


Abstract
Many cellular proteins are methylated on lysine residues and this has been most intensively studied for histone proteins. Lysine methylations on non-histone proteins are also frequent, but in most cases the functional significance of the methylation event, as well as the identity of the responsible lysine (K) specific methyltransferase (KMT), remain unknown. Several recently discovered KMTs belong to the so-called seven-β-strand (7BS) class of MTases and we have here investigated an uncharacterized human 7BS MTase currently annotated as part of the endothelin converting enzyme 2, but which should be considered a separate enzyme. Combining in vitro enzymology and analyzes of knockout cells, we demonstrate that this MTase efficiently methylates K36 in eukaryotic translation elongation factor 1 alpha (eEF1A) in vitro and in vivo. We suggest that this novel KMT is named eEF1A-KMT4 (gene name EEF1AKMT4), in agreement with the recently established nomenclature. Furthermore, by ribosome profiling we show that the absence of K36 methylation affects translation dynamics and changes translation speed of distinct codons. Finally, we show that eEF1A-KMT4 is part of a novel family of human KMTs, defined by a shared sequence motif in the active site and we demonstrate the importance of this motif for catalytic activity.
INTRODUCTION
The addition of a methyl group to a biomolecule represents a very common biochemical reaction within the cell. Methylation reactions are catalyzed by highly specific methyltransferase (MTase) enzymes, most of which use S-adenosylmethionine (AdoMet) as a methyl donor (1). The products of the AdoMet-dependent MTase reactions are the methylated substrate and S-adenosylhomocysteine (AdoHcy). The human genome has been predicted to encode around 200 AdoMet-dependent MTases, which based on their sequences and structures, have been categorized into five different classes (2). The largest class is constituted by the seven-β-strand (7BS) MTases, which share a characteristic structural fold and encompass ∼130 human enzymes (2). The second largest class are the SET (Su(var)3-9, Enhancer-of-zeste, Trithorax) proteins, defined by the presence of a so-called SET-domain and comprising more than 50 characterized and putative human MTases (2).
Lysine methylation is a common post-translational protein modification. The ε-amino group of a lysine side chain can accept up to three methyl groups, resulting in mono- di- or trimethyllysine, and the different methylated species of lysines can exert distinct functions. The functional significance of lysine methylation has been most intensively studied in the context of histone proteins and epigenetics, where the modification constitutes a part of the so called ‘histone code’ and methylation of specific lysines in the histone tails are determinants of gene expression and chromatin state (3). For example, trimethylation of lysine 4 in histone H3 (H3-K4) is often associated with actively transcribed genes whereas trimethylation of H3-K9 or H3-K27 is linked to transcriptional repression (3).
Clearly, many of the human MTases are devoted to protein lysine methylation. In particular, the SET-proteins appear to exclusively encompass lysine (K)-specific MTases (KMTs), several of which specifically target histone tails. However, it has recently become clear that also the 7BS MTases, which methylate substrates ranging from small metabolites to proteins and nucleic acids, also comprise several KMTs. DOT1L was the first human 7BS KMT to be characterized (4). It methylates histone H3, but in contrast to most SET enzymes, targets a residue (K79) located in the globular domain of the protein (4). In recent years, several enzymes of the 7BS class have been shown to methylate specific lysine residues in non-histone substrates (reviewed in (5)). Notably, several of these have been shown to target translation elongation factors. eEF1A-KMT1, eEF1A-KMT2, eEF1A-KMT3 (encoded by the EEF1AKMT1, EEF1AKMT2, EEF1AKMT3 genes, respectively) target individual lysines in eukaryotic translation elongation factor 1 alpha (eEF1A), whereas eEF2-KMT (encoded by the EEF2KMT gene) methylates eukaryotic translation elongation factor 2 (eEF2) (6–9). In addition, several of the members of the so-called MTase Family 16 (MTF16) target various other non-histone substrates (10–15).
eEF1A, as well as its prokaryotic ortholog EF-Tu, perform the essential function of delivering aminoacylated tRNAs to the ribosome, enabling translation of mRNA to protein (reviewed in (16)). However, eEF1A has also been attributed several non-canonical functions, including involvement in protein degradation, apoptosis, organization of the cytoskeleton and nuclear export of tRNAs (reviewed in (17,18)). Humans have two closely related paralogues of eEF1A, denoted eEF1A1 and eEF1A2 (here, collectively referred to as eEF1A) which display distinct expression patterns (19). Mammalian eEF1A is subject to extensive lysine methylation and early studies on rabbit eEF1A identified five methylated lysine residues by peptide sequencing, i.e. K36, K55, K79, K165 and K318. Recent mass spectrometry (MS) studies have confirmed the presence of the corresponding methylated lysines also in human eEF1A (20,21). For three out of these five methylated lysines, the responsible KMT has been identified; eEF1A-KMT1, eEF1A-KMT2 and eEF1A-KMT3 mediate methylation at K79, K318 and K165, respectively (7–9,22,23). However, the KMTs introducing methylation at K36 and K55 remain elusive.
Here, we have characterized a new human MTase, which attracted our attention because it shares sequence similarity with the eEF1A-specific KMT eEF1A-KMT2. This novel MTase is encoded by a set of exons currently annotated as part of the gene encoding the endothelin converting enzyme 2 (ECE2). From analysis of genome and transcriptome data, we conclude that the exons encoding the MTase represent a separate gene. We show that the recombinant MTase methylates a single protein in cell extracts, and the substrate is identified as eEF1A through biochemical purification and protein MS. Since the enzyme represents the fourth eEF1A-specific KMT in humans, we propose that it is named eEF1A-KMT4 (suggested gene name EEF1AKMT4), in keeping with the prevailing nomenclature. Moreover, we demonstrate that eEF1A-KMT4 catalyzes methylation of K36 in recombinant eEF1A in vitro and that the enzyme mediates this modification in cells. Interestingly, we find that global translation is altered in EEF1AKMT4 knockout (KO) cells and that codons specific for asparagine, histidine and tryptophan change their translation speed in the KO relative to wild-type (WT) cells. Finally, we show that eEF1A-KMT4, together with eEF1A-KMT2 and two yet uncharacterized MTases, constitute a novel KMT family, defined by a common sequence motif which is required for MTase activity.
MATERIALS AND METHODS
Bioinformatics analysis
Protein sequence homology was assessed using NCBI’s BLAST algorithm and alignment of protein sequences was performed using the MUSCLE algorithm embedded in Jalview (24). In the reciprocal BLAST analysis, proteins displaying high sequence similarity (expect value <10−20) were classified as putative orthologs and used as queries in reciprocal BLAST searches versus a human protein database. An unrooted phylogenetic tree was generated using the ‘phylogeny.fr’ package (25) and the following protein sequences (referred to by their gene names): EEF1AKMT4 (ref|NP_115707.2|), amino acids 1–214 of METTL13 (ref|NP_057019.3|), EEF1AKMT2 (ref|NP_997719.2|), METTL12 (ref|NP_001036694.1|), METTL21A (ref|NP_660323.3|), EEF1AKMT3 (ref|NP_056248.2|), METTL21C (ref|NP_001010977.1|), VCPKMT (ref|NP_078834.2|), METTL23 (ref|NP_001073979.3|), EEF2KMT (ref|NP_958802.1|), METTL22 (ref|NP_077014.3|), METTL18 (ref|NP_219486.1|), ETFBKMT (ref|NP_776163.1|), CAMKMT (ref|NP_079042.1|), FAM173A (ref|NP_076422.1|), FAM173B (ref|NP_954584.2|), DOT1L (ref|XP_005259716.1|) and EEF1AKMT1 (ref|NP_777588.1|). Tracks of aligned expressed sequence tags (EST) were extracted from the UCSC genome browser (https://genome.ucsc.edu) (Assembly; GRCh37).
Gene cloning and mutagenesis
Plasmid constructs used in this work and the cloning strategy used to generate them, are described in detail in Supplementary Table S1. In brief, relevant open reading frames (ORFs) were amplified using polymerase chain reaction (PCR) and cloned into the indicated vectors using restriction based cloning and T4 ligase (NEB). For restriction based cloning into the pcDNA5/FRT/TO (Life Technologies) vector, DNA fragments encoding eEF1A proteins fused to the calmodulin binding peptide (CBP) and streptavidin binding peptide (SBP) tag from pNTAP-A (Agilent Technologies) were generated by splice overlap extension PCR using using the primers 5′-CTAGGGATCCCCACCATGAAGCGACGATGG-3′ and 5′-GCCCAGCTTGCCCAGCTTGCAG-3′. The identity and integrity of all cloned ORFs was verified by sequencing.
Generation and cultivation of cell lines
HAP-1 EEF1AKMT4 gene KO cells were generated using the CRISPR/Cas9 system as a custom project performed by Horizon Genomics (formerly Haplogen). After transfection, single cells were isolated by limiting dilution and screened for genome editing events by sequencing of the relevant genomic locus<. The analyzed EEF1AKMT4-deficient cell line contains a 17 bp deletion upstream of motifs required for catalytic activity (verified by sequencing) and is now commercially available (Horizon Genomics, HZGHC000530c010). WT and EEF1AKMT4-deficient HAP-1 cells were cultured according to the supplier's instructions in IMDM GlutaMAX™, 10% fetal bovine serum (FBS) and 100 U/ml penicillin-streptomycin.
Cell lines for inducible expression of protein constructs with N-terminal CBP and SBP affinity tags for tandem affinity purification (TAP) were generated as previously described (11). In brief, Flp-In 293 T-REx cells were cultivated in DMEM GlutaMAX™, 10% tetracycline-reduced FBS and 100 U/ml penicillin-streptomycin and transfected with pOG44 together with either pcDNA5/FRT/TO-CBP-SBP-eEF1A1 or pcDNA5/FRT/TO-CBP-SBP-eEF1A2 using Lipofectamine 3000 (Life Technologies) as transfection agent. After 24 h the cells were expanded onto 10 cm diameter dishes and after further 24 h of incubation, 200 μg/ml hygromycin B (Life Technologies) was added to select for successful incorporation of the transgene.
Ribosome profiling and RNA sequencing
Cells for ribosome profiling and RNA sequencing were processed essentially as described (8). In brief, cells were treated with 100 μg/ml cycloheximide (CHX) for 1 min, washed with ice cold phosphate buffered saline (PBS) containing 100 μg/ml CHX and subsequently lysed in lysis buffer (10 mM Tris pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.5 mM dithiothreitol (DTT) and 100 μg/ml CHX). For ribosome profiling, lysates were subsequently treated with 250 U RNase I (Ambion) for 10 min at 22°C and shaking at 1400 rpm. The digestion was stopped by addition of 100 U SUPERase In RNase inhibitor (Ambion). Ribosomes were separated on a 10–50% (w/v) sucrose density gradient in 50 mM Tris pH 7.5, 50 mM NH4Cl, 12 mM MgCl2, 0.5 mM DTT and 100 μg/ml CHX for 3 h at 35 000 rpm and 4°C in a TH-641 rotor (Thermo Scientific). Gradients were fractionated at 0.75 ml/min with continuous monitoring of OD254 on a density gradient fractionator (Isco) coupled to a SYR-101 syringe pump (Brandel). Sodium dodecylsulphate (SDS) was added immediately to 1% and 400 μl fractions were flash-frozen in liquid nitrogen and stored at −80°C until further processing. Monosome fractions were pooled, RNA isolated and separated on 15% polyacrylamide gels (8 M urea, 90 mM Tris-borate, 2 mM ethylenediaminetetraacetic acid, pH 8.0). Ribosome footprints were excised between 28 nucleotide (nt) and 32 nt RNA size markers. Sequencing libraries from ribosome footprints were generated essentially as described (26), except for ligation to a preadenylated 3′-adapter (5′ rAppNNNNCTGTAGGCACCATCAAT/3ddC/ 3′) in a reaction containing 200 000 U T4 RNA ligase 2 (truncated, NEB), 25% polyethylene glycol 8000 and 10 U SUPERase In RNase inhibitor for 4 h at 22°C. The first four bases of the 3′-adapter were randomized to minimize ligation bias (27).
RNASeq libraries were prepared from cells lysed similar to ribosome profiling samples. RNA was isolated from cleared lysates by addition of 1% SDS (w/v) and 5 min incubation at 65°C with intermittent vortexing followed by three rounds of extraction using 10:1 acid–phenol pH 4.3 and bromochloropropane. Total RNA was precipitated and 5 μg used for library generation with the TruSeq Stranded mRNA Library Prep Kit (Illumina) according to the manufacturer's instructions. Libraries were sequenced on an Illumina HiScanSQ generating 50 nt single-end reads. Data were mapped to the hg38 transcriptome (UCSC knownCanonical) and counts generated using a custom script. Differential gene expression analysis for footprints and mRNA was performed using the DESeq2 R-package with a P-adjusted threshold of 0.05 and a log2 fold change threshold of 0.5 (28). Translationally regulated genes were defined as differentially expressed based on the ribosome profiling data and having a log2 fold change of the footprints at least twice as high as in the mRNA data. Genes differentially expressed on the transcript level exclusively were defined accordingly, but taking mRNA reads as the basis. Gene ontology (GO) enrichment was determined using GOrilla (29). For the determination of A-site codon occupancy we used a published strategy (30). Briefly, 29–31 nt reads translating the 0-frame were used with an A-site offset of 15 nt from the start of the read. A-site codon occupancy was normalized to the adjacent unread codons in the +1 +2 and +3 positions relative to the A-site. Ribosome occupancy at the +1 position, was consequently normalized to the neighboring +2 +3 and +4 positions. Ribosome occupancy of the first 15 codons of each transcript was not taken into account for all analyzes to exclude influences of sample preparation. The sequencing data of the ribosome profiling and RNA sequencing experiments are available at the Gene Expression Omnibus (Accession number: GSE97140).
Preparation of cellular extracts
For preparation of cellular extracts for in vitro methylation assays, cells were grown to near confluency on 10 cm diameter dishes. The cells were washed three times with 15 ml ice cold PBS and harvested by centrifugation at 400 × g for 5 min. The resulting pellet was dissolved in ∼300 μl mammalian cell lysis buffer (50 mM Tris pH 7.4, 100 mM NaCl, 1% Triton, 10% glycerol and 1% DTT) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF; Sigma) and 1 × protease inhibitor cocktail (Sigma-Aldrich, P8340) and cell lysis was allowed to proceed for 15 min at 4°C. The lysate was then cleared by centrifugation at 16 100 × g for 30 min. The supernatant was thereafter removed and used as substrate for in vitro methylation assays.
Expression and purification of recombinant proteins
Expression and purification of recombinant hexahistidine (His6) tagged proteins in bacteria was performed using standard and previously described methods (11,31). In brief, relevant ORFs were cloned into the pET28a (Novagen) expression vector and plasmids were transfected into the Escherichia coli BL21-(DE3)-RIPL (Agilent) expression strain. For protein expression, cells were cultured in 500 ml volumes at 37°C with orbital shaking at 250 rpm until the suspension reached an optical density of ∼0.5 at 600 nm. The temperature was then reduced to 18°C and recombinant protein expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside to a final concentration of 100 μM. Protein expression was allowed to proceed for 16 h, whereafter cells were harvested by centrifugation and cell pellets were stored at −20°C until further processing. For protein purification, cell pellets were resuspended in a lysis buffer (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 10% glycerol, 0.5% Nonidet P-40, 30 mM imidazole and 3 mM 2-mercaptoethanol) supplemented with 1 × Complete protease inhibitor mixture (Roche Applied Science), 0.5 mg/ml lysozyme (Sigma) and 25 units/ml Benzonase (Sigma). His6 tagged proteins were incubated with nickel nitrilotriacetic acid-agarose (Qiagen) and allowed to bind for 16 h at 4°C. The resin was extensively washed in lysis buffer and recombinant proteins were eluted with lysis buffer supplemented with 300 mM imidazole. Eluted proteins were then buffer-exchanged with storage buffer (20 mM Tris–HCl pH 6.8, 100 mM NaCl, 5% glycerol and 1 mM DTT) by sequential concentration and dilution using Vivaspin ultracentrifugation devices (Sartorius AG) with a molecular weight cut-off of 10 kDa. Proteins were then aliquoted and stored at −80°C.
In vitro MTase assays
In vitro MTase assays were performed in 50 μl volumes for 1 h at 37°C in assay buffer (50 mM Tris–HCl (pH 7.8 at 25°C), 50 mM KCl and 5 mM MgCl2) supplemented with AdoMet and either 1 mM adenosinetriphosphate (ATP) or 1 mM guanosinetriphosphate (GTP) as indicated. For analysis of methylation events by fluorography, the assay buffer was supplemented with 0.5 μM [3H]AdoMet (Perkin Elmer, specific activity: 78.4 Ci/mmol) and ∼30 μg of protein extracts was incubated with 0.2 μM (10 pmol) recombinant MTase or 0.6 μM (30 pmol or ∼1 ug) of recombinant candidate substrate was incubated with 0.3 μM (15 pmol or ∼0.75 ug) MTase. Methylation reactions were then separated by SDS-polyacrylamide gel electrophoresis (PAGE) and proteins transferred to a polyvinylidene difluoride (PVDF) membrane, which was treated with the scintillation enhancer EN3HANCE (PerkinElmer Life Sciences), and exposed to Carestream Biomax MS film (Sigma-Aldrich) as previously described (11). For MS analysis of in vitro methylated proteins, samples were processed as detailed above with the exception that [3H]AdoMet was replaced with 1 mM non-radioactive AdoMet (NEB) and the relevant gel regions of SDS-PAGE-separated samples were subjected to in-gel proteolytic digestion as described below.
For comparative analysis of different candidate substrates, 200 pmol (4 μM) of each substrate was incubated with varying amounts of MTase and 20 μM AdoMet (0.5 μM [3H]AdoMet and 19.5 μM non-radioactive AdoMet). Reactions were terminated by adding 900 μl ice cold 10% trichloroacetic acid (TCA) and proteins were allowed to precipitate for 30 min at 4°C. Precipitated protein was collected by centrifugation at 16 000 × g for 30 min. The resulting pellet was washed two times with 1 ml acetone and subsequently dissolved in 200 μl 0.1 M KOH, diluted in 4 ml Ultima Gold™ XR (PerkinElmer) liquid scintillation enhancer and subjected to liquid scintillation counting. For comparative analysis of different eEF1A-KMT4 mutant constructs, regions of PVDF membrane corresponding to eEF1A were excised and subjected to liquid scintillation counting as described above.
Chromatography based enrichment of methylated substrates
In vitro methylated protein extract from HAP-1 EEF1AKMT4 KO cells was fractionated using Pierce strong anion-exchange Q or strong cation-exchange S spin columns (Thermo Fisher Scientific). In brief, spin columns were equilibrated with ion exchange (IEX) buffer (50 mM Tris pH 7.4 and 100 mM NaCl) and protein extracts were diluted to a final volume of 400 μl in the same buffer. The diluted sample was then applied to an equilibrated Q column. The flow through was collected and applied to an equilibrated S column and the flow through was, again, collected. Both columns were then washed with 400 μl IEX buffer and proteins were eluted in 75 μl fractions with a stepwise salt gradient (0.15, 0.3, 0.5 and 1.0 M NaCl). All steps were performed at 4°C.
Mass spectrometry
MS analysis was essentially performed as previously described (11). In brief, protein samples were separated by SDS-PAGE, whereafter relevant regions of the gel were excised and subjected to in-gel digestion using trypsin (Sigma-Aldrich) or chymotrypsin (Roche Applied Science). Proteolytic peptides were then analyzed by liquid chromatography coupled to tandem mass spectrometry (MS/MS) setup (LTQ Orbitrap XL mass spectrometer, Thermo Scientific) via nanoelectrospray. Protein identification was performed with an in-house-maintained database of the human proteome and the SEQUEST software. For identification of methylation sites, a single protein database of eEF1A sequences was used and carbamidomethylation was set as a fixed modification and lysine methylation (mono, di and tri), arginine methylation (mono and di), as well as methionine oxidation, were set as dynamic modifications.
Ion chromatograms and tandem mass spectra for described peptides were analyzed and extracted using Qual Browser (v2.0.7). The selective ion setting used to extract chromatograms corresponding to the various methylated forms of the carbamidomethylated chymotryptic peptide encompassing K30-F42 in eEF1A1 and eEF1A2 (KCCarbamidomethylGGIDK(Mex)RTIEKF) were: m/z = 388.7133 (unmethylated), 392.2172 (mono-methylated), 395.7211 (di-methylated) and 399.2250 (tri-methylated) (z = +4) +/− 10 ppm. The signal for the different methylated forms of the peptide was determined as the area under the relevant chromatographic peak using Qual Browser (v2.0.7) and the occupancy of the unique methylated forms was approximated as their relative signal.
Tandem affinity purification
For TAP, Flp-In 293 T-REx derived cell lines inducibly expressing N-terminally CBP-SBP-tagged eEF1A1 or eEF1A2 were seeded into two T175 flasks. Expression of transgenes was induced at a confluency of ∼50% by adding medium containing 1 μg/ml doxycycline and allowed to proceed for 24 h. Cells were harvested by scraping and CBP-SBP-tagged proteins were purified using the InterPlay mammalian TAP system (Agilent) according to the supplier's suggestions, with a few exceptions. In brief, cells were lysed by three consecutive freeze/thaw cycles in the kit's lysis buffer supplemented with 1 mM PMSF and 1 × protease inhibitor cocktail. The supernatant after centrifugation for 1 h at 20 000 × g was then incubated with the SBP affinity resin. Samples were not processed with the CBP resin, as we have occasionally experienced sample loss during TAP and observed adequate enrichment with the SBP resin alone. Instead, protein samples were eluted from the SBP resin by boiling in NuPAGE buffer and stored at −20°C until analyzed by MS.
Preparation of protein extracts from rat organs
Protein extracts from rat organs were prepared as previously described (8). In brief, tissue was washed extensively in ice cold PBS. For each mg of tissue, 3 μl of the mammalian cell lysis buffer described above supplemented with 1 mM PMSF (Sigma-Aldrich) and 1 × protease inhibitor mixture (Sigma-Aldrich, P8340). The tissue was then homogenized in a tissue grinder for 30 min and insoluble material was separated by centrifugation at 16, 100 g for 30 min at 4°C. eEF1A proteins present in the soluble supernatant were enriched by cation (S column) exchange chromatography, and the methylation status of the proteins was assessed by MS as described above.
RESULTS
ECE2/EEF1AKMT4 gene structure
During our efforts to identify novel human KMTs, the protein denoted ECE2 caught our interest, since it contains a putative 7BS MTase domain (Figure 1A) that is somewhat related to eEF1A-KMT2, an eEF1A specific KMT (2,7). Human ECE2 is currently annotated as a multidomain protein encoded by 22 exons localized as two clusters separated by a large intron on human chromosome 3 (Figure 1B). The first cluster, represented by the three 5′ proximal exons, encodes the 7BS MTase domain (32), while the second, downstream exon cluster, consisting of 19 exons, encodes transmembrane and peptidase domains (Figure 1A and B). The ECE2 peptidase is responsible for generating the regulatory peptide endothelin 1 from a larger precursor (33). According to the current gene annotation, the ECE2 gene encodes shorter isoforms that represent the separate MTase or transmembrane/peptidase moieties, in addition to the full-length protein. Intriguingly, while the existence of such shorter isoforms is well supported by EST in human and mouse, the existence of the full-length protein is not supported, since ESTs spanning both the MTase and transmembrane/peptidase regions have not been detected (Figure 1B). Also, in lower animals, such as fruit fly and nematode, potential orthologs of the MTase and peptidase are present, but are encoded by two different genes. Finally, this MTase was detected in a very recent study that used a chemical proteomic profiling approach to identify human MTases at the protein level, but the data did not support the existence of MTase/peptidase fusion proteins (34). Thus, the available data suggest that the current ECE2 gene annotation should be modified, thus considering the exon clusters encoding the MTase and transmembrane/peptidase moieties as two separate genes (Figure 1B). As will become evident from subsequent sections, this MTase represents an eEF1A-specific KMT, which we in agreement with the current nomenclature have named and hereafter refer to, as eEF1A-KMT4 (gene name EEF1AKMT4).
Figure 1.

Gene structure and targeting strategy for the endothelin converting enzyme 2 (ECE2)/EEF1AKMT4 locus. (A) Topology diagram of the 7BS MTase fold. α-helices and β-strands are depicted as grey boxes (denoted Z and A–E) and black arrows (denoted 1–7), respectively, and annotated according to previously established nomenclature (50). (B) Organization and annotation of the human ECE2 locus. Top, current annotation of the ECE2 locus. Regions in the genomic DNA corresponding to annotated exons of ECE2 (located on the forward stand) and CAMK2N2 (located on the reverse strand) are indicated in blue and green, respectively (based on the annotated gene structure according to Ensembl (assembly GRCh37)). Middle, tracks representing spliced expressed sequence tags (ESTs) exported from the UCSC genome browser. Bottom, alternative gene model supported by the EST data. (C) Gene targeting strategy and disruption of the EEF1AKMT4 locus by CRISPR/Cas9 technology. Top, schematic representation of the EEF1AKMT4 gene. Exons and introns are represented by boxes (gray, coding region; white, untranslated regions) and lines, respectively. An arrow indicates the region targeted by CRISPR. Bottom, DNA and protein sequence of targeted region of EEF1AKMT4 locus in HAP-1 wild-type (WT) and EEF1AKMT4 knockout (KO) cells. The conserved motif Post I is indicated by a rectangle and the last residue of the motif, D88, is shown in red. (D) Structural support for an involvement of D88 in coordinating AdoMet binding. The figure is based on a previously published structure (PDB ID: 2PXX) of eEF1A-KMT4 in complex with S-adenosylhomocysteine (AdoHcy; the unmethylated counterpart of AdoMet) (32). AdoHcy and D88 are represented as sticks and the second β-strand (β2) is represented as a cartoon. Possible hydrogen bonds between the carboxyl group of D88 and the ribose moiety of AdoHcy are shown (dashed lines). The figure was generated using the PyMOL Molecular Graphics System, Version 1.3 (Schrodinger, LLC).
Generation of EEF1AKMT4 knockout-cells and demonstration of protein MTase activity
For functional characterization of the eEF1A-KMT4 protein, the EEF1AKMT4 gene was disrupted in HAP-1 cells using the CRISPR/Cas9 technology. To assure abrogation of gene function, guide RNAs were designed to target a segment upstream of a previously described key motif denoted ‘Post I’, localized in the second β-strand in the canonical 7BS topology (Figure 1C). The last residue of motif Post I is universally conserved as an acidic amino acid in the 7BS superfamily and has been suggested to stabilize the interaction with AdoMet by hydrogen bonding (35). The three-dimensional (3D) structure of eEF1A-KMT4 in complex with AdoHcy has been solved (32) and the location and orientation of the corresponding acidic residue (D88), relative to AdoHcy, clearly supports this notion (Figure 1D). A clone that was found (by DNA sequencing) to contain a 17 nt deletion at the target site was used for further work. In this clone, the reading frame of the gene is shifted so that D88 is no longer present (Figure 1C) and a severely truncated version (108 residues) of the eEF1A-KMT4 protein (255 residues) is encoded.
We have previously shown that several recombinant 7BS KMTs can methylate specific proteins when added to a cellular extract and that KMT substrates can be identified by fractionation of the extract (8,14,36). Moreover, such methylation is often strongly enhanced in extracts from cells that lack the relevant KMT, due to an increased level of unmethylated substrate (6,36,37). Thus, to investigate the potential protein MTase activity of eEF1A-KMT4, protein extracts from EEF1AKMT4-deficient and WT HAP-1 cells were incubated with recombinant eEF1A-KMT4 and [3H]AdoMet. Subsequently, proteins were separated by SDS-PAGE and methylation was visualized by fluorography. Several of the characterized human 7BS KMTs modify nucleotide-binding proteins and the addition of nucleotides such as ATP or GTP to in vitro methylation reactions has been shown to enhance methylation (8,11). Therefore, we added ATP to the reaction mixture for eEF1A-KMT4-mediated methylation. Notably, these experiments revealed that recombinant eEF1A-KMT4 specifically methylated ∼50 kDa protein substrate in the WT HAP-1 extract and that methylation was enhanced in EEF1AKMT4-deficient cells (Figure 2A). To exclude the possibility that the observed activity was due to an E. coli MTase co-purifying with eEF1A-KMT4, we assayed the activity of a presumably inactive mutant protein where the key catalytic residue D88 was mutated to alanine (eEF1A-KMT4-D88A). Reassuringly, this single point mutation completely abrogated the activity of the enzyme (Figure 2B). In conclusion, the above experiments establish eEF1A-KMT4 as a protein MTase.
Figure 2.

Identification of eEF1A proteins as candidate substrates for eEF1A-KMT4. (A) eEF1A-KMT4-mediated methylation in protein extracts. Protein extracts from HAP-1 wild-type (WT) or EEF1AKMT4 KO cells were incubated with [3H]-AdoMet and with recombinant eEF1A-KMT4 as indicated. Top panel, Ponceau S-stained membrane of sodium dodecyl sulphate (SDS-PAGE)-separated methylation reactions. Bottom, visualization of methylation by fluorography of membrane in upper panel. (B) Mutation of D88 abrogates the enzymatic activity of eEF1A-KMT4. Protein extract from HAP-1 EEF1AKMT4 KO cells incubated with [3H]-AdoMet in the presence of either recombinant WT eEF1A-KMT4 or the corresponding D88A mutant protein. Reactions were analyzed as in (A). (C) Outline of chromatography based enrichment strategy for eEF1A-KMT4 substrates. In vitro methylated protein extract was loaded onto an anion exchange (Q) column and, in turn, the flow-through (FTQ) was loaded onto a cation exchange (S) column. Proteins were eluted from both columns with a step gradient of increasing NaCl. (D) Enrichment of the ∼50 kDa eEF1A-KMT4 substrate. Proteins were eluted from ion exchange columns with a step gradient ramped from 150 mM (0.15) to 1 M (1.0) NaCl according to the scheme depicted in (C). Collected fractions were analyzed by fluorography as in (A). (E) SDS-PAGE analysis of the main substrate-enriched fraction (0.3S). The fraction containing the bulk of methylated substrate in (D) eluted at 300 mM NaCl from the S column (0.3S). The most prominent proteins in the major ∼50 kDaband were identified as eEF1A1 and eEF1A2 by mass spectrometry (MS) (See Table 1).
Identification of eEF1A proteins as substrates for eEF1A-KMT4
To identify the ∼50 kDa protein targeted by eEF1A-KMT4, we deployed ion chromatography to enrich the methylated substrate, facilitating its subsequent identification by MS. In brief, in vitro [3H]methylated protein extract from EEF1AKMT4-deficient cells was sequentially processed by anion (Q) and cation (S) exchange chromatography, and proteins were eluted with an increasing salt gradient. The signal from the radiolabeled protein was subsequently traced in collected fractions by fluorography (Figure 2C).
The methylated protein was recovered in the flow-through from the Q column (FTQ), which was then applied to the S column (Figure 2D). The radiolabeled eEF1A-KMT4 substrate was found to bind to the S column and mainly elute at 0.3 M NaCl (0.3S) (Figure 2D). SDS-PAGE analysis of a parallel sample, in which [3H]-AdoMet was omitted, revealed that this fraction contained a prominent band matching the molecular mass of the methylated protein (Figure 2E). MS based protein identification revealed the presence of several proteins in this band (Table 1). Interestingly, eEF1A1 and its close paralog eEF1A2 were identified with the highest score and coverage. In addition, the calculated molecular mass and isoelectric point of eEF1A matched the observed electrophoretic mobility and chromatographic behavior of the methylated substrate(s).
Table 1. List of proteins identified in the ∼50 kDa region of the substrate enriched fraction.
| Protein name | Gene identifier | Score | Coverage (%) | Molecular mass (kDa) | Isoelectric point (pI) |
|---|---|---|---|---|---|
| elongation factor 1-α 1 | 4503471 | 80.5 | 23.2 | 50.1 | 9.0 |
| elongation factor 1-α 2 | 4503475 | 66.6 | 12.7 | 50.4 | 9.0 |
| keratin, type II cytoskeletal 1 | 119395750 | 17.9 | 11.2 | 66.0 | 8.1 |
| keratin, type I cytoskeletal 10 | 195972866 | 10.6 | 8.4 | 58.8 | 5.2 |
| keratin, type II cytoskeletal 2 | 47132620 | 8.8 | 6.6 | 65.4 | 8.0 |
| keratin, type II cytoskeletal 6B | 119703753 | 5.3 | 5.3 | 60.0 | 8.0 |
| keratin, type I cytoskeletal 9 | 55956899 | 4.3 | 3.4 | 62.0 | 5.2 |
| protein FAM98B isoform 2 | 109452589 | 2.7 | 3.9 | 37.2 | 6.3 |
| serine hydroxymethyltransferase, mitochondrial isoform 3 | 261862352 | 1.7 | 1.9 | 53.4 | 8.1 |
| α-enolase isoform 1 | 4503571 | 1.7 | 4.6 | 47.1 | 7.4 |
| proliferation-associated protein 2G4 | 124494254 | 1.7 | 2.8 | 43.8 | 6.6 |
To further determine whether eEF1A is a substrate of eEF1A-KMT4, recombinant eEF1A1 was expressed and purified from E. coli and subjected to methylation by recombinant eEF1A-KMT4 in vitro. Since eEF1A proteins bind GTP and since we previously observed nucleotide-dependent methylation of eEF1A by other KMTs (8,31), we investigated the effect of adding GTP or ATP to the reaction mixture for eEF1A-KMT4-mediated methylation. Clearly, eEF1A1 was efficiently methylated in the presence of either ATP or GTP, whereas no detectable methylation occurred in the absence of these cofactors and, notably, the stimulatory effect was more pronounced for GTP than for ATP (Figure 3A). Therefore, GTP was included in all subsequent methylation assays. Similar to previous observations for related enzymes, we observed considerable automethylation of eEF1A-KMT4, but this activity was independent of the presence of nucleotides (Figure 3A).
Figure 3.
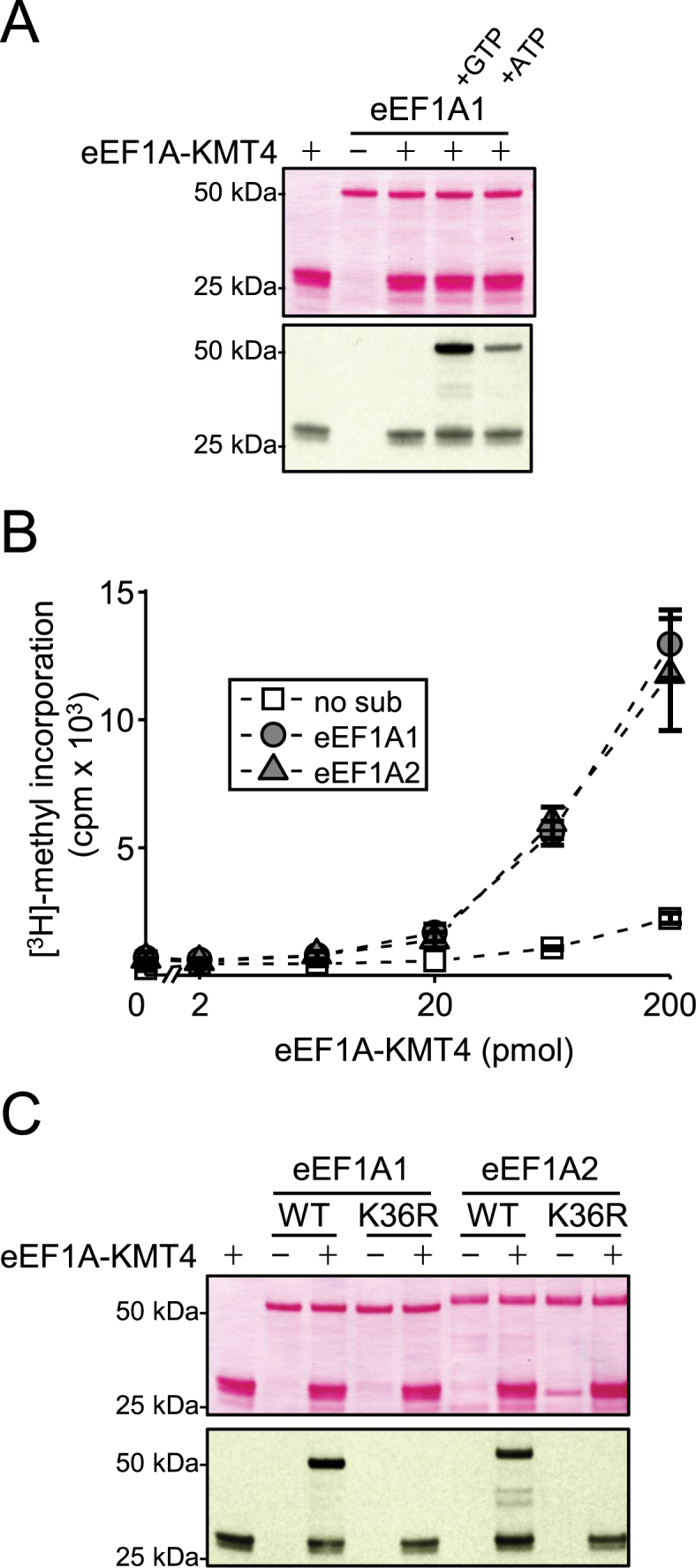
eEF1A-KMT4-mediated methylation of K36 in eEF1A proteins in vitro. (A) Nucleotide-dependent methylation of recombinant eEF1A1 by eEF1A-KMT4. Recombinant eEF1A1 was incubated with [3H]-AdoMet and eEF1A-KMT4, as well as with adenosinetriphosphate (ATP) or GTP as indicated. Top, Ponceau S stained membrane of proteins. Bottom, visualization of methylation by fluorography of membrane in upper panel. (B) Comparison of eEF1A1 and eEF1A2 as substrates for eEF1A-KMT4-mediated methylation. Recombinant eEF1A1 or eEF1A2 was incubated with varying amounts of eEF1A-KMT4. Methylation was quantified as trichloroacetic acid (TCA)-insoluble radioactivity. Error bars represent the standard deviation (n = 3). (C) Evaluation of eEF1A point mutants as substrates for eEF1A-KMT4-mediated methylation. WT eEF1A1 and eEF1A2 or the corresponding Lys36→Arg (K36R) mutant proteins were incubated with recombinant eEF1A-KMT4 in the presence of [3H]-AdoMet. Reactions were analyzed as in (A). Note: In panels (A) and (C), automethylation of recombinant eEF1A-KMT4 is observed, but such automethylation was not observed in the experiments shown in Figure 2A, B and D. This apparent discrepancy is likely because less enzyme and of lower specific activity, was used in Figure 2.
To compare eEF1A1 and eEF1A2 as substrates for eEF1A-KMT4-mediated methylation, these two proteins were incubated with increasing amounts of recombinant eEF1A-KMT4 and a non-limiting amount of radiolabeled AdoMet. Methyl transfer was subsequently quantified by measuring [3H]-methyl groups in TCA insoluble material by liquid scintillation counting. This analysis showed that both paralogs are substrates for eEF1A-KMT4 in vitro and that they are methylated at comparable efficiencies (Figure 3B).
Taken together, the above results demonstrate that recombinant eEF1A-KMT4 is capable of methylating both eEF1A1 and eEF1A2 in vitro and suggest that eEF1A-KMT4-mediated methylation of eEF1A may be modulated by ATP and GTP.
Recombinant eEF1A-KMT4 methylates K36 in eEF1A proteins
To identify the residue(s) targeted by eEF1A-KMT4, recombinant eEF1A1 was treated with eEF1A-KMT4 and non-radioactive AdoMet, followed by proteolytic digestion with chymotrypsin and the resulting peptides were analyzed by MS. In this analysis, peptides with di- or trimethylation on K36 were identified (Supplementary Figure S1). To further support the observed eEF1A-KMT4-mediated methylation of K36, this residue was mutated to arginine in eEF1A1 and eEF1A2, and the resulting proteins (denoted eEF1A1-K36R and eEF1A2-K36R) were assessed for eEF1A-KMT4-mediated methylation. Importantly, neither of the two K36R-mutated eEF1A proteins were subject to eEF1A-KMT4-mediated methylation (Figure 3C), indicating that K36 is the only residue targeted by eEF1A-KMT4. In summary, the above data demonstrate that eEF1A-KMT4 catalyzes methylation of K36 on both eEF1A paralogs in vitro.
Methylation of K36 in eEF1A proteins is dependent on eEF1A-KMT4 in vivo
To investigate the role of eEF1A-KMT4 in the biogenesis of K36 methylation in vivo, we enriched eEF1A proteins from HAP-1 cells and corresponding EEF1AKMT4 KO cells using the previously established purification method (Figure 2C and D) and assessed eEF1A methylation status by MS. Our results showed that all three methylated forms of K36, as well as the unmethylated species, were readily detected in WT cells, whereas K36 was exclusively found in the unmethylated state in EEF1AKMT4 KO cells (Figure 4A and Supplementary Figure S2).
Figure 4.

eEF1A-KMT4-mediated methylation of K36 in eEF1A in vivo. (A) EEF1AKMT4 KO abrogates eEF1A-K36 methylation in human cells. Ion chromatograms representing peptides of the different methylation states of K36 in eEF1A proteins from HAP-1 WT and EEF1AKMT4 knock out (KO) cells are shown. The retention time for analyzed peptides is indicated (arrow) and their relative abundances are indicated (obtained by integration of the area of the relevant peaks; expressed as the percentage of the sum of all peaks). Tandem mass spectra supporting identity of analyzed peptides are shown in Supplementary Figure S2. (B) eEF1A1 and eEF1A2 are both methylated at K36 in vivo. Chromatograms represent the methylation status of K36 in TAP-tagged eEF1A that was affinity-purified from HEK293-derived cells ectopically expressing TAP-tagged eEF1A1 or eEF1A2. (C) Methylation status of eEF1A-K36 in mammalian organs. Chromatograms representing the methylation status of K36 in eEF1A obtained from rat brain, liver, intestine and kidney are shown.
The region surrounding K36 is highly conserved throughout evolution and, consequently, proteolytic peptides from eEF1A1 and eEF1A2 that encompass this residue are indistinguishable by MS. Thus, our observation that a mixture of the various methylated forms of the peptide exists in HAP-1 cells could reflect differences in eEF1A1 versus eEF1A2 methylation at this site in vivo. To investigate this, we over-expressed tagged eEF1A proteins in HEK-293 cells and analyzed the methylation status of K36 in affinity purified proteins. This analysis showed that both eEF1A1 and eEF1A2 are methylated on K36 in vivo and that the trimethylated form is predominant (Figure 4B).
To analyze the occurrence and extent of K36 methylation in different mammalian organs, we enriched eEF1A from rat liver, kidney, intestine and brain, followed by an assessment of the methylation state by MS as above. In these organs, we observed high levels of K36 methylation (Figure 4C), similarly to our observations from HAP-1 WT cells. In all analyzed organs, the bulk (>90%) of eEF1A was methylated and the trimethylated form was predominant.
Thus, we have demonstrated that methylation of eEF1A on K36 is dependent on eEF1A-KMT4 and occurs in a wide range of mammalian cell types. Taken together with our findings on eEF1A-KMT4-mediated methylation of eEF1A in vitro, these results indicate that eEF1A-KMT4 is necessary and sufficient for methylation of K36 in eEF1A1 and eEF1A2 in vivo.
The absence of eEF1A-KMT4 activity changes cellular mRNA translation
Since eEF1A-KMT4 targets eEF1A, a key component of the translational apparatus, we sought to explore its impact on translation. To this end, we performed quantitative mRNA translation analysis by ribosome profiling (38), comparing the EEF1AKMT4 KO cells to the WT HAP-1 cells. When analyzing mRNA abundance and mRNA translation, we found that translation levels were changed for a large number of genes. While the expression of many genes was mainly changed at the level of transcription, 823 out of 1317 differentially translated genes displayed changes that could not be subscribed solely to altered transcription (Figure 5A, blue circles and Supplementary Table S2). Interestingly, genes encoding ribosomal proteins were enriched amongst the upregulated genes, reflected by an over-representation of translation-associated GO terms (e.g. ER/SRP-associated translation; Figure 5B). Furthermore, genes associated with several basic metabolic processes showed increased translation in the EEF1AKMT4 KO. Downregulated genes show a less clearly defined GO-term enrichment. Processes such as cell migration and organization of the extracellular matrix were most significantly downregulated (Figure 5C). Similarly, genes associated with protein folding in the ER and tRNA aminoacylation were enriched among the downregulated ones, thus showing a functional link to the upregulated processes.
Figure 5.

The effect of EEF1AKMT4 KO on the translational landscape. (A) Differential expression plot showing the log2-fold changes on the mRNA and ribosome footprint level in EEF1AKMT4 KO relative to WT cells. Indicated are: not significantly changed genes (DEseq2 padj >0.05) (gray), genes differentially expressed at the mRNA level only (green), genes with differential ribosome footprinting only (blue) and genes that were changed at both the mRNA and ribosome footprinting levels (red). (B) GO analysis (Cellular Process) for genes significantly upregulated on the footprint level only. n = 322, numbers in brackets indicate genes matching this category. (C) GO analysis (Cellular Process) for genes significantly downregulated on the footprint level only. n = 501, numbers in brackets indicate genes matching this category. (D) Ribosome occupancy in EEF1AKMT4 KO cells compared with WT cells. A-site occupancy and +1 occupancy are normalized to untranslated downstream codons (mean ± SD, n = 3). Symbol size indicates the relative frequency of codons.
Since the EEF1AKMT KO affected translation genes and aminoacyl tRNA synthetases and since eEF1A-KMT4-mediated methylation of eEF1A may influence its ability to deliver aminoacyl-tRNA to the ribosome, we compared the transcriptome-wide codon occupancy at A-site in the ribosome in the KO versus the WT HAP-1 cells (Figure 5D). We found that ribosome occupancy, which is a proxy for translation speed, was altered for several codons in the EEF1AKMT4 KO relative to the WT. Interestingly, codons that encode the same amino acid generally showed the same trend. This was most apparent for ACC and AAT, which encode asparagine and showed decreased occupancy in the KO cells. Conversely, CAC and CAT, which encode histidine, showed increased A-site occupancy. Furthermore, the effect was observed to a lesser extent for codons corresponding to several other amino acids (decreased A-site occupancy: Asp, Ala, Val, Tyr; increased A-site occupancy: Pro, Leu; Figure 5D). In addition, the single Trp-encoding codon TGG showed reduced occupancy in the KO cells. However, aminoacyl tRNA synthetases with altered expression or translation levels do not correlate with codons that show changes in codon occupancy. Importantly, when we analyzed the relative codon occupancy of the +1 site, representing the unread codon that follows the A-site, no such effect was observed. Most codons displayed 0.9- to 1.1-fold changes, corresponding to the experimental error of the method (Figure 5D). Furthermore, the +1 codons did, in contrast to the A-site codons, not show similar effects for synonymous codons. This indicates that the observed effect of the EEF1AKMT KO on A-site occupancy is specific and that methylation of eEF1A contributes to the fine-tuning of translation rates for a subset of tRNAs.
eEF1A-KMT4 belongs to a novel family of established and putative KMTs
eEF1A-KMT4 belongs to a subfamily of four related proteins within the human 7BS superfamily (2), as indicated in a phylogenetic tree representing established and putative human 7BS KMTs (Figure 6A). In addition to eEF1A-KMT4, this family encompasses eEF1A-KMT2, METTL12 and METTL13. eEF1A-KMT2 and eEF1A-KMT4 are single-domain proteins consisting of a core 7BS-MTase domain with short flanking sequences (Figure 6B). METTL12 has been reported as a mitochondrial protein and, accordingly, carries an N-terminal mitochondrial targeting sequence (15). METTL13 has a unique domain structure, as it consists of two unrelated 7BS MTase domains. The N-terminal MTase domain of METTL13 is closely related to eEF1A-KMT4, whereas the closest human sequence homolog of the C-terminal domain is spermidine synthase, which uses decarboxylated AdoMet as a co-substrate for aminopropylation of putrescine to spermidine (39). eEF1A-KMT2 and eEF1A-KMT4 have now been established as KMTs (present study and (9)), suggesting that also METTL12 and METTL13 may possess KMT activity. To gain further insights into the MTase family constituted by these four proteins, we performed a bioinformatics analysis.
Figure 6.

Identification of a protein family of eEF1A-KMT4-like KMTs. (A) Phylogenetic tree of human 7BS KMTs. MTF16 members (green), the family of eEF1A-KMT4-like proteins (red) and the remaining putative or characterized 7BS KMTs (blue) are represented. (B) Domain organization of the human eEF1A-KMT4-like proteins. The MTase domain of eEF1A-KMT4 is closely related to the 7BS domains of eEF1A-KMT2, METTL12 and the N-terminal domain of METTL13 (indicated in red). METTL13 contains a second 7BS MTase domain (yellow) which is not related to eEF1A-KMT4 and its closest human homologs is spermidine synthase (SPDS). (C) eEF1A-KMT4 homologs share motif Post II. The region encompassing the β-strand 4 in the canonical 7BS structure, as well as motifs ‘II’ and ’Post II" (red rectangle) are shown. Hs, Homo sapiens; Ce, Caenorhabditis elegans; At, Arabidopsis thaliana; Dm, Drosophila melanogaster; Dr, Danio rerio; Nv, Nematostella vectensis; Sc, Saccharomyces cerevisiae. (D) Localization of the residues of motif Post II in the active site of eEF1A-KMT4. Top, cartoon representation of the eEF1A-KMT4 structure (PDB 2PXX). β-strands and α-helices of the 7BS core fold are colored black and gray, respectively. The N-terminal extension preceding the core 7BS fold is indicated in orange, whereas, motif Post II is shown in red. The co-substrate AdoHcy is shown in stick representation. Bottom, surface representation of structure in upper panel, reoriented to display residues in motif Post II. Inset, magnification of the structure illustrating the presence of a tunnel, constituted by motif Post II and the N-terminal extension, ending in proximity of the sulfur atom (yellow) of AdoHcy. (E) Mutational analysis of motif Post II in eEF1A-KMT4. Mutant eEF1A-KMT4 proteins with single amino acid substitutions in motif Post II were tested for MTase activity on recombinant eEF1A1. Activities of mutant enzymes are represented relative to that of the WT enzyme. Error bars represent standard deviation (n = 3). Representative results from a corresponding fluorography experiment are shown in Supplementary Figure S3.
First, we identified putative orthologs of the four human family members in various organisms. Proteins that retrieved the original human query as the primary hit were categorized as likely orthologs. This analysis revealed that eEF1A-KMT2 has the widest evolutionary distribution of the family members with likely orthologs in budding yeast (Saccharomyces cerevisiae), plants (Arabidopsis thaliana), nematodes (Caenorhabditis elegans) and insects (Drosophila melanogaster) (Table 2). Orthologs of METTL13 and eEF1A-KMT4 were identified exclusively in multicellular eukaryotes, but eEF1A-KMT4 showed a scattered distribution within this group, being present in vertebrates, plants and nematodes, but absent in e.g. insects. Putative METTL12 orthologs were mainly confined to vertebrates, where they showed a rather scattered evolutionary distribution, but were also found in a few invertebrate species.
Table 2. Human eEF1A-KMT4-like proteins and their orthologs in common models organisms.
| Human protein query | Accession # | Identity (%)* | Putative orthologs (expect value) | |||
|---|---|---|---|---|---|---|
| D. melanogaster | C. elegans | A. thaliana | S. cerevisiae | |||
| eEF1A-KMT4 | NP_115707.2 | 100 | NP_001021001.1 (1e-36) | NP_195162.2 (4e-42) | ||
| METTL13 | NP_057019.3 | 41 | NP_001260634.1 (6e-169) | NP_501024.1 (2e-92) | NP_850171.1 (7e-102) | |
| METTL12 | NP_001036694.1 | 31 | ||||
| eEF1A-KMT2 | NP_997719.2 | 27 | NP_608733.1 (3e-45) | NP_500612.1 (2e-28) | NP_176841.1 (2e-38) | NP_012200.1 (1e-25) |
*To eEF1A-KMT4. Determined by EBI-needle using region corresponding to core 7BS domain of eEF1A-KMT4 (I62-S176) as query.
Second, we generated a sequence alignment of the putative orthologs from various organisms. Strikingly, all these orthologs contain a conserved sequence motif, i.e. [D/E]-K-G-T-X-D (consensus) located directly downstream of the fourth β-strand of the 7BS fold (Figure 6C). The corresponding segment has been referred to as motif ’Post II’ in other 7BS KMTs and has been linked to substrate specificity. For example, the human members of MTF16, consisting of several established KMTs, have a conserved [D/E]-X-X-[Y/F] motif at the corresponding position (12). Also other 7BS KMTs, such as eukaryotic Dot1/DOT1L and bacterial PrmA, display a conserved sequence motif at this position, and residues within this motif have been implicated in catalysis and substrate recognition (5). Analysis of the previously reported structure of eEF1A-KMT4 revealed that the [D/E]-K-G-T-X-D motif and the N-terminal extension preceding the core 7BS fold form the walls of a tunnel leading up to the sulfur atom of AdoHcy (corresponding to the methyl bearing sulfur atom of AdoMet) (Figure 6D). Thus, the location of this motif in the structure of eEF1A-KMT4 suggests that it is important for the catalytic function of the enzyme.
To investigate the functional importance of the [D/E]-K-G-T-X-D motif, we individually mutated the relevant side-chain-containing residues in eEF1A-KMT4 (i.e. G131 was excluded) to alanine and assayed the activity of the mutant enzymes on recombinant eEF1A1 (Figure 6E and Supplementary Figure S3). We found that mutation of the charged residues in this motif (i.e. E129, D134 or K130) abrogated enzymatic activity, whereas mutation of remaining residues (i.e. T132 or L133) reduced methylation.
In conclusion, the above bioinformatics analyzes demonstrate that eEF1A-KMT4, together with its paralogs eEF1A-KMT2, METTL12 and METTL13, constitute a subfamily of human MTases with homologs in multiple eukaryotic organisms. Furthermore, we demonstrate the presence of a shared [D/E]-K-G-T-X-D motif, corresponding to the previously described and specificity-associated motif Post II. We find that this motif is crucial for enzymatic activity of eEF1A-KMT4, suggesting that also METTL12 and METTL13 are KMTs.
DISCUSSION
MTases of the 7BS superfamily have been shown to methylate a wide range of substrates and it has in recent years become clear that several of these enzymes target lysine residues in proteins (5). Through the present work, we have established the biochemical function of the yet uncharacterized 7BS MTase eEF1A-KMT4, showing that this enzyme is a KMT specifically targeting K36 in eEF1A, both in vitro and in cells. Interestingly, EEF1AKMT4 KO caused alterations in the translation of several mRNAs, as well as in the translation rates of codons corresponding to specific amino acids. Moreover, we have noted that eEF1A-KMT4, together with the eEF1A-specific KMT eEF1A-KMT2 and the yet uncharacterized MTases METTL12 and METTL13, form a 7BS MTase family that shares a functionally important sequence motif, indicating that also METTL12 and METTL13 are KMTs.
Besides possessing a common structural fold, the members of the 7BS class of MTases share several conserved sequence motifs, denoted ‘I’, ‘Post I’, ‘II’ and ‘III’. In addition to these general motifs, more closely related 7BS MTases with similar substrate specificities typically share a motif denoted ‘Post II’, localized immediately after β-strand 4 of the 7BS fold. For example, several MTases acting on DNA and RNA have a so-called DPPY-motif (consensus [S/N/D]-P-P-[Y/W/F]) at this location (40). Also, it has been demonstrated that the ten human MTF16 members share a so-called DXXY-motif (consensus [D/E]-X-X-[Y/F]) at the Post II position (12), and several of these enzymes have recently been established as KMTs (6,8,10–15,41).
Moreover, motif Post II is generally conserved between orthologous 7BS KMT enzymes from different organisms. For example, the bacterial PrmA KMTs have a conserved A-N-[I/L] motif at this position, whereas the eukaryotic Dot1 KMTs share a Post II motif with sequence N-N-F-X-F (5). Interestingly, the previously characterized 7BS KMTs for which the 3D structure has been determined have an aspartate, glutamate, asparagine or glutamine residue of motif Post II at an equivalent position within the structure and in the immediate vicinity of the methyl group to be transferred from AdoMet to the substrate (5). In the case of PrmA and Dot1, the only two 7BS KMTs for which structures of enzyme/substrate complexes are available, this residue has indeed been implicated in substrate recognition (5,42–44). Thus, one may speculate that acidic residues within the [D/E]-K-G-T-X-D motif found in the family of KMTs described here (eEF1A-KMT2, eEF1A-KMT4, METTL12 and METTL13) play a similar catalytically important function, since individual replacement of these residues in eEF1A-KMT4 completely abrogated enzymatic activity.
A ternary complex formed by eEF1A, GTP and aminoacyl-tRNA binds to the ribosome, ensuring the sampling of the anticodon to match the codon in the ribosomal A-site. Subsequently, GTP is hydrolyzed and the correct aminoacyl-tRNA becomes accommodated in the A-site (reviewed in (45)). During translation tRNAs are recognized by shape and not by sequence. Thus, eEF1A needs to present all tRNAs to the ribosome in a similar manner despite the tRNAs having different primary sequence and being charged with different amino acids. Here, methylation may provide a cellular strategy to tune eEF1A for specific subsets of aminoacyl-tRNAs. We found that the absence of K36 methylation caused a change in the translation rates of specific codons. Interestingly, not all codons respond in a similar way. For example, the codons AAC and AAT (encoding asparagine) showed a lower codon occupancy in the EEF1AKMT4 KO cells than in WT, indicating faster translation in the mutant. In contrast, CAC and CAT (encoding histidine) showed an increased A-site occupancy in the KO cells. While for asparagine, aspartic acid, histidine, one isoacceptor dominates the cellular pool, this is not the case for phenylalanine. Furthermore, we did not find a direct link between differential regulation of aminoacyl tRNA synthetases and codons with altered occupancy in the EEF1AKMT4 KO suggesting that the relationship is more complex. Importantly, translation is a highly optimized process that depends on consistent kinetics; therefore it is likely that its key players were fine-tuned during evolution. Intriguingly, post-transcriptional tRNA modifications show parallels to this theme: the absence of tRNA anticodon modifications alters codon-specific translation dynamics similar to eEF1A methylation (30,46). This suggests that evolution has generated different and independent mechanisms to fine-tune translation and to optimize its kinetics. Nevertheless, the translational responses that we observed in the KO are likely a combination of direct perturbations and downstream effects of these defects. Thus, it will be important to analyze these processes in the future to obtain a detailed molecular understanding of how the different players in translation ensure its optimal dynamics.
We observed that EEF1AKMT4 inactivation had interesting effects on the translation of certain codons and consider it likely that these effects reflect the lack of methylation on K36 in eEF1A. However, one cannot completely rule out the possibility that the EEF1AKMT4 KO cells have undergone changes during their generation and cultivation, relative to the ‘WT’ HAP-1 cells, that are independent of, or secondary to, the EEF1AKMT4 gene KO. Thus, to more firmly establish that the observed translation phenotypes are directly caused by the lack of eEF1A-KMT4 activity, one would ideally test if complementation of the KO cells with an ectopically expressed EEF1AKMT4 gene would reverse these phenotypes. However, we have experienced that it is difficult to obtain full restoration of methylation by complementation of MTase-deficient HAP-1 cells. For instance, in the case of eEF1A-KMT3 (gene name EEF1AKMT3; previously known as METTL21B) methylation was only restored to ∼50% of WT levels in the complemented cells (8).
The methylated lysines on the highly abundant eEF1A protein typically contain several methyl groups each and eEF1A is therefore one of most heavily lysine methylated proteins in the cell, similar to histone proteins. However, despite the recent identification of several eEF1A-specific KMTs, the molecular function(s) of eEF1A methylation remains largely enigmatic, and it has not been firmly established that such methylations are involved in regulatory mechanisms. Interestingly, recent findings suggest that eEF1A methylation may indeed be subject to active regulation. The eEF1A-specific KMT Efm6 from S. cerevisiae was shown to mediate methylation on K390 and methylation was only present at low levels during normal growth conditions, but could be dramatically increased by EFM6 over-expression (31). Moreover, EFM6 was recently reported to be upregulated during hypoxia (47). Taken together, these observations indicate that K390 methylation in yeast is subject to active regulation, and likely modulates eEF1A function during hypoxia and possibly under other stress conditions. Similarly, it was very recently shown that methylation of human eEF1A on K165 was modulated in response to ER stress and altered growth conditions, accompanied by corresponding changes in the expression of the responsible KMT, eEF1A-KMT3 (8). Here we report that K36 in eEF1A is mostly trimethylated in various tissues, but interestingly a substantial fraction (∼40%) was un-, mono- or dimethylated in the cancer-derived HAP-1 cells. Future research will likely investigate the potential role of K36 methylation in regulating eEF1A function. However, a first insight may already stem from our ribosome profiling data that show that removal of K36 methylation affects ER related processes, suggesting that methylation at both K36 and K165 are linked to the cellular response to ER stress.
eEF1A is a highly connected protein (48) with several non-canonical functions beyond its role in delivering aminoacyl-tRNA to the ribosome. Besides being methylated on a number of lysine residues, eEF1A has also been reported to be subject to acetylation, ubiquitination and SUMOylation of lysine as well as phosphorylation of serine, threonine and tyrosine (49). For histone proteins, it is well established that combinations of dynamic post-translational modifications (PTMs) constitute a so-called ‘histone code’, which influences chromatin state and gene activity. Considering the emerging evidence that eEF1A methylation is dynamic and that the protein is extensively modified by other PTMs, a similar ‘eEF1A code’ that actively regulates eEF1A function may conceivably exist.
In recent years, several eukaryotic 7BS KMTs targeting eEF1A have been discovered and these enzymes typically target a single lysine residue. In S. cerevisiae, the enzymes responsible for all the known lysine methylations on eEF1A have now been identified and most of these enzymes are 7BS KMTs. The S. cerevisiae 7BS KMTs Efm7, Efm5, Efm4 and Efm6 introduce methylations at K3, K79, K316 and K390, respectively, in eEF1A (7,22,23,31). Two of these enzymes are conserved from yeast to humans. eEF1A-KMT2 is the human ortholog of Efm4 and is responsible for methylation of K318 in human eEF1A (corresponding to K316 in yeast eEF1A) (9). eEF1A-KMT1 catalyzes methylation at K79 and is the human ortholog of Efm5 (7). In addition, the human MTF16 member eEF1A-KMT3 was recently shown to catalyze methylation of K165 in eEF1A (8). In keeping with this emerging nomenclature for human 7BS KMTs we therefore propose that the protein investigated herein is referred to as eEF1A-KMT4, and is encoded by the novel EEF1AKMT4 gene that was until now considered to be part of the ECE2 gene.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Marianne Fyhn for providing the rat organs. We thank Helge Andre Dahl for assistance with DNA cloning and recombinant protein expression. We thank Claudia Gräf for technical support during preparation and sequencing of ribosome-profiling and RNASeq libraries. We are most grateful to Dr Elspeth Bruford from the Human Genome Organisation (HUGO) Gene Nomenclature Committee (HGNC) for useful input on gene annotation.
Footnotes
Present address: Magnus E. Jakobsson, Department of Proteomics, Novo Nordisk Foundation Center for Protein Research (NNF-CPR), Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Norwegian Cancer Society [107744-PR-2007-0132]; Research Council of Norway [FRIMEDBIO-240009]; Max Planck Society; European Research Council [ERC-2012-StG 310489- tRNAmodi to S.A.L.]; University of Oslo; International Max Planck Research School-Molecular Biomedicine (to B.S.N.); Cells in Motion Graduate School. Funding for open acess charge: Research Council of Norway [FRIMEDBIO-240009].
Conflict of interest statement. None declared.
REFERENCES
- 1. Schubert H.L., Blumenthal R.M., Cheng X.. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 2003; 28:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrossian T.C., Clarke S.G.. Uncovering the human methyltransferasome. Mol. Cell Proteomics. 2011; 10:doi:10.1074/mcp.M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greer E.L., Shi Y.. Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012; 13:343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feng Q., Wang H., Ng H.H., Erdjument-Bromage H., Tempst P., Struhl K., Zhang Y.. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002; 12:1052–1058. [DOI] [PubMed] [Google Scholar]
- 5. Falnes P.O., Jakobsson M.E., Davydova E., Ho A.Y., Malecki J.. Protein lysine methylation by seven-β-strand methyltransferases. Biochem. J. 2016; 473:1995–2009. [DOI] [PubMed] [Google Scholar]
- 6. Davydova E., Ho A.Y., Malecki J., Moen A., Enserink J.M., Jakobsson M.E., Loenarz C., Falnes P.O.. Identification and characterization of a novel evolutionarily conserved lysine-specific methyltransferase targeting eukaryotic translation elongation factor 2 (eEF2). J. Biol. Chem. 2014; 289:30499–30510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamey J.J., Winter D.L., Yagoub D., Overall C.M., Hart-Smith G., Wilkins M.R.. Novel N-terminal and lysine methyltransferases that target translation elongation factor 1A in yeast and human. Mol. Cell Proteomics. 2015; 15:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malecki J., Aileni V.K., Ho A.Y., Schwarz J., Moen A., Sorensen V., Nilges B.S., Jakobsson M.E., Leidel S.A., Falnes P.O.. The novel lysine specific methyltransferase METTL21B affects mRNA translation through inducible and dynamic methylation of Lys-165 in human eukaryotic elongation factor 1 alpha (eEF1A). Nucleic Acids Res. 2017; 45:4370–4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimazu T., Barjau J., Sohtome Y., Sodeoka M., Shinkai Y.. Selenium-based S-adenosylmethionine analog reveals the mammalian seven-beta-strand methyltransferase METTL10 to be an EF1A1 lysine methyltransferase. PLoS One. 2014; 9:e105394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cloutier P., Lavallee-Adam M., Faubert D., Blanchette M., Coulombe B.. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. 2013; 9:e1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jakobsson M.E., Moen A., Bousset L., Egge-Jacobsen W., Kernstock S., Melki R., Falnes P.O.. Identification and characterization of a novel human methyltransferase modulating Hsp70 function through lysine methylation. J. Biol. Chem. 2013; 288:27752–27763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kernstock S., Davydova E., Jakobsson M., Moen A., Pettersen S., Maelandsmo G.M., Egge-Jacobsen W., Falnes P.O.. Lysine methylation of VCP by a member of a novel human protein methyltransferase family. Nat. Commun. 2012; 3:1038. [DOI] [PubMed] [Google Scholar]
- 13. Magnani R., Dirk L.M., Trievel R.C., Houtz R.L.. Calmodulin methyltransferase is an evolutionarily conserved enzyme that trimethylates Lys-115 in calmodulin. Nat. Commun. 2010; 1:1–6. [DOI] [PubMed] [Google Scholar]
- 14. Malecki J., Ho A.Y., Moen A., Dahl H.A., Falnes P.O.. Human METTL20 is a mitochondrial lysine methyltransferase that targets the beta subunit of Electron Transfer Flavoprotein (ETFbeta) and modulates its activity. J. Biol. Chem. 2015; 290:423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rhein V.F., Carroll J., He J., Ding S., Fearnley I.M., Walker J.E.. Human METTL20 methylates lysine residues adjacent to the recognition loop of the electron transfer flavoprotein in mitochondria. J. Biol. Chem. 2014; 289:24640–24651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dever T.E., Green R.. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb. Perspect. Biol. 2012; 4:a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abbas W., Kumar A., Herbein G.. The eEF1A proteins: at the crossroads of oncogenesis, apoptosis, and viral Infections. Front. Oncol. 2015; 5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mateyak M.K., Kinzy T.G.. eEF1A: thinking outside the ribosome. J. Biol. Chem. 2010; 285:21209–21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kahns S., Lund A., Kristensen P., Knudsen C.R., Clark B.F., Cavallius J., Merrick W.C.. The elongation factor 1 A-2 isoform from rabbit: cloning of the cDNA and characterization of the protein. Nucleic Acids Res. 1998; 26:1884–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cao X.J., Arnaudo A.M., Garcia B.A.. Large-scale global identification of protein lysine methylation in vivo. Epigenetics. 2013; 8:477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo A., Gu H., Zhou J., Mulhern D., Wang Y., Lee K.A., Yang V., Aguiar M., Kornhauser J., Jia X. et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics. 2014; 13:372–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dzialo M.C., Travaglini K.J., Shen S., Loo J.A., Clarke S.G.. A new type of protein lysine methyltransferase trimethylates Lys-79 of elongation factor 1A. Biochem. Biophys. Res. Commun. 2014; 455:382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lipson R.S., Webb K.J., Clarke S.G.. Two novel methyltransferases acting upon eukaryotic elongation factor 1A in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 2010; 500:137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J.. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009; 25:1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M. et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008; 36:W465–W469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S.. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012; 7:1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lecanda A., Nilges B.S., Sharma P., Nedialkova D.D., Schwarz J., Vaquerizas J.M., Leidel S.A.. Dual randomization of oligonucleotides to reduce the bias in ribosome-profiling libraries. Methods. 2016; 107:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z.. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009; 10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nedialkova D.D., Leidel S.A.. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell. 2015; 161:1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jakobsson M.E., Davydova E., Malecki J., Moen A., Falnes P.O.. Saccharomyces cerevisiae Eukaryotic Elongation Factor 1A (eEF1A) is methylated at Lys-390 by a METTL21-like methyltransferase. PLoS One. 2015; 10:e0131426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tempel W., Wu H., Dombrovsky L., Zeng H., Loppnau P., Zhu H., Plotnikov A.N., Bochkarev A.. An intact SAM-dependent methyltransferase fold is encoded by the human endothelin-converting enzyme-2 gene. Proteins. 2009; 74:789–793. [DOI] [PubMed] [Google Scholar]
- 33. Emoto N., Yanagisawa M.. Endothelin-converting enzyme-2 is a membrane-bound, phosphoramidon-sensitive metalloprotease with acidic pH optimum. J. Biol. Chem. 1995; 270:15262–15268. [DOI] [PubMed] [Google Scholar]
- 34. Horning B.D., Suciu R.M., Ghadiri D.A., Ulanovskaya O.A., Matthews M.L., Lum K.M., Backus K.M., Brown S.J., Rosen H., Cravatt B.F.. Chemical proteomic profiling of human methyltransferases. J. Am. Chem. Soc. 2016; 138:13335–13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Katz J.E., Dlakic M., Clarke S.. Automated identification of putative methyltransferases from genomic open reading frames. Mol. Cell Proteomics. 2003; 2:525–540. [DOI] [PubMed] [Google Scholar]
- 36. Malecki J., Dahl H.A., Moen A., Davydova E., Falnes P.O.. The METTL20 homologue from Agrobacterium tumefaciens is a dual-specificity protein-lysine methyltransferase that targets ribosomal protein L7/L12 and the β; subunit of Electron Transfer Flavoprotein (ETFβ;). J. Biol. Chem. 2016; 291:9581–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fusser M., Kernstock S., Aileni V.K., Egge-Jacobsen W., Falnes P.O., Klungland A.. Lysine methylation of the valosin-containing protein (VCP) Is dispensable for development and survival of mice. PLoS One. 2015; 10:e0141472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S.. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009; 324:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu H., Min J., Ikeguchi Y., Zeng H., Dong A., Loppnau P., Pegg A.E., Plotnikov A.N.. Structure and mechanism of spermidine synthases. Biochemistry. 2007; 46:8331–8339. [DOI] [PubMed] [Google Scholar]
- 40. Bheemanaik S., Reddy Y.V., Rao D.N.. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem. J. 2006; 399:177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jakobsson M.E., Moen A., Falnes P.O.. Correspondence: on the enzymology and significance of HSPA1 lysine methylation. Nat. Commun. 2016; 7:11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Demirci H., Gregory S.T., Dahlberg A.E., Jogl G.. Recognition of ribosomal protein L11 by the protein trimethyltransferase PrmA. EMBO J. 2007; 26:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Min J., Feng Q., Li Z., Zhang Y., Xu R.M.. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell. 2003; 112:711–723. [DOI] [PubMed] [Google Scholar]
- 44. Sawada K., Yang Z., Horton J.R., Collins R.E., Zhang X., Cheng X.. Structure of the conserved core of the yeast Dot1p, a nucleosomal histone H3 lysine 79 methyltransferase. J. Biol. Chem. 2004; 279:43296–43306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zaher H.S., Green R.. Quality control by the ribosome following peptide bond formation. Nature. 2009; 457:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zinshteyn B., Gilbert W.V.. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS. Genet. 2013; 9:e1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bendjilali N., MacLeon S., Kalra G., Willis S.D., Hossian A.K., Avery E., Wojtowicz O., Hickman M.J.. Time-course analysis of gene expression during the Saccharomyces cerevisiae hypoxic response. G3. (Bethesda). 2017; 7:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stark C., Breitkreutz B.J., Reguly T., Boucher L., Breitkreutz A., Tyers M.. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006; 34:D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M.. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012; 40:D261–D270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin J.L., McMillan F.M.. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 2002; 12:783–793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.