


Abstract
Covalent attachment of a synthetic triantennary N-acetylagalactosamine (GalNAc) ligand to chemically modified siRNA has enabled asialoglycoprotein (ASGPR)-mediated targeted delivery of therapeutically active siRNAs to hepatocytes in vivo. This approach has become transformative for the delivery of RNAi therapeutics as well as other classes of investigational oligonucleotide therapeutics to the liver. For efficient functional delivery of intact drug into the desired subcellular compartment, however, it is critical that the nucleic acids are stabilized against nucleolytic degradation. Here, we compared two siRNAs of the same sequence but with different modification pattern resulting in different degrees of protection against nuclease activity. In vitro stability studies in different biological matrices show that 5′-exonuclease is the most prevalent nuclease activity in endo-lysosomal compartments and that additional stabilization in the 5′-regions of both siRNA strands significantly enhances the overall metabolic stability of GalNAc–siRNA conjugates. In good agreement with in vitro findings, the enhanced stability translated into substantially improved liver exposure, gene silencing efficacy and duration of effect in mice. Follow-up studies with a second set of conjugates targeting a different transcript confirmed the previous results, provided additional insights into kinetics of RISC loading and demonstrated excellent translation to non-human primates.
INTRODUCTION
Advances in the siRNA design and chemistry combined with suitable delivery platforms have spurred the development of investigational RNAi therapeutics as a potential new class of therapeutics against a wide variety of human diseases (1–3). In particular, the use of a synthetic triantennary N-acetylgalactosamine (GalNAc) ligand has transformed the field by enabling safe and efficient delivery of chemically modified siRNAs to the liver (4). The GalNAc ligand is designed to bind with high affinity to the asialoglycoprotein receptor (ASGPR) expressed on hepatocytes (5–8) and engineered to be covalently attached to siRNA utilizing various strategies for conjugation either during solid phase oligonucleotide synthesis or via post-synthetic coupling (9–12). This approach has now widely been adopted for the efficient delivery of different classes of investigational oligonucleotide therapeutics to the liver (13–16).
En route from the subcutaneous site of administration to the cytosol of the hepatocytes in the liver, GalNAc–siRNA conjugates encounter a wide variety of extra- and intracellular nucleases that can rapidly metabolize insufficiently protected siRNAs. The gene silencing activity, therefore, largely depends on design features, which enhance their ability to withstand extra- and intracellular nucleases while maintaining the ability to get recruited by the RNA-induced silencing complex (RISC) (17).
Chemical modifications in the sugar-phosphate backbone play a critical role in stabilizing siRNA conjugate against nucleases. In our siRNA designs, the ribosugar moieties of each nucleotide are generally modified with either 2′-deoxy-2′-fluoro (2′-F) or 2′-O-methyl (2′-OMe). These are common and widely used nucleic acid modifications, which have shown to provide some protection against nucleolytic degradation compared to natural RNA and DNA, particularly in the context of double-stranded siRNA (18–22). We have previously demonstrated that placement of additional phosphorothioate (PS) linkages at the 5′-end of both sense and antisense strands can substantially improve the in vivo potency of GalNAc–siRNA conjugates with median effective dose (ED50) of ≤1 mg/kg in rodents, following a single subcutaneous (SC) administration (4).
The goal of the present work was to further study the impact of additional PS modifications on the metabolic stability of GalNAc–siRNA conjugates in various biological matrices and to determine the potential correlation of the in vitro findings with in vivo pharmacokinetics (PK) and pharmacodynamics (PD). Further, translation of the findings across siRNAs and species was investigated. The results illustrate how relatively small changes in siRNA chemistry can significantly impact the performance of GalNAc–siRNA conjugates, a discovery, which has fundamentally improved the platform technology and enabled the advancement of multiple programs into clinical development (23–25).
MATERIALS AND METHODS
siRNA synthesis
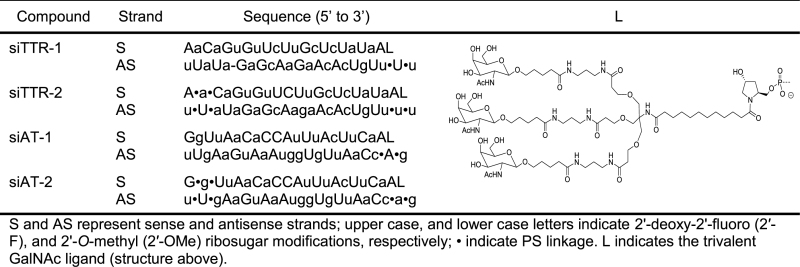
All oligonucleotides were prepared at scales between 1–10 μmol on a MerMade 192 or an ABI 394 synthesizer using commercially available 5′-O-DMT-3′-O-(2-cyanoethyl-N,N-diisopropyl) phosphoramidite monomers by following standard protocols for solid phase synthesis and deprotection (26,27). The GalNAc ligand was introduced at the 3′-end of the sense strand of the siRNA using a functionalized solid support as described (4). The PS linkages were introduced by the oxidation of phosphite utilizing 0.1 M N,N-dimethyl-N′-(3-thioxo-3H-1,2,4-dithiazol-5-yl)methanimidamide (DDTT) in pyridine (28). After deprotection, single strands were purified by ion-exchange HPLC followed by desalting and annealing of equimolar amounts of complementary strands to provide the desired GalNAc–siRNA conjugates shown in Table 1.
Table 1. Designs and sequences of the GalNAc–siRNA conjugates.
|
In vitro stability assay
Stability of siRNA duplexes siTTR-1 and siTTR-2 were studied in three biological matrices namely, cynomolgus monkey plasma, rat liver cytosol and rat liver tritosomes (Xenotech LLC) as reported earlier (29).
PK studies
All procedures using animals were conducted by certified laboratory personnel using protocols consistent with local, state and federal regulations. Experimental protocols were approved by the Institutional Animal Care and Use Committee, the Association for Assessment and Accreditation of Laboratory Animal Care International (accreditation number: 001345). C57BL/6 male mice, aged 6–8 weeks, acquired from Charles River Laboratories were administered siTTR-1, siTTR-2 or siAT-2 conjugates subcutaneously or intravenously with a volume of 10 μl/g of body weight (n = 2 per time point) at a dose of 1, 2.5, 5 or 10 mg/kg. The study designs are depicted in Table 2. Two mice from each study group were sacrificed at desired time points to harvest plasma, liver and kidney samples for analysis. Mice were perfused with saline following blood collection prior to organ harvest.
Table 2. Study designs for siTTR-1 and 2 PK and siAT-2 PK/PD studies.
| Group | Test article | Dose (mg/kg) | Route | No. of animals | Blood and tissues collection time points |
|---|---|---|---|---|---|
| 1 | siTTR-1 | 10 | SC | 24 | 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, 168 and 336 h post-dose |
| 2 | siTTR-2 | 10 | IV | 22 | 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, 168 h post-dose |
| 3 | siTTR-2 | 10 | SC | 28 | 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, 168 and 336 h post-dose |
| 4 | PBS | - | SC | 2 | 24 h post-dose |
| 1 | siAT-2 | 1 | SC | 44 | 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, 168, 336, 408, 504, 576, 672, 744, 840, 912 and 1008 h post-dose |
| 2 | siAT-2 | 2.5 | SC | 44 | 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, 168, 336, 408, 504, 576, 672, 744, 840, 912 and 1008 h post-dose |
| 3 | siAT-2 | 5 | SC | 44 | 0.083, 0.25, 0.5, 1, 2, 4, 8, 24, 48, 96, 168, 336, 408, 504, 576, 672, 744, 840, 912 and 1008 h post-dose |
| 4 | PBS | - | SC | 2 | 24 h post-dose |
To analyze and quantify siRNA from plasma and tissue samples, two different assays were used, an HPLC-based hybridization assay using fluorescent probes for detection of antisense strand termed Atto-probe assay and an assay based on stem-loop quantitative polymerase chain reaction (SL-qPCR). The selection of the assay was based on the dose levels used in the respective studies. For high dose (10 mg/kg and greater) studies, such as the siTTR and siAT comparative PK studies, the Atto-probe assay with its better resolution of parent full length antisense strand was used. For the lower dose PK/PD study including quantification of RISC-loaded siRNA, the SL-qPCR assay with its substantially higher sensitivity was used (see also Supplementary Table S1 summarizing the lower limits of siRNA quantification for both assays).
Sample preparation for Atto-probe assay
Plasma samples were diluted 1:3 in Epicentre Tissue and Cell Lysis Solution for MasterPure Kit (Epicentre, Madison, WI, USA) containing 2% (v/v) proteinase K solution and digested at 50°C at 1000 rpm in micro tube shaker (VWR, Radnor, PA, USA). Whole frozen liver and kidney tissues were pulverized using 2010 Geno/Grinder (Spex, Metuchen, NJ, USA). Pre-cooled Stainless Steel Grinding Balls (Spex, Metuchen, NJ) were added to 15 ml Nalgene polycarbonate grinding jars or 5 ml polyethylene vials (Thermo Scientific, Waltham, MA, USA) containing frozen tissue and were ground at 1200 rpm for 2 min. 40–60 mg of tissue powder was weighed on dry ice and transferred to 1.5 ml centrifuge tubes. Proteinase K lysis buffer was added to the weighed tissue at final concentration of 100 mg/ml for liver and 50 mg/ml for kidney. Proteinase K lysis buffer was prepared by adding 2% (v/v) proteinase K (Life Technologies Carlsbad, CA) to Epicentre Tissue and Cell Lysis Solution for MasterPure Kit (Epicentre, Madison, WI, USA). The samples were incubated overnight at 50°C at 1000 rpm in a microtube shaker (VWR, Radnor, PA) for cell lysis, which were then used for siRNA analysis by the assay described below.
Analysis of siTTR PK study using Atto-probe assay
All samples from the siTTR PK study were analyzed by an Atto-probe assay. The concentration of the conjugates in plasma and tissue samples was detected by annealing at 95°C a fluorescent RNA probe (Atto-probe) complementary to the antisense strand of siTTR-1 and siTTR-2 (see also Supplementary Table S2). A Shimadzu HPLC instrument equipped with a RF-20A xs fluorescence detector, LC-20AD liquid chromatography and ion exchange (IEX) mobile phase was used for analysis. The samples were injected on SIL-20AC HT autosampler with a DNAPac-PA-200 analytical column (4.0 × 250 mm, Thermo Fisher Scientific). All devices were controlled through the Lab Solution version 1.25 data system from Shimadzu. A portion of the blank matrix lysate was spiked with siTTR-1 and siTTR-2 compounds to generate the standard curves and quality control (QC) samples. The standard curve range of siTTR-1 was 15–1000 ng/ml, 80–20 000 ng/g and 160–40000 ng/g, for plasma, liver and kidney tissues, respectively. The standard curve range of siTTR-2 was 15–1000 ng/ml, 200–20 000 ng/g and 160–40 000 ng/g, for plasma, liver and kidney tissues, respectively. The lower limit of quantification (LLOQ) for plasma, liver and kidney were 8 ng/ml, 80 ng/g and 160 ng/g respectively for siTTR conjugates.
Analysis of siAT-2 PK/PD study using SL-RT-qPCR
Total siAT-2 in liver samples was quantified using SL-RT-qPCR by reconstituting liver powder at 10 mg/ml in PBS containing 0.25% Triton-X 100. The tissue suspension was further ground with 5-mm steel grinding balls at 50 cycles/s for 5 min in a tissue homogenizer (Qiagen Tissue Lyser LT) at 4°C. Homogenized samples were then heated at 95°C for 5 min, briefly vortexed and allowed to rest on ice for 5 min. Samples were then centrifuged at 21000 × g for 5 min at 4°C. The siRNA-containing supernatants were transferred to new vials; sense and antisense strand levels were quantified by stem loop reverse transcription followed by Taqman PCR (SL-RT qPCR) based on previously published methods (30,31). Primers and probe used for SL-RT qPCR assay are shown in Supplementary Table S2. The siRNA concentrations were determined using a standard curve generated by spiking the siRNA into blank matrix of the same concentration and source. The lower limit of quantitation of siAT-2 was 0.4 ng/g.
Ago2-bound siRNA from mouse liver was quantified by preparing liver lysates at 100 mg/ml in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 2mM EDTA, 0.5% Triton-X 100) supplemented with freshly added protease inhibitors (Sigma-Aldrich, P8340) at 1:100 dilution and 1 mM PMSF (Life Technologies). Total liver lysate (10 mg) was used for each Ago2 immunoprecipitation (IP) and control IP. Anti-Ago2 antibody and control mouse IgG were purchased from Wako Chemicals (Clone No.: 2D4) and Santa Cruz Biotechnology (sc-2025), respectively. Protein G Dynabeads (Life Technologies) were used to precipitate antibodies. Ago2-associated siRNAs were eluted by heating (50 μl PBS, 0.25% Triton; 95°C, 5 min) and quantified by SL-RT qPCR as described (30,31).
Pharmacodynamic studies in mice
Frozen mouse livers were ground and 10 mg from each sample was resuspended in 500 μl Qiazol and passed through a QIAshredder column (Qiagen, 79654) to homogenize the tissue. Chloroform (100 μl) was added to each sample, which was mixed and then incubated at room temperature for 10 min. Samples were spun at 12 000 × g at 4°C for 15 min. The aqueous phase was transferred to a new tube followed by the addition of 1.5 vol. of 100% ethanol. RNA was purified using miRNeasy Mini kits (Qiagen, 217004). RNA concentration and purity were measured with a NanoDrop (ThermoFisher Scientific). cDNA was synthesized from 1.5 μg of total RNA per sample using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, 4374967). Real-time qPCR was performed using Light Cycler 480 Probes Master (2× mix, Roche, 04887301001) and TaqMan Gene Expression Assays (20×, Thermo Fisher Scientific) on a Roche 480 Light Cycler Real Time PCR System (31). The Dual Color Hydrolysis Probe pre-set program was used: pre-incubation for 10 min at 95°C, 45 cycles of 10 s at 95°C, 30 s at 60°C and 1 s at 72°C, followed by a 40°C cooling step for 30 s. The relative abundance of transthyretin (Ttr, 20× TaqMan Gene Expression Assay, Mm00443267_m1, Thermo Fisher Scientific) and antithrombin (At3, 20× TaqMan Gene Expression Assay, Mm00446570_m1, Thermo Fisher Scientific) mRNA was determined by comparison with the internal reference gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh, 20x TaqMan Endogenous Control, 4352339E, Thermo Fisher Scientific) using the ΔΔCt method.
Pharmacodynamic studies in NHPs
Cynomolgus monkeys (n = 3 per group, males) received a single SC administration (1 ml/kg) of siAT-1 (10 mg/kg) or siAT-2 (1 and 10 mg/kg). At various time points, serum was collected and analysed for AT protein levels. AT protein levels were measured using the AssayMax Human ATIII ELISA Kit (AssayPro, St. Charles, MO, USA) as per manufacturer's instructions (25). Protein levels from individual animals at each time point were normalized to their respective individual pre-dose level (average of three pre-dose values).
Statistical analysis
Due to the limited sample size (n = 2 per time point), differences in mean plasma or tissue concentrations in the comparative PK studies were tested using individual concentration-time data points pooled between 2 and 24 h post dose (N = 8). An unpaired student's t-test was performed while testing differences in means between two groups. For comparing more than two groups, one-way analysis of variance (ANOVA) with post hoc (Tukey) test for multiple comparisons was performed. P < 0.05 was regarded as statistically significant.
RESULTS AND DISCUSSION
In vitro metabolic stability comparison
In a comparative study involving two siRNAs of the same sequence with very similar 2′-F and 2′-OMe ribosugar modification pattern but different degree of PS modification, we have previously observed significant potency differences in mice (4). The first compound, here called siTTR-1, only contained two consecutive PS linkages at the 3′-end of the antisense strand and the GalNAc ligand moiety at the 3′-end of the sense strand (Table 1). The second compound, here called siTTR-2, contained two additional consecutive PS linkages at the 5′-end of both sense and antisense strands.
The potency difference was confirmed by direct comparison of the dose-response for both GalNAc-conjugates in mice 4 days post a single SC administration (Figure 1A). Fitting an inhibitory effect sigmoidal Emax model to the data, the effective dose levels that produced 50% of maximum drug induced inhibitory effect of liver TTR mRNA (ED50) for siTTR-1 and siTTR-2 were calculated to be approximately 5 and 1 mg/kg, respectively.
Figure 1.

Comparison siTTR-1 and siTTR-2 conjugates: (A) dose response in C57BL/6 female mice 4 days post a single SC administration; (B) metabolic stability in cynomolgus monkey plasma, rat liver tritosomes and rat liver cytosol; compounds were incubated with different matrices for 24 h and remaining full-length sense and antisense strands were quantified by HPLC.
To simulate the diverse nucleases activities, which the GalNAc–siRNA conjugates may encounter along their route from the injection site to the cytoplasm of hepatocytes, we utilized different biological matrices including cynomolgus plasma, rat liver tritosomes (enriched lysosomal extract) and rat liver cytosol. siRNA conjugates (0.1 mg/ml in PBS) were incubated with plasma, tritosome and cytosol for 24 h and analysed by HPLC. The percentage of the full-length sense and antisense strands at a given time point was used as a measure of nuclease stability in a particular matrix. In Figure 1B, the stability of the two TTR-siRNA conjugates after 24 h incubation at 37°C is shown in all three matrices, cynomolgus plasma, rat liver tritosomes and rat liver cytosol. Across all matrices, the siTTR-2 conjugate exhibited excellent stability with very little degradation detectable. While the antisense strand of the siTTR-1 conjugate was also very stable in both, plasma and cytoplasm, a significant fraction of the sense strand underwent degradation. The difference between the two compounds, however, was most pronounced in the tritosome matrix, with virtually no sense strand and less than 40% of the full length antisense strand of the siTTR-1 conjugate detectable after 24 h.
It is worth noting that, for both conjugates, we observed sense strand metabolites in the tritosome assay, which matched the deglycosylated sense strand, i.e. the loss of all three GalNAc sugars from the ligand. Based on the strong glycosidase activity present in the endo-lysosomal compartment, this was expected and is consistent with observations in vivo with loss of one, two or three GalNAc moieties observed within minutes of uptake into hepatocytes by ASGPR (data not included) as well as reported data by other groups (15,16). Therefore, for the purpose of this stability analysis, we considered deglycosylated sense strands as full-length material.
Pharmacokinetics and pharmacodynamics in mice
Having established the impact of additional PS linkages at the 5′-end of the duplex on in vitro metabolic stability, the effect on pharmacokinetics was studied after a single 10 mg/kg dose of each compound to non-fasted wild-type C57BL/6 male mice. While siTTR-1 was administered by SC administration only, siTTR-2 was administered both intravenously and subcutaneously. Following SC administration, the plasma time concentration profiles of the two conjugates were found to be similar with comparable mean peak concentration (Cmax), time to reach plasma Cmax (Tmax) and exposure expressed by the total area under the curve (AUC) (Table 3 and Figure 2 A). When comparing intravenous (IV) versus subcutaneous (SC) administration for siTTR-2, higher Cmax at earlier Tmax was observed for IV, as expected with an overall ∼3-fold higher plasma exposure.
Table 3. Summary of plasma, liver and kidney PK parameters for full length GalNAc–siRNA conjugates in mice following single SC or IV administration.
| PK parameters | Plasma | Liver | Kidney | ||||||
|---|---|---|---|---|---|---|---|---|---|
| siTTR-1 | siTTR-2 | siTTR-1 | siTTR-2 | siTTR-1 | siTTR-2 | ||||
| SC | SC | IV | SC | SC | IV | SC | SC | IV | |
| Apparent t1/2β (h) | NR | 0.47 | 1.9 | 3.06 | 21.8 | 24.4 | 5.54 | 31.5 | 27.5 |
| t max (h) | 0.5 | 0.25 | 0.08 | 2 | 2 | 4 | 1 | 4 | 0.08 |
| Cmax (μg/g) | 3.80 | 2.30 | 45.2 | 37.3 | 87.8 | 35.9 | 3.25 | 8.13 | 36.1 |
| AUC0–t (h*μg/g) | 4.60 | 3.60 | 13.58 | 337 | 2071 | 1266 | 34.3 | 258 | 453 |
| AUC0–24 (h*μg/g) | NR | 0.47 | 1.9 | 337 | 1105 | 706 | 34.3 | 139 | 226 |
NR = not reportable due to insufficient data points in the terminal phase for slope estimation.
Figure 2.

Concentration-time profiles of siTTR-1 and siTTR-2 in plasma (A), liver (B) and kidney (C) after a single SC or IV administration of 10 mg/kg in C57BL/6 male mice; shown are the individual data points (from two animals) at each time point as well as their mean (dotted lines). The insert in (B) shows the first 24 h post dose.
Rapid plasma clearance is associated with efficient uptake into liver with peak tissue levels (Cmax) observed between 2 and 4 h after a single IV or SC administration (Table 3). Based on those peak tissue levels, at least 33 and 77% of the administered dose distributed to liver for siTTR-1 and siTTR-2 conjugates, respectively following a single SC administration of 10 mg/kg. Significantly (P = 0.0012) higher liver levels and exposure observed for siTTR-2 compared to siTTR-1 (Figure 2B) can be attributed to the enhanced metabolic stability, particularly in the endo-lysosomal compartment (as shown in Figure 1B), due to the presence of terminal PS stabilization. This is also reflected in the more rapid clearance of the siTTR-1 with an apparent t1/2β of ∼3 h compared to around 24 h for siTTR-2. Comparing IV versus SC dosing, significantly higher (P = 0.019) liver concentrations and AUC0–t were achieved by the SC route of administration, which indicates that the higher peak concentration levels observed in plasma for the IV dose may have breached the limit of ASGPR capacity in the liver.
Compared to liver, kidney PK parameters indicate a substantially lower distribution to kidney for both conjugates (P = 0.006 for siTTR-1, P < 0.0001 for siTTR-2) with an ∼10-fold lower peak concentration and overall exposure. Based on peak concentration levels, this amounts to only ∼2% of the total administered dose and underlines the efficient delivery to liver mediated by the GalNAc ligand (Table 3 and Figure 2C). The approximately four-fold higher kidney Cmax and 2-fold higher kidney exposure (AUC0–t) observed for IV compared to SC administration of siTTR-2 (P = 0.026) aligns well with the differences in liver exposure discussed above and indicates that saturation of ASGPR capacity in the liver increases distribution to the kidney and likely results in higher urinary excretion. Consistent with the lower metabolic stability and the liver PK, the siTTR-1was cleared from kidney at a faster rate with an ∼6-fold lower β-elimination half-life.
The profound effect of the additional PS linkages on metabolic stability and PK suggested a similar impact on duration of RNAi-mediated gene silencing. The impact on conjugate PD as measured by their ability to reduce TTR mRNA levels in liver over time is illustrated in Figure 3 showing the levels of TTR transcript remaining in liver after a single SC dose of 10 mg/kg of siTTR-1 and siTTR-2. As expected, efficacy and durability of TTR mRNA silencing was substantially improved for the enhanced stability conjugate siTTR-2 compared to siTTR-1. This is in good agreement with the liver PK results and confirms the strong correlation between metabolic stability and in vivo performance, both in terms of efficacy and duration of effect.
Figure 3.

Ttr mRNA knockdown as a function of time in mice treated with 10 mg/kg siTTR-1 or siTTR-2 SC relative to PBS-treated animals at 24 h and normalized to mouse Gapdh mRNA; shown are the individual data points (from two animals) at each time point as well as their mean (dotted lines).
To evaluate the generalizability of the findings, another pair of GalNAc–siRNA conjugates targeting antithrombin (AT) was subjected to a similar comparison (siAT-1 and siAT-2, Table 1). Same as for the siTTR pair, both siRNAs were identical in sequence and chemistry except that the conjugate with enhanced stabilization (siAT-2) contained additional PS linkages in the 5′-region of both strands, which were absent in the parent (siAT-1). The comparative study included plasma and liver PK as well as mRNA silencing after a single 25 mg/kg dose in mice (Supplementary Data, PK and PD comparison of siAT-1 and siAT-2 in mice). In good agreement with the previous results, both conjugates showed very similar plasma PK profiles while liver exposure was ∼30-fold higher for siAT-2 compared to siAT-1 (Supplementary Figure S1, P < 0.0001). This translated into a marked improvement in efficacy and duration of effect for siAT-2 as shown in Supplementary Figure S2.
Taken together, the results confirm across different sequences that, even in the context of chemically modified GalNAc–siRNA conjugates, further increase in metabolic stability can substantially improve the activity in mice. The placement of additional PS linkages in the 5′-region of both strands increases in liver exposure thereby underlining the importance of adequate stabilization against 5′-exonuclease activity, which is predominant in the endo-lysosomal compartment. It is worth noting, however, that the magnitude of the effect can vary across sequences. In case of the siTTR pair, an approximately 6-fold increase in liver exposure translated into a 5-fold increase in potency. For siAT-2 compared to siAT-1, a 30-fold increase in liver exposure was observed. Comparing the DR from the PK/PD study of siAT-2 (Supplementary Figure S3) with the mRNA knockdown observed with the 25 mg/kg dose of siAT-1 (Supplementary Figure S2), the increase in potency appears to be approximately 25-fold. In both cases, however, the increase in potency correlated well with the increase in liver exposure.
PK/PD relationship
To investigate the PK/PD relationship in more detail, a separate study was performed, in which mice were administered a single SC dose of siAT-2 at 1, 2.5 or 5 mg/kg. In addition to the total levels of siRNA in liver and the levels of AT mRNA (Supplementary Figure S3), the concentration-time profiles of RISC-loaded siRNA were measured using Ago2-immunoprecipitation followed by stem-loop qPCR (Figure 4). While the maximal concentration of siRNA in the liver occurs much earlier (tmax ∼ 4 h) than maximal silencing activity (nadir around 168 h, i.e. 7 days post dose), the levels of RISC-loaded siRNA correlate well with the silencing activity. This indicates that after entering the cells via ASGPR-mediated endocytosis, it takes several days to build up levels of RISC-loaded siRNA for maximal target silencing suggesting slow endosomal trafficking and release of the siRNA conjugates into the cytosol where the RISC is active.
Figure 4.

Time-concentration profiles of siAT-2 after a single SC dose of 2.5 mg/kg in mice overlaid with the gene silencing profile as measured by reduction of AT mRNA. siRNA concentration in (A) total liver and (B) RISC after Ago2 immunoprecipitation, measured by stem-loop RT-qPCR; shown are the individual data points (from two animals) at each time point as well as their mean (dotted lines).
The relationship of total liver siRNA and RISC-loaded siRNA (31) with AT gene silencing is depicted in Figure 5 with time points of ≥120 h post dose plotted across all dose levels. During this period, essentially covering the time from nadir through recovery back to baseline, the total siRNA levels in liver correlate well with mRNA knockdown with R2 of 0.86 (Figure 5A). The effective concentration of full length siRNA in total liver required for 50% knockdown of AT mRNA (EC50) was calculated to be approximately 50 ng/g. Similarly, mRNA silencing correlates well with siRNA concentrations in RISC with R2 of 0.71 and an EC50 of ∼1.5 ng/g of RISC-loaded antisense strand in liver (Figure 5B). Taken together, the data suggests that, for this particular conjugate, total liver concentrations to sustain 50% knockdown of the target mRNA are about 30–40-fold higher than the corresponding concentration of RISC-loaded siRNA required supporting the notion that only a small fraction of the total siRNA gets loaded into RISC and contributes to functional activity.
Figure 5.

Silencing activity (as measured by reduction of AT mRNA in liver tissue) as a function of (A) siRNA concentration in total liver and (B) concentration of RISC-loaded siRNA.
Translation to nonhuman primates
While the results in rodents were encouraging, it was important to investigate the translation to higher species as a critical milestone towards clinical development of RNAi therapeutics with enhanced metabolic stability. Figure 6 shows the reduction of serum AT after a single SC injection of siAT-1 or siAT-2 in nonhuman primates (NHP). Comparing the protein reduction achieved with siAT-2 conjugate at 1 mg/kg to the 10 mg/kg dose of siAT-1 suggests an improvement in potency of at least 10-fold with a concomitant extension of duration. Despite the 10-fold lower dose, AT protein levels return back to baseline around day 50 post dose for siAT-2 compared to day 20 for the less stable parent siAT-1.
Figure 6.

AT protein levels were measured following single SC injection of siAT-1 (10 mg/kg) or siAT-2 (1 and 10 mg/kg) in cynomolgus monkeys (n = 3 males per group). AT protein was measured in serum for ∼60 days and protein levels from individual animals were normalized to their respective individual pre-dose serum protein level at each time point. Each data point represents the remaining AT for the group average of three animal samples assayed in technical triplicates ± the standard deviation of the group.
The results confirm that, similar to rodents, stability against 5′-exonuclease-mediated degradation is a key factor contributing to GalNAc–sRNA conjugate performance in higher species. Further, the changes in siRNA chemistry using additional PS linkages in the 5′-region of both strands were effective across species resulting in similar improvements in vivo performance for the enhanced stability design in rodent and non-human primates.
CONCLUSION
The present work aimed to further elucidate the effect of chemical modifications, such as the phosphorothioates used in this work, which can impart additional stabilization against nuclease activity when positioned appropriately within the siRNA, on the in vivo performance of GalNAc conjugates. Therefore, pairs of conjugates with the same sequence and 2′-modification pattern but with different degrees of 5′-stabilization were subjected to a comprehensive in vitro and in vivo assessment. Major metabolic stability differences were detected in lysosomal extracts (tritosomes) with prevalent 5′-exonuclease activity suggesting that the observed in vivo differences were primarily due to rapid endo-lysosomal degradation of the conjugate without enhanced stabilization. The results demonstrate that the additional protection against 5′-exonuclease attack via additional PS linkages in the 5′-region had little impact on plasma PK but resulted in higher liver exposure, substantial improvements in potency and prolonged duration of gene silencing. The improvements observed in rodents translated well to nonhuman primates highlighting the importance of enhanced 5′-stabilization for robust gene silencing with GalNAc–siRNA conjugates across species. The more detailed understanding of the relationship between siRNA chemistry and metabolic stability led to the discovery of enhanced stability designs, which have fundamentally transformed the siRNA conjugate platform and helped to progress multiple programs into pre-clinical and clinical development. Efforts are currently ongoing to further refine conjugate design by carefully balancing enhanced stability with the need to maintain optimal compatibility with the endogenous RNAi machinery.
Supplementary Material
Footnotes
Present addresses:
Minggeng Gao, Shire, Lexington, MA 02421, USA.
Renta Hutabarat, Independent Consultant, Dracut, MA 01826, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Alnylam Pharmaceuticals. Funding for open access charge: Alnylam Pharmaceuticals.
Conflict of interest statement. All authors are, or have been during the time this work was conducted, employees of Alnylam Pharmaceuticals.
REFERENCES
- 1. Fitzgerald K., White S., Borodovsky A., Bettencourt B.R., Strahs A., Clausen V., Wijngaard P., Horton J.D., Taubel J., Brooks A. et al. A highly durable RNAi therapeutic inhibitor of PCSK9. N. Engl. J. Med. 2017; 376:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zimmermann T.S., Karsten V., Chan A., Chiesa J., Boyce M., Bettencourt B.R., Hutabarat R., Nochur S., Vaishnaw A., Gollob J.. Clinical proof of concept for a novel hepatocyte-targeting GalNAc–siRNA conjugate. Mol. Ther. 2017; 25:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wittrup A., Lieberman J.. Knocking down disease: a progress report on siRNA therapeutics. Nat. Rev. Genet. 2015; 16:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nair J.K., Willoughby J.L., Chan A., Charisse K., Alam M.R., Wang Q., Hoekstra M., Kandasamy P., Kel’in A.V., Milstein S. et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014; 136:16958–16961. [DOI] [PubMed] [Google Scholar]
- 5. Rensen P.C., Sliedregt L.A., Ferns M., Kieviet E., van Rossenberg S.M., van Leeuwen S.H., van Berkel T.J., Biessen E.A.. Determination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepatocytes in vitro and in vivo. J. Biol. Chem. 2001; 276:37577–37584. [DOI] [PubMed] [Google Scholar]
- 6. Rensen P.C., van Leeuwen S.H., Sliedregt L.A., van Berkel T.J., Biessen E.A.. Design and synthesis of novel N-acetylgalactosamine-terminated glycolipids for targeting of lipoproteins to the hepatic asialoglycoprotein receptor. J. Med. Chem. 2004; 47:5798–5808. [DOI] [PubMed] [Google Scholar]
- 7. Biessen E.A., Noorman F., van Teijlingen M.E., Kuiper J., Barrett-Bergshoeff M., Bijsterbosch M.K., Rijken D.C., van Berkel T.J.. Lysine-based cluster mannosides that inhibit ligand binding to the human mannose receptor at nanomolar concentration. J. Biol. Chem. 1996; 271:28024–28030. [DOI] [PubMed] [Google Scholar]
- 8. Biessen E.A., Vietsch H., van Berkel T.J.. Induction of hepatic uptake of lipoprotein(a) by cholesterol-derivatized cluster galactosides. Arterioscler. Thromb. Vasc. Biol. 1996; 16:1552–1558. [DOI] [PubMed] [Google Scholar]
- 9. Huang X., Leroux J.C., Castagner B.. Well-defined multivalent ligands for hepatocytes targeting via asialoglycoprotein receptor. Bioconj. Chem. 2017; 28:283–295. [DOI] [PubMed] [Google Scholar]
- 10. Rajeev K.G., Nair J.K., Jayaraman M., Charisse K., Taneja N., O'Shea J., Willoughby J.L., Yucius K., Nguyen T., Shulga-Morskaya S. et al. Hepatocyte-specific delivery of siRNAs conjugated to novel non-nucleosidic trivalent N-acetylgalactosamine elicits robust gene silencing in vivo. ChemBioChem. 2015; 16:903–908. [DOI] [PubMed] [Google Scholar]
- 11. Matsuda S., Keiser K., Nair J.K., Charisse K., Manoharan R.M., Kretschmer P., Peng C.G., A V.K.i., Kandasamy P., Willoughby J.L. et al. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015; 10:1181–1187. [DOI] [PubMed] [Google Scholar]
- 12. Migawa M.T., Prakash T.P., Vasquez G., Wan W.B., Yu J., Kinberger G.A., Ostergaard M.E., Swayze E.E., Seth P.P.. A convenient synthesis of 5′-triantennary N-acetyl-galactosamine clusters based on nitromethanetrispropionic acid. Bioorg. Med. Chem. Lett. 2016; 26:2194–2197. [DOI] [PubMed] [Google Scholar]
- 13. Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F. et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014; 42:8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prakash T.P., Yu J., Migawa M.T., Kinberger G.A., Wan W.B., Ostergaard M.E., Carty R.L., Vasquez G., Low A., Chappell A. et al. Comprehensive structure-activity relationship of triantennary N-acetylgalactosamine conjugated antisense oligonucleotides for targeted delivery to hepatocytes. J. Med. Chem. 2016; 59:2718–2733. [DOI] [PubMed] [Google Scholar]
- 15. Yu R.Z., Graham M.J., Post N., Riney S., Zanardi T., Hall S., Burkey J., Shemesh C.S., Prakash T.P., Seth P.P. et al. Disposition and pharmacology of a GalNAc3-conjugated ASO targeting human lipoprotein (a) in mice. Mol. Ther.-Nucleic Acids. 2016; 5:e317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu R.Z., Gunawan R., Post N., Zanardi T., Hall S., Burkey J., Kim T.W., Graham M.J., Prakash T.P., Seth P.P. et al. Disposition and pharmacokinetics of a GalNAc3-conjugated antisense oligonucleotide targeting human lipoprotein (a) in monkeys. Nucleic Acid Ther. 2016; 26:372–380. [DOI] [PubMed] [Google Scholar]
- 17. Mukherjee D., Fritz D.T., Kilpatrick W.J., Gao M., Wilusz J.. Analysis of RNA exonucleolytic activities in cellular extracts. Methods Mol. Biol. 2004; 257:193–212. [DOI] [PubMed] [Google Scholar]
- 18. Cummins L.L., Owens S.R., Risen L.M., Lesnik E.A., Freier S.M., McGee D., Guinosso C.J., Cook P.D.. Characterization of fully 2′-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res. 1995; 23:2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Layzer J.M., McCaffrey A.P., Tanner A.K., Huang Z., Kay M.A., Sullenger B.A.. In vivo activity of nuclease-resistant siRNAs. RNA. 2004; 10:766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takahashi M., Minakawa N., Matsuda A.. Synthesis and characterization of 2′-modified-4′-thioRNA: a comprehensive comparison of nuclease stability. Nucleic Acids Res. 2009; 37:1353–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allerson C.R., Sioufi N., Jarres R., Prakash T.P., Naik N., Berdeja A., Wanders L., Griffey R.H., Swayze E.E., Bhat B.. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005; 48:901–904. [DOI] [PubMed] [Google Scholar]
- 22. Prakash T.P., Kinberger G.A., Murray H.M., Chappell A., Riney S., Graham M.J., Lima W.F., Swayze E.E., Seth P.P.. Synergistic effect of phosphorothioate, 5′-vinylphosphonate and GalNAc modifications for enhancing activity of synthetic siRNA. Bioorg. Med. Chem. Lett. 2016; 26:2817–2820. [DOI] [PubMed] [Google Scholar]
- 23. Chan A., Liebow A., Yasuda M., Gan L., Racie T., Maier M., Kuchimanchi S., Foster D., Milstein S., Charisse K. et al. Preclinical development of a subcutaneous ALAS1 RNAi therapeutic for treatment of hepatic porphyrias using circulating RNA quantification. Mol. Ther.-Nucleic Acids. 2015; 4:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liebow A., Li X., Racie T., Hettinger J., Bettencourt B.R., Najafian N., Haslett P., Fitzgerald K., Holmes R.P., Erbe D. et al. An investigational RNAi therapeutic targeting glycolate oxidase reduces oxalate production in models of primary hyperoxaluria. J. Am. Soc. Nephrol. 2017; 28:494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sehgal A., Barros S., Ivanciu L., Cooley B., Qin J., Racie T., Hettinger J., Carioto M., Jiang Y., Brodsky J. et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat. Med. 2015; 21:492–497. [DOI] [PubMed] [Google Scholar]
- 26. Beaucage S.L. Solid-phase synthesis of siRNA oligonucleotides. Curr. Opin. Drug Discov. Dev. 2008; 11:203–216. [PubMed] [Google Scholar]
- 27. Mueller S., Wolf J., Ivanov S.A.. Current strategies for the synthesis of RNA. Curr. Org. Synth. 2004; 1:293–307. [Google Scholar]
- 28. Guzaev A.P. Reactivity of 3H-1,2,4-dithiazole-3-thiones and 3H-1,2-dithiole-3-thiones as sulfurizing agents for oligonucleotide synthesis. Tetrahedron Lett. 2011; 52:434–437. [Google Scholar]
- 29. Parmar R., Willoughby J.L., Liu J., Foster D.J., Brigham B., Theile C.S., Charisse K., Akinc A., Guidry E., Pei Y. et al. 5′-(E)-Vinylphosphonate: A Stable Phosphate Mimic Can Improve the RNAi Activity of siRNA-GalNAc Conjugates. Chembiochem. 2016; 17:985–989. [DOI] [PubMed] [Google Scholar]
- 30. Elkayam E., Parmar R., Brown C.R., Willoughby J.L., Theile C.S., Manoharan M., Joshua-Tor L.. siRNA carrying an (E)-vinylphosphonate moiety at the 5′ end of the guide strand augments gene silencing by enhanced binding to human Argonaute-2. Nucleic Acids Res. 2017; 45:3528–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pei Y., Hancock P.J., Zhang H., Bartz R., Cherrin C., Innocent N., Pomerantz C.J., Seitzer J., Koser M.L., Abrams M.T. et al. Quantitative evaluation of siRNA delivery in vivo. RNA. 2010; 16:2553–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.