


Abstract
The analysis of RNA tertiary structure is hindered by the fact that not too many structural data are available and a significant amount of them are in low resolution. Due to the atomic coordinate errors posed by the limitations of low-resolution RNA three-dimensional structures, it becomes a critical challenge to extract key geometric characteristics of RNA, particularly, the interaction of bases. To address this issue, we have devised a comparative method, named CompAnnotate, that utilizes more precise structural information of high-resolution homologs to annotate the base-pairing interactions in the low-resolution structures, by aligning and making comparative geometric assessments. The benchmarking results show that our method can improve the annotations of the existing methods significantly. We have achieved different levels of improvements for various methods and datasets, including an example of significant sensitivity and precision enhancement from 28 to 57% and from 53 to 82%, respectively.
INTRODUCTION
Non-coding RNAs (ncRNAs) play various important roles in biological processes (1,2). The information of ncRNAs is encoded in both the primary sequences and the three-dimensional (3D) structures, which are interlinked by base-pairing patterns with tertiary interactions (3,4). Studies of the RNA 3D structures can provide essential insights into their functionalities. With the increasing number of RNA structures deposited in Protein Data Bank (PDB) (5), analyzing their 3D structures, particularly the interactions between nucleotides, are attracting increasing research focus (6–8).
Base-pairing interactions are the primary building blocks of RNA structure. These interactions stabilize local conformations and eventually determine how the whole RNA folds and shapes (9,10). Various research and their consequent observations for RNA depend on the different characteristics of these interactions (11–13). Finding and categorizing RNA structural motifs is one significant example of such research. Tools such as FR3D (9), RNA Bricks (14), RNAMotifScan (15), RNAMotifScanX (8) deal with this issue and are largely built upon base-pairing annotation. Another relevant research is aligning RNA structures. Base-pairing annotation plays a key role in many alignment tools such as DIAL (16), R3D Align (6), ARTS (17), SARA (18) and STAR3D (19). Moreover, methods dealing with RNA–protein interactions also rely on base-pairing annotations (20).
RNA bases can interact with each other in different ways and form diverse geometric conformations. Leontis and Westhof proposed a generalization of base-pairing interaction types, combining canonical (Watson–Crick A–U, G–C or G–U wobble) and non-canonical base pairs that involve the interactions among different combinations of Watson–Crick edges, hoogsteen edges and sugar edges of bases (21). Lemieux and Major did further work on the geometric issues focusing on the sliding of the bases along the interacting faces and proposed an extended set of interactions, using their new set of edges (22). Leontis et al. have done more work to define additional types of base-pairing interactions (9). From all these work, we acquired a very good understanding of the different types of base-pairing interactions that can possibly happen among RNA bases. However, for a given PDB, determining which particular bases are interacting with each other and what specific types of interactions they are forming is not so obvious. It is a challenging task to do it correctly for the various molecules with various levels of data quality.
There are different existing methods for annotating base pairs of a particular RNA from the 3D information available in PDB formatted data. Some of the well known methods are MC-Annotate (7), RNAView (23), FR3D (9), DSSR (24) and ClaRNA (10). These methods focus on a single PDB file while determining their annotations. Even though these tools are implemented using sophisticated approaches to detect base-pairing interactions, their performance is limited by the quality of the PDB data they are dealing with. These methods can perform well in detecting base pairs when the resolution of the structure is high, but cannot detect base pairs accurately enough when the resolution is low. Due to the expensive procedure, generating high-resolution 3D structure information is not always affordable (25) and there are many molecules for which only low-resolution structures are available. A low-resolution 3D structure data have inherent errors in it from the experiment that produced the structure information; these data are expected to be relatively poor in preciseness. This lack of preciseness creates some unavoidable geometric obstacles for the computational methods that use these structure data to annotate the interactions. As a result, low-resolution PDB causes all the existing annotation methods to suffer from the wrong detection of interactions.
To solve this issue, we introduce CompAnnotate, a comparative tool capable of compensating for geometric limitations in the low-resolution 3D structure data. Instead of dealing only with the low-resolution RNA structure independently, an additional higher resolution homologous RNA structure is involved in CompAnnotate to assist the annotation. The necessary geometric information, which may not be available at the required accuracy level in low-resolution PDB, can be available in other structurally similar high-resolution PDB. It is particularly the case for the RNAs that have highly conserved local structures, such as ribosomal RNAs. Here, we can use the assumption that homologous RNAs are likely to have the same type of base pairs in homologous positions. This assumption is utilized in CompAnnotate to map geometric information from the available homologous high-resolution RNA structure to the low-resolution RNA structure. Using alignment and analysis of the local geometry of base pairs, the inferability of geometric information in two different RNA structures is determined. Then, using the inferable geometric information, a better base-pairing list is selected for the given low-resolution RNA.
We benchmarked CompAnnotate by analyzing the correctness of base-pairing annotations for different pairs of high and low-resolution PDBs. Annotated base-pairing lists from the existing methods (MC-Annotate, RNAView, FR3D, DSSR and ClaRNA) are used as input for CompAnnotate and the corresponding modified base-pairing lists come as output. The CompAnnotate annotation is then compared with a high-resolution benchmark data for each method independently. We have found significant improvement and overall better sensitivity and precision of base-pairing annotation for all the methods. In addition, we have checked the specific impact of this enhanced annotation, considering the regions expected to be known motifs. We have identified a lot of important base-pairing interactions that are essential for those regions to be classified as motifs, but the existing methods can not detect them. CompAnnotate has successfully annotated a significant amount of those missing base-pairing interactions. These improvements in the base-pairing annotation, made by CompAnnotate, are expected to boost the scope and accuracy of the study involving RNA structure and function.
MATERIALS AND METHODS
The CompAnnotate takes and processes the sequence information, structure data and existing base-pairing annotations for a given pair of RNA structures, namely reference RNA and target RNA (will be defined in the next section). It first generates an alignment of these two RNAs. The alignment allows to categorize the annotated base pairs into three subsets. One subset includes base pairs from target RNA that can not be aligned with two bases in the reference RNA. This subset of base pairs is directly included. The second subset includes base pairs from reference RNA that can not be aligned with two bases in the target RNA. This subset of base pairs is directly excluded. The final subset includes base pairs from both reference and target RNA that can be aligned with corresponding bases. This subset goes through further vetting before getting included in the final annotation. At the end of this process, the set of output base pairs is generated. This CompAnnotate workflow is shown in Figure 1 and the details are described in the following sections.
Figure 1.

The workflow of CompAnnotate.
Data processing
The basic input to CompAnnotate is the sequence and 3D structural data from PDB and the corresponding base-pairing annotation output from any annotation tool, for a pair of RNAs. One in the pair is a target RNA, T, for which the annotation improvement will be made. The target RNA structure can be a very low-resolution PDB and the annotations for this structure from the existing annotation tools can have an erroneous list of base pairs. The other RNA in the pair is a higher resolution reference RNA, R. The structural information and base-pairing annotation from the reference RNA are used to improve the base-pairing annotation in the target RNA. The reference RNA is expected to be phylogenetically close to the target RNA as much as possible—the closer the RNAs are, the better the annotation should be.
The target and reference RNAs are aligned to get the correspondence of bases. STAR3D (19) has been used as the preferred alignment tool in CompAnnotate. But other structure alignment or sequence alignment with proper affine gap penalty can also be used as the input to CompAnnotate (result improvements for sequence alignment are shown in Supplementary Tables S6 and 7). With the support of input alignment, CompAnnotate does further aligning of base pairs considering the annotations from the existing tool (MC-Annotate, RNAView, FR3D, DSSR or ClaRNA). For each base pair of one RNA, a compatible base pair in the other RNA is searched, using the base correspondence as a guide. The resulting fine-tuned alignment of base pairs reflects the potential areas of similarity and dissimilarity in the RNAs.
Base-pairing annotation inferability
CompAnnotate determines which annotation in the reference RNA can be utilized to infer the annotation of target RNA. Based on the alignment, it defines inferability of annotation to decide the potential pair of bases that should be considered or discarded. The annotations used here include both canonical and non-canonical base pairs, representing the tertiary structure of RNA. These set of annotated base pairs for the reference and target structure, R and T can be denoted as SR and ST. Let the alignment of reference and target sequences be defined by a 2*m size matrix, where the 1st row represents the reference sequence r[1…m] and the second row represents the target sequence t[1…m] (see Figure 2). Each row contains a string of bases and gaps, but no column will contain two gaps aligned. The gaps are denoted by ‘—’.
Figure 2.

Example of alignment and base pairing annotation inferability. The solid arcs imply annotated base pairs and the dashed arcs imply corresponding aligned pairs of bases that are not members of SR or ST.
For any two columns i and j in the alignment, a relationship can be defined for the pairs (ri, rj) and (ti, tj), based on the given base-pairing annotations in SR and ST. In defining this relationship, we consider 2 bp are of the same type if they have the same participating edges which include Watson–Crick, hoogsteen or sugar edge (21), along with the same cis/trans orientation. If (ri, rj) ∈SR, (ti, tj) ∈ ST and both are the same type of base pairs, they are treated as common base pairs. Aligned bases are not required to be identical; as long as they are mapped to each other by the alignment module and have the same type of interacting edges and the same cis/trans orientation in the base-pairing annotation, they are treated as common base pairs. If neither of (ri, rj) and (ti, tj) is annotated as a base pair, they are denoted as, unrelated. If one of the pairs is annotated as a base pair and any of ri, rj, ti and tj is ‘—’, then they are considered as non-inferable. There are two cases of non-inferable depending on whether the base pair is from the reference or the target RNA. Finally, where all of ri, rj, ti and tj are bases but did not form common base pairs relationship, it is denoted as inferable—case 1 if (ri, rj) ∈ SR and inferable—case 2 if (ti, tj) ∈ ST. There is one special relationship situation that is worth mentioning separately. If (ri, rj) ∈ SR and (ti, tj) ∈ ST, but the base pairs are not of same type, they are categorized as inferable—case 1. All these relationships are summarized in Table 1 and some of the corresponding examples are given in Figure 2.
Table 1. The relationship between (ri, rj) and (ti, tj) for the base-pairing annotation inferability.
| Relationship | Description |
|---|---|
| Common base pairs | (ri, rj) ∈ SR and (ti, tj) ∈ ST, and both |
| have the same type of interacting edges and | |
| cis/trans orientation | |
| Unrelated | Neither of (ri, rj) or (ti, tj) is annotated |
| as a base pair | |
| Non-inferable—case 1 | (ri, rj)∈SR, but one or both of ti and tj is ‘—’ |
| Non-inferable—case 2 | (ti, tj)∈ST, but one or both of ri and rj is ‘—’ |
| Inferable—case 1 | (ri, rj)∈SR, and ti, tj are bases (not gap) |
| Inferable—case 2 | (ti, tj)∈ST, and pair of bases (ri, rj) ∉ SR |
These relationships define how the annotation information can be inferred for the base pairs, from the reference RNA to the target RNA. For the case of common base pairs relationship, both (ti, tj) and (ri, rj) verify each other as properly annotated base pairs. For the unrelated and non-inferable relationship, there is no compatible information for a valid comparative analysis and consequently, no modification attempt is considered. As a result, the annotated target base pair (ti, tj) in the non-inferable—case 2 relationship are directly included by CompAnnotate to the accepted list of base pairs for the target RNA. On the other hand, the annotated reference base pair (ri, rj) in the non-inferable—case 1 relationship are directly excluded. Eventually, the inferable—case 1 and inferable—case 2 become the primary focus. In inferable—case 1 relationship, for each base pair (ri, rj), there is a corresponding pair of bases ti and tj in the target RNA that is a potential candidate for being annotated as a base pair. On the other hand, in the inferable—case 2 relationship, for each base pair (ti, tj), there is a corresponding pair of bases ri and rj in the reference RNA. Those pair of bases can aid to decide which of the (ti, tj) should be kept as base pairs and which one should be discarded. All the further geometric observations and analysis for inclusion–exclusion of base pairs are done based on the comparison of the pairs ((ri, rj), (ti, tj)) for the inferable—case 1 and inferable—case 2 relationship.
Comparative geometric analysis
The annotation of base pairs relies on the distances among atoms in the bases (26). But the proper measurement of the distance becomes a challenging concern as the exact coordinates of the atoms are not found. The coordinates in the structure data are affected by the quality of the experiment. The exact coordinates are not observed, only a close approximation; the resolution along with some other quality measurement parameters of the structure data describes in a statistical sense how displaced the coordinates can be from the actual coordinates. The expected displacement of the coordinates from the actual position increases in low-resolution compared to the high-resolution counterpart. The base-pairing annotation can be affected by this coordinate displacement issue in general, but the situation becomes extremely challenging in the low resolution data.
To get an estimation of how displaced the coordinates can be, we have considered the following Cruickshank’s equation for dispersion precision indicator (DPI) (27,28),
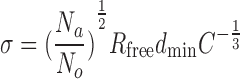 |
(1) |
where C is completeness, Rfree is free R-value, dmin is maximum resolution, Na is the number of atoms and No is number of observations (reflections) included in the refinement. This equation corresponds to the approximate overall standard uncertainties (28). We use σ as the measure of potential displacement of atomic coordinates in a given PDB.
Now, let’s consider two atoms a and b in any RNA structure where the actual distance between them is D(a, b). Now, applying the uncertainty of bond length between two atoms (27), the observed distance DH(a, b) in a high-resolution PDB, H, with a potential coordinate displacement σH, is likely to be:
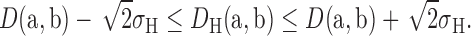 |
(2) |
Similarly, the observed distance DL(a, b) in a low-resolution PDB, L, with a potential coordinate displacement σL, is likely to be:
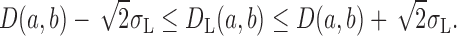 |
(3) |
Equations 2 and 3 show that the observed distances can vary from one PDB to another PDB. As σL is expected to be relatively higher, the range of variation of distances increases accordingly. Here, we can imply
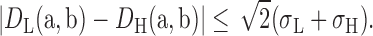 |
(4) |
To improve base-pairing interactions in the low-resolution target RNA, T, Equation 4 provides a platform to make the comparative geometric assessment for the aligned bases between T and the high-resolution reference RNA, R. For the inferable relationships defined in the previous section, we can use the atoms in the pairs of bases (ri, rj) and (ti, tj) to infer the annotation from R to T. Three to five pairs of relevant atoms in (ri, rj) is mapped to the corresponding relevant atoms in (ti, tj). If the mapped pairs of relevant atoms are considered compatible on account of potential displacement, we can annotate the base-pairing interaction for (ti, tj) based on the interaction in (ri, rj). The list of considered atoms for the different types of base pairs (Supplementary Tables S1–5), along with how the relevant atoms are chosen is described in the Supplementary Data.
For the inferable—case 1 relationship, (ri, rj) is annotated as a base pair by an existing tool, but (ti, tj) is not. The coordinate displacement issue can cause this difference. In this case, if the observed distances between the relevant atoms in (ri, rj) and (ti, tj) satisfy the condition in Equation 4, it suggests that (ti, tj) can be annotated as a base pair, on account of observed coordinate displacement. We use the base-pairing annotation for (ri, rj) as the annotation for (ti, tj). On the other hand, for the inferable—case 2 relationship, (ti, tj) is annotated as a base pair by an existing tool, but (ri, rj) is not. In this case, two scenarios will be considered. For scenario 1, if the observed distances satisfy the condition in Equation 4, we assume that the annotation difference is due to the coordinate displacement. In this scenario, we discard the annotation for (ti, tj). For scenario 2, if the observed distances do not satisfy the condition in the Equation 4, we assume that the annotation difference is due to other reasons than the coordinate displacement. The reference and the target RNA being from different organisms, one plausible reason is the inherent structural difference between them. In this scenario, the base-pairing annotation for (ti, tj) is kept unchanged.
RESULTS
Base-pairing annotation
In this section, we will measure the improvement of CompAnnotate over five tools, including MC-Annotate, RNAView, FR3D, DSSR and ClaRNA. To conduct our experiments, some 16S and 23S rRNAs, whose 3D structures are the largest ones in the PDB, are used as target RNAs. The high-resolution structures of their homologs in other species are chosen as reference RNAs. In addition, we also used benchmark structures which are the high-resolution entry of the same RNAs as the target RNAs. The annotation performance of CompAnnotate on the target RNAs are evaluated by comparing with the base-pairing annotation of the benchmark structures. In the PDB database, we found a few 16S and 23S rRNAs that have both high-resolution and low-resolution 3D structures. Table 2 lists the details of the datasets for organisms Thermus thermophilus, Escherichia coli and Deinococcus radiodurans, which has been used in our experiments for benchmarking the performance.
Table 2. Chains, organisms, PDB IDs and resolutions of the ribosomal RNAs used as benchmarking datasets.
| Target | Reference | Benchmark | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Chain | Organism | PDB ID | Res. | Organism | PDB ID | Res. | Organism | PDB ID | Res. |
| 23S | T. thermophilus | 2B9N | 6.76 Å | E. coli | 3R8S | 3.0 Å | T. thermophilus | 3V2F | 2.7 Å |
| D. radiodurans | 2ZJR | 2.91 Å | |||||||
| 16S | T. thermophilus | 1YL4 | 5.5 Å | E. Coli | 4GD1 | 3.0 Å | T. thermophilus | 2VQE | 2.5 Å |
| 2B9M | 6.76 Å | ||||||||
‘Res.’ is representing the resolution of the corresponding PDB.
For the target RNA with PDB ID: 2B9N, we have used two different reference RNAs, 3R8S and 2ZJR, to show that CompAnnotate achieves improvement independent of the reference RNA and also to show that different reference RNAs can cause different levels of improvement. It also emphasizes the fact that choosing a proper reference RNA can make the results better. On the other hand, for the target RNAs 1YL4 and 2B9M, the same reference RNA, 4GD1 is used. For benchmarking 23S chain, the PDB ID: 3V2F is used and for 16S chain, PDB ID: 2VQE is used. We had to make some special considerations while addressing FR3D annotation data, collected from NDB server. The NDB server uses data from the PDB server and reflects the recent changes the PDB server made. The annotation for the PDB IDs: 2B9N, 2B9M, 3R8S, 4GD1, 3V2F and 1YL4 were not directly found there. Rather, we had to deal with the annotation for compatible new versions of PDB IDs: 4V4S, 4V9D, 4V8I and 4V4P. We have done the necessary mapping to extract the corresponding base-pairing list for the original versions of the PDBs we are working with for benchmarking purposes.
MC-Annotate, RNAView, FR3D, DSSR and ClaRNA have different approaches of annotating base pairs. While canonical base-pairing annotations are quite similar across all these methods, non-canonical base-pairing annotations are very different from each other. By comparing with the annotation of benchmark PDB, we found that many base pairs detected in the high-resolution benchmark PDBs are not detected for the low-resolution target PDBs. Moreover, many base pairs are annotated in low-resolution target PDB, that are not supported by the annotation of the high resolution benchmark PDB. This implies the lower sensitivity and precision of these methods. CompAnnotate works on the annotation output of the existing methods to combine and filter the reference and target PDB annotation data. It can be considered as an extension tool, that uses the annotations for multiple PDBs from existing methods and generates better annotation results of the particular low-resolution PDBs. We work on each existing method independently, and generate a CompAnnotate version of annotation for each method separately. As a result, depending on which annotation method is being used for input data, CompAnnotate can be used as different annotation tools, such as CompAnnotate (MC-Annotate), CompAnnotate (RNAView), CompAnnotate (FR3D), CompAnnotate (DSSR) and CompAnnotate(ClaRNA).
Before going into the overall performance benchmarking, let’s first get some insight on how the CompAnnotate method works, by using an example pair of the target and reference RNA. The inferable base pairs are the source of improvement in CompAnnotate. Common and non-inferable base pairs are kept the same. They do not contribute to the overall improvement. Table 3 shows the details of annotations acceptance for the target RNA structure 2B9N with the aiding reference RNA structure 3R8S while using the CompAnnotate (MC-Annotate) version of the tool. Here, the results for canonical and non-canonical base pairs are shown separately. We can see that everything is accepted for common base pairs and non-inferable—case 2. On the other hand, everything is rejected for non-inferable—case 1. But, for the inferable cases, base pairs are being selected based on geometric observations. Among the 149 bp in canonical inferable—case 1 relationship and 322 bp in non-canonical inferable—case 1 relationship, 101 and 193 bp are accepted, respectively. These newly added base pairs by CompAnnotate were not included for the target RNA by the existing annotation, which is the most significant part of the improvement. For the inferable target base pairs, almost all the base pairs are excluded. The rejection of erroneously annotated base pairs for this inferable target base pair case accounts for the other part of the improvement. The annotation acceptance details using the CompAnnotate (RNAView), CompAnnotate (FR3D), CompAnnotate (DSSR), CompAnnotate (ClaRNA) versions are shown in the Supplementary Tables S8–11.
Table 3. Acceptance ratios of different inferability relationship for MC-Annotate annotation, using reference structure in PDB ID: 3R8S and target structure in PDB ID: 2B9N.
| Target (Ref.) | Relationship | Total # of bp | Accepted # of bp | Accept % |
|---|---|---|---|---|
| Canonical | Common | 597 | 597 | 100.00 |
| Inferable—case 1 | 149 | 101 | 67.79 | |
| Inferable—case 2 | 24 | 1 | 4.17 | |
| Non-inferable—case 1 | 46 | 0 | 0.00 | |
| Non-inferable—case 2 | 17 | 17 | 100.00 | |
| Non-canonical | Common | 83 | 83 | 100.00 |
| Inferable—case 1 | 322 | 193 | 59.94 | |
| Inferable—case 2 | 106 | 0 | 0.00 | |
| Non-inferable—case 1 | 31 | 0 | 0.00 | |
| Non-inferable—case 2 | 4 | 4 | 100.00 |
Now, we are going to analyze the overall impact of these inclusion and exclusion of base pairs. To make the comparison, we have addressed another issue related to PDB structure data. Even though the target and benchmark RNA structure are from the same organism, there are some regions with missing residues. For the purpose of fair performance comparison, we have excluded the annotated base pairs involving those regions, from both the existing method and CompAnnotate. The comparison shows annotation improvement for both canonical and non-canonical base pairs of the target RNAs. The sensitivity and precision of canonical base pair detection by regular methods (existing methods without CompAnnotate extension) are already quite good, even in low-resolution. However, the sensitivity and precision of CompAnnotate for canonical base pairs are similar or better. On the other hand, the non-canonical base pair detection is largely affected by the coordinate errors. As a result, there are more opportunities for the comparative methods to address and improve non-canonical base-pairing annotations. CompAnnotate has achieved a significant improvement particularly in annotating the non-canonical base pairs. The precision and sensitivity improvement of both the total canonical and non-canonical base-pairing annotations for the given datasets are shown in Tables 4 and 5 along with the corresponding bar chart in Figure 3 (Note that, the results shown for non-canonical base pairs here include only those base pairs whose interacting edges are Watson–Crick, hoogsteen or sugar edges, but not both edges are Watson–Crick). The broke down performance details of all the datasets for each method are given in the Supplementary Tables S12–21.
Table 4. The improvements in base-pairing annotation sensitivity for five tools by using CompAnnotate.
| Regular | CompAnnotate | |||||
|---|---|---|---|---|---|---|
| Method | Base pair type | # of bp in benchmark | # of bp detected | % | # of bp detected | % |
| MC-Annotate | Canonical | 2578 | 2055 | 79.71 | 2180 | 84.56 |
| Non-canonical | 1224 | 338 | 27.61 | 702 | 57.35 | |
| RNAView | Canonical | 2634 | 2242 | 85.12 | 2274 | 86.33 |
| Non-canonical | 2782 | 1009 | 36.27 | 1466 | 52.70 | |
| FR3D | Canonical | 2622 | 2322 | 88.56 | 2333 | 88.98 |
| Non-canonical | 2788 | 1390 | 49.86 | 1691 | 60.65 | |
| DSSR | Canonical | 2618 | 2269 | 86.67 | 2293 | 87.59 |
| Non-canonical | 1640 | 670 | 40.85 | 961 | 58.60 | |
| ClaRNA | Canonical | 2518 | 2079 | 82.57 | 2183 | 86.70 |
| Non-canonical | 1456 | 500 | 34.34 | 865 | 59.41 | |
The better performances are shown in bold.
Table 5. The improvements in base-pairing annotation precision for five tools by using CompAnnotate.
| Regular | CompAnnotate | |||||
|---|---|---|---|---|---|---|
| Method | Base pair type | # of bp in benchmark | Match/conflict | % | Match/conflict | % |
| MC-Annotate | Canonical | 2578 | 2055/51 | 97.58 | 2180/31 | 98.60 |
| Non-canonical | 1224 | 338/303 | 52.73 | 702/154 | 82.01 | |
| RNAView | Canonical | 2634 | 2242/155 | 93.53 | 2274/73 | 96.89 |
| Non-canonical | 2782 | 1009/1286 | 43.97 | 1466/560 | 72.36 | |
| FR3D | Canonical | 2622 | 2322/157 | 93.67 | 2333/58 | 97.57 |
| Non-canonical | 2788 | 1390/1113 | 55.53 | 1691/459 | 78.65 | |
| DSSR | Canonical | 2618 | 2269/114 | 95.22 | 2293/44 | 98.12 |
| Non-canonical | 1640 | 670/665 | 50.19 | 961/313 | 75.43 | |
| ClaRNA | Canonical | 2518 | 2079/31 | 98.53 | 2183/29 | 98.69 |
| Non-canonical | 1456 | 500/193 | 72.15 | 865/124 | 87.46 | |
The ‘match’ is representing the true positives and the ‘conflict’ is representing the false positives, against the positive data represented by ‘# of bp in benchmark’. The better performances are shown in bold.
Figure 3.

Comparison of sensitivity and precision for regular and CompAnnotate methods. The top panel shows the sensitivity and precision for canonical base-pairing annotations. The bottom panel shows the sensitivity and precision for non-canonical base-pairing annotations.
Annotation in RNA structural motifs
To analyze the performance of the CompAnnotate base-pairing annotation more closely, we have observed the interactions inside the motifs. For our observations, we considered a few known regions of C-loop, kink-turn and sarcin-ricin motifs. Initially, we picked the location of known motifs for PDB ID: 1S72 from the result of RNAMotifScanX (8). Then we mapped the locations, to get the regions in PDB ID: 2B9N that are expected to be motifs. The considered motifs in 2B9N are 908–913/863–869 (C-loop), 1234–1238/1208–1215 (kink-turn), 2653–2657/2664–2667 (sarcin-ricin), 456–460/469–472 (sarcin-ricin) and 2724–2728/2679–2685 (C-loop). Then for these regions, we analyzed how the known expected base pairs are annotated for the regular and CompAnnotate method.
In this low-resolution RNA structure, the regular annotation methods miss many base pairs that are necessary for a region to be considered as a motif. CompAnnotate can successfully include proper base pairs in such cases. The count of base pairing annotation for our five sample motifs is shown in Table 6. Here we show the improvement of annotation in target PDB ID: 2B9N, considering both of the reference PDB IDs: 3R8S and 2ZJR. CompAnnotate has the same or more expected annotations for all the methods in these motif regions. We show detailed 3D and 2D representations (drawn using PyMOL and Xfig) of motifs and base-pairing annotation in Figure 4, choosing one example for each method. The 3D structures in the figure show the structural characteristics for the motifs and visual justification why these regions are being considered as motifs. The consensus structures (15) show the expected base-pairing interactions for a given motif. For the C-loop 908–913/863–869, the regular MC-Annotate method detects four out of six known base pairs and the CompAnnotate (MC-Annotate) can detect all six. For the kink-turn 1234–1238/1208–1215, the regular RNAView method detects three out of seven known base pairs and the CompAnnotate (RNAView) can detect six. For the sarcin-ricin 2653–2657/2664–2667, the regular FR3D method detects zero out of five known base pairs and the CompAnnotate (FR3D) detects four. For the sarcin-ricin 456–460/469–472, the regular DSSR method detects zero out of five known base pairs and the CompAnnotate (DSSR) detects four. For the C-loop 2724–2728/2679–2685, the regular ClaRNA method detects three out of six known base pairs and the CompAnnotate (ClaRNA) can detect all six.
Table 6. Comparison of base-pairing annotations between the regular and CompAnnotate methods for known motifs in a 23S rRNA (PDB ID: 2B9N).
| Annotated base pair count | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MC-Annotate | RNAView | FR3D | DSSR | ClaRNA | |||||||
| Motif (Type: expected # of bp) | Target (Ref) | Regular | CompA | Regular | CompA | Regular | CompA | Regular | CompA | Regular | CompA |
| 908–913/863–869 (C-loop: 6) | 2B9N (3R8S) | 4 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 6 |
| 2B9N (2ZJR) | 4 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 5 | 6 | |
| 1234–1238/1208–1215 (Kink-turn: 7) | 2B9N (3R8S) | 3 | 6 | 3 | 5 | 5 | 5 | 4 | 5 | 3 | 5 |
| 2B9N (2ZJR) | 3 | 5 | 3 | 6 | 5 | 5 | 4 | 5 | 3 | 5 | |
| 2653–2657/2664–2667 (Sarcin-ricin: 5) | 2B9N (3R8S) | 1 | 3 | 1 | 3 | 0 | 4 | 1 | 3 | 0 | 3 |
| 2B9N (2ZJR) | 1 | 2 | 1 | 3 | 0 | 4 | 1 | 3 | 0 | 2 | |
| 456–460/469–472 (Sarcin-ricin: 5) | 2B9N (3R8S) | 0 | 4 | 0 | 4 | 2 | 4 | 0 | 4 | 0 | 4 |
| 2B9N (2ZJR) | 0 | 4 | 0 | 4 | 2 | 4 | 0 | 4 | 0 | 4 | |
| 2724–2728/2679–2685 (C-loop: 6) | 2B9N (3R8S) | 5 | 5 | 4 | 6 | 5 | 6 | 5 | 6 | 3 | 5 |
| 2B9N (2ZJR) | 5 | 6 | 4 | 6 | 5 | 6 | 5 | 6 | 3 | 6 | |
‘CompA’ represents CompAnnotate version of the tools, compared to the regular version.The improvements are shown in bold.
Figure 4.

The base-pairing annotations of CompAnnotate and other five tools for five RNA structure motifs in a 23S rRNA (PDB ID: 2B9N). (A) C-loop (908–913/863–869) structure and consensus with MC-Annotate and CompAnnotate annotation. (B) Kink-turn (1234–1238/1208–1215) structure and consensus with RNAView and CompAnnotate annotation. (C) Sarcin-ricin (2653–2657/2664–2667) structure and consensus with FR3D and CompAnnotate annotation. (D) Sarcin-ricin (456–460/469–472) structure and consensus with DSSR and CompAnnotate annotation. (E) C-loop (2724–2728/2679–2685) structure and consensus with ClaRNA and CompAnnotate annotation.
DISCUSSION
In this paper, we have presented a novel tool, named CompAnnotate, to predict base-pairing interactions in the low-resolution RNA 3D structures. It adopts a high-resolution homolog as the reference RNA to infer the plausible base pairs in the target RNA by using a comparative method. The target and the reference RNAs are aligned first and then the different annotations of base pairs in them are analyzed to make better prediction for the target RNA. The distances of these inferred base pairs are restricted to specific ranges determined by the quality of two RNA structure data to improve the accuracy. CompAnnotate has been implemented as a C program and benchmarked with other five tools, including MC-Annotate, RNAView, FR3D, DSSR and ClaRNA. The experimental results show that CompAnnotate improves the performance of these state-of-the-art tools in the base-pairing annotation for low-resolution RNA structures, especially the prediction of non-canonical base pairs. The tool is also tested by applying its results to RNA structural motif identification. It can be seen that CompAnnotate is better in detecting the potential base pairs in the conserved motifs for the low-resolution RNAs. In conclusion, CompAnnotate helps to explore the plausible base pair interactions in RNAs whose high-resolution 3D structural information is unavailable. These newly detected interactions can be applied to the study of RNA tertiary structures, which may aid to uncover their functions in the cell.
The approach used in CompAnnotate is a significant step toward including the coordinate uncertainties in the context of base-pairing annotations. Based on this work, further research can be done to improve the annotation result for PDB data. One possible extension of CompAnnotate is to use multiple high-resolution RNA structures as reference RNAs, when more RNA structures will be deposited in the PDB database. Future work may also focus on the PDB data that does not have necessary parameters to calculate DPI. According to the statistics of attributes usage in the PDBx/mmCIF dictionary, around 80% of PDB data have necessary attributes for DPI calculation. How the uncertainties of the coordinates can be calculated for rest of the 20% PDBs still remains as a challenge to solve. Another future work can include devising an unified geometric platform to combine annotation results from multiple tools together. It is noticed that none of the annotation tools can uncover all the base pairs and on the other hand, many predicted base pairs of one tool are not shared by the others. With an algorithm to compare the prediction results of multiple tools, there are opportunities to combine all the annotation of base pairs to provide a more comprehensive solution.
AVAILABILITY
CompAnnotate is publicly accessible and available on http://genome.ucf.edu/CompAnnotate.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Chenghua Shao, Wen Zhang and Zukang Feng for their guidance in understanding the biochemical aspect of coordinate uncertainty issue.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences of the National Institutes of Health (NIH NIGMS) [R01GM102515]. Funding for open access charge: NIH NIGMS [R01GM102515].
Conflict of interest statement. None declared.
REFERENCES
- 1. Eddy S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001; 2:919–929. [DOI] [PubMed] [Google Scholar]
- 2. Storz G. An expanding universe of noncoding RNAs. Science. 2002; 296:1260–1263. [DOI] [PubMed] [Google Scholar]
- 3. Carter A.P., Clemons W.M., Brodersen D.E., Morgan-Warren R.J., Wimberly B.T., Ramakrishnan V.. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000; 407:340–348. [DOI] [PubMed] [Google Scholar]
- 4. Woodson S.A. Compact intermediates in RNA folding. Annu. Rev. Biophys. 2010; 39:61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rahrig R.R., Leontis N.B., Zirbel C.L.. R3D Align: global pairwise alignment of RNA 3D structures using local superpositions. Bioinformatics. 2010; 26:2689–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gendron P., Lemieux S., Major F.. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001; 308:919–936. [DOI] [PubMed] [Google Scholar]
- 8. Zhong C., Zhang S.. RNAMotifScanX: a graph alignment approach for RNA structural motif identification. RNA. 2015; 21:333–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sarver M., Zirbel C.L., Stombaugh J., Mokdad A., Leontis N.B.. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J. Math. Biol. 2008; 56:215–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Waleń T., Chojnowski G., Gierski P., Bujnicki J.M.. ClaRNA: a classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic Acids Res. 2014; 42:e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leontis N.B., Lescoute A., Westhof E.. The building blocks and motifs of RNA architecture. Curr. Opin. Struct. Biol. 2006; 16:279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Halder S., Bhattacharyya D.. RNA structure and dynamics: a base pairing perspective. Prog. Biophys. Mol. Biol. 2013; 113:264–283. [DOI] [PubMed] [Google Scholar]
- 13. Lee J.C., Gutell R.R.. Diversity of base-pair conformations and their occurrence in rRNA structure and RNA structural motifs. J. Mol. Biol. 2004; 344:1225–1249. [DOI] [PubMed] [Google Scholar]
- 14. Chojnowski G., Walen T., Bujnicki J.M.. RNA Bricks–a database of RNA 3D motifs and their interactions. Nucleic Acids Res. 2014; 42:D123–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhong C., Tang H., Zhang S.. RNAMotifScan: automatic identification of RNA structural motifs using secondary structural alignment. Nucleic Acids Res. 2010; 38:e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferre F., Ponty Y., Lorenz W.A., Clote P.. DIAL: a web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res. 2007; 35:W659–W668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dror O., Nussinov R., Wolfson H.. ARTS: alignment of RNA tertiary structures. Bioinformatics. 2005; 21(Suppl. 2):47–53. [DOI] [PubMed] [Google Scholar]
- 18. Capriotti E., Marti-Renom M.A.. SARA: a server for function annotation of RNA structures. Nucleic Acids Res. 2009; 37:W260–W265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ge P., Zhang S.. STAR3D: a stack-based RNA 3D structural alignment tool. Nucleic Acids Res. 2015; 43:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iwakiri J., Kameda T., Asai K., Hamada M.. Analysis of base-pairing probabilities of RNA molecules involved in protein-RNA interactions. Bioinformatics. 2013; 29:2524–2528. [DOI] [PubMed] [Google Scholar]
- 21. Leontis N.B., Westhof E.. Geometric nomenclature and classification of RNA base pairs. RNA. 2001; 7:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lemieux S., Major F.. RNA canonical and non-canonical base pairing types: a recognition method and complete repertoire. Nucleic Acids Res. 2002; 30:4250–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang H., Jossinet F., Leontis N., Chen L., Westbrook J., Berman H., Westhof E.. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003; 31:3450–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu X.J., Bussemaker H.J., Olson W.K.. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015; 43:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parisien M., Major F.. Determining RNA three-dimensional structures using low-resolution data. J. Struct. Biol. 2012; 179:252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leontis N.B., Stombaugh J., Westhof E.. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res. 2002; 30:3497–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cruickshank D. Remarks about protein structure precision. Acta Crystallogr. D Biol. Crystallogr. 1999; 55:583–601. [DOI] [PubMed] [Google Scholar]
- 28. Murshudov G.N., Dodson E.J.. Simplified error estimation a la Cruickshank in macromolecular crystallography. CCP4 Newsl. Protein Crystallogr. 1997; 33:31–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.