CLIP-Seq dataset and derived, for the first time, a potential sequence-structure RNA binding motif for YY1. ssHMM revealed two major preferred sequence-structure contexts for YY1: a strong CU-rich motif in a multiloop context and a G-rich stem motif (Table 2). Although MEMERIS was able to retrieve the CU-rich sequence motif, it was not able to reveal the stem motif, even when run with different sets of parameters and searching for multiple motifs simultaneously. In addition, MEMERIS was not able to determine the location of the CU-rich motif in the multiloop context, simply because it does not differentiate between different single-stranded structure types. Both RNAcontext and GraphProt recovered a motif which merges both sequence motifs recovered by ssHMM (Additional File 2). However, GraphProt located the motif in the stem while RNAcontext located it in a hairpin loop context.
The Fisher exact test analysis for YY1 shows that the motif learned by ssHMM is not merely an artifact, but represents the genuine sequence-structure preference of YY1 for RNA sequences (Section 3.6 in Additional File 1). The motif is significantly (corrected p-value <1e-16) enriched compared to background sites in a set of YY1 binding site sequences that have not been used for training.
The learned YY1 motif model is also able to classify binding site versus background site sequences on a separate test set with an Area under the Precision-Recall curve of 0.83 (see Figure 4).
Figure 4.

Recovered YY1 motif is confirmed by classification analysis. Precision-Recall curve for ssHMM in a classification setting for the YY1 dataset. The three curves represent three independent runs of ssHMM. In each run, a model was trained on the training set before classification performance was measured on the separate test set. The average Area under the Precision-Recall curve over all three runs is 0.83.
The identification of new RBP motifs like the one for YY1 can contribute to shedding light on RNA functional characterization. For example, lncRNAs harbouring a YY1 motif might be involved in gene silencing, similarly to Xist, and this function might be mediated by YY1. In addition, this example shows that our approach can retrieve more than one preferred sequence motif, can determine their respective structural contexts, and can help characterizing protein preferences where other approaches return ambiguous results.
ssHMM reliably recovers motifs from synthetic sequences
We evaluated the performance of ssHMM on randomly generated synthetic data with varying characteristics and compared the results with MEMERIS and RNAcontext. Because the different tools use different structure representations, the produced structure motifs could not be compared directly. RNAcontext, for instance, produces only a single set of relative structural context affinities over the entire motif and MEMERIS does not output a structure motif at all. Therefore, we confined our comparison to an evaluation of how well the tools recover implanted sequence motifs from the synthetic datasets. We are aware that this comparison might confer an advantage on models that optimize a sequence-only model, such as MEMERIS.
To produce the datasets, we implanted fuzzy sequence motifs into background sequences at random locations. 24 datasets were generated which varied in average per-position motif information content (1.0 and 0.5), background sequence type (random and 3′UTR), and type and prevalence of a specific structural context for the implanted motifs (hairpin or stem; 10%, 50% and 100%). After running the tools on the datasets, Tomtom was used to evaluate the motif recovery performance (see Materials and Methods).
Figure 5 contains the results of the analysis. The first important observation is that both ssHMM and MEMERIS are able to perfectly recover 100% of the motifs with an information content of 1.0, while RNAcontext reached results considerably below 100%. The second observation is that, for an information content of 0.5, ssHMM’s recovery rate increases when the percentage of motifs located in a specific structural context grows from 10% to 100%. This shows that the structural context helps our tool to better identify a fuzzy sequence motif.
Figure 5.

Comparison of recovery rates of ssHMM, MEMERIS, and RNAcontext on synthetic data. Shown is the fraction of successfully recovered sequence motifs from 24 synthetic datasets (y-axis) for different tools. The upper panel shows results for motifs of information content 1.0 while the lower panel shows results for motifs of information content 0.5. The left two panels show results for synthetic motifs planted into a hairpin context of random or 3′UTR background sequences, respectively. The right two panels show results for synthetic motifs planted into a stem context of random or 3′UTR background sequences, respectively. The x-axis denotes the fraction of motifs (10%, 50% or 100%) that were implanted into a hairpin or stem context. For MEMERIS, two different values for the pi parameter are plotted (pi=0 and pi=100). The pi parameter determines the importance of the single-stranded context relative to the sequence. For pi=1, all results lay between the results for these two parameter values and are therefore not plotted. To avoid overlapping lines, results for MEMERIS (pi=100) and ssHMM were shifted slightly up by 2 and 1 percent, respectively. The fraction of successfully recovered motifs was computed with a q-value threshold of 0.05 on the highest ranking match between the original and the recovered motif.
GraphProt displayed a very weak performance over all synthetic datasets (data not shown). This might be due to the synthetic nature of the generated sequence-structure motifs, which do not form biologically valid secondary structures and cannot be represented in a valid graph structure. As a comparison of our tool to GraphProt in this test setting would not be completely fair, we excluded GraphProt from this analysis.
The recovery of fuzzy motifs with an information content of 0.5 was influenced by the type of background sequences they were implanted in. For random background sequences, ssHMM reached slightly lower recovery rates than MEMERIS but higher rates than RNAcontext when at least 50% of the motifs were implanted in a specific structure. For 3′UTR background, all tested tools reached recovery rates below those for random background sequences, especially for hairpin motifs. This is probably due to strong confounding regulatory signals, other than the implanted motif, in the 3′UTRs.
In a detailed analysis of dataset H.K, we assessed the impact of background GC content on the motif recovery performance of ssHMM. We found that motifs implanted into low-GC (GC < 40%) and high-GC (GC > 60%) 3′UTRs could be better recovered than motifs implanted into medium-GC (40% ≤ GC ≤ 60%) 3′UTRs (motif recovery rates of 49%, 17%, and 4%, respectively). This suggests that differences between the motif GC content and the GC content of the background sequences actually have a significant impact on motif retrieval. The synthetic motifs with a mean GC content of 50% stand out against low-GC or high-GC background sequences and are consequently easier to recover.
MEMERIS’s recovery rate was in general strongly influenced by the choice of the pi parameter. It must be chosen in advance, but is, in most of the cases, unknown to the user. MEMERIS performed well in retrieving motifs from a hairpin loop, with an average recovery rate of 89.5% for pi = 0 (strong prior for hairpin loop) and 79.9% for pi = 100 (no prior) versus 77.4% and 69.2% for ssHMM and RNAcontext, respectively. For stem motifs, MEMERIS’s average recovery rate is 99% for pi = 100 but drops dramatically to 58.6% for pi = 0 versus 95% and 93% for ssHMM and RNAcontext, respectively.
To evaluate ssHMM’s ability to accurately detect the structural context of a binding site besides its sequence motif, we analyzed the synthetic datasets with hairpin motif. We found a striking Pearson correlation of 0.91 between the hairpin fraction recovered by ssHMM and the estimated hairpin fraction of the synthetic dataset (Section 2.4 in Additional File 1). This confirms that ssHMM is able to recover both accurate sequence and structure motifs.
ssHMM can distinguish between real binding sites and background
With our motif finder, we analyzed 25 different PAR-CLIP, HITS-CLIP and iCLIP datasets for 27 different RBPs from various sources. With the exception of two mouse datasets, all datasets stemmed from human HEK293 and HeLa cells.
Fisher’s exact test on the likelihood of positive RBP sites versus background sites under the trained model yielded adjusted p-values below the significance threshold of 0.05 for all CLIP-Seq datasets (Supplementary Table S12, Additional File 1). This demonstrates that our trained motif model can significantly distinguish between real binding sites and background sites.
ssHMM outperforms MEMERIS on the majority of proteins in a classification setting
As described in Materials and Methods, we compared ssHMM with the three other tools in a classification setting. Across all datasets, the discriminative classifiers RNAcontext and GraphProt reached a substantially higher Area under the Precision-Recall curve (AUCPR) than the generative motif finders ssHMM and MEMERIS (Additional File 4). Due to the differences between generative and discriminative approaches, this is expected and confirms that RNAcontext and GraphProt are far more suitable than the motif finders for a classification task.
When comparing only the motif finders, ssHMM outperformed MEMERIS on at least 15 out of 23 datasets across three settings, while MEMERIS slightly outperformed ssHMM only on 7 dataset (Supplementary Table S8 in Additional File 1). On average, the increases in AUCPR of ssHMM over MEMERIS were considerably larger than the decreases, with several gains higher than 10%. These results demonstrate that the full sequence-structure model of ssHMM yields a benefit over the MEMERIS sequence-only model.
To confirm the usefulness of sampling over several secondary structures during training, we further analyzed the differences between sampling over all structures versus only the optimal one as determined by the structure prediction tool. Training on all shapes led to models that are better fit to the training data and perform slightly better in a classification setting for some of the analyzed proteins (see Section 3.4, Additional File 1).
Motifs identified by ssHMM possess high information content
While ssHMM is not as powerful as RNAcontext and GraphProt in a classification setting, it is well suited for motif retrieval, the primary applications of a motif finder. We measured the ability of ssHMM to retrieve informative motifs given a set of binding site sequences by computing the information content (IC) of the retrieved motif models. The higher the information content, the more specific is the motif and the lower is the chance to find it in a sequence by chance. Information content values for all 25 RBP retrieved motif models are reported in Additional File 2 together with the values computed from the MEMERIS, RNAcontext and GraphProt models.
As expected, for almost all proteins ssHMM finds more expressive motifs than GraphProt and RNAcontext. For all proteins, the motifs retrieved by ssHMM had a higher sequence-structure information content than those by GraphProt (mean IC of 2.55 versus 1.66). This points to the fact that a large structural context, as the one encoded in the GraphProt model, might play a role in discriminating RBP sites versus non-RBP sites. The motifs obtained directly from ssHMM were also generally more expressive than those by RNAcontext (see Additional File 2). On average, ssHMM and RNAcontext motifs had a sequence information content of 0.64 and 0.39, respectively. Only for five proteins did RNAcontext produce a more expressive motif than ssHMM. For three of those, DGCR8, FXR2 and PUM2, the motif recovered by RNAcontext was different from the one retrieved by both MEMERIS and ssHMM. For PUM2, the RNAcontext motif does not agree with the literature motif. Another example is PTBP1, where RNAcontext fails to retrieve any motif. For PUM2 and NOVA, GraphProt also produced motifs which do not entirely agree with the literature knowledge. MEMERIS-derived motifs had a higher sequence information content than the motifs derived from all the three other tools. This is expected given that MEMERIS optimizes a sequence-only model, rather than a joint sequence-structure model.
Negative correlation between sequence and structure specificity in RBP binding sites
We investigated in more detail the relationship between sequence and structure in RBP binding sites. Over all CLIP-Seq datasets, we observe a significant negative correlation of –0.451 (Spearman’s rank correlation, P-value 0.02) between sequence and structure information content (Figure 6). This observation is concordant to what Schneider et al. described in 1986: binding sites tend to contain approximately as much information as is necessary for them to be recognized (48). In RNA-RBP binding, sequence and structure specificity seem to complement each other so that RBPs with a strong sequence preference tend to exhibit only a weak or no structure preference and vice versa.
Figure 6.

Sequence and structure information content of motif models trained on CLIP-Seq datasets are negatively correlated. Shown are the sequence information content (x-axis) and structure information content (y-axis) of ssHMM motifs for all 25 CLIP-Seq datasets (average per position). Both are negatively correlated with a significant Spearman’s rank correlation coefficient of -0.451 (p-value 0.02). Plotted is also the linear regression line.
ssHMM can efficiently handle large datasets
In order to be suitable for scientific use, modern motif finders are required to process large datasets in reasonable time. CLIP-Seq is a high-throughput protocol and produces datasets that commonly hold tens of thousands of RBP binding sites. It is therefore vital that motif finding algorithms scale well with input datasets of increasing size.
To assess the runtimes of GraphProt, RNAcontext, MEMERIS and ssHMM, we took runtime measurements on datasets of increasing size (see Section 4, Additional File 1). Figure 7 illustrates that the four different motif finders did not scale equally well on the datasets. While GraphProt and ssHMM showed the best runtime performance, MEMERIS and RNAcontext do not scale sufficiently well to be applied to large datasets. Although RNAcontext’s runtime increases linearly with the size of the dataset, it was the slowest tool, mainly due to its secondary structure prediction algorithm Sfold, which amounted to more than 90% of its runtime. For MEMERIS, we observed a quadratic runtime progression. While it was the fastest of the four approaches on the smallest dataset, it was quickly overtaken by ssHMM and GraphProt on larger datasets, where MEMERIS’s runtime increased by a factor of 243. Consequently, MEMERIS exceeded 7 days of runtime on several of our CLIP-Seq datasets. Our motif finder and GraphProt scale best with increasing size of the input dataset. Both tools showed a linear runtime progression and processed 2000 sequences in less than 3 min on a single CPU.
Figure 7.

Runtime comparison between GraphProt, MEMERIS, RNAcontext, and our motif finder (ssHMM). The CPU time in seconds (y-axis) is plotted against the number of input sequences (x-axis).
All in all, GraphProt and ssHMM are the only methods able to process large datasets in reasonable time.
DISCUSSION
Knowing the sequence-structure specificity of RNA-binding proteins is essential for understanding RNA post-transcriptional regulatory processes. While several tools have been developed to extract de novo sequence motifs from sets of DNA sequences, the function and activity of RNA sequences is additionally influenced by the secondary structure around their binding site (3,49,50).
We developed ssHMM, a de novo motif finder capable of extracting sequence-structure RNA binding motifs from large sets of RNA sequences generated from genome-wide experiments such as CLIP-Seq. ssHMM estimates binding sequence and structure specificities simultaneously for every individual position of an RBP binding site. The model incorporates five different structural contexts and therefore gives a specific description of the preferred structural context of an RBP. MEMERIS on the other hand uses single-strand propensities for guiding the motif search and outputs only sequence logos. It can neither associate sequence motifs to a stem context nor distinguish hairpin loops from multi-loops. The motif models generated by ssHMM are visualized by a graph of state nodes with sequence logos representing sequence preferences and arrows representing likely transitions between structural contexts. The graphs are easy to interpret and visualize direct interdependencies between sequence and structure.
The analysis of CLIP-Seq datasets from RBPs with previously known binding motifs confirmed that ssHMM recovers correct motifs from biological data. We have demonstrated the ability of ssHMM to derive novel sequence-structure motifs for RBPs such as DGCR8 and YY1. For YY1, ssHMM captured a mixture of two sequence motifs, one in stem and the other one in multiloop. To our knowledge, this motif mixture has not been discovered before and could not be fully retrieved by the other tools. In addition, the analysis of CLIP-Seq datasets with ssHMM revealed an interesting anti-correlation between a protein’s sequence and structure preference. Although this has been observed for single proteins, a systematic relationship between the two information contents has never been shown so far.
The evaluation of our motif finder on synthetic data revealed its ability to retrieve 75–100% of the implanted motifs in almost all settings, in the absence of other confounding signals (see Figure 5). ssHMM’s motif recovery rate progressively increased with the fraction of weak sequence motifs (information content 0.5) located in a specific structural context. This demonstrates that incorporating the structure into the model greatly helps the tool to identify weaker sequence motifs, regardless of their location. In particular, ssHMM slightly outperformed all tools in recovering weak sequence motifs located in a strong stem context. MEMERIS was in some cases superior to ssHMM in recovering weak sequence motifs, but its performance strongly depends on the choice of the pi parameter, the prior for a single-stranded context. As the structural context of a motif is not known in advance, an unfavorable choice of the pi value might lead to poor motif recovery. Notably, RNAcontext performed better than both MEMERIS and ssHMM in recovering hairpin loop motifs from 3′UTRs.
ssHMM could significantly discriminate real binding sites from background sites for all 25 analyzed datasets, confirming that the produced motif models capture true signals in the data. In a classification setting, ssHMM outperformed MEMERIS on the majority of the proteins, indicating that a sequence-structure model is, in many cases, a win over a sequence-only motif model. RNAcontext and GraphProt reached considerably higher classification accuracy than both ssHMM and MEMERIS in discriminating real RBP from background sites for all proteins. However, while having a very high classification accuracy (AUCPR), they learned more abstract features and produced motifs with lower sequence-structure information content than ssHMM. Furthermore, one may argue that their strong classification performance is to be attributed, at least for some proteins, to GC content bias between the positive and negative sets.
Given their lower classification performance, ssHMM and MEMERIS are not the most suitable tools for classifying RBP binding sites versus non-binding sites. For such a task, GraphProt and RNAcontext are to be preferred.
However, if the goal of an analysis is motif discovery or biological interpretation, the fuzzy motifs derived by GraphProt and RNAcontext are unsuitable and harder to interpret. In this case, a motif finder, such as ssHMM, is more appropriate because it is designed to extract the most informative sequence-structure pattern from the data.
Finally, ssHMM is faster than other tools. It scales linearly on the input size and was the fastest tool in our runtime analysis (on a par with GraphProt). While MEMERIS exhibited quadratic runtime, and RNAcontext was impeded by a slow structure prediction tool, our approach was even able to analyze datasets with more than 20 000 sequences in a reasonable time. In contrast, we had large difficulties processing these larger CLIP-Seq datasets with MEMERIS and RNAcontext. Although not shown in this study, ssHMM can in principle be applied to any set of RNA sequences generated from experimental techniques other than CLIP-Seq (for example SELEX experiments). As future perspective, the motif models generated by ssHMM can be stored and used to search for sequence-structure motif hits in query RNA sequences. For example, one could look for RBP motifs in long non-coding RNAs, a class of RNAs whose full spectrum of possible functions is still poorly understood. ssHMM can also help annotating long non-coding RNA functions based on their most likely interaction partners.
CONCLUSIONS
We have developed a new efficient algorithm to determine the most probable sequence-structure motif, or combination of motifs, given a large set of RNA sequences. As RNA secondary structure is required for the specificity of RBP binding, and large-scale assays are becoming more and more popular in studying RNA–RBPs interactions, our method will contribute to the systematic understanding of such interactions.
DATA AVAILABILITY
The ssHMM software is available for download at github.molgen.mpg.de/heller/ssHMM. Documentation of the tool is provided at sshmm.readthedocs.io. Synthetic and CLIP-Seq datasets used in this study can be found at github.molgen.mpg.de/heller/ssHMM_data.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the developers of the GHMM Python package that is used by ssHMM. They also wish to thank Donald Buczek for testing ssHMM and Brian Caffrey, Alina Munteano and Edgar Steiger for proofreading of the manuscripts. Furthermore, they would like to thank the reviewers for their constructive and helpful comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Freie Universität Berlin; Deutsche Forschungsgemeinschaft [SFB-TR 84]. Funding for open access charge: Institutional funding from the Max Planck Institute for Molecular Genetics, Berlin.
Conflict of interest statement. None declared.
REFERENCES
- 1. Glisovic T., Bachorik J.L., Yong J., Dreyfuss G.. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008; 582:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M. et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012; 149:1393–1406. [DOI] [PubMed] [Google Scholar]
- 3. Fukunaka T., Ozaki H., Terai G., Asai K., Iwasaki W., Kiryu H.. CapR: revealing structural specificities of RNA-binding protein target recognition using CLIP-seq data. Genome Biol. 2014; 15:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kazan H., Ray D., Chan E.T., Hughes T.R., Morris Q.. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput. Biol. 2010; 6:e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. D’haeseleer P. How does DNA sequence motif discovery work?. Nat. Biotechnol. 2006; 24:959–961. [DOI] [PubMed] [Google Scholar]
- 6. Sinha S., Tompa M.. YMF: a program for discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res. 2003; 31:3586–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thomas-Chollier M., Sand O., Turatsinze J., Janky R., Defrance M., Vervisch E., Brohée S., van Helden J.. RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 2008; 36:W119–W127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Georgiev S., Boyle A.P., Jayasurya K., Ding X., Mukherjee S., Ohler U.. Evidence-ranked motif identification. Genome Biol. 2010; 11:R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D’haeseleer P. What are DNA sequence motifs?. Nat. Biotechnol. 2006; 24:423–425. [DOI] [PubMed] [Google Scholar]
- 10. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 11. Lawrence C.E., Altschul S.F., Boguski M.S., Liu J.S., Neuwald A.F., Wootton J.C.. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993; 262:208–214. [DOI] [PubMed] [Google Scholar]
- 12. Maerkl S.J., Quake S.R.. A systems approach to measuring the binding energy landscapes of transcription factors. Science. 2007; 315:233–237. [DOI] [PubMed] [Google Scholar]
- 13. Roider H.G., Kanhere A., Manke T., Vingron M.. Predicting transcription factor affinities to DNA from a biophysical model. Bioinformatics. 2007; 23:134–141. [DOI] [PubMed] [Google Scholar]
- 14. Stormo G.D., Zhao Y.. Determining the specificity of protein–DNA interactions. Nat. Rev. Genet. 2010; 11:751–760. [DOI] [PubMed] [Google Scholar]
- 15. Manley J.L. SELEX to identify protein-binding sites on RNA. Cold Spring Harb. Protoc. 2013; 2013:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ray D., Kazan H., Chan E.T., Castillo L.P., Chaudhry S., Talukder S., Blencowe B.J., Morris Q., Hughes T.R.. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009; 27:667–670. [DOI] [PubMed] [Google Scholar]
- 17. Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B.. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003; 302:1212–1215. [DOI] [PubMed] [Google Scholar]
- 18. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M., Jungkamp A., Munschauer M. et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J.. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010; 17:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uren P.J., Bahrami-Samani E., Burns S.C., Qiao M., Karginov F.V., Hodges E., Hannon G.J., Sanford J.R., Penalva L.O., Smith A.D.. Site identification in high-throughput RNA–protein interaction data. Bioinformatics. 2012; 28:3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corcoran D.L., Georgiev S., Mukherjee N., Gottwein E., Skalsky R.L., Keene J.D., Ohler U.. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011; 12:R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friedersdorf M.B., Keene J.D.. Advancing the functional utility of PAR-CLIP by quantifying background binding to mRNAs and lncRNAs. Genome Biol. 2014; 15:R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Helden J., André B., Collado-Vides J.. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J. Mol. Biol. 1998; 281:827–842. [DOI] [PubMed] [Google Scholar]
- 24. Dao P., Hoinka J., Takahashi M., Zhou J., Ho M., Wang Y., Costa F., Rossi J., Backofen R., Burnett J., Przytycka T.. AptaTRACE Elucidates RNA Sequence-Structure Motifs from Selection Trends in HT-SELEX Experiments. Cell Syst. 2016; 3:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brazma A., Jonassen I., Vilo J., Ukkonen E.. Predicting Gene Regulatory Elements in Silico on a Genomic Scale. Genome Res. 1998; 8:1202–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bailey T.L., Williams N., Misleh C., Li W.W.. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006; 34:W369–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Foat B.C., Morozov A.V., Bussemaker H.J.. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006; 22:e141–e149. [DOI] [PubMed] [Google Scholar]
- 28. Thijs G., Marchal K., Lescot M., Rombauts S., De Moor B., Rouzé P., Moreau Y.. A Gibbs Sampling Method to Detect Overrepresented Motifs in the Upstream Regions of Coexpressed Genes. J. Comput. Biol. 2002; 9:447–464. [DOI] [PubMed] [Google Scholar]
- 29. Liu X., Brutlag D.L., Liu J.S.. BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. 2001; 127–138. [PubMed] [Google Scholar]
- 30. Shida K. GibbsST: a Gibbs sampling method for motif discovery with enhanced resistance to local optima. BMC Bioinformatics. 2006; 7:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hiller M., Pudimat R., Busch A., Backofen R.. Using RNA secondary structures to guide sequence motif finding towards single-stranded regions. Nucleic Acids Res. 2006; 34:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bahrami-Samani E., Penalva L.O., Smith A.D., Uren P.J.. Leveraging cross-link modification events in CLIP-seq for motif discovery. Nucleic Acids Res. 2015; 43:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maticzka D., Lange S.J., Costa F., Backofen R.. GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol. 2014; 15:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sigova A.A., Abraham B.J., Ji X., Molinie B., Hannett N.M., Guo Y.E., Jangi M., Giallourakis C.C., Sharp P.A., Young R.A.. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015; 350:978–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steffen P., Voß B., Rehmsmeier M., Reeder J., Giegerich R.. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics. 2006; 22:500–503. [DOI] [PubMed] [Google Scholar]
- 36. Reuter J.S., Mathews D.H.. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010; 11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blin K., Dieterich C., Wurmus R., Rajewsky N., Landthaler M., Akalin A.. DoRiNA 2.0 - upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2015; 43:D160–D167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological). 1995; 57:289–300. [Google Scholar]
- 39. Jensen K.B., Dredge B.K., Stefani G., Zhong R., Buckanovich R.J., Okano H.J., Yang Y.Y.L., Darnell R.B.. Nova-1 Regulates Neuron-Specific Alternative Splicing and Is Essential for Neuronal Viability. Neuron. 2000; 25:359–371. [DOI] [PubMed] [Google Scholar]
- 40. Chénard C.A., Richard S.. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 2008; 86:233–242. [DOI] [PubMed] [Google Scholar]
- 41. Vermeulen A., Behlen L., Reynolds A., Wolfson A., Marshall W.S., Karpilow J., Khvorova A.. The contributions of dsRNA structure to Dicer specificity and efficiency. RNA. 2005; 11:674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Macias S., Plass M., Stajuda A., Michlewski G., Eyras E., Cáceres J.F.. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat. Struct. Mol. Biol. 2012; 19:760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roth B.M., Ishimaru D., Hennig M.. The core microprocessor component DiGeorge syndrome critical region 8 (DGCR8) is a nonspecific RNA-binding protein. J. Biol. Chem. 2013; 288:26785–26799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Han J., Lee Y., Yeom K.-H., Kim Y., Jin H., Kim V.N.. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004; 18:3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Auyeung V.C., Ulitsky I., McGeary S.E., Bartel D.P.. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013; 152:844–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Conrad T., Marsico A., Gehre M., Ørom U.A.. Microprocessor activity controls differential miRNA biogenesis in vivo. Cell Rep. 2014; 9:542–554. [DOI] [PubMed] [Google Scholar]
- 47. Jeon Y., Lee e.T.. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011; 146:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schneider T.D., Stormo G.D., Gold L., Ehrenfeucht A.. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 1986; 188:415–431. [DOI] [PubMed] [Google Scholar]
- 49. Shao Y., Chan C.Y., Maliyekkel A., Lawrence C.E., Roninson I.B., Ding Y.. Effect of target secondary structure on RNAi efficiency. RNA. 2007; 13:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kertesz M., Iovino N., Unnerstall U., Gaul U., Segal E.. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007; 39:1278–1284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ssHMM software is available for download at github.molgen.mpg.de/heller/ssHMM. Documentation of the tool is provided at sshmm.readthedocs.io. Synthetic and CLIP-Seq datasets used in this study can be found at github.molgen.mpg.de/heller/ssHMM_data.