

Abstract
Characterizing and quantifying genome size variation among organisms and understanding if genome size evolves as a consequence of adaptive or stochastic processes have been long-standing goals in evolutionary biology. Here, we investigate genome size variation and association with transposable elements (TEs) across lepidopteran lineages using a novel genome assembly of the common wood-white (Leptidea sinapis) and population re-sequencing data from both L. sinapis and the closely related L. reali and L. juvernica together with 12 previously available lepidopteran genome assemblies. A phylogenetic analysis confirms established relationships among species, but identifies previously unknown intraspecific structure within Leptidea lineages. The genome assembly of L. sinapis is one of the largest of any lepidopteran taxon so far (643 Mb) and genome size is correlated with abundance of TEs, both in Lepidoptera in general and within Leptidea where L. juvernica from Kazakhstan has considerably larger genome size than any other Leptidea population. Specific TE subclasses have been active in different Lepidoptera lineages with a pronounced expansion of predominantly LINEs, DNA elements, and unclassified TEs in the Leptidea lineage after the split from other Pieridae. The rate of genome expansion in Leptidea in general has been in the range of four Mb/Million year (My), with an increase in a particular L. juvernica population to 72 Mb/My. The considerable differences in accumulation rates of specific TE classes in different lineages indicate that TE activity plays a major role in genome size evolution in butterflies and moths.
Keywords: butterfly, Lepidoptera, Leptidea, genome expansion, transposable elements, population
Introduction
Causes and consequences of variation in genome size across taxa have been a matter of debate in the field of evolutionary biology (Petrov 2001; Cavalier-Smith 2005; Gregory 2005; Lynch 2007; Oliver etal. 2007). The observations that variation in genome size in extant species is on the level of ten- to hundred thousand folds (Gregory 2004) and that genome size correlates only weakly with the amount of coding sequence and the organism complexity (the C-value paradox; Thomas Jr. 1971) have rendered a lot of attention and attempts to understand the underlying mechanisms generating this variation (Petrov 2001; Cavalier-Smith 2005; Gregory 2005; Lynch 2007; Oliver etal. 2007; Fontdevila 2011). Key mutation classes that contribute to genome expansion are predominantly transposable element (TE) proliferations (e.g., Pritham 2009; Tenaillon etal. 2010), gene- (e.g., Ohno 1970; Lu etal. 2012) or genome duplications (Fontdevila 2011) and replication slippage of tandem repeat sequences (Ellegren 2004). Quantification of the rate and prevalence of these mutation types is essential to get a comprehensive understanding of the forces that shape genome size evolution in different lineages. Two major lines of argument have been put forward to explain the variation in genome size. In essence, these can be sorted under adaptive processes on the one hand (Gregory and Hebert 1999; Cavalier-Smith 2005; Gregory 2005), and stochastic gain or loss of genomic regions on the other (Petrov 2001, 2002a, b; Lynch 2007). The former make a case that addition of DNA can influence cell size and rate of cell division, which might have an effect on organism development, or that selection for increased cell size drives genome expansion to increase stability and/or enhance molecular transport from nucleus to cytoplasm (Cavalier-Smith 2005). Non-adaptive models rather focus on mutation and fixation biases of insertions and deletions as a consequence of random genetic drift driving genome expansion or contraction (Petrov 2001; Lynch 2007)—for example, that genome expansion is mainly driven by proliferation of TEs, ‘selfish-DNA’ that either copy and paste themselves passing through an RNA intermediary step (retrotransposons), or cut and paste themselves (DNA transposons) within the genome of the host organism (Orgel and Crick 1980; Kazazian 2004; Kidwell 2005). The role of TE proliferation for genome size variation is supported by the universal existence of TEs in virtually all eukaryotic genomes (Elliott and Gregory 2015) and genomic gigantism in salamanders is for example a result of hyperactivity of specific long terminal repeats (LTRs) (Sun etal. 2012). However, also amounts of non-repetitive DNA (non-coding, non-TE) tend to co-vary with genome size indicating that non-TE insertion/deletion forces might play an important role in genome size variation (Lynch 2007). Prokaryotes have compact genomes almost entirely consisting of protein coding genes, while the amount of non-coding DNA is higher in unicellular eukaryotes and constitutes the major part of the genome in multicellular eukaryotes (Lynch 2007). This has been attributed to be a consequence of an increased cost of replication of excess DNA that is more efficiently selected against in prokaryotes and unicellular eukaryotes that generally have very large effective population sizes, although there are currently no data at hand that directly verify this assumption (Lynch 2007). Interestingly, TE insertions have also been shown to be hindered by epigenetic mechanisms underlying inhibition of homologous recombination in prokaryotes (Fedoroff 2012).
In insects, observed genome size variation ranges from 0.09 to 16 Gb (Gregory etal. 2007; Hanrahan and Johnston 2011; Maumus etal. 2015), but sampling is sparse and sporadic with low representation of particular taxonomic groups, and there is indecisive support for natural selection on the one hand (Arnqvist etal. 2015), and/or genetic drift on the other (Lefébure etal. 2017) in driving genome size evolution (Sessegolo etal. 2016). Within the order Lepidoptera (butterflies and moths) there is considerable genome size variation (up to 7-fold) but, although these insects are among the most diverse groups of invertebrates, so far very few species have been studied in detail (Gregory and Hebert 2003). To investigate the impact of TEs on genome size variation in this understudied group of organisms, we here quantify the abundance of different classes of TEs across species and populations with considerable variation in genome sizes. We then use a phylogenetic approach to estimate genome expansion rates as a consequence of bursts of TE proliferation. Our main study system consists of three cryptic species of butterflies from the Eurasian genus Leptidea (Pieridae), namely the wood-whites Leptidea sinapis, L. reali, and L. juvernica. This triplet represents one of the most striking examples of cryptic species in Eurasian butterflies (Dincă etal. 2011, 2013) and has been widely studied in terms of ecology (e.g., Friberg and Wiklund 2010; Friberg etal. 2011), behavior (e.g. Wiklund 1977; Friberg and Wiklund 2007) and chromosome evolution (e.g., Lukhtanov etal. 2011; Šíchová etal. 2016). The results from these studies call for a better understanding of the genomic particularities of Leptidea, but there are very limited data on genome architecture and genetic variation within and between species, both in this genus and as compared with other lepidopterans. Preliminary data suggest that there is considerable nucleus size differences between species in the Leptidea genus (Šíchová etal. 2015), potentially indicating that there is substantial genome size variation between the species despite recent estimated divergence times (Dincă etal. 2011). To quantify the presence and accumulation of TEs, we assessed the interspersed repeat content of six populations from three cryptic Leptidea species and compared the results with lepidopteran taxa with previously available genome assemblies. Our results indicate that genome expansion can occur rather quickly [∼70 Mb/Million years (My)] as a result of activity of specific repeat elements in certain lineages, potentially as a consequence of increased rate of TE proliferation during periods with reduced effective population size.
Materials and Methods
Genome Assembly
Offspring from one mated L. sinapis female collected in Sweden during June 2013 were recurrently inter-crossed to generate a five-generation full-sib inbred line. Larvae of the fifth generation were kept until the ultimate (fifth) instar before being harvested for DNA extraction. Genomic DNA was extracted using a standard phenol–chloroform protocol (Sambrook etal. 1989) from three full siblings from the fifth generation. Illumina paired-end libraries with insert sizes 180 and 650 bp were generated using genomic DNA obtained from one of the sampled individuals. Two Illumina mate-pair libraries with insert sizes 4 and 8 kb, respectively, were generated using the remaining two individuals (one for each library). All libraries were sequenced to deep coverage (>150X paired-end coverage, >100X mate-pair coverage) using Illumina HiSeq 2500 technology (Illumina, Inc., San Diego, USA) with 125 bp read length. The sequences were assembled using ALLPATHS-LG (Butler etal. 2008) to obtain a draft reference genome assembly that was 650 Mb in total length and consisted of 7,096 scaffolds. The draft assembly was then screened for potential contaminants using the NCBI BLAST database (Altschul etal. 1990). Five scaffolds were identified as potential contaminants based on high sequence similarity (e-value < e−10) to non-lepidopteran taxa and deviating overall GC content and were removed from the assembly. The removed scaffolds were in total 134,034 bp long with four shorter (2 kb each) and one 128 kb long. The longest and three of the shorter removed scaffolds were likely remaining host plant material since they had high sequence similarity to plant species Trifolium pratense and Lotus japonicus (Leptidea butterfly larvae feed on plants from the pea family, Fabaceae). One of the three shorter removed scaffolds was 100% identical to a plasmid (suicide vector pCD-RAsl1). We assembled the L. sinapis mtDNA genome by mapping all L. sinapis reads to the previously available mitochondrial genome of the close relative Leptidea amurensis (Hao etal. 2014; Šíchová etal. 2016) using BWA (Li and Durbin 2010), and assembling the reads using ABySS (Simpson etal. 2009). The process was repeated once with the de novo assembled L. sinapis mtDNA genome as reference for mapping. The final mtDNA assembly matched one scaffold from the genome assembly to 100%. The mtDNA assembly was 15,171 bp long and was annotated using the Mitos web server (Bernt etal. 2013). After removing likely contaminant sequences and the mtDNA-derived scaffold, the final nuclear genome assembly used for downstream analysis consisted of 7,090 scaffolds spanning in total 643 Mb. The scaffold N50 was 857.2 kb and 95% of the assembly was covered by 1,083 scaffolds. The quality of the assembly was further assessed by reciprocal similarity searches of scaffolds with BLAST (Altschul etal. 1990) and to identify potentially false duplicated regions we screened the assembly for conserved genes using BUSCO (Simao etal. 2015). Since PCR-based sequencing techniques may be GC-biased (Kozarewa etal. 2009), we did a GC-corrected assembly size estimate following the procedure by Warr etal. (2015) and Davey etal. (2016). In addition, K-mer analyses were performed for all libraries used for the genome assembly using the K-mer Analysis Toolkit, KAT (Mapleson etal. 2017). The rationale behind this was to visually inspect the distribution of k-mers to assess completeness, heterozygosity and potential collapsed repeats in the genome assembly. To get an additional and independent estimate of the genome size in L. sinapis, we also generated a Supernova genome assembly (Weisenfeld etal. 2017) from 10X Genomics Chromium linked-read data. Paired-reads of 150 bp were sequenced on the Illumina HiSeq X technology (Illumina, Inc., San Diego, USA) using a Chromium library with maximized insert sizes from the same DNA as used for paired-end libraries of the Allpaths-LG assembly. The size of the Supernova assembly was the same as the ALLPATHS-LG assembly, further supporting that the genome size of L. sinapis is in the range of 650 Mb. The genome statistics and comparisons with other previously available butterfly and moth genomes are provided in table 1 and the comparison of the ALLPATHS-LG and Supernova assemblies are presented in supplementary table 1 and figure 1, Supplementary Material online [both assemblies are available at the European Nucleotide Archive (ENA) under project accession numbers ERZ468508 and ERS1830260, respectively].
Table 1.
Genome Assembly Statistics of L. sinapis (boldface) and Comparisons to 12 Previously Available Lepidopteran Genomes (http://www.lepbase.org; Challis etal. 2017)
| Species | Assembly | #Scaff | N50 | LS | GC | BUSCO |
|---|---|---|---|---|---|---|
| B. mori | 482 | 43, 463 | 4.0 | 16.2 | 41.7 | 54 |
| C. cecrops | 729 | 60, 049 | 0.23 | 2.0 | 37.1 | 56 |
| D. plexippus | 249 | 5, 397 | 0.715 | 6.2 | 28.0 | 67 |
| H. Melpomene | 275 | 795 | 2.1 | 9.4 | 33.1 | 55 |
| L. sinapis | 643 | 7, 090 | 0.857 | 6.9 | 31.7 | 54 |
| L. accius | 298 | 29, 988 | 0.525 | 3.1 | 35.6 | 57 |
| M. sexta | 419 | 20, 871 | 0.664 | 3.3 | 35.2 | 63 |
| M. cinxia | 390 | 8, 261 | 0.119 | 0.7 | 31.0 | 34 |
| P. glaucus | 376 | 68, 029 | 0.23 | 2 | 37.4 | 56 |
| P. polytes | 227 | 3, 874 | 3.7 | 9.9 | 35.0 | 53 |
| P. xuthus | 243 | 15, 362 | 3.4 | 13.8 | 33.5 | 62 |
| P. sennaea | 345 | 20, 800 | 0.256 | 2.2 | 33.4 | 63 |
| P. rapaea | 246 | 7, 348 | 0.617 | 3.3 | 32.7 | 57 |
NOTE.—Assembly size (Assembly), N50 for scaffolds (N50) and longest scaffold (LS) are given in Mb, and GC-content (GC) and fraction of completely covered conserved arthropod genes (BUSCO) are given in %
The genome assemblies for these species were downloaded from the web repository: http://prodata.swmed.edu/LepDB/, last accessed June 9, 2017 (Cong etal. 2016; Shen etal. 2016).
Fig. 1.

—Phylogenetic relationship and divergence time estimates for 15 Lepidoptera species with available genome assemblies including a detailed representation of the Leptidea clade with one representative accession for each of the 6 populations analysed. The phylogeny is constructed using 224 conserved, single-copy orthologous arthropod genes with previously available anchoring points for divergence time estimates. Error bars on nodes indicate the 95% confidence interval for divergence time estimates (scale bar at the bottom) and the tree is colored according to genome size of included taxa using Phytools (Revell 2012) (scale bar at bottom right). Bootstrap support values for nodes are given when <100.
Population Sampling and Re-Sequencing
Ten individuals from each of six different populations [L. sinapis central Sweden (LsSwe), L. sinapis northern Spain (LsSpa), L. sinapis eastern Kazakhstan (LsKaz), L. reali northern Spain (LrSpa), L. juvernica eastern Kazakhstan (LjKaz), and L. juvernica Ireland (LjIre)] were sampled in the field during seasons 2013–2015. DNA was extracted from head and thorax combined using standard phenol–chloroform procedures (Sambrook etal. 1989). Each sample was prepared for sequencing by generating individually barcoded, 380-bp paired-end Illumina libraries. Samples were multiplexed and sequenced using Illumina HiSeq technology (Illumina, Inc., San Diego, USA). Library preparations and sequencing were done by the SNP&SEQ Technology Platform at the Science for Life Laboratory (SciLife, Stockholm and Uppsala). Sequencing was performed twice, the first run included LsSpa, LsKaz, LrSpa, and LjKaz and was run on the Illumina HiSeq 2000 instrument (100 bp read length), multiplexing on two separate lanes (20 samples per lane). The second bout of sequencing was performed using updated chemistry (125 bp read length) and included all populations; LsSpa, LsKaz, LrSpa, and LjKaz were multiplexed on two lanes (20 samples per lane) and LsSwe and LjIre were multiplexed on two lanes (10 samples per lane) and ran on the Illumina HiSeq 2500 instrument. The final sequencing reads obtained for each individual were trimmed for remaining adapter sequences and low quality bases (Q-score <30) using the tool Cutadapt (Martin 2011). The overall coverage of quality filtered reads was estimated to be 8–15X per individual given a L. sinapis reference genome size of 650 Mb (see above). All sequence reads for genome assembly and population re-sequencing have been deposited at the ENA under project accession number PRJEB21838 (for details see supplementary tables 1 and 2, Supplementary Material online).
Table 2.
Counts of Categories of Highly Conserved Arthropod Genes Present in the L. sinapis Assembly Assessed by the BUSCO Gene Search Tool (Simao etal. 2015)
| Category | Arthropod | Eukaryote |
|---|---|---|
| Total searched | 2,718 | 434 |
| Complete | 1,447 | 284 |
| Duplicated | 43 | 5 |
| Fragmented | 818 | 27 |
| Missing | 410 | 118 |
K-mer-Based Genome Size Estimates
To estimate the approximate genome size of each species/population, mean sequencing depth was estimated for each individual using the K-mer counting tool JELLYFISH (Marcais and Kingsford 2011). Histograms were generated using the “-histo” command in JELLYFISH to identify the distinct K-mer peak by plotting multiplicity of unique K-mers versus the total number of K-mers (cf. Guo etal. 2015). All uneven K-mer lengths from 15 to 35 were assessed and the optimal K-mer length was estimated to be 17 (most distinct peaks). Sequencing reads were then randomly sub-sampled to the coverage of 5X for each individual to reduce potential coverage bias on the genome size estimate. The randomly sampled reads were used to generate K-mer histograms (multiplicity of unique K-mers with size 17 vs. the total number of K-mers) using JELLYFISH (Marcais and Kingsford 2011). The genome size of each individual was estimated by dividing the total number of nucleotides in the read set with the estimated coverage. This procedure has previously been used to estimate genome size of, for example, Bemisia tabaci and the conclusion has been that K-mer-based estimates are inflated compared with what was expected from flow cytometry (Guo etal. 2015). To correct for this bias, we scaled the K-mer-based estimates for each sample with the genome size information we have from our L. sinapis reference genome (see above). All in-house developed scripts for the genome size estimation are provided in a GitHub repository (https://github.com/venta380/Leptidea-Genome-size-scripts) and a detailed description of the steps and K-mer distributions for L. sinapis genome assembly libraries (180 bp PE, 650 bp PE, 3 kb MP, 8 kb MP) and all individuals from each of the six Leptidea populations (380 bp PE) are provided in supplementary figure 2, Supplementary Material online. The distribution graphs were generated using KAT (Mapleson etal. 2017) for high-coverage (genome assembly) libraries and in-house developed R-scripts for low-coverage (population re-sequencing) libraries. Assessment of genome-size variation across Leptidea populations and pin-pointing the deviating population was done by applying the rank-based Kruskal–Wallis test followed by the Nemenyi‘s test of multiple comparisons as implemented in R (https://www.r-project.org/).
Phylogenetic Reconstruction
Genome assemblies of ten butterfly and moth species (Bombyx mori, Calycopis cecrops, Danaus plexippus, Heliconius melpomene, Lerema accius, Manduca sexta, Melitaea cinxia, Papilio glaucus, Papilio polytes, and Papilio xuthus) were obtained from the Lepbase database (http://www.lepbase.org; last accessed June 9, 2017, Challis etal. 2017) and the genome sequences of two additional species, Phoebis sennae and Pieris rapae, were obtained from the web repository: http://prodata.swmed.edu/LepDB/, last accessed June 9, 2017, (Cong etal. 2016; Shen etal. 2016). Our L. sinapis reference genome was used for the Swedish L. sinapis population and whole-genome consensus sequences were generated for the individuals with the highest coverage from each of the remaining five Leptidea populations using the “mpileup” command in SAMtools (Li etal. 2009). The set of 18 genome sequences was screened for core arthropod genes using BUSCO v.1.22 (Simao etal. 2015). We found 224 complete single copy genes common to all the 18 scanned genomes in the arthropod gene set. The protein sequences for each of these genes were obtained for each species and aligned using MAFFT (Katoh and Standley 2013). A global phylogeny was generated using RAxML v.8.2.4 (Stamatakis 2006) with the amino acid substitution model “PROTGAMMAGTR” to allow for rate variation among sites and calculate the most accurate likelihood scores (Izquierdo-Carrasco etal. 2011), and node support was estimated using 10,000 bootstrap iterations.
Estimating Divergence Times
Protein coding nucleotide sequences were obtained using genomic coordinates for the previously identified 224 common single-copy genes and the sequences were aligned using PRANK (Löytynoja and Goldman 2005). The alignments were corrected by taking codon constraints into account using MACSE (Ranwez etal. 2011). The corrected alignments were used to estimate the divergence time between the focal (Leptidea) group and the most closely related taxa in the previously available genome dataset (P. sennae, P. rapae; Pieridae), and the divergence time between clades within the Leptidea species set using BEAST v.1.7.5 (Drummond etal. 2012). Prior probabilities were set using previously available divergence time estimates (Hedges and Kumar 2009). The prior for the time to most recent common ancestor (TMRCA) of families Papilionidae and Nymphalidae was set at 122 million years (My) with a standard deviation of 12 My (Espeland etal. 2015), the priors for the internal TMRCAs within these families were set to 31 ± 10 and 103 ± 12 My, respectively (Hedges and Kumar 2009; Wahlberg etal. 2009; Nazari etal. 2011; Condamine etal. 2012), and the prior for TMRCA of Pieridae was set to 79 ± 12 My (Heikkilä etal. 2012). All priors were set to represent normal distributions with a log-normal relaxed clock and multiple hits were corrected for using the GTR substitution model as implemented in the software. The phylogeny established using RAxML (see above) was given as a guide tree. The divergence time estimates for Leptidea—Phoebis/Pieris and for LjKaz—LjIre were used to estimate genome expansion rates in Leptidea in general after the split from the other Pieridae butterflies (Phoebis and Pieris), and in LjKaz after the split from LjIre.
RepeatModeler and RepeatMasker Analyses
We conducted RepeatModeler version 1.0.8 (Smit and Hubley 2010) de-novo predictions of repetitive elements in each of the following 12 lepidopteran genome assemblies: B. mori, C. cecrops, D. plexippus, H. melpomene, L. accius, M. sexta, M. cinxia, P. glaucus, P. polytes, P. rapae, P. xuthus, and P. sennae. We merged the resultant raw libraries with curated in-depth repeat annotations of H. melpomene (Lavoie etal. 2013), Heliconius erato (Ray DA, unpublished data), and all Hexapoda repeats available in Repbase (Bao etal. 2015). Redundancies between the eleven RepeatModeler libraries and the existing curated libraries were removed using the ReannTE_mergeFasta.pl script available via https://github.com/4ureliek/ReannTE/, last accessed June 12, 2017 (Kapusta etal. 2017) while giving priority to retaining curated consensus sequences. We then annotated all sampled lepidopteran genome assemblies via RepeatMasker version 4.0.6 with the “ncbi” search algorithm (Smit etal. 1996–2010) using this specific library (supplementary data file 1, Supplementary Material online). Landscapes of relative TE activity were generated using the calcDivergenceFromAlign.pl and createRepeatLandscape.pl scripts of the RepeatMasker packages. Ages of TE copies were inferred by dividing each TE copy‘s Kimura 2-parameter distance to respective TE consensus by the neutral substitution rate. Lineage-specific neutral substitution rates were based on 4-fold degenerate sites in 224 coding genes that were also used to reconstruct the phylogenetic relationship between all taxa in the study (see below). For plot readability, we grouped TE families into the subclasses short interspersed elements (SINEs), long interspersed elements (LINEs), cut-and-paste DNA transposons (DNA elements), LTR elements, and “Unclassified”.
dnaPipeTE Analyses
To estimate within-population differences in TE abundances, we analyzed the Leptidea spp. re-sequencing data using dnaPipeTE (Goubert etal. 2015). DnaPipeTE performs de novo assembly of TEs from a low-coverage subsample of re-sequencing reads in Trinity (Grabherr etal. 2011), followed by automatic quantification and annotation of TEs in the re-sequencing reads together with Repbase repeats. DnaPipeTE thus allows the quantification of recently active repeat elements in re-sequencing data, unlike RepeatMasker which quantifies repeats with a wide range of ages across genome assemblies. To optimize the amount of re-sequencing data for dnaPipeTE subsampling of each population, we selected one individual per Leptidea spp. population and ran dnaPipeTE on subsamples ranging between 200,000 and 1,200,000 reads in intervals of 100,000 reads (11 runs). For each of the 11 runs per individual, we selected the subsample yielding the highest contig N50 metric in the Trinity assembly step of dnaPipeTE, as a measure of optimized read subsampling. The optimized read subsample (LsSwe = 500,000 reads, LsKaz = 400,000 reads, LsSpa = 500,000 reads, LrSpa = 600,000 reads, LjIre = 300,000 reads, LjKaz = 700,000 reads) was then used to run dnaPipeTE on the remaining nine individuals of each population, respectively. Similar to the aforementioned RepeatMasker annotations, TE families were grouped into TE subclasses “DNA elements”, “SINEs”, “LINEs”, “LTRs”, and “Unclassified”.
To get time estimates of variation in TE activity over the course of butterfly divergence, the divergence levels between repeats within each class were time scaled using neutral mutation rate estimates (2.9×10−9 mutations per site per generation) from H. melpomene (Keightley etal. 2015), assuming one generation per year in general. Current generation times vary considerably across lepidopteran taxa (Boggs etal. 2003) and many species show regional variation in voltinism dependent on climatic conditions. Within Leptidea for example, populations inhabiting the regions in central and southern Europe are usually multivoltine while populations in the northern part of the distribution range are univoltine (Friberg and Wiklund 2007). In addition, over evolutionary time scales the generation times may have varied in lineages due to variation in climatic conditions and distribution ranges (Altermatt 2010). The estimated divergence times of different repeat classes may thus not be seen as absolute values comparable across all lineages but rather as relative temporal variation in repeat proliferation rates within lineages.
Results
The L. sinapis Genome Assembly
The size of the L. sinapis genome assembly was 643 Mb (table 1), considerably larger than the size of the closest relatives with sequenced and well-characterized genome, the cloudless sulphur P. sennae (Cong etal. 2016) and the small cabbage white P. rapae (Shen etal. 2016). Reciprocal similarity searches of all repeat masked scaffolds using BLAST (Altschul etal. 1990) did not reveal any significantly similar scaffolds indicating false duplication during the assembly process. At a sequence similarity level of ≥ 95%, the average scaffold proportion that aligned to other scaffolds was only 1.55%, similar to what is observed when running an identical analysis for H. melpomene scaffolds (1.69%). The GC corrected assembly estimate was 569 Mb, which is smaller than the genome assembly (643 Mb), but this method excludes gaps (N:s) which constitute 10.5% of the L. sinapis assembly—gaps included, the GC corrected estimate hence matches the de novo assembly estimate very well (637 Mb). The assembly contained large proportions of single-copy, highly conserved eukaryotic [n = 434 entries in total, 311 (72%) identified in L. sinapis] and arthropod [2,718 entries, 2,265 (83%) identified in L. sinapis] gene sets, and we found only a minor fraction of duplicated genes [43 (1.6%) and 5 (1.8%) in each respective gene set, table 2]. The BUSCO scores (Simao etal. 2015) also indicated that the fractions of genes that were entirely missing [118 (27%) and 410 (15%) for each class, respectively, table 2] were similar to what has been observed in previous high-quality butterfly genome assemblies (The Heliconius Genome Sequencing Consortium 2012; Cong etal. 2016; Davey etal. 2016). In addition, the additional assembly effort using a different sequencing approach (10X Genomics Chromium linked-reads) and assembly technique (“Supernova”, Weisenfeld etal. 2017) resulted in the same total assembly length (643 Mb, supplementary fig. 1 and table 1, Supplementary Material online). The k-mer analysis of genome assembly libraries consistently showed a distinct peak with limited heterozygosity and ∼50% repeat content and no obvious high-coverage fraction indicating collapsed repeats (supplementary fig. 2, Supplementary Material online). This indicates that the genome assembly of L. sinapis is of high quality and can be used to infer the underlying reasons for the significantly larger genome size of this species compared with most previously characterized lepidopteran taxa.
Reconstruction of Phylogenies and Rate Estimates
Phylogenetic reconstruction using the concatenated 224 core single-copy arthropod genes from the 16 lepidopteran genomes verified previously established topologies (fig. 1), both for global Lepidoptera species relationships (Cong etal. 2016), and for the Leptidea cryptic species complex (Dincă etal. 2011). The phylogenetic analysis also supported considerable genetic structuring within the Leptidea clade, LsSpa being reciprocally monophyletic to the combined populations of LsSwe and LsKaz, and LjIre being reciprocally monophyletic as compared to LjKaz (fig. 1). Our estimated divergence times corresponded well with previous estimates of divergence times within Lepidoptera. For instance, the split between H. melpomene and M. cinxia was estimated to ca. 68.1 My (95% CI 56.8–79.6 My) in our analysis, matching previous estimates well (64.7–78.5 My), and the divergence time between P. xuthus and P. polytes has previously been estimated to 23–40 My which agrees with our estimate of ca. 28.8 My (95% CI 23.7–34.5 My) (Hedges and Kumar 2009; Wahlberg etal. 2009; Nazari etal. 2011). Our main interest was to get approximate divergence time estimates for the Leptidea clade as compared with other Pieridae (represented by P. sennae and P. rapae) and for the species within the Leptidea genus to estimate the rate of genome size changes. We found that the TMRCA of the Leptidea and the P. sennae/P. rapae lineage was 80.6 My (95% CI 67.6–93.2 My) and the TMRCA for the whole Leptidea species complex was ∼3.0 My (95% CI 2.5–3.6 My). Within the Leptidea clade, the estimates of TMRCA for L. sinapis (LsSwe, LsKaz, LsSpa) and L. juvernica (LjKaz, LjIre) populations were 1.5 My (95% CI 1.1–1.8 My) and 1.6 My (95% CI 1.3–2.0 My), respectively (fig. 1).
Association between Genome Size and Repeat Content in Lepidoptera
To assess the fractions of interspersed repeat elements, we scanned 13 representative lepidopteran genome assemblies for interspersed repeat content. There was a very strong association (Pearson‘s; n = 13, r = 0.800, P = 0.001) between genome size and proportion of repeat elements across species and L. sinapis was at the extreme end with a considerably larger proportion of repeats in the genome than other lepidopteran species (38%; fig. 2). To further investigate the contribution of specific repeat groups to genome size variation across Lepidoptera we counted the abundance of SINEs, LINEs, DNA elements, LTR elements, and unclassified interspersed repeats using the RepeatModeler/RepeatMasker tools and found a difference in occurrence of specific repeat classes between taxa (fig. 3, supplementary table 3, Supplementary Material online). Specifically, the L. sinapis genome contained a larger proportion of LINEs (5.4%), DNA elements (5.7%), and unclassified repeats (22.8%) than most other taxa, while fractions of other repeat subclasses (SINEs and LTRs) were within the main range of other lepidopterans (fig. 3, supplementary table 3, Supplementary Material online). When counting the number of repeat subfamilies identified by RepeatModeler (Smit and Hubley 2010) and Repeat Masker (Smit etal. 1996–2010), present with ≥1,000 copies in a specific genome, the range was between six (D. plexippus) and 235 (L. sinapis) (supplementary table 4a, Supplementary Material online). L. sinapis had the highest LINE subfamily repeat count of any species, 4.8 times higher than the average across all other lineages, and comparatively high counts of DNA element (3.8 times higher than the mean in L. sinapis, highest in C. cecrops), LTR (3.4 times higher than mean, highest in C. cecrops), and unclassified subfamily repeats (2.5 times higher than the mean, highest in C. cecrops). The number of SINE subfamily repeats observed in L. sinapis was more similar to the range observed in other lineages (1.8 times higher than mean, highest in B. mori) (supplementary table 4b, Supplementary Material online).
Fig. 2.
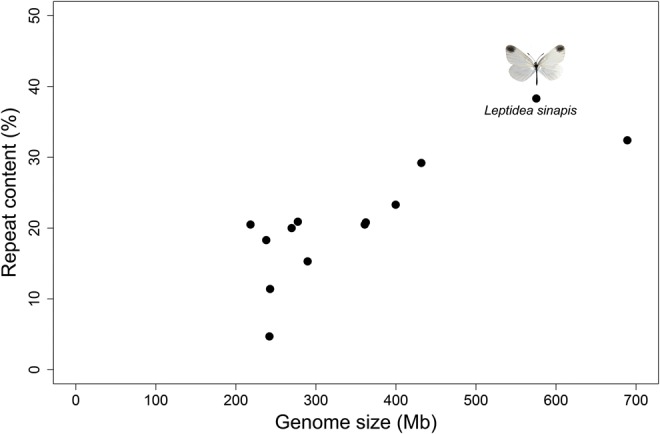
—Illustration of the strong relationship (Pearson‘s, n = 13, r = 0.800, P-value = 0.001) between genome size and repeat content in L. sinapis and other Lepidoptera species with assembled genomes. Note that genome size in this figure is the portion of the genome analyzed for repeat content and not the genome size estimate based on the assembly.
Fig. 3.

—Cumulative barplot illustrating the fractions of specific TE classes (color coded) and the total repeat content in L. sinapis and the 12 additional genome assemblies included in the comparative analysis. The top panel shows the total amount (in 100 Mb) and the bottom panel shows the genomic fractions (in %) of each specific TE class in each genome assembly.
Association between Genome Size and Repeat Content within Leptidea
The k-mer-based estimates of genome sizes within Leptidea showed that LjKaz had considerably larger genome size [776.7 Mb, ∼13% larger, Kruskal–Wallis test: χ2= 22.97, df = 5, P < 0.001, see supplementary table 5, Supplementary Material online, for details on the multiple population comparisons] than any other Leptidea population (on average 686.3 Mb), including the conspecific Irish population LjIre (656.0 Mb; fig. 4). We applied the dnaPipeTE pipeline (Goubert etal. 2015) on optimized subsampled re-sequencing reads (300,000–700,000 reads per individual) to de novo assemble the “repeatome” for each individual. This permitted the estimation of the abundance and relative age of recently active repeat elements in the re-sequencing data. There was a strong correlation between the fraction of both LINEs (Pearson’s: n = 6, r = 0.42, P < 0.001) and LTRs (n = 6, r = 0.57, P <0.001) with genome size within Leptidea but the correlation was driven only by the considerably higher repeat content and larger genome size in LjKaz (fig. 4). Upon removal of the LjKaz samples from the dataset we observed no significant correlations (Pearson’s: n = 6, r = 0.029, P = 0.84, and n = 6, r = 0.085, P = 0.56, for LINEs and LTRs, respectively) between specific repeat fractions and genome sizes estimates, indicating that the genome size and repeat content is similar across all other Leptidea populations (fig. 4). The fractions of LINEs and SINEs were negatively correlated in Leptidea (Pearson’s: n = 60, r = −0.36, P < 0.005), predominantly as an effect of a higher than average LINE content and lower than average SINE content in LjKaz (supplementary fig. 3, Supplementary Material online). However, the total amount of SINEs in LjKaz is at the same level as in other Leptidea populations, indicating that the genome expansion in LjKaz did not involve SINEs (supplementary fig. 3, Supplementary Material online). To further illustrate the discrepancy between LjKaz and other Leptidea populations, we performed a principal component analysis (PCA) based on the repeat fractions in the genome of all ten individuals in each respective population. The results clearly show that LjKaz deviates from the other populations (fig. 5).
Fig. 4.
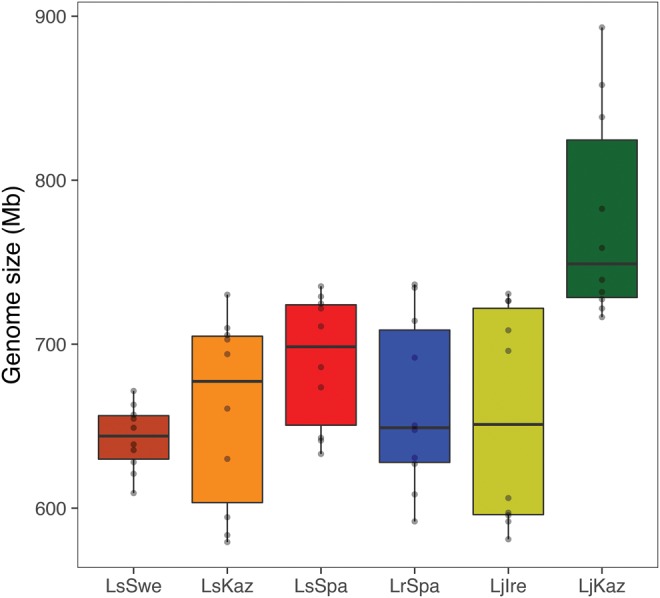
—Distribution of genome size estimates based on K-mer distributions (Jellyfish, Marcais and Kingsford 2011) for the ten samples within each of the six Leptidea populations. LjKaz (dark green), which has been observed to have a larger cell nucleus than other Leptidea species (Šíchová etal. 2015), has a larger genome size (mean = 777 Mb) than all other Leptidea populations/species (mean range = 643–690 Mb). The box in the box-plot illustrates first to third quartiles (Q1 and Q3 = box borders), median (horizontal line within box) and whiskers illustrate the interquartile range (Q1−1.5(Q1–Q3) and Q3 + (Q1−Q3)). Each data point is given as a single grey dot.
Fig. 5.
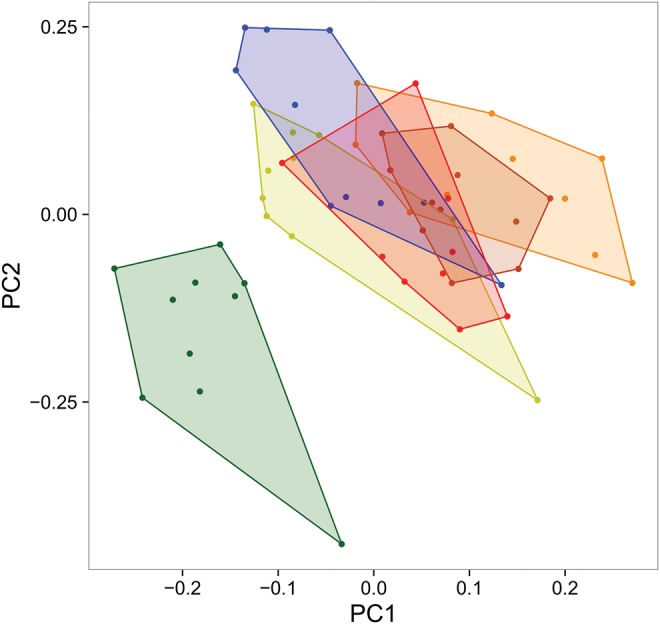
—A PCA analysis of all repeat subclasses identified in the ten individuals in each of the six Leptidea populations. The colors represent the populations (LsSwe = brown, LsKaz = orange, LsSpa = red, LrSpa = blue, LjIre = light green, LjKaz = dark green).
Reconstruction of Transposable Element Activity in Lepidoptera Lineages
To obtain further information about timing and rates of proliferation of specific repeat elements, the aforementioned 13 genome assemblies were scanned for repeat content and sequence divergence between individual copies of repeat subclasses LINEs, SINEs, LTRs, DNA transposons, and unclassified TEs using RepeatMasker. By applying a neutral mutation rate of 2.9×10−9, estimated from de novo mutations in H. melpomene (Keightley etal. 2015), and the previously established tree topology, we modeled the divergence time of specific repeats in each lineage from the per-copy distance to consensus and visualized the activities of each respective repeat class in the butterfly tree of life. We found that the activity of TEs has been modest in butterflies in general but that particular elements have experienced a higher proliferation rate in the lineage leading to the Leptidea clade after the split from the other pierids around 80 Mya, with a considerable burst of activity in the time period 10–20 Mya (fig. 6). A notable observation is that the patterns of proliferation vary extensively between lineages (fig. 6) with particular repeat element classes being active at different time points during the radiation. Here, we point to some examples to illustrate this phenomenon. In almost all sampled lineages, for the identified LINE repeat families we observed a more or less constant rate of proliferation, but with an overall higher rate in the Leptidea lineage after the split from Phoebis/Pieris (fig. 6). SINEs show very varying patterns with an activity peak coinciding with the general TE activity peak in the Leptidea lineage and a similar activity pattern in the Bombyx lineage, while there is a more constant low rate in Calycopis, Pieris, Phoebis, Melitaea, and P. glaucus and more ancestral activity peaks in P. xuthus and P. polytes (fig. 6). LTRs are generally found at low frequency and the only observable pattern is a recent activity increase in B. mori and a higher ancestral activity in the Papilio lineage prior to the split of the three species (fig. 6). Unclassified repeats are abundant in several lineages, except in H. melpomene, the only lepidopteran species with a completely curated in-depth TE annotation (Lavoie etal. 2013), and in D. plexippus, which has a minor proportion of the genome consisting of repeats in general. Unclassified repeats also show varying patterns of activity across lineages where they are abundant (figs. 3 and 6). The most obvious pattern observed is the comparatively recent activity burst in B. mori that contrasts with constant, low rates in P. xuthus, P. glaucus, P. sennae, M. cinxia, L. accius, P. rapae, and M. sexta, comparatively high rate over the past 50 My in Calycopis and peaks of activity ∼10 and 30 Mya in L. sinapis and P. glaucus, respectively (fig. 6). It should be noted that the observed general low rate of repeat activity far back in time (>50 My) in TE classes and lineages at least partly reflects the systematic problems in inferring activity for old repeats as a consequence of difficulties in identification when sequence divergence gets too high.
Fig. 6.

—Illustration of activity of different subclasses of TEs in the 13 lineages under study. The x-axis represents divergence time (Million years) estimated from copy to consensus differences in repeats (DNA elements, SINEs, LINEs, LTRs, and unknown) scaled by the neutral mutation rate, and the y-axis shows the total amount (Megabases) of repeats for each time interval. The phylogenetic tree shows the relationship between species as estimated using a set of 224 conserved nuclear genes.
The frequency analyses of different repeat subclasses across Leptidea populations indicate that the activity of especially LINEs, LTRs, and DNA elements has continued or even increased in the LjKaz lineage after the split from LjIre (supplementary fig. 3, Supplementary Material online). The divergence time estimates for Leptidea versus P. sennae/P. rapae and for species within the Leptidea group allowed us to quantify rates of genome size expansion in the Leptidea lineage in general and within the species L. juvernica in particular. These estimates point towards a mean expansion rate of 4.3 Mb/My in the Leptidea lineage after the split from other pierid butterflies ca. 80.6 Mya, and a mean expansion rate of 72 Mb/My in the LjKaz lineage after the split from LjIre ca. 1.6 Mya. These estimates can be translated to roughly 4 bp expansion rates per year in Leptidea in general, and 72 bp per year in LjKaz after the split from LjIre.
Discussion
The L. sinapis Genome Assembly
In this study, we quantify the prevalence of TEs in butterflies and moths with extensive variation in genome size and estimate the rate of genome expansion in the Leptidea lineage in general and between different populations in the species L. juvernica in particular. The underlying observation, a considerably larger genome size in Leptidea than in other butterflies with assembled genomes, spurred the interest to investigate the mechanistic underpinnings of such dramatic variation and to try relating that to theories of adaptive versus neutral scenarios for gain and loss of genetic material. The genome assembly of L. sinapis was found to be 643 Mb which is one of the largest genome assembly of any lepidopteran taxon currently available (Challis etal. 2017), for example 2.8 times larger than the smallest (227 Mb, P. polytes), and almost two times larger than the average lepidopteran genome (356.6 Mb) among the 12 investigated species for which relatively high-quality genome assemblies are available. In perspective, the 80 Lepidoptera species (91 entries in total) listed in the animal genome size database (www.genomesize.com; last accessed July 4, 2017, 2017-07-04; Gregory 2006) have a mean C-value of 0.62 pg [±0.29 s.d.; i.e., 0.61 ± 0.28 Gb following the conversion of Doležel etal. (2003) and the range varies from 0.23 to 1.94 pg (i.e., 0.22–1.90 Gb), with B. mori at a C-value of 0.52 pg (i.e., 0.51 Gb; genome assembly size = 482 Mb)]—this indicates that the genome size of L. sinapis is by no means extreme within the group. Furthermore, comparisons between genome size estimates based on flow-cytometry and DNA sequence assemblies consistently points toward that the latter tend to underestimate the genome size since repetitive sequences (especially centromeric and telomeric regions and non-recombining parts of sex-limited sex chromosomes) are always difficult to assemble (Chaisson etal. 2015; Kapusta and Suh 2017). This general feature of the assembly process, in combination with the observed high repeat content (38%) indicates that the L. sinapis genome might be larger than our current estimate—preliminary data from flow-sorting actually points toward a genome size of >800 Mb in L. sinapis, and even larger in L. juvernica, although these are estimates based on one single male specimen from each species, respectively (Petr Nguyen, personal communication).
To rule out the possibility that the comparatively large genome assembly of L. sinapis was an assembly artefact generating false duplication of a potentially highly heterozygous genome (Zhang and Backström 2014), we scanned the assembly for highly conserved eukaryotic (n = 434 entries in total) and arthropod (2,718 entries) gene sets and found only a minor fraction of duplicated genes (5 and 43 in each respective gene set which corresponds to 1.1 and 1.6% of all genes). Furthermore, since the gene counts based on conserved gene sets were very similar between our L. sinapis assembly and previously sequenced lepidopterans, the genome expansion in L. sinapis is very unlikely a result of whole or partial genome duplications.
Previous observations indicate that cell nucleus size in L. juvernica is considerably larger than in L. sinapis and L. reali (Šíchová etal. 2015). In agreement with that, our analyses showed that the Kazakhstan population LjKaz (776.7 Mb) had considerably larger genome size (∼13% larger) than any other Leptidea population (on average 686.3 Mb), even the Irish population LjIre (656.0 Mb). This indicates that there is genome size variation also within L. juvernica and allows for estimating rates of genome size change over comparatively short divergence times (see below).
Phylogenetic Analyses
When comparing our nuclear gene-based divergence time estimates to previously available data, there was good agreement regarding the deeper nodes corresponding to the split between Papilionidae and Nymphalidae, and nodes within these families associated with the splits between H. melpomene and M. cinxia and between P. xuthus and P. polytes, respectively (Hedges and Kumar 2009; Wahlberg etal. 2009; Nazari etal. 2011). However, the nuclear gene-based divergence time estimates within the Leptidea complex (e.g., TMRCA = 2.4–3.6 My) were considerably higher than previous estimates based on a small set of mitochondrial and nuclear genes (TMRCA = 0.3 My; Dincă etal. 2011). Given the comparatively small data set used by Dincă etal. (2011), the extensive variation in the ratio of mtDNA to nuclear mutation rates (Allio etal. 2017), and the good agreement between ours and previous estimates of other divergence times in the Lepidoptera phylogeny (Hedges and Kumar 2009; Wahlberg etal. 2009; Nazari etal. 2011; Condamine etal. 2012; Heikkilä etal. 2012; Espeland etal. 2015; ), the divergence times within Leptidea presented in this study are likely more robust.
Variation in TE Activity, Genome Size and Karyotype Structure in Lepidoptera
To get an amalgamated view of the causes and consequences of genome size variation it is crucial to have detailed information about the genomic architecture of the organisms (Petrov 2001; Gregory 2005). Therefore, we set out to investigate if the genome size differences in Lepidoptera could be explained by the expansion of specific selfish genetic elements. In line with previous research from a wide range of taxonomic groups—for example pufferfish (Aparicio etal. 2002); rice and thale cress (Bennetzen etal. 2005); three-spine stickleback (Blass etal. 2012); birds/mammals (Kapusta etal. 2017); Norway spruce (Nystedt etal. 2013); Hordeum grasses (Vicient etal. 1999), and migratory locust (Wang etal. 2014)— our analyses in Lepidoptera show that genome size variation across lineages can be largely explained by differences in overall content of TEs. Across all lineages, the investigated portion of the genomes contained between seven and 38% interspersed repeats, and different lineages displayed considerable differences in proportions and numbers of specific elements and TE subfamilies, with L. sinapis containing both the highest proportion of TEs and the largest number of TE subfamilies. The overall largest group of repeats in our data was in the partition that could not be accurately classified in automatic de novo repeat predictions. This has also been observed in a recently developed genome assembly of the squinting bush brown butterfly (Bicyclus anyana) that contained 18% unclassified and 7% classified repeats (Nowell etal. 2017). A recent study in birds where unclassified repeats were manually curated showed that those predominantly represented LTRs (Kapusta and Suh 2017), but it is not clear if this also applies to Lepidoptera and tedious manual curation of unclassified repeats will be necessary to get a detailed picture of the composition of TEs in this category. There was a negative relationship between the fractions of LINEs and SINEs when comparing LjKaz to the other populations in the Leptidea clade but the amount of SINE sequences in all Leptidea genomes were similar. This suggests that the activity of SINEs has been very low in LjKaz after the split from the other Leptidea populations. This is in line with the observation that SINEs mainly proliferate by hijacking the enzymatic macherinery of LINEs and, if so, often outnumber their LINE counterparts (Ohshima etal. 1996; Ohshima and Okada 2005).
By comparing the average genome sizes of Leptidea to other lepidopteran lineages we could estimate the rate of genome size increase over time. The closest relatives to Leptidea available in the sample set is P. sennae and P. rapae. We estimated these two lineages (both within family Pieridae) to have a divergence time of ∼80 My and this translates to an overall net expansion rate of four Mb/My which is ∼4 bp/year. If we assume a generation time of one generation per year, this yields a net expansion rate in Leptidea of four bp per generation (see the Methods section for comments on assumed generation times and how that may affect rate estimates). Since the proliferation mechanism for TEs involves entire repeat sequences ranging from ∼100 bp in SINEs to over 10 kb in LTRs (Sotero-Caio etal. 2017) and repeat sequences underlie the overwhelming part of the observed genome size differences, the increase in genome size should however rather be seen as a punctuated process rather than a steady, stepwise addition of small DNA fragments. Nonetheless, the low overall expansion rate in Leptidea suggests hyperactivity of TEs and that there is limited power for natural selection to act against their proliferation unless TE insertions occur in functional regions.
Our phylogenetic analysis based on nuclear genes clearly separates L. sinapis populations with distinct karyotypes into monophyletic clades, a novel finding that has not previously been detected with smaller marker sets (Dincă etal. 2011, 2013). It is tempting to speculate about the association between a high repeat content and the high fission/fusion rate resulting in dramatic variation in chromosome numbers within and across species within the Leptidea species complex (Dincă etal. 2011; Lukhtanov etal. 2011; Šíchová etal. 2015). Previous analyses indicate that interspersed repeats may mediate chromosomal rearrangements via for example non-homologous recombination (Völker etal. 2010; Zhang etal. 2011) which means that TE activity may not only have an impact on genome size, but also on the karyotype evolution. Interestingly, several Lepidoptera lineages show evidence for recurrent chromosome number changes. Within the subgenus Agrodiaetus chromosome numbers vary from 2n = 20 to 2n = 268 (Lukhtanov etal. 2005) and the species Lysandra coridon displays a chromosome number cline similar to L. sinapis but less dramatic (Talavera etal. 2013). In addition, Polymmatus atlanticus has the highest number of chromosomes (2n = 448–452) recorded in any metazoan organism (Lukhtanov 2015). This highlights the suitability of Lepidoptera in general, and Leptidea butterflies with their extreme karyotype variation in particular (Dincă etal. 2011; Lukhtanov etal. 2011; Šíchová etal. 2015), as a study system for investigating the role of repetitive sequences on karyotype structure, why chromosome number changes seem to be spurting in specific lineages and how rearrangements affect patterns of genome differentiation, adaptation and speciation.
Genome Size Variation and TE Activity in Leptidea
An intriguing observation related to the observed larger genome size in Leptidea as compared with other lepidopteran lineages was that cell nucleus size varies across Leptidea species with a considerably larger nucleus in L. juvernica than in L. reali and L. sinapis (Šíchová etal. 2015). The L. juvernica populations sampled in the comparison were collected in the Czech Republic and samples from other parts of the distribution range were not included. Previous analyses have not detected any morphological differences, including male or female genitalia, that suggest the existence of a cryptic species within L. juvernica (Dincă etal. 2011), and no prezygotic barriers among LjKaz, LjIre, and LjSwe populations have been found (which exist between L. sinapis, L. reali, and L. juvernica because of female choice) (Dincă etal. 2013). Genetic analyses based on the generally fast evolving mtDNA genes (COI and ND1) and a handful of nuclear genes have shown that the LjKaz population is undistinguishable from all other populations studied from European mainland (Dincă etal. 2011, 2013; Lukhtanov etal. 2011; Šíchová etal. 2015). Given our results, showing that LjKaz has a considerably larger genome size than LjIre (and all other sampled Leptidea populations), it is plausible that a genome size expansion has happened in “continental” Eurasian L. juvernica after the split from LjIre. This could potentially be a consequence of lower effective population size and less efficient purging of slightly deleterious TE insertions in LjKaz (Lynch 2007). In support of that, genome-wide analyses of nucleotide diversity (π) show that L. juvernica in general have a reduced level of polymorphism (π = 0.11–0.17%) compared with other Leptidea populations (π = 0.33–0.39%), and the reduction in LjKaz (π = 0.11%) is particularly pronounced (Talla V, Dincă V, Vila R, Wiklund C, Backström N, Unpublished data). Since the divergence time between LjKaz and LjIre was estimated to 1.6 My and the genome size difference was roughly 115 Mb, the net genome expansion rate is in the range of 72 Mb/My. Again, when translated to per generation (year) the expansion rate is rather modest (<100 bp/year). Recent, detailed proliferation rate analyses of LTRs in Drosophila melanogaster (one insertion in 1,000 to 1,000,000 generations; Huang etal. 2012) and LINEs (one insertion in 212 births; Xing etal. 2009) and Alu-elements (one insertion in 20 births; Cordaux etal. 2006) in humans indicate that proliferation rates vary across taxa and between types of repeats; at the extreme, novel insertions of TEs have been observed to occur with up 20–100 transposition events (0.1–1 Mb in total) in a single generation in some taxa (Petrov 2001 and references therein). Hence, the observed overall genome expansion rates as a consequence of TE activity in Leptidea in general and in LjKaz in particular are not spectacular, but high enough to generate considerable genome size differences over short evolutionary time. Again, we cannot specifically rule out that the increase in genome size is an adaptive response in LjKaz as compared to other Leptidea lineages, but the observation of stochastic variation in TE proliferation rates, likely accompanied by reduced efficiency of purifying selection during periods of limited population size, suggests that genetic drift has contributed considerably to genome size evolution in Leptidea (Lynch 2007). Notably, differences in TE proliferation rates might even be exacerbated under environmental stress (Kim etal. 2014; Migicovsky and Kovalchuk 2014), indicating that variation in environmental conditions during population isolation—for example, under allopatric separation during glacial periods—may also contribute to genome size evolution.
Perspectives
Even if accumulation of repetitive DNA might be associated with a cost due to insertion in functional regions and potentially increased energetic demands for the replication machinery, novel inserted DNA may also provide a template for evolution of novel functions. It has, for example, been suggested that the gain of introns in eukaryotes might have had a non-adaptive origin, but once present they have allowed for more diverse transcript sets due to potential for alternative splicing of genes (Lynch 2007) and TEs have been shown to mediate expression regulatory functions (Rebollo etal. 2012; Elbarbary etal. 2016; Chuong etal. 2017). A striking finding involving a classic textbook example of a rapid evolutionary response due to natural selection is that a large TE insertion in the first intron of the gene cortex underlies the melanistic phenotype in Biston betularia, the peppered moth (Van't Hof etal. 2016). This is potentially just a beginning of what will be unearthed in the upcoming years. Although we are still far from having a detailed understanding of potential functional gains or deleterious effects of proliferation of repetitive sequences in the Lepidoptera, the increasing availability of high-quality genome assemblies and accumulating annotation information and functional genomic studies will likely shed light on costs and benefits of repetitive DNA proliferation in general and in specific cases also in this group of organisms.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This study was supported by a Junior Research Grant from the Swedish Research Council [VR 2013-4508 to N.B.], MINECO and AEI/FEDER, UE project grants [CCGL2013-48277-P; CGL2016-76322-P to R.V.], and a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme [project no. 625997 to V.D.]. We acknowledge additional funding for library preparation and sequencing from Kungliga Fysiografiska Sällskapet i Lund (Nilsson-Ehle Donations) and the Science for Life Laboratory (SciLife Sweden) Biodiversity Program. The SNP&SEQ Technology Platform and Uppsala Genome Center performed the library preparations and the sequencing supported by Science for Life Laboratory (SciLife, Stockholm); a national infrastructure funded by the Swedish Research Council (VR-RFI) and the Knut and Alice Wallenberg Foundation. The computations were performed on resources provided by SNIC through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX; Lampa etal. 2013) under Project # b2014034. We thank Remi-Andre Olsen for help with the Allpaths-LG assembly, Roy N. Platt II and David A. Ray for providing the TE library for H. erato, Aurélie Kapusta for providing the Perl script to merge TE libraries, Clément Goubert for help with optimizing dnaPipeTE and for commenting on an earlier version of the manuscript, and Brian Nelson and Catherine Bertrand for advice and help with sampling L. juvernica in Ireland. We thank the associate editor and four anonymous reviewers for constructive and insightful suggestions and comments that helped improve and clarify the manuscript.
Literature Cited
- Allio R, Donega S, Galtier N, Nabholz B.. 2017. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals—implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Mol Biol Evol. doi:10.1093/molbev/msx197 [DOI] [PubMed] [Google Scholar]
- Altermatt F. 2010. Climatic warming increases voltinism in European butterflies and moths. Proc R Soc B: Biol. 277(1685):1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Aparicio S, et al. 2002. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297(5585):1301–1310. [DOI] [PubMed] [Google Scholar]
- Arnqvist G, et al. 2015. Genome size correlates with reproductive fitness in seed beetles. Proc R Soc B: Biol. 282(1815):e20151421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima K, Kohany O.. 2015. Repbase update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6:11.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Ma J, Devos KM.. 2005. Mechanisms of recent genome size variation in flowering plants. Ann Bot. 95(1):127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt M, et al. 2013. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phyl Evol. 69(2):313–319. [DOI] [PubMed] [Google Scholar]
- Blass E, Bell M, Boissinot S.. 2012. Accumulation and rapid decay of non-LTR retrotransposons in the genome of the three-spine stickleback. Genome Biol Evol. 4(5):687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs CL, Watt WB, Ehrlich PR.. 2003. Butterflies: ecology and evolution taking flight. Chicago: The University of Chicago Press. [Google Scholar]
- Butler J, et al. 2008. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res. 18(5):810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. 2005. Economy, speed and size matter: evolutionary forces driving nuclear genome miniaturization and expansion. Ann Bot. 95(1):147–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaisson MJ, et al. 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517(7536):608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis RJ, Kumar S, Dasmahapatra KK, Jiggins CD, Blaxter M.. 2017. Lepbase—the lepidopteran genome database. BioRxiv. [Google Scholar]
- Chuong EB, Elde NC, Feschotte C.. 2017. Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet. 18(2):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condamine FL, Sperling FA, Wahlberg N, Rasplus JY, Kergoat GJ.. 2012. What causes latitudinal gradients in species diversity? Evolutionary processes and ecological constraints on swallowtail biodiversity. Ecol Lett. 15(3):267–277. [DOI] [PubMed] [Google Scholar]
- Cong Q, et al. 2016. Speciation in cloudless sulphurs gleaned from complete genomes. Genome Biol Evol. 8(3):915–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordaux R, Hedges DJ, Herke SW, Batzer MA.. 2006. Estimating the retrotransposition rate of human Alu elements. Gene 373:134–137. [DOI] [PubMed] [Google Scholar]
- Davey JW, et al. 2016. Major improvements to the Heliconius melpomene genome assembly used to confirm 10 chromosome fusion events in 6 Million years of butterfly evolution. G3: Genes, Genomes Genet. 6:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincă V, Lukhtanov VA, Talavera G, Vila R.. 2011. Unexpected layers of cryptic diversity in wood white Leptidea butterflies. Nat Commun. 2:e324.. [DOI] [PubMed] [Google Scholar]
- Dincă V, et al. 2013. Reproductive isolation and patterns of genetic differentiation in a cryptic butterfly species complex. J Evol Biol. 26(10):2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doležel J, Barto s. J, Voglmayr H, Greilhuber J.. 2003. Nuclear DNA content and genome size of trout and human. Cytometry A 51(2):127–128. [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29(8):1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary RA, Lucas BA, Maquat LE.. 2016. Retrotransposons as regulators of gene expression. Science 351(6274):aac7247.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H. 2004. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 5(6):435–445. [DOI] [PubMed] [Google Scholar]
- Elliott TA, Gregory TR.. 2015. Do larger genomes contain more diverse transposable elements? BMC Evol Biol. 15:69.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espeland M, et al. 2015. Ancient Neotropical origin and recent recolonisation: phylogeny, biogeography and diversification of the Riodinidae (Lepidoptera: Papilionoidea). Mol Phyl Evol. 93:296–306. [DOI] [PubMed] [Google Scholar]
- Fedoroff. 2012. Transposable elements, epigenetics, and genome evolution. Science 338:758–767. [DOI] [PubMed] [Google Scholar]
- Fontdevila A. 2011. The dynamic genome: a darwinian approach. New York: Oxford University Press, Inc. [Google Scholar]
- Friberg M, Aalberg Haugen IM, Dahlerus J, Gotthard K, Wiklund C.. 2011. Asymmetric life-history decision-making in butterfly larvae. Oecologia 165:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg M, Wiklund C.. 2007. Generation dependent female choice: behavioral polyphenism in a bivoltine butterfly. Behav Ecol. 18(4):758–763. [Google Scholar]
- Friberg M, Wiklund C.. 2010. Host-plant-induced larval decision-making in a habitat/host-plant generalist butterfly. Ecology 91(1):15–21. [DOI] [PubMed] [Google Scholar]
- Goubert C, et al. 2015. De novo assembly and annotation of the Asian tiger mosquito (Aedes albopictus) repeatome with dnaPipeTE from raw genomic reads and comparative analysis with the yellow fever mosquito (Aedes aegypti). Genome Biol Evol. 7(4):1192–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29(7):644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR. 2006. The Animal Genome Size Database. (http://www.genomsize.com; last accessed July 4, 2017)
- Gregory TR. 2005. The evolution of the genome. Boston (MA: ): Elsevier Academic Press. [Google Scholar]
- Gregory TR. 2004. Macroevolution, hierarchy theory, and the C-value enigma. Paleobiology 30(2):179–202. [Google Scholar]
- Gregory TR, Hebert PDN.. 2003. Genome size variation in lepidopteran insects. Can J Zool. 81(8):1399–1405. [Google Scholar]
- Gregory TR, Hebert PDN.. 1999. The modulation of DNA content—proximate causes and ultimate consequences. Genome Res. 9(4):317–324. [PubMed] [Google Scholar]
- Gregory TR, et al. 2007. Eukaryotic genome size databases. Nucl Acids Res. 35(Database issue):D332–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo LT, et al. 2015. Flow cytometry and K-mer analysis estimates of the genome sizes of Bemisia tabaci B and Q (Hemiptera: Aleyrodidae). Front Physiol. 6:e144.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan SJ, Johnston JS.. 2011. New genome size estimates of 134 species of arthropods. Chrom Res. 19(6):809–823. [DOI] [PubMed] [Google Scholar]
- Hao J-J, Hao J-S, Sun X-Y, Zhang L-L, Yang Q, Park Y.. 2014. The complete mitochondrial genomes of the Fenton‘s wood white, Leptidea morsei, and the lemon emigrant, Catopsilia pomona. J Insect Sci. 14(1):e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Kumar S.. 2009. The timetree of life. New York: Oxford University Press. [Google Scholar]
- Heikkilä M, Kaila L, Mutanen M, Peña C, Wahlberg N.. 2012. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc R Soc B: Biol. 279(1731):1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CRL, Burns KH, Boeke JD.. 2012. Active transposition in genomes. Ann Rev Genet. 46:651–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo-Carrasco F, Smith SA, Stamatakis A.. 2011. Algorithms, data structures, and numerics for likelihood-based phylogenetic inference of huge trees. BMC Bioinformatics 12(1):e470.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Suh A.. 2017. Evolution of bird genomes—a transposon's-eye view. Ann NY Acad Sci. 1389(1):164–185. [DOI] [PubMed] [Google Scholar]
- Kapusta A, Suh A, Feschotte C.. 2017. Dynamics of genome size evolution in birds and mammals. Proc Nat Acad Sci U S A. 114:E1460–E1469.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian JHH. 2004. Mobile elements: drivers of genome evolution. Science 303(5664):1626–1632. [DOI] [PubMed] [Google Scholar]
- Keightley PD, et al. 2015. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol. 32(1):239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell M. 2005. Transposable elements In: Gregory TR, editor. The evolution of the genome. Burlington (MA: ): Elsevier Academic Press; p. 165–221. [Google Scholar]
- Kim YB, et al. 2014. Divergence of Drosophila melanogaster repeatomes in response to a sharp microclimate contrast in Evolution Canyon, Israel. Proc Natl Acad Sci U S A. 111(29):10630–10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarewa I, et al. 2009. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat Methods 6(4):291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampa S, Dahlö M, Olason PI, Hagberg J, Spjuth O.. 2013. Lessons learned from implementing a national infrastructure in Sweden for storage and analysis of next-generation sequencing data. Gigascience 2(1):9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie C, Platt R, Novick P, Counterman B, Ray D.. 2013. Transposable element evolution in Heliconius suggests genome diversity within Lepidoptera. Mobile DNA 4(1):21.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefébure T, et al. 2017. Less effective selection leads to larger genomes. Genome Res. http://genome.cshlp.org/content/early/2017/04/19/gr.212589.116 and the DOI is 10.1101/gr.212589.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transformation. Bioinformatics 26(5):589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N.. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 102(30):10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Peatman E, Tang H, Lewis J, Liu Z.. 2012. Profiling of gene duplication patterns of sequenced teleost genomes—evidence for rapid lineage-specific genome expansion mediated by recent tandem duplications. BMC Genomics 13:246.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukhtanov VA. 2015. The blue butterfly Polyommatus (Plebicula) atlanticus (Lepidoptera, Lycaenidae) holds the record of the highest number of chromosomes in the non-polyploid eukaryotic organisms. Comp Cytogenet. 9(4):683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukhtanov VA, Dincă V, Talavera G, Vila R.. 2011. Unprecedented within-species chromosome number cline in the wood white butterfly Leptidea sinapis and its significance for karyotype evolution and speciation. BMC Evol Biol. 11(1):e109.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukhtanov VA, et al. 2005. Reinforcement of pre-zygotic isolation and karyotype evolution in Agrodiaetus butterflies. Nature 436(7049):385–389. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland (MA: ): Sinauer Associates. [Google Scholar]
- Mapleson D, Garcia Accinelli G, Kettleborough G, Wright J, Clavijo BJ.. 2017. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33:574–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcais G, Kingsford C.. 2011. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27(6):764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1):10–12. [Google Scholar]
- Maumus F, Fiston-Lavier A-S, Quesneville H.. 2015. Impact of transposable elements on insect genomes and biology. Curr Opin Insect Sci. 7:30–36. [DOI] [PubMed] [Google Scholar]
- Migicovsky Z, Kovalchuk I.. 2014. Transgenerational changes in plant physiology and in transposon expression in response to UV-C stress in Arabidopsis thaliana. Plant Signal Behav. 9(11):e976490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazari V, et al. 2011. Phylogenetic systematics of Colotis and associated genera (Lepidoptera: Pieridae): evolutionary and taxonomic implications. J Zool Syst Evol Res. 49(3):204–215. [Google Scholar]
- Nowell RW, et al. 2017. A high-coverage draft genome of the mycalesine butterfly Bicyclus anynana. Gigascience 6(7):1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystedt B, et al. 2013. The Norway spruce genome sequence and conifer genome evolution. Nature 497(7451):579–584. [DOI] [PubMed] [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. Berlin: Springer. [Google Scholar]
- Ohshima K, Hamada M, Terai Y, Okada N.. 1996. The 3' ends of tRNA-derived short interspersed repetitive elements are derived from the 3' ends of long interspersed repetitive elements. Mol Cell Biol. 16(7):3756–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K, Okada N.. 2005. SINEs and LINEs: symbionts of eukaryotic genomes with a common tail. Cytogenet Genome Res. 110(1–4):475–490. [DOI] [PubMed] [Google Scholar]
- Oliver MJ, Petrov D, Ackerly D, Falkowski P, Schofield OM.. 2007. The mode and tempo of genome size evolution in eukaryotes. Genome Res. 17(5):594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel LE, Crick FHC.. 1980. Selfish DNA: the ultimate parasite. Nature 284(5757):604–607. [DOI] [PubMed] [Google Scholar]
- Petrov DA. 2002a. DNA loss and evolution of genome size in Drosophila. Genetica 115(1):81–91. [DOI] [PubMed] [Google Scholar]
- Petrov DA. 2001. Evolution of genome size- new approaches to an old problem. Trends Genet. 17(1):23–28. [DOI] [PubMed] [Google Scholar]
- Petrov DA. 2002b. Mutational equilibrium model of genome size evolution. Theoret Pop Biol. 61(4):531–544. [DOI] [PubMed] [Google Scholar]
- Pritham EJ. 2009. Transposable elements and factors influencing their success in eukaryotes. J Hered. 100(5):648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez V, Harispe S, Delsuc F, Douzery EJ.. 2011. MACSE: multiple alignment of coding sequences accounting for frameshifts and stop codons. PLoS One 6:e22594.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R, Romanish MT, Mager DL.. 2012. Transposable elements: an abundant and natural source of regulatory sequences for host genes. Ann Rev Genet. 46:21–42. [DOI] [PubMed] [Google Scholar]
- Revell LJ. 2012. Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol Evol. 3(2):217–223. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T.. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor (NY: ): Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sessegolo C, Burlet N, Haudry A.. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biol Lett. 12(8):e20160407.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, et al. 2016. Complete genome of Pieris rapae, a resilient alien, a cabbage pest, and a source of anti-cancer proteins. F1000 Res. 5:e2631.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šíchová J, et al. 2016. Fissions, fusions, and translocations shaped the karyotype and multiple sex chromosome constitution of the northeast-Asian wood white butterfly, Leptidea amurensis. Biol J Linn Soc. 118:457–471. [Google Scholar]
- Šíchová J, et al. 2015. Dynamic karyotype evolution and unique sex determination systems in Leptidea wood white butterflies. BMC Evol Biol. 15:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Simpson JT, et al. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19(6):1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R.. 2010. RepeatModeler Open-1.0. http://www.repeatmasker.org/, last accessed June 12, 2017.
- Smit A, Hubley R, Green P.. 1996-2010. RepeatMasker Open-3.3.0. http://www.repeatmasker.org/, last accessed June 9, 2017.
- Sotero-Caio CG, Platt RN, Suh A, Ray DA.. 2017. Evolution and diversity of transposable elements in vertebrate genomes. Genome Biol Evol. 9(1):161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688–2690. [DOI] [PubMed] [Google Scholar]
- Sun C, et al. 2012. LTR retrotransposons contribute to genomic gigantism in plethodontid salamanders. Genome Biol Evol. 4(2):168–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Lukhtanov VA, Rieppel L, Pierce NE, Vila R.. 2013. In the shadow of phylogenetic uncertainty: the recent diversification of Lysandra butterflies through chromosomal change. Mol Phyl Evol. 69(3):469–478. [DOI] [PubMed] [Google Scholar]
- Tenaillon MI, Hollister JD, Gaut BS.. 2010. A triptych of the evolution of plant transposable elements. Trends Plant Sci. 15(8):471–478. [DOI] [PubMed] [Google Scholar]
- The Heliconius Genome Sequencing Consortium 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CA., Jr. 1971. The genetic organization of chromosomes. Ann Rev Genet. 5:237–256. [DOI] [PubMed] [Google Scholar]
- Van't Hof AE, et al. 2016. The industrial melanism mutation in British peppered moths is a transposable element. Nature 534(7605):102–105. [DOI] [PubMed] [Google Scholar]
- Vicient CM, Suoniemi A, Anamthawat-Jónsson K, Tanskanen J, Beharav A, Nevo E.. 1999. Retrotransposon BARE-1 and its role in genome evolution in the genus Hordeum. Plant Cell. 11(9):1769–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völker M, et al. 2010. Copy number variation, chromosome rearrangement and recombination during avian evolution. Genome Res. 20(4):503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlberg N, et al. 2009. Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. Proc R Soc B: Biol. 276(1677):4295–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. 2014. The locust genome provides insight into swarm formation and long-distance flight. Nat Commun. 5:e2957.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr A, et al. 2015. Identification of low-confidence regions in the pig reference genome (Sscrofa10.2). Front Genet. 6:e338.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Kumar V, Shah P, Church D, Jaffe DB.. 2017. Direct determination of diploid genome sequences. BioRxiv https://doi.org/10.1101/070425, last accessed August 8, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklund C. 1977. Oviposition, feeding and spatial separation of breeding andforaging habitats in a population of Leptidea sinapis. Oikos 28(1):56–68. [Google Scholar]
- Xing J, et al. 2009. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 19(9):1516–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yu C, Krishnaswamy L, Peterson T.. 2011. Transposable elements as catalysts for chromosome rearrangements. Methods Mol Biol. 701:315–326. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Backström N.. 2014. Assembly errors cause false tandem duplicate regions in the chicken (Gallus gallus) genome sequence. Chromosoma 123(1–2):165–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.