

Abstract
Organelle genomes exhibit remarkable diversity in content, structure, and size, and in their modes of gene expression, which are governed by both organelle- and nuclear-encoded machinery. Next generation sequencing (NGS) has generated unprecedented amounts of genomic and transcriptomic data, which can be used to investigate organelle genome transcription. However, most of the available eukaryotic RNA-sequencing (RNA-seq) data are used to study nuclear transcription only, even though large numbers of organelle-derived reads can typically be mined from these experiments. Here, we use publicly available RNA-seq data to assess organelle genome transcription in 59 diverse plastid-bearing species. Our RNA mapping analyses unraveled pervasive (full or near-full) transcription of mitochondrial, plastid, and nucleomorph genomes. In all cases, 85% or more of the organelle genome was recovered from the RNA data, including noncoding (intergenic and intronic) regions. These results reinforce the idea that organelles transcribe all or nearly all of their genomic material and are dependent on post-transcriptional processing of polycistronic transcripts. We explore the possibility that transcribed intergenic regions are producing functional noncoding RNAs, and that organelle genome noncoding content might provide raw material for generating regulatory RNAs.
Keywords: mitochondrial transcription, noncoding RNA, organelle gene expression, pervasive transcription, plastid transcription
Introduction
Organelle genomes can be extreme at both the DNA and RNA levels (Smith and Keeling 2015; Smith and Keeling 2016). Gene fragmentation (Barbrook etal. 2010), gene and chromosome number variation (Shao etal. 2012; Janouškovec etal. 2013), diverse genome topology (e.g., circular or linear with telomeres; Bendich 2007), and genome size range (Sloan etal. 2012) are some of the many examples of organelle genomic diversity. Similarly, the expression of organelle genomes can be unconventional, including noncanonical genetic codes (Burger etal. 2003), substitutional or insertion/deletion RNA-editing (Castandet and Araya 2011), trans-splicing followed by polyadenylation (Vlcek etal. 2011), and even translational bypassing (Masuda etal. 2010; Lang etal. 2014). In many instances, unraveling these complicated genomic and transcriptional architectures took years of laborious investigation, using a wide range of molecular biology techniques (Sanitá Lima etal. 2016).
More recently, next generation sequencing (NGS) has allowed researchers to take a genome-wide approach to investigating organelle genomes and transcriptomes (Ruwe etal. 2013). For instance, high-throughput RNA sequencing (RNA-seq) of isolated organelles helped uncover pervasive transcription in the human mitochondrial genome and barley plastid genome (Mercer etal. 2011; Zhelyazkova etal. 2012). Given the popularity of NGS, organelle transcription can now easily be explored using publicly available RNA-seq data from whole-cell experiments (Smith 2013). Indeed, such an approach revealed full transcription of plastid DNAs (ptDNAs) from various land plants (Shi etal. 2016) and in the mitochondrial DNAs (mtDNAs) of Polytomella green algae (Tian and Smith 2016).
Most of the researchers that generate whole-cell eukaryotic RNA-seq data are not necessarily interested in organelle transcription, and many treat the organelle-derived reads as contamination, filtering them out before downstream analyses. Consequently, public databases, such the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA), are increasingly becoming an untapped source for organelle transcriptomic data from eukaryotic RNA-seq experiments, regardless of the NGS sequencing protocol that was used (Smith and Sanitá Lima 2016).
RNA-seq data alone are rarely enough to uncover the full complexity of organelle gene expression, but they are a fast, efficient, and cost-effective first approach to studying transcription (Dietrich etal. 2015). Although pervasive transcription has been extensively demonstrated in nuclear and bacterial systems (Berretta and Morillon 2009; Wade and Grainger 2014), it is not yet known how common this process is among organelle genomes. Most of the reports of genome-wide transcription in organelles come solely from model species (Hotto etal. 2012; Ro etal. 2013; Ross etal. 2016), suggesting that this strategy is the norm, rather than the exception, in mitochondria and plastids, and perhaps inherited from their bacterial progenitors (Shi etal. 2016). So, is pervasive transcription a common theme among mtDNAs and ptDNAs across the eukaryotic domain? And do compact versus bloated organelle genomes differ in their transcriptional patterns?
Here, by taking advantage of publicly available eukaryotic RNA-seq data, we investigate the transcriptional architecture of diverse plastid-bearing species, and show that pervasive transcription is a widespread phenomenon across the eukaryotic domain, including in very large organelle genomes with high noncoding contents. We speculate about the potential function roles (if any) of organelle noncoding RNAs (ncRNAs), particularly with respect to land plants and mixotrophs. If anything, these data highlight the utility of freely accessible RNA-seq data for organelle gene expression studies.
Materials and Methods
Using the NCBI Taxonomy Browser (https://www.ncbi.nlm.nih.gov/taxonomy, last accessed July 10, 2017), we identified 59 plastid-bearing species for which complete mitochondrial, plastid, and/or nucleomoprh genome sequences (>100 kb) and ample RNA-seq data sets were available. We limited our search to species with organelle genomes that were 100 kb or greater. Previously, we explored the prevalence of pervasive transcription in small and compact organelle genomes (≤105 kb; Sanitá Lima and Smith 2017), and here we wanted to see if the same trends held for larger organelle DNAs with long intergenic regions.
The 59 species we identified include land plants and other members of the Archaeplastida as well as various species with “complex” plastids, such as cryptophytes and stramenopiles (supplementary table S1, Supplementary Material online). The organelle genomic architectures of these species span the gamut of size (∼104–980 kb), coding content (∼0.6–82%), structure (circular vs. linear), and chromosome number (intact vs. fragmented). The RNA-Seq data were downloaded from the NCBI SRA (Kodama etal. 2012), and the genome sequences from GenBank. See supplementary table S1, Supplementary Material online for detailed information on the RNA-seq and organelle genome data we collected, including accession numbers, read counts, sequencing technologies, organelle genome features (e.g., GC content, genome topology, and percent protein-coding), and the strains used for genome and transcriptome sequencing.
We ensured that the RNA-seq and corresponding organelle genome data came from the same species, but sometimes they came from different strains of the same species (supplementary table S1, Supplementary Material online). Also, the RNA-seq experiments we sourced were often generated using very different protocols and experimental conditions (supplementary table S1, Supplementary Material online). Nevertheless, these caveats did not hinder the mapping analyses (see below).
Mapping analyses were performed using Geneious v9.1.6 (Biomatters Ltd., Auckland, NZ; Kearse etal. 2012). Briefly, raw whole-cell RNA-seq reads were mapped to the corresponding organelle genomes with Bowtie 2 (Langmead and Salzberg 2012) using the default settings, the highest sensitivity option, and a min/max insert size of 50 nt/750 nt. We allowed each read to be mapped up to two locations to account for repeated regions, which are common in organelle genomes (Smith and Keeling 2015). The mapping histograms were extracted from Geneious.
Results
Pervasive Transcription Is Widespread across Organelle and Nucleomorph Genomes
For each of the organelle genomes studied here, RNA-seq reads covered 85% or more of the reference sequence (RefSeq), regardless of the genome size, noncoding content, or taxonomic grouping (fig. 1, and supplementary table S1 and fig. S1, Supplementary Material online). In 24 cases, >99% of the organelle DNA sequence was present at the RNA level. In other words, all of the genomes exhibited pervasive, genome-wide transcription. The mean RNA-seq read coverage was consistently high across the different genomes, varying from ∼30 to >2,300,000 reads/nt.
Fig. 1.

—Occurrence of pervasive transcription in mitochondrial, plastid and nucleomorph genomes across plastid-bearing species. Unscaled phylogenetic relationships were adapted accordingly from different studies: overall angiosperm phylogeny (Stevens 2001), Millettioid sensu late clade phylogeny (Wojciechowski 2006), major eukaryotic groups phylogeney (Burki 2014), ferns phylogeny (Plackett etal. 2015) and Cucurbitaceae phylogeny (Renner and Schaefer 2016) mt, mitochondrion; pt, plastid; cy, cyanelle; nm, nucleomorph; RefSeq %, percentage of the reference organelle genome covered by one or more transcripts; Coding %, percentage of the amount of coding sequences (tRNA-, rRNA-, and protein coding genes) in the organelle genome. The coding % was manually determined by extracting tRNA-, rRNA- and coding sequences (CDS) annotations and then subtracting spurious annotations using Geneious v9.1.6 (Kearse etal. 2012).
Together, these data indicate that noncoding regions from disparate organelle genomes are broadly transcribed, which can be clearly deduced from the RNA-seq mapping histograms (supplementary fig. S1, Supplementary Material online). This was true for relatively compact genomes, such as the ptDNA of the stramenopile alga Nannochloropsis oceanica (82% coding; RefSeq coverage 94%) as well as for the highly bloated organelle genomes (fig. 1 and supplementary table S1 and fig. S1, Supplementary Material online). For instance, RNA-seq coverage exceeded 90% for the very large mitochondrial genomes of the land plants Salvia miltiorrhiza (∼499 kb, ∼9.5% coding), Capsicum annum (∼507 kb, ∼12% coding), Rhazya stricta (∼548 kb, ∼8% coding), Asclepias syriaca (∼682 kb, ∼5% coding), Phoenix dactylifera (∼715 kb, ∼5% coding), and Cucurbita pepo (∼982 kb, ∼15% coding; fig. 2). This implies that hundreds of thousands of nucleotides of ncRNAs are being generated in these mitochondria, and within distinct groups of angiosperm (e.g., asterids, commelinids, and rosids).
Fig. 2.
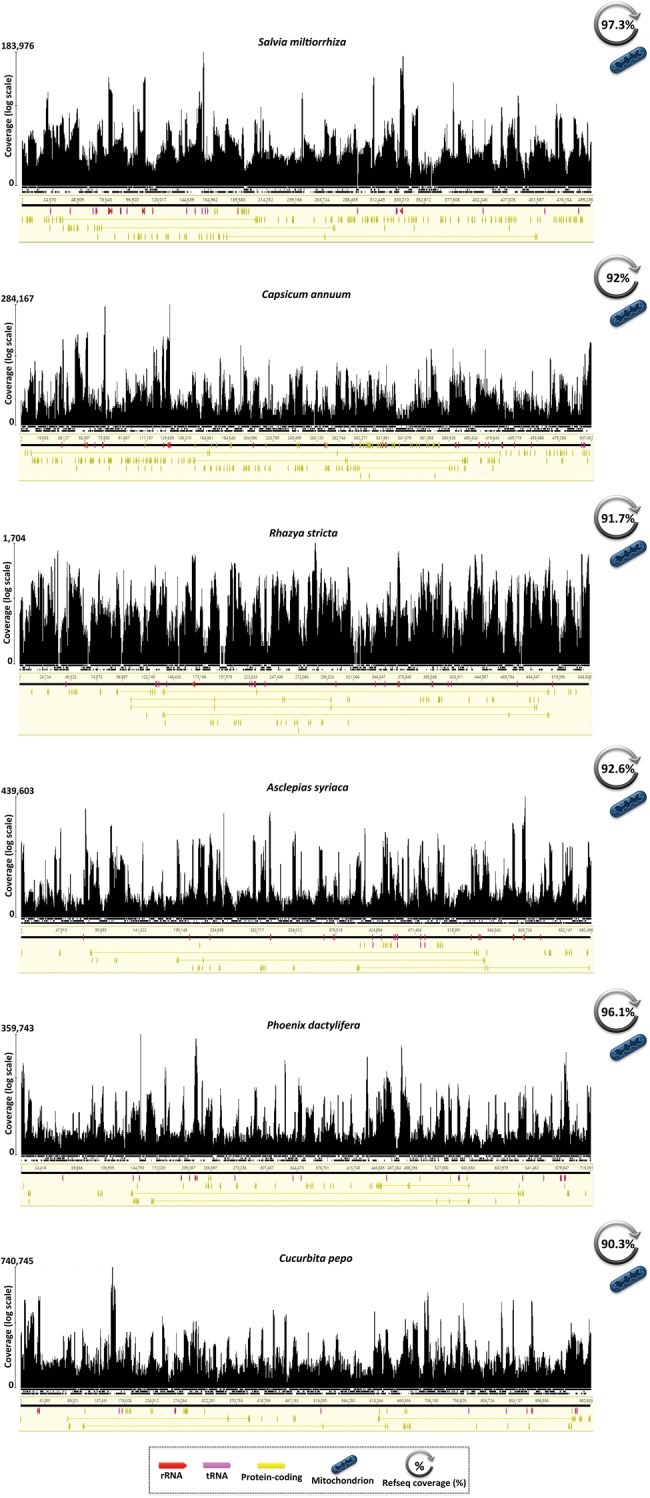
—Full transcription of bloated mitochondrial genomes in land plants. Mapping histograms show coverage depth (transcripts mapped per nucleotide) on a log scale. Organelle genome annotations are from genome assemblies deposited in GenBank (accession numbers provided in supplementary table S1, Supplementary Material online). Mapping contigs are not to scale and direction of transcription is given by the arrows of the annotated genes. Mapping histograms were extracted from Geneious v9.1.6 (Kearse etal. 2012).
In fact, pervasive transcription of mitochondrial and plastid genomes appears to be the norm rather than the exception across plastid-bearing species as a whole. We found that it was common throughout the Archaeplastida, including in land plants, green algae, red algae, and glaucophytes, as well as in species with eukaryote-eukaryote derived plastids. Complete or nearly complete transcription is also found in organisms coming from very different habitats and ecosystems, such as deserts (e.g., Welwitschia mirabilis), irrigated cultures (e.g., Zea mays and Glycine max), freshwater (e.g., Tetradesmus obliquus), and seawater (e.g., Pyropia spp.).
Among the most impressive examples of pervasive organelle transcription comes from the mtDNA of the dinoflagellate alga Symbiodinium minutum, a coral symbiont (Coffroth and Santos 2005). This ∼326 kb genome is made up of >99% noncoding DNA, all of which appears to be transcriptionally active (fig. 1 and supplementary table S1 and fig. S1, Supplementary Material online). This result is consistent with a previous report of full mitochondrial transcription of the S. minutum mitochondrial genome using a different data set (Shoguchi etal. 2015). We also observed full transcription in the nucleomorph genomes of Cryptomonas paramecium and Hemiselmis andersenii (fig. 3).
Fig. 3.
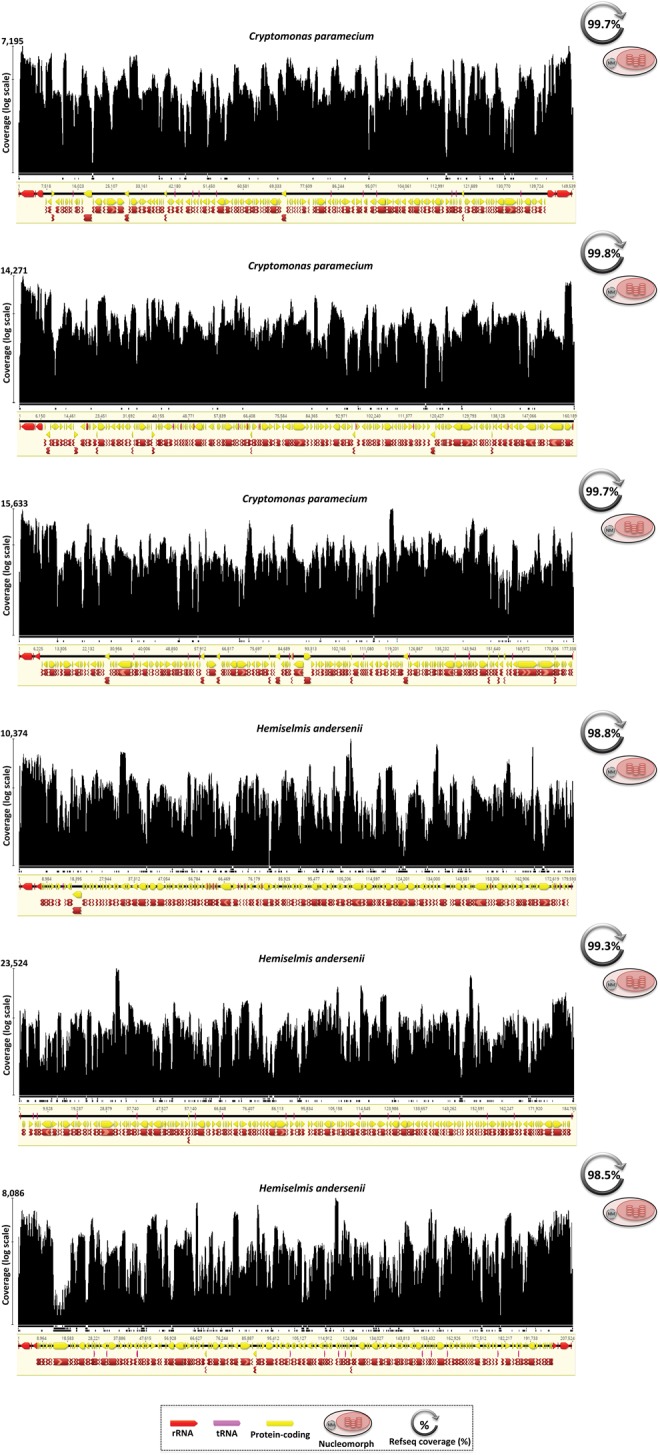
—Full transcription of nucleomorph genomes in cryptophytes. Cryptomonas paramecium and Hemiselmis andersenii had full transcription for each one of the three chromosomes in their nucleomorph genomes, including telomeric regions. Mapping histograms follow the same structure as in figure 2; mapping contigs are not to scale.
Discussion
Our RNA mapping analyses provide various insights into organelle transcription and how it can be investigated using publicly available RNA-seq data. First, the size of the RNA-seq data sets we employed did not always positively correlate with the overall organelle genome read coverage (supplementary table S1, Supplementary Material online). This was to be expected given that the RNA-seq data we used came from different experiments and laboratory groups and were produced under varying conditions and sequencing protocols. Poly-A selection, for example, can lead to an enrichment in highly AT-rich organelle transcripts, and in some lineages, including land plants, organelle polyadenylation is a target for transcript degradation (Small etal. 2013). But we quickly overcame any issues associated with biased or underrepresented organelle reads by combining multiple RNA-seq data sets from different experiments (supplementary table S1, Supplementary Material Online).
We also found differences in the RNA-seq coverage statistics for plastid and mitochondrial genomes. For the species which we had complete sequence data for both the mitochondrial and plastid genomes, the latter tended to have higher overall and mean coverage rates than the former. This could be connected to transcript abundance or genome copy number of plastids versus mitochondria, or perhaps the half-life of mitochondrial transcripts is shorter than that of plastid RNAs, or merely that mitochondria are responding to the experimental treatments differently than plastids.
In some instances, organelle genome intergenic regions were not completely represented in the RNA-seq data (i.e., RefSeq coverage <100%). This is possibly a consequence of post-transcriptional processing resulting in the cleavage of those regions, thus, preventing them from being captured in the transcriptomic sequencing experiment. But even when considering these few missing regions, there is no denying that organelle genomes typically go full transcription no matter their structure, size, content, or taxonomic grouping.
Many of the genomes we analyzed undergo minor to moderate amounts of substitutional RNA editing (Shoguchi etal. 2015; Shi etal. 2016). We did not set out to specifically study post-transcriptional editing, but we were able to easily identify edited sites from our mapping analyses, reinforcing the utility of freely available RNA-seq for quantifying and categorizing RNA editing in organelle systems (Smith 2013; Moreira etal. 2016; Shi etal. 2016). Micro-RNA (miRNA) analyses were also beyond the scope of our work, but nevertheless we covered 4.5% of the Citrullus lanatus (watermelon) mitochondrial genome using only a few micro-RNA NGS data sets (data not shown). Telomeric RNA can be studied using RNA-seq: we found widespread telomeric transcription of the nucleomorph genomes from C. paramecium and H. andersenii, which is in line with previous work on the mitochondrial telomeres of Polytomella spp. (Tian and Smith 2016) and apicomplexan parasites (Raabe etal. 2010). The significance of organelle telomeric transcription is not known, but in the nuclei of humans, mice, yeast, and zebrafish, telomeres can be transcribed into regulatory long ncRNAs called TERRA (telomeric repeat-containing RNA; Arora etal. 2012; Maicher etal. 2012; Cusanelli and Chartrand 2015).
The utility of RNA-seq for scrutinizing organelle gene expression has its limitations and drawbacks. For example, nuclear mitochondrial-like and nuclear plastid-like DNA (NUMTs and NUPTs)—and even mitochondrial plastid-like DNA (MTPTs)—could be mistaken as bona fide organelle genome sequences in RNA-seq mapping experiments, and this is of particular concern for species with multiple mitochondria and/or plastids per cell (Smith 2011; Smith etal. 2011). Another downside to the approach used here is contamination. Genomic DNA (local or foreign) can persist in RNA-seq libraries even after treatments to eliminate it (Haas etal. 2012), but this is an issue affecting all types of RNA-seq analyses and not just those focusing on organelle transcription. Even RNA-seq data derived from isolated organelles can have contamination: we were able to recover ∼97% of the Euglena gracilis plastid genome with RNA-seq data sets produced from isolated mitochondria (supplementary table S1 and fig. S1, Supplementary Material online). Clearly, plastids and plastid RNA passed through the isolation protocol.
Although accepting the shortcomings of RNA-seq, the mapping data presented here do support the idea that organelle genomes are pervasively transcribed in a wide array of species. Again, this is not the first report of genome-wide organelle transcription. More than 25 years ago, Finnegan and Brown (1990) characterized the transcription of noncoding DNA in maize mitochondria. More recently, organelle ncRNAs have been described from animals and plants, some of which are candidates for gene regulation (Hotto etal. 2012; Ro etal. 2013; Ross etal. 2016). And every month brings more and more examples of complete organelle genome transcription from disparate groups throughout the eukaryotic tree of life, but the functional relevance of this is poorly understood (Vendramin etal. 2017). Similar trends are emerging from studies of nuclear genomes, where accounts of pervasive transcription are widespread, so much so that the expressions “noncoding RNA revolution” and “eukaryotic genome as an RNA machine” are now commonplace (Amaral etal. 2008; Cech and Steitz 2014). However, there are ongoing and heated debates about whether noncoding RNAs are functional (Ponjavic etal. 2007; Struhl 2007; Doolittle 2013). No matter where you stand on the debate, there is no denying that at least some noncoding RNAs are functional and participate in major biological process (Louro etal. 2009; Cabili etal. 2011; Esteller 2011), from synaptic plasticity (Smalheiser 2014) to cancer development (Fang and Fullwood 2016).
Given the prevalence of pervasive transcription, many are questioning/exploring its evolutionary origins (Ulitsky 2016). Pervasive genome-wide transcription is standard fare for bacteria, including alphaproteobacteria and cyanobacteria (Landt etal. 2008; Georg etal. 2009; Schlüter etal. 2010; Mitschke etal. 2011a, 2011b; Voigt etal. 2014). Therefore, its widespread occurrence in organelles is arguably an ancestral trait (Shi etal. 2016). But the prevalence of full genome transcription in organelles is made more impressive by the fact that it can occur in systems with massive noncoding DNA contents (>90%), much larger than those of most bacteria. Could some of this noncoding organelle RNA have a regulatory role? And, if so, do large and bloated organelle genomes have more regulatory RNAs than their smaller, more compact counterparts?
Recent data have supported the hypothesis that ncRNAs (both long and short) carry out crucial functions within mitochondria and plastids (Vendramin etal. 2017). For example, mitochondria can produce miRNAs (Smalheiser etal. 2011) and act as a reservoir for nuclear-encoded ones (Bandiera etal. 2011), which can respond to environmental cues and regulate both cytosolic and organelle transcription (Duarte etal. 2014). Likewise, nuclear long noncoding RNAs appear to mediate crosstalk between the nucleus and mitochondrion (Vendramin etal. 2017). The nature and function of plastid and nuclear-encoded plastid-targeted noncoding RNAs are poorly understood (Zhelyazkova etal. 2012), but likely perform similar roles to those in the mitochondrion. That ncRNAs can move between organelles raises interesting questions about the transport machinery mediating this movement, most of which remain a mystery (Dietrich etal. 2015; Vendramin etal. 2017). The transport of RNA is even more complicated in the case of complex plastids (Keeling 2013), cyanelles (Steiner and Löffelhardt 2002), and nucleomorphs (Moore and Archibald 2009).
Pervasive organelle transcription might also be involved in plastid development (and its putative link to land plant terrestrialization) as well as in trophic mode determination in mixotrophs. Plastid-specific traits, such as high-light tolerance and ptDNA architectural features, might have had a fundamental role in the evolutionary transition from water to land (de Vries etal. 2016). If true, variation in the number and types of ncRNA could have helped shape and regulate the characteristics that allowed for the terrestrialization of land plants. Land plants, for example, have an array of plastids (e.g., proplastids, chloroplasts, chromoplasts, and amiloplasts; Jarvis and López-Juez 2013), which could likely be generated and regulated in part by ncRNAs. Similar arguments can be made for the evolution of mixotrophic algae, which can switch between heterotrophy and photoautotrophy (Jassey etal. 2015). Although speculative, the mechanisms for trophic mode determination could be partly controlled by organelle (or nuclear) ncRNAs generated via pervasive transcription. It would be interesting to explore the hypothesis that organelle genome size variation (together with organelle number) played a role in the evolution of mixotrophy. After all, noncoding sequences can be used as the raw material for generating new regulatory pathways (Libri 2015).
Although not the first account of pervasive organelle transcription, this is the first report to show such widespread occurrence of this phenomenon. Most of the data used in our work came from whole-cell RNA-seq experiments in which the organelle reads were ignored. That we could use these data to assemble complete or near-complete organelle transcriptomes highlights the value of publicly available RNA-seq experiments (and the SRA) for organelle research. This work also emphasizes the ease at which one can assemble a complete organelle genome from RNA-seq data alone. A quick scan through the SRA reveals many species for which there are whole-cell RNA-seq data but no or minimal organelle DNA sequence data (Smith and Sanitá Lima 2016). Some of these species are poorly studied marine protists of great ecological importance, which had their transcriptomes sequenced as part of the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP; Keeling etal. 2014). As a proof of concept, 14 land plant plastid genomes were recently de novo assembled from transcriptomic data coming from SRA (Shi et al 2016). Clearly, publicly available whole-cell RNA-seq data are a goldmine for organelle genomics and transcriptomics (Smith 2013). We just need to start digging.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was supported by a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada to D.R.S.
Literature Cited
- Amaral PP, Dinger ME, Mercer TR, Mattick JS.. 2008. The eukaryotic genome as an RNA machine. Science 319(5871): 1787–1789. [DOI] [PubMed] [Google Scholar]
- Arora R, Brun CM, Azzalin CM.. 2012. Transcription regulates telomere dynamics in human cancer cells. RNA 18(4): 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandiera S, et al. 2011. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One 6(6): e20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbrook AC, Howe CJ, Kurniawan DP, Tarr SJ.. 2010. Organization and expression of organelle genomes. Philos Trans R Soc Lond B Biol Sci. 365(1541): 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendich AJ. 2007. The size and form of chromosomes are constant in the nucleus, but highly variable in bacteria, mitochondria and chloroplasts. BioEssays 29(5): 474–483. [DOI] [PubMed] [Google Scholar]
- Berretta J, Morillon A.. 2009. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 10(9): 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger G, Gray MW, Lang BF.. 2003. Mitochondrial genomes: anything goes. Trends Genet. 19(12): 709–716. [DOI] [PubMed] [Google Scholar]
- Burki F. 2014. The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb Perspect Biol. 6(5): a016147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabili MN, et al. 2011. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25(18): 1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castandet B, Araya A.. 2011. RNA editing in plant organelles. Why make it easy?. Biochemistry 76(8): 924–931. [DOI] [PubMed] [Google Scholar]
- Cech TR, Steitz JA.. 2014. The noncoding RNA revolution—trashing old rules to forge new ones. Cell 157(1): 77–94. [DOI] [PubMed] [Google Scholar]
- Coffroth MA, Santos SR.. 2005. Genetic diversity of symbiotic dinoflagellates in the genus Symbiodinium. Protist 156(1): 19–34. [DOI] [PubMed] [Google Scholar]
- Cusanelli E, Chartrand P.. 2015. Telomeric repeat-containing RNA TERRA: a noncoding RNA connecting telome biology to genome integrity. Front Genet. 6: 143.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries J, Stanton A, Archibald JM, Gould SB.. 2016. Streptophyte terrestrialization in light of plastid evolution. Trends Plant Sci. 21(6): 467–476. [DOI] [PubMed] [Google Scholar]
- Dietrich A, Wallet C, Iqbal RK, Gualberto JM, Lotfi F.. 2015. Organellar non-coding RNAs: emerging regulation mechanisms. Biochimie 117: 48–62. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. 2013. Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci U S A. 110(14): 5294–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte FV, Palmeira CM, Rolo AP.. 2014. The role of microRNAs in mitochondria: small players acting wide. Genes 5(4): 865–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12(12): 861–874. [DOI] [PubMed] [Google Scholar]
- Fang Y, Fullwood MJ.. 2016. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genomics Proteomics Bioinformatics 14(1): 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan PM, Brown GG.. 1990. Transcriptional and post-transcriptional regulation of RNA levels in maize mitochondria. Plant Cell 2(1): 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georg J, et al. 2009. Evidence for a major role of antisense RNAs in cyanobacterial gene regulation. Mol Syst Biol. 5: 305.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Chin M, Nusbaum C, Birren BW, Livny J.. 2012. How deep is deep enough for RNA-Seq profiling of bacterial transcriptomes?. BMC Genomics 13: 734.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotto AM, Germain A, Stern DB.. 2012. Plastid non-coding RNAs: emerging candidates for gene regulation. Trends Plant Sci. 17(12): 737–744. [DOI] [PubMed] [Google Scholar]
- Janouškovec J, et al. 2013. Evolution of red algal plastid genomes: ancient architectures, introns, horizontal gene transfer, and taxonomic utility of plastid markers. PLoS One 8(3): e59001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis P, López-Juez E.. 2013. Biogenesis and homeostasis of chloroplasts and other plastids. Nat Rev Mol Cell Biol. 14(12): 787–802. [DOI] [PubMed] [Google Scholar]
- Jassey VEJ, et al. 2015. An unexpected role for mixotrophs in the response of peatland carbon cycling to climate warming. Sci Rep. 5: 16931.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12): 1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, et al. 2014. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12(6): e1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ. 2013. The number, speed, and impact of plastid endosymbioses in eukaryotic evolution. Annu Rev Plant Biol. 64: 583–607. [DOI] [PubMed] [Google Scholar]
- Kodama Y, Shumway M, Leinonen R.. 2012. The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res. 40(D1): D54–D56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, et al. 2008. Small non-coding RNAs in Caulobacter crescentus. Mol Microbiol. 68(3): 600–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, et al. 2014. Massive programmed translational jumping in mitochondria. Proc Natl Acad Sci U S A. 111(16): 5926–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL.. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri D. 2015. Sleeping beauty and the beast (of pervasive transcription). RNA 21(4): 678–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louro R, Smirnova AS, Verjovski-Almeida S.. 2009. Long intronic noncoding RNA transcription: expression noise or expression choice?. Genomics 93(4): 291–298. [DOI] [PubMed] [Google Scholar]
- Maicher A, Kastner L, Dees M, Luke B.. 2012. Deregulated telomere transcription causes replication-dependent telomere shortening and promotes cellular senescence. Nucleic Acids Res. 40(14): 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda I, Matsuzaki M, Kita K.. 2010. Extensive framshift at all AGG and CCC codons in the mitochondrial cytochrome c oxidase subunit 1 gene of Perkinsus marinus (Alveolata; Dinoflagellata). Nucleic Acids Res. 38(18): 6186–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, et al. 2011. The human mitochondrial transcriptome. Cell 146(4): 645–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke J, et al. 2011a. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A. 108: 2124–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitschke J, Vioque A, Haas F, Hess WR, Muro-Pastor AM.. 2011b. Dynamics of transcriptional start site selection during nitrogen stress-induced cell differentiation in Anabaena sp. PCC7120. Proc Natl Acad Sci U S A. 108: 20130–20135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CE, Archibald JM.. 2009. Nucleomorph genomes. Annu Rev Genet. 43: 251–264. [DOI] [PubMed] [Google Scholar]
- Moreira S, Valach M, Aoulad-Aissa M, Otto C, Burger G.. 2016. Novel modes of RNA editing in mitochondria. Nucleic Acids Res. 44(10): 4907–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plackett ARG, Di Stilio VS, Langdale JA.. 2015. Ferns: the missing link in shoot evolution and development. Front Plant Sci. 6: 972.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponjavic J, Ponting CP, Lunter G.. 2007. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 17(5): 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe CA, et al. 2010. A global view of the nonprotein-coding transcriptome in Plasmodium falciparum. Nucleic Acids Res. 38(2): 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner SS, Schaefer H.. 2016. Phylogeny and evolution of the Cucurbitaceae In: Grumet R, Katzir N, Garcia-Mas J, editors. Genetics and genomics of Cucurbitaceae. New York (NY: ): Springer International Publishing. [Google Scholar]
- Ro S, et al. 2013. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. 23(6): 759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross E, Blair D, Guerrero-Hernández C, Sánchez Alvarado A.. 2016. Comparative and transcriptome analyses uncover key aspects of coding- and long noncoding RNAs in flatworm mitochondrial genomes. G3 (Bethesda) 6(5): 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruwe H, Castandet B, Schmitz-Linneweber C, Stern DB.. 2013. Arabidopsis chloroplast quantitative editotype. FEBS Lett. 587(9): 1429–1433. [DOI] [PubMed] [Google Scholar]
- Sanitá Lima M, Smith DR.. 2017. Pervasive, genome-wide transcription in the organelle genomes of diverse plastid-bearing protists. G3. doi: 10.1534/g3.117.300290. [DOI] [PMC free article] [PubMed]
- Sanitá Lima M, Woods LC, Cartwright MW, Smith DR.. 2016. The (in)complete organelle genome: exploring the use and non-use of available technologies for characterizing mitochondrial and plastid chromosomes. Mol Ecol Resour. 16(6): 1279–1286. [DOI] [PubMed] [Google Scholar]
- Schlüter JP, et al. 2010. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 11: 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R, Zhu XQ, Barker SC, Herd K.. 2012. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol Evol. 4(11): 1088–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, et al. 2016. Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci Rep. 6: 30135.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoguchi E, Shinzato C, Hisata K, Satoh N, Mungpakdee S.. 2015. The large mitochondrial genome of Symbiodinium minutum reveals conserved noncoding sequences between dinoflagellates and apicomplexans. Genome Biol Evol. 7(8): 2237–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, et al. 2012. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 10(1): e1001241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Lugli G, Thimmapuram J, Cook EH, Larson J.. 2011. Mitochondrial small RNAs that are up-regulated in hippocampus during olfactory discrimination training mice. Mitochondrion 11(6): 994–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR. 2014. The RNA-centred view of the synapse: non-coding RNAs and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 369(1652): 20130504.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small ID, Rackham O, Filipovska A.. 2013. Organelle transcriptomes: products of a deconstructed genome. Curr Opin Microbiol. 16(5): 652–658. [DOI] [PubMed] [Google Scholar]
- Smith DR. 2011. Extending the limited transfer window hypothesis to inter-organelle DNA migration. Genome Biol Evol. 3: 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR. 2013. RNA-Seq data: a goldmine for organelle research. Brief Funct Genomics 12(5): 454–456. [DOI] [PubMed] [Google Scholar]
- Smith DR, Crosby K, Lee RW.. 2011. Correlation between nuclear plastid DNA abundance and plastid number supports the limited transfer window hypothesis. Genome Biol Evol. 3: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ.. 2015. Mitochondrial and plastid genomes architecture: reoccurring themes, but significant differences at the extremes. Proc Natl Acad Sci U S A. 112(33): 10177–10184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Keeling PJ.. 2016. Protists and the wild, wild west of gene expression: new frontiers, lawlessness, and misfits. Annu Rev Microbiol. 70: 161–178. [DOI] [PubMed] [Google Scholar]
- Smith DR, Sanitá Lima M.. 2016. Unraveling chloroplast transcriptomes with ChloroSeq, an organelle RNA-seq bioinformatics pipeline. Brief Bioinform. DOI: 10.1093/bib/bbw088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JM, Löffelhardt W.. 2002. Protein import into cyanelles. Trends Plant Sci. 7(2): 72–77. [DOI] [PubMed] [Google Scholar]
- Stevens PF. (2001). Angiosperm phylogeny website. Available from: http://www.mobot.org/MOBOT/Research/APweb/, last accessed July 10, 2017.
- Struhl K. 2007. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 14(2): 103–105. [DOI] [PubMed] [Google Scholar]
- Tian Y, Smith DR.. 2016. Recovering complete mitochondrial genome sequences from RNA-seq: a case study of Polytomella non-photosynthetic green algae. Mol Phylogenet Evol. 98: 57–62. [DOI] [PubMed] [Google Scholar]
- Ulitsky I. 2016. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat Rev Genet. 17(10): 601–614. [DOI] [PubMed] [Google Scholar]
- Vendramin R, Marine JC, Leucci E.. 2017. Non-coding RNAs: the dark side of nuclear-mitochondrial communication. EMBO J. 36(9): 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlcek C, Marande W, Teijeiro S, Lukeš J, Burger G.. 2011. Systematically fragmented genes in a multipartite mitochondrial genome. Nucleic Acids Res. 39(3): 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt K, et al. 2014. Comparative transcriptomics of two environmentally relevant cyanobacteria reveals unexpected transcriptome diversity. ISME J. 8(10): 2056–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JT, Grainger DC.. 2014. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat Rev Microbiol. 12(9): 647–653. [DOI] [PubMed] [Google Scholar]
- Wojciechowski MF. (2006). Millettioid sensu lato clade. Available from: http://tolweb.org/Millettioid_sensu_lato_clade/60341/2006.06.14.
- Zhelyazkova P, et al. 2012. The primary transcriptome of barley chloroplasts: numerous noncoding RNAs and the dominating role of the plastid-encoded RNA polymerase. Plant Cell 24(1): 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.