


Abstract
Pluripotency, the ability of embryonic stem cells to differentiate into specialized cell types, is determined by ESC-specific gene regulators such as transcription factors and chromatin modification factors. It is not well understood how ESCs are poised for differentiation, however, and methods are needed for prognosis of the molecular changes in the differentiation of ESCs into specific organs. We describe a new approach to infer cell-type specific gene regulatory programs based on gene regulatory interactions in ESCs. Our method infers the molecular logic of gene regulatory mechanisms by mapping the position-specific combinatory patterns of numerous regulators in ESCs into cell-type specific gene regulations. We validate the proposed approach by recapitulating the RNA-seq and microarray data of neuronal progenitor cells, adult liver cells, and ESCs from the integrated patterns of diverse gene regulators in ESCs. We find that the collective functions of diverse gene regulators in ESCs represent distinct gene regulatory programs in specialized cell types. Our new approach expands our understanding of the differential gene regulatory information in developments encoded in regulatory networks of ESCs.
INTRODUCTION
Embryonic stem cells are distinguished by their ability to differentiate into any cell type and by their ability to propagate (1). The pluripotency and the totipotency of embryonic stem cells are determined by ESC-specific gene-regulators (2). Therefore, understanding the pluripotency of ESCs requires us to understand the gene regulatory mechanisms in ESCs. The aim of this study is to understand how embryonic stem cells poise for differentiation into specialized cell types. Gene regulatory networks are composed of gene regulating protein factors and target genes. The gene expression program encoded in the genome is executed by transcription factors that bind to cis-regulatory sequences and modulate gene expression in response to environmental and developmental cues. Chromatin modifications, chromatin modification factors, and transcription factors are simultaneously involved in gene regulation. Inside a nucleus, genomic DNAs are packed into 3D structures. Chromatin modifications (3), transcription regulating protein factors (3), and RNA Pol II complexes (4) mediate the configurations of the chromatin 3D structures that bring regulatory elements, even in distant DNA segments, to the target genes for transcription regulation (5–8). Chromatin modification factors reversibly change chromatin modification status (9,10).
The integrative effects of the diverse gene regulators have not been well investigated, although the gene regulatory functions of individual factors continue to be studied intensively. Various gene regulating factors have been shown to play crucial roles in gene regulation and developmental processes. For example, previous studies find that the loss of function mutations of individual genes encoding gene regulators, such as DNA methyltransferases (11), histone modification factors (12), chromatin remodeling factors (13) and transcription factors (14), result in developmental defects. In other words, the collective action of numerous gene regulators is essential for normal development from ESCs. The complex interactions among genes and gene-regulating factors imply that only by understanding the combinatory and sequential logic of gene regulators can we acquire full regulatory information about the genes.
Inside a cell, a gene interacts with diverse factors simultaneously, and the interactions among multiple gene regulating factors produce differential gene regulation. So far, gene expression in ESCs is inferred using only a single factor (15), or a subset of factors, such as restricting factors into chromatin modifications (16), or transcription factors (17). Inference models considering only a single gene regulator or subsets of gene regulators, however, do not suffice to explain gene regulation, even in ESCs. Therefore, to integrate prior studies of the functions of individual gene regulating factors into regulatory mechanisms, we propose a new approach to infer cell-type specific gene regulation. We study computational models to predict differential gene expression in specialized cell types based on the integrative patterns of the known gene regulators in embryonic stem cells. The gene regulators we consider include transcription factors, chromatin modifications, DNA methylations, chromatin modification enzymes, and DNA-binding factors associated with chromatin domains. We develop and apply a more realistic model to explain the experimental measurements of gene regulation in diverse cell types. When position specific enrichments of comprehensive gene regulators in ESCs are integrated into patterns, the integrative interaction patterns are efficiently mapped to distinct gene regulatory programs in diverse cell types. We validate the model by explaining the experimental measurements of differential gene regulation in diverse cell types. The results show that position-specific and combinatory operations of diverse gene regulators in ESCs poise for differential gene regulation in specialized cell types as well as encoding gene expression information in ESCs.
MATERIALS AND METHODS
ChIP-seq data sources and mapping to the mouse genome
Chromatin modification ChIP-seq data for H3K4me1, H3K27ac, H3, H3K4me3, p300 for mESCs, and mouse adult liver cells are obtained from Creyghton et al. (GSE 24165) (18). DNA methylations ChIP-seq data for mC, 5hmC, 5caC, 5fC in mESCs are obtained from Shen et al. (GSE42250) (19). H3.3 ChIP-seq data in mESCs are obtained from a previous study (10). H2AZ and acetylated H2HAZ ChIP-seq data in mESCs are obtained from Hu et al. (GSE34483) (20). Transcription factor ChIP-seq data for Nanog, Oct4, Sox2, Smad1, E2F1, Tcfcp2I1, CTCF, Zfx, STAT3, KLF4, Esrrb, n-Myc and p300 in mESCs are obtained from Chen et al. (GSE11431) (21). H3, H4K20me3 H3K9me3, and H3K36me3 ChIP-seq data in mES are obtained from Mikkelsen et al. (GSE12241) (22). KDM2A ChIP-seq data in mESCs are obtained from Neil P. Blackledge et al. (GSE21202) (23). SUZ12, EZH2 and RING1B ChIP-seq data in mESCs are obtained from Ku et al. (GSE13084) (24). Med12, Smc1/2/3 Med1, Nipbl and CTCF ChIP-seq data in mESCs are obtained from Kagey et al. (25). HDAC1, HDAC2, LSD1, REST (transcription repressor of neuronal genes in non-neuronal cells), COREST and Mi2b ChIP-seq data are obtained from Whyte et al. (GSE27844) (26).
The raw ChIP-seq data in SRA format are transformed into fastq files and mapped to the reference genome (mm9). The 30–50 bp sequences from the ChIP-seq data are mapped to the mouse reference genome (mm9) by perfect and unique matching without allowing any mismatch or gap. The reads are then extended to 150 bp from their 5′ end.
Analysis of RNA-seq data
The raw RNA-seq data of mESCs are obtained from a previous study (10). The RNA-seq analysis is performed using the Tuxedo software package with default settings. RNA-seq reads are mapped to the mouse genome (NCBI37/mm9) using Bowtie2. Tophat with default settings is used to detect splice sites. The Cufflinks software package is used to assemble transcripts based on the Refseq mRNA sequence database (mm9). A total of 48 228 transcripts are detected from two RNA-seq replicate experiments and their mean values are used for further analysis. log2 values of the FPKM are used as the target transcription levels of the prediction models. Silenced transcripts are defined as having expression levels between 0 and 1 FPKM. The processed ChIP-seq and RNA-seq data are in the supplementary material.
Binary encoding of ChIP-seq signals of gene regulators
For each ChIP-seq experiment for a factor, the number of ChIP-seq reads mapped to a 200bp window is counted and then a P-value <10−5 is used as a cutoff to statistically detect significant enrichment of a factor at a locus in the whole genome. The P-value, 10−5 indicates that the false positive rate is 10−5. Then for each position around the genes, the ChIP-seq signals are position-specifically normalized to consider the position-specific effects. For each 200bp region, the distributions of the ChIP-seq reads from ∼26 500 genes are normalized to z-scores so that the mean is zero and the standard deviation is 1. The loci with position-specific z-scores greater than 1 are considered to interact with a factor.
Therefore, the loci considered to interact with a factor satisfy two criteria: (i) the locus at a position from a gene start site specifically interacts with a factor with a position-specific z-score >1 and (ii) the locus is significantly enriched with ChIP-seq reads with a genome-wide P-value <10−5.
The enrichment patterns of 52 gene regulators at individual positions are transformed to a binary code of 0s and 1s, where 1 denotes each enrichment signal and 0 denotes no significant enrichment.
Gaussian process models
By using Gaussian process models and Jaccard coefficients as kernel functions, binary codes of arrangements of gene regulators are mapped to gene expression levels. We infer the distribution of the gene expression levels (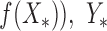 of gene regulator patterns which are not used for model training,
of gene regulator patterns which are not used for model training,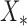 as the test data, based on the experimental measurements, and Y for gene regulator interactions X as the training data, which can be represented as
as the test data, based on the experimental measurements, and Y for gene regulator interactions X as the training data, which can be represented as 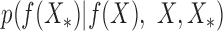 with means and standard deviations.
with means and standard deviations.
We calculate the conditional distribution 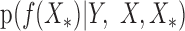 based on the experimental data. With the assumption of a Gaussian distribution, the distribution of target gene expression levels follows:
based on the experimental data. With the assumption of a Gaussian distribution, the distribution of target gene expression levels follows:
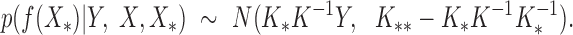 |
The best estimate for 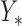 is the mean of this distribution, the expected value:
is the mean of this distribution, the expected value:
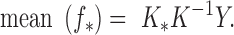 |
The uncertainty or variance is estimated by its variance: 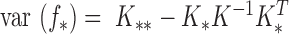
We evaluate the performance of the GP models using the correlation coefficients between the predicted and measured values of the test data. We conduct 1000 runs and calculate 1000 correlation coefficients per round of model training and test. The R code and the data implementing this study are in the supplementary material.
RESULTS
Generating gene-regulator codes from ChIP-seq data
To understand the integrative effects of diverse gene regulators associated with genomic DNAs, we investigate predictive models as a function of the patterns of gene regulators around genes. Clearly, the performance of any probabilistic inference models depends on the features chosen. Previous studies have identified factors which play crucial roles in gene regulation and have measured genome-wide interactions between genomic DNAs and individual gene regulating factors by ChIP-seq experiments in high resolution. Based on the previous studies on gene regulatory functions in ESCs of protein factors, we use the binding patterns of 52 protein factors as the explanatory variables of gene expression levels, measured by RNA-seq and microarrays. The gene regulators include 21 chromatin modifications and DNA methylations, 15 transcription factors, 10 chromatin modifiers and 6 chromatin conformation regulators.
An array of position-specific enrichment signals of diverse gene regulators is converted to a binary bitmap code and the interaction patterns are used as a 1D input variable of the Gaussian process models (27), which are non-parametric and non-linear Bayesian probabilistic models (Figure 1). The models infer gene regulation levels based on the integrative interaction patterns of gene regulators in ESCs.
Figure 1.

Unified models predicting gene regulation based on landscapes of gene-regulating factors. For each gene, position specific combinatorial patterns of 52 gene regulators in promoter regions in ESCs are used to predict cell type specific gene regulation using Gaussian process models.
The interaction patterns are generated from the enrichment signals of ChIP-seq reads of 52 factors at 21 positions spanning the 2 kb region around the transcription start sites in a 200 bp window. The enrichment levels of 52 gene regulators at individual positions are transformed to a simple binary code of 0s and 1s, where 1 denotes an enrichment signal and 0 denotes no significant enrichment. By approximating continuous enrichment values into binary values, 0 and 1, the binary vectors encode large dimensional logical operations of cells, i.e. complex combinations of AND, OR, NOT, NAND, NOR and XOR logic are approximated to bitmap vectors. The binary patterns represent the complex relationships among diverse factors at individual positions, such as the complementary, co-operative, competitive, and antagonistic interactions affecting gene regulation.
The binary patterns are used to build circuits of any complexity from the building blocks. The patterns of gene regulators in individual genes are used as explanatory variables of gene regulation. Gaussian process models are used to estimate the summation of the logical operations in cells and generate distributions of target gene expression levels based on the integrative gene regulator patterns, and to map individual gene regulator patterns to gene expression levels. The Gaussian process models consider statistical variations of experimental data and uncertainty of specific models. The models are validated by using patterns of gene regulators of unseen genes.
Integrative interaction patterns of gene regulators at a promoter region in mESCs significantly explain the gene expression level in mESCs
We first examine the coordinated effects of arrangements of the gene regulators on gene regulation in embryonic stem cells. Using the patterns of the 52 factors and 21 binding site combinations around the transcription start sites, we infer mRNA levels of the genes in mESCs. We find that the patterns of gene regulating factors in mESCs efficiently predict the mRNA levels in mESCs (Figure 2A). The result validates that the coordinated interactions among gene regulators are indicators of gene expression of the genes (Figure 2A). Because of experimental variations, however, it is not possible to match the predicted values and experimentally measured levels perfectly, but the predictive model based on integrative gene regulator patterns significantly explains the gene expression levels measured by RNA-seq experiments.
Figure 2.

The integrative patterns of transcription factors and chromatin modifications significantly explain gene expression levels in mouse embryonic stem cells. (A) Integrative patterns of 52 gene regulators in ESCs efficiently predict mRNA levels in ESCs measured in mRNA-sequencing experiments. The models are trained by using 15 000 genes and tested by using the remaining 10,000 genes. The correlation coefficients between measured and predicted mRNA levels are around 0.8, P-value ≈ 0. (B) Predicting gene expression levels in ESC from the patterns of a single gene regulator and the classes of gene regulators: six chromatin domain associated factors, 10 chromatin modifiers, 15 transcription factors, 21 chromatin modifications, and all 52 factors. Boxplots show distributions of the correlation coefficients between the predicted and measured values of 1000 performance tests of model training by using randomly selected 5000 genes for the respective functional classes of gene regulators.
Next, we examine the efficiency of predictive models by arrangements of a single gene regulator around transcription start sites. For each gene regulator, we build models inferring gene expression levels from the arrangements of a single gene regulator around promoter regions and validate them by using independent sets of genes. The correlation coefficients between the predicted and measured values are low, less than 0.2 for the entire gene regulator examined, i.e. our models using a single gene regulator do not show significant prediction performance in inferring gene expression levels in mESCs (Figure 2B). The correlation coefficients between the predicted and the measured values for individual gene regulators are statistically significant, which confirms that the individual gene regulators are significantly associated with gene expression levels, although individual gene regulators alone are not predictive features of gene expression.
We classify the gene regulators into functional classes: chromatin modifications and DNA methylations; transcription factors; chromatin modification enzymes; and chromatin domain associated factors. For each class, we use the coordinated arrangements of multiple gene regulators in the class to infer gene expression levels in mESCs measured in RNA-seq experiments. We find that the coordinated arrangement patterns of chromatin modifications and DNA methylations show the highest prediction performance of gene expression levels in ESCs among the four classes; in fact, the number of factors is greater than the other three classes (Figure 2B). As the number of gene regulators in a class increases, the model performance using the patterns of the gene regulators also increases. To examine whether the number of factors affects the performance of the models based on arrangements of chromatin modifications, we randomly select 15 chromatin modifications and validate the model performance. We find that the predictive model of gene expression levels performs better than the models using 15 ESC-specific transcription factors. The results suggest that the arrangements of various chromatin modifications around the promoter play complementary roles in gene expression in ESCs.
Considering vast amounts of gene regulators in ESCs, our model infers gene expression in mESC with high accuracy and low variations in the model's performance. The results suggest that gene expression regulation involves the collective action of numerous gene regulators.
Patterns of the gene regulators in mESCs represent differential gene regulation in NPCs
Beyond predicting accurate gene expression levels in ESCs, we investigate whether the interaction patterns of gene regulators around genes in mESCs can predict differential gene regulation after fate determination and embryogenesis. Differentiation of ESCs to neuronal progenitor cells (NPCs) reflects critical developmental fate determination in the embryo to become a neuroectoderm (18). Therefore, we examine the differential gene regulation of neuronal progenitor cells from embryonic stem cells.
Distinct molecular mechanisms are involved in gene regulation. Chromatin modifications and DNA methylations restrict interactions among DNA-binding factors and regulatory elements. Chromatin modifiers change chromatin modification status. In particular, chromatin modifications in ESCs mark the poised genes that are differentially regulated in differentiated cell types. For example, double histone modifications of H3K27me3 and H3K4me3 are enriched in genes in ESCs that are up-regulated in NPCs. Therefore, we examine whether a specific molecular mechanism is preferentially involved in differential gene regulation in NPCs from ESCs.
First, we examine whether the interaction patterns of a single regulator in ESCs associates with gene expression differentiation in NPCs. We estimate gene expression differentiation in NPCs from ESCs by the change of mRNA levels using RNA-seq data of NPCs and ESCs, respectively. We build a model inferring gene expression differentiation in neuronal progenitor cells by using the gene regulator patterns in mESCs as explanatory variables (Figure 3C) and validate the model by applying it to the remaining 50% of the gene sets that are not used for modeling. We find that some gene regulating factors in ESCs are highly related with differential gene expression in NPCs. In particular, the patterns of H3K27me3 in mESCs significantly associate with differential gene regulation in NPCs. The top ten highly associated factors are H3K27me3, H3K36me3, E2F1, H3K9ac, SUZ12, EZH2, H3K27ac, LSD1, c-MYC and H3K4me3.
Figure 3.

The integrated patterns of gene regulators in ESCs fully explain differential gene expression between NPCs and ESCs. (A) Inferring differential gene expression in NPCs from patterns of 52 gene regulating factors in ESCs. (B) Inferring differential gene regulation in NPCs from patterns of six chromatin domain associated factors, 10 chromatin modifiers, 15 transcription factors, 21 chromatin modifications, and all 52 factors in ESCs. Negative control shows correlation coefficients between the predicted NPC-specific gene regulations based on whole 52 factors and measured adult liver-specific gene regulation. (C) Inferring differential gene expression levels in NPCs from patterns of a single gene regulator in ESCs. The top 10 highly predictive factors are marked.
To understand the differential contribution of gene regulators on inferring differential gene regulation, we categorize the 52 gene regulating factors by chromatin modifications and DNA methylations, transcription factors, chromatin modifiers, and chromatin domain associated factors. We examine the regulators in each class that predict the differential gene regulation in NPCs (Figure 3B). In contrast to ESCs, binding patterns transcription factors in ESCs significantly associate with differential gene regulation in NPCs and ESCs, whereas the models predicting gene expression change based on chromatin modifications in ESCs, which show instability in multiple repeated tests, do not significantly explain gene expression differentiation in NPCs. Binding patterns of transcription factors and chromatin modifiers in ESCs significantly explain the differential gene regulation in NPCs. The results suggest that transcription factors and chromatin modifiers are involved in cell-type specific gene regulation, whereas chromatin modifications tend to be associated with invariant gene regulation. The results also suggest that the interactions among genes and diverse gene regulators in mESCs represent regulatory programs in embryogenesis and cellular differentiation and imply that the integrative interaction patterns of gene regulators in ESCs are important features of pluripotency and totipotency of ESCs. The results support that the mESC specific-transcription factors are preferentially involved in pluripotency of ESCs.
Next, we investigate whether the combinatory patterns of diverse gene regulators in mESCs predict differential gene regulation in NPCs. Our model based on the integrative interaction patterns of 52 gene regulators in mESCs significantly explains gene expression differentiation in NPCs (Figure 3A). To validate the specificity of the model inferring NPC-specific gene regulation, we examine the model performance inferring gene regulation in other tissues. Applying the NPC-specific model to liver-specific gene regulation does not show any significant correlation between the predicted and measured liver-specific gene regulation. The result shows that the NPC-specific gene regulatory networks encoded in ESCs are highly specific to NPC-specific gene regulation. In summary, the results suggest that the integrative interaction patterns of the gene regulators in mESCs significantly associate differentiation of gene regulatory program in embryogenesis.
Interaction patterns of the gene regulators in mESCs represent gene regulatory program in B cell progenitor cells and adult liver cells
We further investigate whether gene regulator patterns in mESCs predict differential gene regulatory programs in adult liver cells and B cell progenitor cells. We estimate the levels of gene expression differentiation in B cell progenitor cells and adult liver cells by analyzing cDNA microarray data of mESCs, proB cells and liver cells.
We build a model inferring gene expression differentiation in adult liver cells and B progenitor cells, respectively, by using the gene regulator codes in mESCs as explanatory variables (Figure 4A) and validate the model by applying it to the remaining gene sets that are not used for modeling. We measure the differential gene regulation levels in the adult cells from liver cells by using microarray data in mESCs, adult liver cells, and B cell progenitors (proB). Our models based on the integrative interactions in ESCs significantly explain gene expression differentiation in B cell progenitor cells and adult liver cells.
Figure 4.

The differential gene regulation in adult liver cells and B cell progenitors are effectively predicted by the integrative pattern of the gene regulators in ESCs. (A) The integrative patterns of gene regulators in ESCs efficiently predict differential gene expression in adult liver cells and B cell progenitors from ESCs measured by using cDNA microarray experiments. The correlation coefficients between measured and predicted differential levels are 0.71 and 0.72 for adult liver cells and B cell progenitors, respectively. The models are trained by using 15 000 genes and tested by using the remaining 10 000 genes. (B) The differential gene regulation levels in both adult liver cells and B cell progenitors are not sufficiently explained by using a subset of gene regulators in functional classes, i.e. chromatin domain factors, chromatin modifiers, transcription factors, and chromatin modifications including DNA methylations. Boxplots show distributions of the correlation coefficients between predicted and measured values of 1000 performance tests of model training by using randomly selected 5000 genes for the respective functional classes of gene regulators.
Next, we examine whether the four regulator categories are associated with gene expression differentiation in liver and proB cells. For each category, we build models predicting gene expression change in liver and proB cells, respectively (Figure 4B). We find that transcription factor binding patterns in mESCs associate with gene expression differentiation in adult liver cells or B cell progenitor cells, whereas the models predicting gene expression change based on chromatin modifications and chromatin modifiers in ESCs do not significantly explain gene expression differentiation in both adult liver cells and B cell progenitor cells. Using a single gene regulator in ESCs does not significantly explain the differential gene regulations in both liver and B cell progenitors. The results suggest that the integrative interaction patterns of diverse gene regulators in mESCs are involved in mESC-specific expression and gene expression differentiation in adult cell types.
DISCUSSION
New algorithmic approaches are necessary to understand the diverse factors regulating a gene in a cell and the resulting gene regulatory programs. This paper provides an efficient, data-driven computational approach to explain the experimental measurements of gene expression and enable molecular insights into gene regulatory mechanisms. Predictive models are built to infer condition-specific gene expression from the integrated patterns of known gene regulators and to validate that the coordinated interactions of the diverse factors recapitulate mRNA levels measured by using mRNA-seq and microarrays. The integrated gene regulatory codes in embryonic stem cells significantly explained differential gene regulation in embryogenesis and fate determination. Distinct interactions of diverse gene regulators were associated with distinct gene regulation in diverse cell types. The combinatory patterns of diverse gene regulators in ESCs efficiently represent gene regulatory programs in diverse cell types as well as ESCs.
The combinatory patterns of numerous gene regulating factors represent the molecular logic of gene regulatory mechanisms. Integrating diverse gene regulators into patterns and the pattern recognitions can improve the ability of predictive models inferring gene regulation over only using subsets of principal gene regulators. The results of this study show that distinct arrangements of diverse gene regulators in ESCs ally to specific gene expression programs in diverse cell types. We show that the collective actions of numerous gene regulators as a whole in ESCs are required for ESCs to poise for differential gene regulation in specialized cell types generating infinite combinatorial modes of gene expression. The results imply that the collective action of numerous gene regulating factors controls gene expression and poises for ESCs to differentiate into specialized cell types. It also implies that discovering new regulatory functions of gene regulators and updating the inference models by integrating the studies can improve the accuracy of the predictive models and further our understanding of the molecular mechanisms of developments. In summary, the results demonstrate that gene regulatory machineries in ESCs poise for differentiation into specialized cells in response to environmental and developmental cues. The cell-type specific gene regulatory programs can be inferred from the integrative interaction patterns of gene-regulators in ESCs. The new approach to predicting the gene regulatory programs in diverse cell types from ESCs will be useful to develop prognosis systems for normal developments from stem cells.
The cell-type specific gene regulation is more efficiently explained by complex regulatory interactions of numerous factors than a molecular mechanism or a specific gene regulator. The complexity of gene regulating functions among numerous factors may make biological systems robust to genetic polymorphisms and environmental changes.
Furthermore, gene regulatory mechanisms are adaptive control systems. While the gene regulatory interactions in ESCs can explain ∼70% of gene expression differentiation in NPCs in the early stage of development, poised information in ESCs can explain ∼60% of the fully developed adult liver cell specific gene regulation. The inference of cell-type specific gene regulation from poised gene regulatory patterns in ESCs is less efficient in adult liver cells than NPCs, which is an early stage of development. The results suggest that gene regulatory programs are modulated in response to environmental stimuli and developmental cues.
In conclusion, this study expands our understanding of the molecular mechanisms of gene regulation in ESCs and emphasizes that gene regulatory mechanisms are complex adaptive systems regulated by the interaction of diverse factors.
DATA AVAILABILITY
The R code and the raw data replicating this study are in the supplementary material.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: M.H. conceived and designed the study and conducted the data analyses. M.H. and S.H. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Pusan National University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M.. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282:1145–1147. [DOI] [PubMed] [Google Scholar]
- 2. Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S.. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–872. [DOI] [PubMed] [Google Scholar]
- 3. Jenuwein T., Allis C.D.. Translating the histone code. Science. 2001; 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 4. Stasevich T.J., Hayashi-Takanaka Y., Sato Y., Maehara K., Ohkawa Y., Sakata-Sogawa K., Tokunaga M., Nagase T., Nozaki N., McNally J.G. et al. . Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature. 2014; 516:272–275. [DOI] [PubMed] [Google Scholar]
- 5. Jin F., Li Y., Dixon J.R., Selvaraj S., Ye Z., Lee A.Y., Yen C.A., Schmitt A.D., Espinoza C.A., Ren B.. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. et al. . An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chepelev I., Wei G., Wangsa D., Tang Q., Zhao K.. Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 2012; 22:490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ha M. Understanding the chromatin remodeling code. Plant Sci. 2013; 211:137–145. [DOI] [PubMed] [Google Scholar]
- 9. Clapier C.R., Cairns B.R.. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009; 78:273–304. [DOI] [PubMed] [Google Scholar]
- 10. Ha M., Kraushaar D.C., Zhao K.. Genome-wide analysis of H3.3 dissociation reveals high nucleosome turnover at distal regulatory regions of embryonic stem cells. Epigenet. Chromatin. 2014; 7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li E., Bestor T.H., Jaenisch R.. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992; 69:915–926. [DOI] [PubMed] [Google Scholar]
- 12. Yao T.P., Oh S.P., Fuchs M., Zhou N.D., Ch’ng L.E., Newsome D., Bronson R.T., Li E., Livingston D.M., Eckner R.. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998; 93:361–372. [DOI] [PubMed] [Google Scholar]
- 13. Ho L., Jothi R., Ronan J.L., Cui K., Zhao K., Crabtree G.R.. An embryonic stem cell chromatin remodeling complex, esBAF, is an essential component of the core pluripotency transcriptional network. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:5187–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nichols J., Zevnik B., Anastassiadis K., Niwa H., Klewe-Nebenius D., Chambers I., Scholer H., Smith A.. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998; 95:379–391. [DOI] [PubMed] [Google Scholar]
- 15. Ha M., Hong S.. DNA context represents transcription regulation of the gene in mouse embryonic stem cells. Sci. Rep. 2016; 6:24343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong X., Greven M., Kundaje A., Djebali S., Brown J., Cheng C., Gingeras T., Gerstein M., Guigo R., Birney E. et al. . Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012; 13:R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beer M.A., Tavazoie S. Predicting gene expression from sequence. Cell. 117:185–198. [DOI] [PubMed] [Google Scholar]
- 18. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. et al. . Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen L., Wu H., Diep D., Yamaguchi S., D’Alessio A.C., Fung H.L., Zhang K., Zhang Y.. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell. 2013; 153:692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu G., Cui K., Northrup D., Liu C., Wang C., Tang Q., Ge K., Levens D., Crane-Robinson C., Zhao K.. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013; 12:180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. et al. . Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008; 133:1106–1117. [DOI] [PubMed] [Google Scholar]
- 22. Mikkelsen T.S., Ku M., Jaffe D.B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T.K., Koche R.P. et al. . Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007; 448:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blackledge N.P., Zhou J.C., Tolstorukov M.Y., Farcas A.M., Park P.J., Klose R.J.. CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell. 2010; 38:179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ku M., Koche R.P., Rheinbay E., Mendenhall E.M., Endoh M., Mikkelsen T.S., Presser A., Nusbaum C., Xie X., Chi A.S. et al. . Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008; 4:e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kagey M.H., Newman J.J., Bilodeau S., Zhan Y., Orlando D.A., van Berkum N.L., Ebmeier C.C., Goossens J., Rahl P.B., Levine S.S. et al. . Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010; 467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Whyte W.A., Bilodeau S., Orlando D.A., Hoke H.A., Frampton G.M., Foster C.T., Cowley S.M., Young R.A.. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012; 482:221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rasmussen C.E., Williams C.K.I.. Gaussian Processes for Machine Learning. 2006; Cambridge: MIT Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The R code and the raw data replicating this study are in the supplementary material.