


Abstract
A current question in the high-order organization of chromatin is whether topologically associating domains (TADs) are distinct from other hierarchical chromatin domains. However, due to the unclear TAD definition in tradition, the structural and functional uniqueness of TAD is not well studied. In this work, we refined TAD definition by further constraining TADs to the optimal separation on global intra-chromosomal interactions. Inspired by this constraint, we developed a novel method, called HiTAD, to detect hierarchical TADs from Hi-C chromatin interactions. HiTAD performs well in domain sensitivity, replicate reproducibility and inter cell-type conservation. With a novel domain-based alignment proposed by us, we defined several types of hierarchical TAD changes which were not systematically studied previously, and subsequently used them to reveal that TADs and sub-TADs differed statistically in correlating chromosomal compartment, replication timing and gene transcription. Finally, our work also has the implication that the refinement of TAD definition could be achieved by only utilizing chromatin interactions, at least in part. HiTAD is freely available online.
INTRODUCTION
Recent years have seen rapid development in exploring high-order organization of chromatin due to the chromosome conformation capture (3C) technique (1) and its derivatives, such as 4C (2,3), 5C (4), ChIA-PET (5), Hi-C (6), TCC (7), Capture Hi-C (8) and in situ Hi-C (9), etc. It is now known that chromatin is neatly packed in nucleus, in which topologically associating domain (TAD) is a kind of structural unit in linking chromatin organization and biological functions, at least in drosophila (10) and mammalian genomes (11,12). It was reported that TAD could constrain enhancer-promoter targeting in gene regulation (13,14), shape replication timing (15) and determine pathogenicity of genomic duplications (16). The switch of TAD boundary was observed in mouse limb development (17), and the boundary knockout on mouse model directly proved that the disruption of TAD boundary led to development disease (18). The studies on cancer genomes also revealed that mutations occurred on TAD boundaries could contribute to oncogene activation (19,20), implying the association of TAD disruption with tumorigenesis.
TAD itself is a hierarchical organization which needs to be further clarified. TAD is traditionally defined as a continuous chromatin region in which the loci interact with each other more frequently than the loci outside the region (11,12,21). However, different levels of chromatin domains satisfy this criterion more or less, especially with the improvement of data quality and sequencing depth. By thoroughly investigating specific chromatin regions with 5C, Phillips-Cremins et al. found that there existed smaller chromatin domains (called sub-TADs) inside the traditional TADs (22). Further comparisons revealed that TADs were stable across cell types, whereas sub-TADs could vary greatly to facilitate gene regulation. By improving Hi-C experimental pipeline and sequencing depth, Rao et al. observed hierarchical overlapping among different chromatin domains in the genome-wide scale (9). A recent work also revealed that TADs exhibited structural heterogeneity and functional diversity in mammalian genomes (23). These phenomena suggest the existence of hierarchical domains in chromatin, which cannot be explained by traditional TADs.
Several methods have been proposed to identify hierarchical chromatin domains from Hi-C chromatin interactions. Rao et al. proposed an Arrowhead transformation on bias-corrected chromatin interaction matrix and then used dynamic programming to identify chromatin domains at multiple scales simultaneously (9). TADtree proposed by Weinreb and Rahpael used a weighted interval scheduling with multiplicities to find TAD forest (24). However, the high computational complexity in this algorithm limits its available resolutions. Matryoshka (bioRxiv https://doi.org/10.1101/032953) proposed by Malik and Patro identified various chromatin domains at different resolutions, and then the consensus hierarchy through domain clustering was used to generate hierarchical chromatin domains. Recently, a network modularity based method was proposed to identify hierarchical chromatin domains by utilizing different resolution parameter values (bioRxiv https://doi.org/10.1101/089011). The method CaTCH identified large levels of hierarchical chromatin domains by using only a single parameter, reciprocal insulation (25). However, it did not point out the TAD positions in the hierarchical domains. Instead, additional data, such as CCCTC-binding factor (CTCF) enrichment, were needed as references to identify the TADs. Similarly, HBM identified hierarchical domains until the given chromatin was merged into a single cluster, without pointing out the TAD positions (26). Finally, some methods, such as Armatus (27), spectral method (28), MrTADFinder (bioRxiv https://doi.org/10.1101/097345) and IC-Finder (29), could identify overlapped or hierarchical domains across different parameter values, but they did not automatically reconcile the hierarchy and consensus among these domains.
The aforementioned methods mainly treat TADs as local insulations but neglect their global properties, making it hard to judge where the TADs stand in the hierarchical domains. In this work, except the local insulations, we further constrain TADs to the optimal separation on intra-chromosomal interactions. To facilitate representation, TAD and its smaller chromatin domains are together called hierarchical TAD in this work. Inspired by our TAD constraint, we developed an iterative optimization procedure, called HiTAD, to detect hierarchical TADs from Hi-C chromatin interactions, and then applied HiTAD to analyzing Hi-C and in situ Hi-C datasets with different sequencing depths involving several human and mouse cell types (Supplementary Table S1). Compared to the selected two methods (Arrowhead and TADtree), HiTAD can detect more hierarchical TADs with higher replicate reproducibility and inter cell-type conservation. With a novel domain-based alignment strategy, we defined several change types of hierarchical TADs which were not systematically studied. Our analyses on these hierarchical TADs show that TADs and sub-TADs differ in correlating chromosomal compartment, replication timing domain and transcriptional regulation.
MATERIALS AND METHODS
Data sources, processing and representation
The in situ Hi-C datasets of human cell types GM12878, IMR90 and K562 were downloaded from NCBI with accession number GSE63525 (9). For traditional Hi-C, two independently generated datasets of human cell type GM12878 were downloaded from NCBI with accession numbers GSE48592 (30) and GSE63525 (9) respectively. The datasets of human cell type IMR90 and mouse cell types mESC and Cortex were downloaded from NCBI with accession numbers GSE43070 (14) and GSE35156 (11) respectively. The dataset of human cell type Panc1 was downloaded from ENCODE (31). Raw Hi-C data were processed and corrected by using software hiclib (32). The bins located in gap regions were removed from calculation but included in visualization. The summary of Hi-C datasets is listed in Supplementary Table S1. With respect to ChIP-Seq and RNA-Seq datasets, the processed data were downloaded from ENCODE (31), including epigenomic and binding peaks from ChIP-Seq and expression of long RNA contigs from RNA-Seq. The called domain boundaries of replication timing were downloaded from public website (http://mouseencode.org/publications/mcp05/) (33). Human CTCF motif was downloaded from a database for ENCODE transcription factors (http://compbio.mit.edu/encode-motifs/) (34), and mouse CTCF motif was scanned in Fimo (35) by using the same human PWM as input. The human and mouse reference genomes hg19 and mm10 were used in sequence alignments.
HiTAD overview
The idea behind HiTAD is that TADs are optimal domains to separate intra-chromosomal interactions in global level. Combining the fact that TADs can also be divided into smaller domains in a hierarchical way, the detection of hierarchical TADs can be transformed into an iterative optimization procedure by defining appropriate objective functions from interaction frequencies (Figure 1). In this work, the objective function is defined as the enrichment between intra-domain interaction frequencies and inter-domain interaction frequencies in a way to reduce the impact of genomic distance. To speed up the calculation, an adaptive directionality index (DI)-based Hidden Markov Model (HMM) is proposed to sensitively generate a genome-wide pool of bottom domains by using only local insulation. Then these bottom domains are used as basic elements to detect TADs by using global intra-chromosomal interactions under given objective function. To better perform TAD detection, a recursive formula is used to solve the optimization problem. These detected TADs are next used to generate corresponding sub-TADs in a similar way, but with the bottom domains localized within the TAD as initial domain pools. Similar procedure is applied to subsequent-level domain detection until bottom domains are met. Finally, under a domain-based alignment proposed by us, the reproducible domains from two replicates are maintained to guarantee the accuracy in the highly variable Hi-C chromatin interactions. HiTAD is a fast and memory-saving method that can be implemented in PC (Supplementary Table S2). Next, we will introduce the detailed implementation of HiTAD.
Figure 1.

HiTAD workflow. First, the bottom domains are detected by using adaptive DI-based HMM. Second, under the objective function derived from chromatin interactions, a recursive formula is applied to searching all possible TADs by using bottom domains as input. Third, TADs are generated by maximizing the objective function from the searching space. Fourth, sub-TADs and other level domains are generated in a similar way but with localized bottom domains as input to optimization problem. Finally, the hierarchical domains reproducible from two replicates are maintained. The calculations were performed at 20 kb resolution on in situ Hi-C dataset IMR90.
Bottom boundaries detected by adaptive directionality index
To sensitively detect the boundaries with various domain sizes, we proposed adaptive DI by modifying traditional DI (11,22). Let  represent the matrix of chromatin interaction frequencies after bias correction, where
represent the matrix of chromatin interaction frequencies after bias correction, where 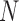 is the number of bins at the given resolution. For every selected bin
is the number of bins at the given resolution. For every selected bin  , the adaptive DI is defined as:
, the adaptive DI is defined as:
 |
(1) |
where 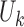 denotes the upstream interaction frequency between bin
denotes the upstream interaction frequency between bin  and bin
and bin 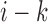 ,
, 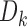 denotes the corresponding downstream interaction frequency and
denotes the corresponding downstream interaction frequency and  is the window size on bin
is the window size on bin  . Since domain sizes vary from tens of kilobases to several megabases at currently available resolutions, the window size
. Since domain sizes vary from tens of kilobases to several megabases at currently available resolutions, the window size 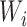 is determined adaptively based on local interaction environment (Supplementary Figure S1). Specifically, let
is determined adaptively based on local interaction environment (Supplementary Figure S1). Specifically, let  denote the interaction bias when comparing upstream bin
denote the interaction bias when comparing upstream bin  to downstream bin
to downstream bin 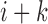 , in which 1 represents upstream bias and 0 represents downstream bias. Then four kinds of state transitions from
, in which 1 represents upstream bias and 0 represents downstream bias. Then four kinds of state transitions from  to
to  can be observed (
can be observed (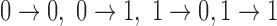 ). Generally, the state transitions should be statistically same from
). Generally, the state transitions should be statistically same from 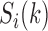 to
to 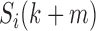 if bin
if bin 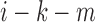 or bin
or bin 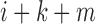 is located in the same domain with bin
is located in the same domain with bin 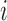 . Let
. Let 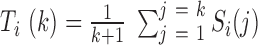 , and then the maximum or minimum values of
, and then the maximum or minimum values of  are selected as candidate window sizes. Since
are selected as candidate window sizes. Since 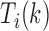 is discrete, the maximum or minimum is determined locally by comparing its 10 nearest neighbor values, i.e. five neighbors in each side. To further guarantee the best selection on window size, a chi-square statistics is constructed as
is discrete, the maximum or minimum is determined locally by comparing its 10 nearest neighbor values, i.e. five neighbors in each side. To further guarantee the best selection on window size, a chi-square statistics is constructed as  , where
, where 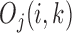 is the observed frequency in one of the four kinds of transitions,
is the observed frequency in one of the four kinds of transitions, 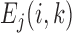 is the expected frequency which is set to be
is the expected frequency which is set to be 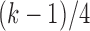 and
and  represents the candidate window sizes selected above. Then the minimum value of
represents the candidate window sizes selected above. Then the minimum value of 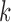 satisfying
satisfying 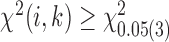 is selected as the final window size
is selected as the final window size 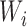 . Next, the calculated adaptive DIs from Equation 1 are used as input in HMM to detect bottom boundaries. Five states (start, upstream bias, no bias, downstream bias and end) and corresponding state transitions are set in HMM (Supplementary Table S3). Three-distribution Gaussian mixture is used to emit state and Baum–Welch algorithm is used to perform training on data. The detected boundaries are reused to further improve sensitivity through following procedure. Let
. Next, the calculated adaptive DIs from Equation 1 are used as input in HMM to detect bottom boundaries. Five states (start, upstream bias, no bias, downstream bias and end) and corresponding state transitions are set in HMM (Supplementary Table S3). Three-distribution Gaussian mixture is used to emit state and Baum–Welch algorithm is used to perform training on data. The detected boundaries are reused to further improve sensitivity through following procedure. Let 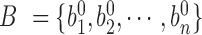 denote the initial boundaries from the adaptive DI-based HMM, where
denote the initial boundaries from the adaptive DI-based HMM, where  is the genomic position of the
is the genomic position of the  boundary and
boundary and  is the total boundary number. Let
is the total boundary number. Let 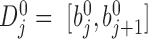 denote the domain where bin
denote the domain where bin 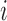 localizes. The new window size of bin
localizes. The new window size of bin 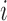 is set to
is set to 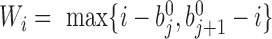 , and the corresponding adaptive DI is recalculated by using Equation 1. All recalculated adaptive DIs are used as input in HMM to detect new bottom boundaries and bottom domains. This recalculation is repeated until more than 95% boundaries detected from two neighbor steps can be aligned to each other (see ‘Domain and boundary alignment’ section). Generally, the convergence is achieved by only one to three iterations. We also performed comparison between adaptive DI-based HMM and traditional DI-based HMM. Since traditional DI depends on the fixed window sizes, different values were selected for thorough comparisons. The results show that most boundaries from traditional DI-based HMM can be detected by adaptive DI-based HMM. By contrast, many boundaries from adaptive DI-based HMM cannot be detected by traditional one (Supplementary Figure S2 and Table S4). The major reason underlying this difference is that traditional DI cannot reconcile the various domain sizes on boundary detection by using only one fixed window size.
, and the corresponding adaptive DI is recalculated by using Equation 1. All recalculated adaptive DIs are used as input in HMM to detect new bottom boundaries and bottom domains. This recalculation is repeated until more than 95% boundaries detected from two neighbor steps can be aligned to each other (see ‘Domain and boundary alignment’ section). Generally, the convergence is achieved by only one to three iterations. We also performed comparison between adaptive DI-based HMM and traditional DI-based HMM. Since traditional DI depends on the fixed window sizes, different values were selected for thorough comparisons. The results show that most boundaries from traditional DI-based HMM can be detected by adaptive DI-based HMM. By contrast, many boundaries from adaptive DI-based HMM cannot be detected by traditional one (Supplementary Figure S2 and Table S4). The major reason underlying this difference is that traditional DI cannot reconcile the various domain sizes on boundary detection by using only one fixed window size.
Hierarchical TAD detection
The finally generated boundary set above is denoted as 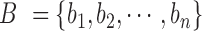 . The TAD identification is transformed into selecting a best subset
. The TAD identification is transformed into selecting a best subset 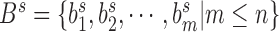 from set
from set  to generate corresponding TAD set
to generate corresponding TAD set  . To eliminate the impact of genomic distance on interaction frequency, the arrowhead transformation proposed by Rao et al. (9) is used to measure the interaction-frequency difference between intra-domain interactions and inter-domain interactions:
. To eliminate the impact of genomic distance on interaction frequency, the arrowhead transformation proposed by Rao et al. (9) is used to measure the interaction-frequency difference between intra-domain interactions and inter-domain interactions:
 |
where bin 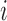 is the genomic position and
is the genomic position and  is the genomic distance from bin
is the genomic distance from bin  . Under this calculation, the square region of TAD
. Under this calculation, the square region of TAD 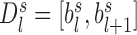 should be separated to form an upper-triangle region
should be separated to form an upper-triangle region  and a lower-triangle region
and a lower-triangle region 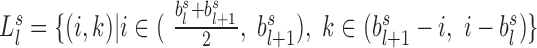 , where
, where 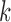 is the genomic distance dependent on bin
is the genomic distance dependent on bin 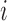 (Supplementary Figure S3). The objective function is defined as:
(Supplementary Figure S3). The objective function is defined as:
 |
(2) |
where 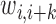 and
and 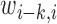 are the weights on the interaction-frequency differences and
are the weights on the interaction-frequency differences and 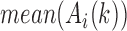 represents the average of
represents the average of 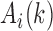 in the corresponding region. Since
in the corresponding region. Since 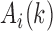 in the upper-triangle region represents the difference between upstream inter-domain interaction and intra-domain interaction, it tends to be negative in the TAD region. By contrast,
in the upper-triangle region represents the difference between upstream inter-domain interaction and intra-domain interaction, it tends to be negative in the TAD region. By contrast, 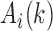 in the lower-triangle region represents the difference between intra-domain interaction and downstream inter-domain interaction, and tends to be positive. Then the values in the upper-triangle region are set to
in the lower-triangle region represents the difference between intra-domain interaction and downstream inter-domain interaction, and tends to be positive. Then the values in the upper-triangle region are set to 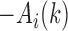 .
.
The weight in Equation 2 is set to fold change between the observed and expected interaction frequencies, in which a previous procedure is used to calculate the expected interaction frequency by considering both genomic distance and local interaction background (23). Briefly, let 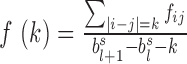 denote the average interaction frequency at genomic distance
denote the average interaction frequency at genomic distance  in TAD
in TAD 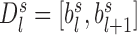 , and then the smoothed interaction frequency
, and then the smoothed interaction frequency 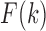 is obtained by using B-Spline approximation. Since the number of chromatin interactions decreases with the genomic distance
is obtained by using B-Spline approximation. Since the number of chromatin interactions decreases with the genomic distance 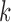 ,
,  may fluctuate when
may fluctuate when  increases gradually. To alleviate the impact of these fluctuations, the expected interaction frequency is defined as
increases gradually. To alleviate the impact of these fluctuations, the expected interaction frequency is defined as 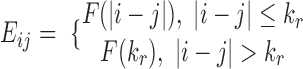 , where
, where  is the first turning point. The final expected interaction frequency at position
is the first turning point. The final expected interaction frequency at position 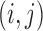 is calculated by following a previous window strategy to take local interaction background into consideration (9):
is calculated by following a previous window strategy to take local interaction background into consideration (9):
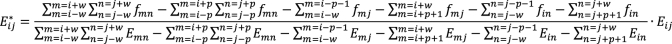
where 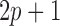 and
and 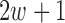 are the two square-window sizes centered at
are the two square-window sizes centered at 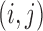 with
with 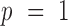 and
and 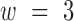 . Then the corresponding weight is defined as
. Then the corresponding weight is defined as  . Finally, all weights in the TAD
. Finally, all weights in the TAD  are normalized by using a piecewise function:
are normalized by using a piecewise function:
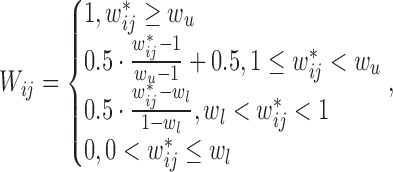 |
(3) |
where,  and
and 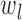 are the 99 percentile and 1 percentile respectively to reduce the impact of outliers. If
are the 99 percentile and 1 percentile respectively to reduce the impact of outliers. If 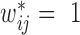 in Equation 3, the corresponding weight is set to 0.5. The same value (0.5) is set to the chromatin interactions within bottom domains to avoid their over impacts on objective function since
in Equation 3, the corresponding weight is set to 0.5. The same value (0.5) is set to the chromatin interactions within bottom domains to avoid their over impacts on objective function since  in bottom domains are generally large due to very high interaction frequencies. The chromatin interactions with
in bottom domains are generally large due to very high interaction frequencies. The chromatin interactions with  are excluded from weight calculation and objective function.
are excluded from weight calculation and objective function.
Under the objective function defined in Equation 2, TAD detection is transformed into searching the solution with maximum value. Then a recursive formula is used to solve this optimization problem. Specifically, for the first bottom domain 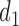 , the objective function is calculated by:
, the objective function is calculated by:
 |
where  represents the set of all kinds of TAD selections on domain
represents the set of all kinds of TAD selections on domain 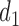 in the first step, and
in the first step, and 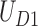 and
and  are the corresponding upper-triangle and lower-triangle regions defined previously. Actually, the start domain
are the corresponding upper-triangle and lower-triangle regions defined previously. Actually, the start domain  can only be an independent TAD or merged with consecutive downstream bottom domains (Supplementary Figure S4a). The set of TAD selections on domain
can only be an independent TAD or merged with consecutive downstream bottom domains (Supplementary Figure S4a). The set of TAD selections on domain  in the initial step is used in next step. In step
in the initial step is used in next step. In step 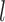 on domain
on domain 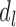 , the objective function is calculated:
, the objective function is calculated:
 |
where  and
and 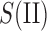 represent two subsets for domain
represent two subsets for domain 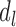 . In subset
. In subset  , domain
, domain 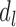 is independent on the candidate TADs generated in previous steps, whereas in subset
is independent on the candidate TADs generated in previous steps, whereas in subset  ,
, 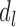 has already been contained in previous steps and should be re-evaluated (Supplementary Figure S4b). The ultimate goal is to find the boundary subset
has already been contained in previous steps and should be re-evaluated (Supplementary Figure S4b). The ultimate goal is to find the boundary subset 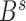 to maximize objective function
to maximize objective function 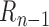 , which can be written as
, which can be written as 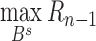 . To fasten the calculation, the maximum TAD size is limited to 4 Mb in the aforementioned search procedure in this work. However, different selections on TAD-size limitation only slightly influence the accuracy of TAD detection (Supplementary Table S5).
. To fasten the calculation, the maximum TAD size is limited to 4 Mb in the aforementioned search procedure in this work. However, different selections on TAD-size limitation only slightly influence the accuracy of TAD detection (Supplementary Table S5).
The sub-TADs in each TAD 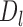 are detected by using the same procedure as TAD detection except that the input boundary set is replaced by the boundary subset
are detected by using the same procedure as TAD detection except that the input boundary set is replaced by the boundary subset 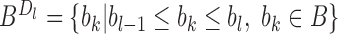 , where
, where  and
and  are the upstream and downstream boundaries of TAD
are the upstream and downstream boundaries of TAD  respectively. The subsequent level domains in each sub-TAD are detected in the similar way until bottom domains are met. Finally, the TADs and other domains reproducible from two replicates are maintained to form hierarchical TADs. The reproducibility is calculated by using following alignment strategy.
respectively. The subsequent level domains in each sub-TAD are detected in the similar way until bottom domains are met. Finally, the TADs and other domains reproducible from two replicates are maintained to form hierarchical TADs. The reproducibility is calculated by using following alignment strategy.
Domain and boundary alignment
Traditionally, domain and boundary are aligned by matching boundaries with nearest genomic positions between two Hi-C datasets. This strategy generally assigns a threshold in advance, but neglects the usage of domain themselves. To facilitate the domain and boundary matching for hierarchical TADs, a domain-based alignment strategy is proposed in this work by considering all domains in the same time.
Specifically, let  and
and  denote two boundary sets. For chromatin region
denote two boundary sets. For chromatin region 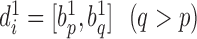 in set
in set  and chromatin region
and chromatin region 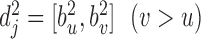 in set
in set 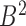 , let
, let 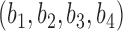 represent the ascending order of corresponding genomic positions
represent the ascending order of corresponding genomic positions  . The overlap ratio between these two regions is defined as:
. The overlap ratio between these two regions is defined as:
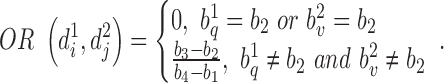 |
Correspondingly, for region 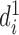 in set
in set 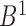 , the best mapping in set
, the best mapping in set 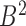 is defined as
is defined as 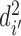 satisfying
satisfying 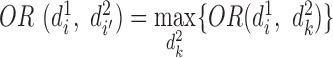 , where
, where 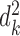 represents any chromatin region in set
represents any chromatin region in set 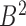 . This directional mapping is denoted as
. This directional mapping is denoted as 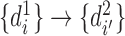 . The reverse mapping
. The reverse mapping 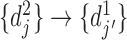 from set
from set 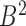 to set
to set 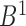 can be defined in the same way. If
can be defined in the same way. If 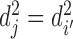 and
and 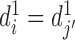 , then chromatin regions
, then chromatin regions  and
and  are defined as bidirectional mapping, i.e.
are defined as bidirectional mapping, i.e.  . When performing alignment, the selected bottom domain in one boundary set can be mapped to a chromatin region composed of several consecutive bottom domains in another boundary set. By integrating the two directional mappings, the consecutive bottom domains in the same boundary set are combined to form the combinatorial bidirectional mapping. As shown in Supplementary Figure S5, there are three bottom domains
. When performing alignment, the selected bottom domain in one boundary set can be mapped to a chromatin region composed of several consecutive bottom domains in another boundary set. By integrating the two directional mappings, the consecutive bottom domains in the same boundary set are combined to form the combinatorial bidirectional mapping. As shown in Supplementary Figure S5, there are three bottom domains  and
and 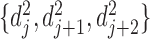 in boundary set
in boundary set 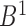 and set
and set  respectively. According to the mapping definition, the domain mapping from set
respectively. According to the mapping definition, the domain mapping from set 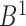 to set
to set 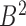 is
is 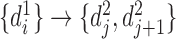 ,
, 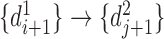 and
and 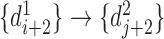 , and the reverse mapping is
, and the reverse mapping is 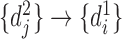 ,
, 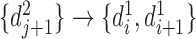 and
and 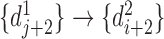 . Combing these two directional mappings yields the combinatorial bidirectional mapping:
. Combing these two directional mappings yields the combinatorial bidirectional mapping:  and
and 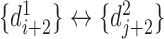 .
.
The hierarchical TAD alignment is based on the mapping strategy defined above. First, bidirectional mapping is performed for all bottom domains in two boundary sets  and
and 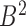 , including the combinatorial bidirectional mapping. Second, for each chromosome, starting from the first TAD in the domain set
, including the combinatorial bidirectional mapping. Second, for each chromosome, starting from the first TAD in the domain set 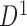 generated from boundary set
generated from boundary set 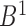 , the bottom domains of current TAD are extracted and bidirectional mapping is searched in the other domain set
, the bottom domains of current TAD are extracted and bidirectional mapping is searched in the other domain set 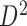 generated from boundary set
generated from boundary set  . If not all bottom domains are bidirectionally mapped, the TAD and its downstream TAD are merged to search bidirectional mapping again. This TAD merging and mapping iteration is repeated until bidirectional mapping is achieved. Then the hierarchical levels of mapped bottom domains in domain set
. If not all bottom domains are bidirectionally mapped, the TAD and its downstream TAD are merged to search bidirectional mapping again. This TAD merging and mapping iteration is repeated until bidirectional mapping is achieved. Then the hierarchical levels of mapped bottom domains in domain set 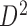 are extracted and recorded for corresponding TADs in domain set
are extracted and recorded for corresponding TADs in domain set 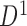 . All level sub-domains in current TAD or merged TADs are extracted and the same mapping procedure is performed. Third, the next TAD without performing mapping is selected as a new start to repeat above procedure until all TADs in the selected chromosome undergo hierarchical mapping, including all level sub-domains. Fourth, the next chromosome is selected to perform the same hierarchical mapping until all chromosomes are done.
. All level sub-domains in current TAD or merged TADs are extracted and the same mapping procedure is performed. Third, the next TAD without performing mapping is selected as a new start to repeat above procedure until all TADs in the selected chromosome undergo hierarchical mapping, including all level sub-domains. Fourth, the next chromosome is selected to perform the same hierarchical mapping until all chromosomes are done.
Other calculations in HiTAD analysis
In method comparison, the hierarchical TAD detection in TADtree is performed by using recommended parameters. Reproducibility is defined as the number of reproducible domains dividing the maximum domain number in two replicates. Conservation ratio between two cell types is defined in the similar way by using the domain numbers in two cell types to replace those in two replicates. In method evaluation, the random hierarchical TADs are generated by using the following iterative shuffling procedure. To preserve original hierarchies, the TAD sizes and corresponding sub-domain sizes are recorded for each chromosome. The TAD positions are shuffled according to the recorded TAD sizes in each chromosome, and then the sub-TAD positions are shuffled in each TAD according to the recorded sub-TAD sizes. The above shuffling is repeated until no hierarchical levels are recorded in the original hierarchical TADs. As for boundary analysis, the enrichments of epigenomic peaks, CTCF peaks and CTCF motifs are calculated by following previous works (11,36) and the compartments are calculated at 200 Kb resolution by using package hiclib (32).
RESULTS
Method comparison
To investigate the performance of HiTAD, we first compared HiTAD to two other methods with available software, Arrowhead implemented in Juicer (37) and TADtree (24). Matryoshka and the network modularity based method were excluded from comparisons due to software-installation difficulty and code unavailability respectively. The methods CaTCH (25) and HBM (26) were excluded since they do not explicitly point out TAD positions. The methods for traditional domains, such as DI based HMM (11), HiCseg (38), TopDom (39), TADbit (bioRxiv https://doi.org/10.1101/036764), HiCExplorer (bioRxiv https://doi.org/10.1101/115063), Armatus (27), spectral method (28), MrTADFinder and IC-Finder (29), were also excluded since they don not explicitly detect or automatically output hierarchical TADs. The selected methods were applied to both traditional and in situ Hi-C datasets under 40 kb resolution since it is pretty hard for TADtree to be run at higher resolutions. To simplify the statement, traditional and in situ Hi-C datasets are denoted by different suffixes, such as GM12878-T and GM12878-I respectively. To clarify hierarchical levels, TAD is denoted as level 0, sub-TAD is denoted as level 1 and subsequent domain level is denoted as level 2, etc.
HiTAD outperforms the other two methods in domain sensitivity, replicate reproducibility and inter cell-type conservation (Figure 2). HiTAD detects more domains in all levels for both in situ and traditional Hi-C datasets, especially in level 0, level 1 and level 2 domains (Figure 2A). This sensitivity improvement mainly arises from the fact that HiTAD successfully detects domains in more chromatin regions compared to the other two methods (Supplementary Figure S6a). Besides, the different domain size distributions can also contribute to differences in domain numbers. Generally, the domain sizes from HiTAD are smaller than those from Arrowhead but larger than or comparable to those from TADtree (Supplementary Figure S6b). Next GM12878-I and GM12878-T with four biological replicates available (Supplementary Table S1) were selected to evaluate reproducibility since HiTAD generally needs two replicates to detect hierarchical TADs. The four replicates were divided into two groups to independently generate two sets of hierarchical TADs for HiTAD. As for the other two methods, the two replicates in the same group were merged in hierarchical TAD detection. Our calculation shows that HiTAD outperforms the other two methods in the replicate reproducibility in all hierarchical levels on both GM12878-I and GM12878-T (Figure 2B). Similarly, the hierarchical TADs detected by HiTAD are more conserved across cell types than those detected by the other two methods in level 0, level 1 and level 2 domains (Figure 2C).
Figure 2.

Performance comparison. Three methods, Arrowhead, TADtree and HiTAD, are included to perform comparisons. The left and right figures illustrate the results calculated from in situ and traditional Hi-C datasets respectively. (A) Domain number. (B) Replicate reproducibility. (C) Inter cell-type conservation.
Finally, it should be noted that the metrics used in this work just reflect parts of algorithmic performance. Generally, higher domain number and chromatin coverage could only indicate better sensitivity, reproducibility is an important aspect of algorithms, and conservation ratio reflects the biological aspect of hierarchical TADs. We think these metrics together can reflect the algorithmic performance in hierarchical TAD detection, but they are not necessarily equal to algorithmic accuracy. However, there is currently no golden standard to evaluate the accuracy of hierarchical TADs. Compared to traditional TADs, it is more difficult to evaluate hierarchical TADs since this evaluation contains both chromatin domains and the hierarchies (Supplementary Figure S7). Better metrics or standards may be developed in the future.
Method evaluation
To obtain better details, hierarchical TADs were detected at 20 kb resolution for in situ Hi-C datasets in the following analyses. Since the numbers of level 2 and level 3 domains were quite limited, these domains were combined in next calculations. The shared boundary among different level domains was classified as lower-level boundary.
We first evaluated HiTAD by measuring the insulation effects of detected hierarchical boundaries. Two histone modifications, H3K36me3 and H3K27me3, were selected to represent active and inactive signals. The boundary insulation was calculated by following a previous procedure (9). Briefly, in each level of hierarchical TADs, every domain was divided into 10 bins, and the strength of histone modification was recorded for every bin. Then an 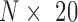 matrix was generated, where 20 columns represent the signal strengths of two consecutive domains and rows represent all possible consecutive domains. The correlations of the columns of this matrix reflect how the epigenomic signals in any two bins are correlated. Our calculations show that the signals in the same domain are highly correlated to each other, but the signal correlations between two consecutive domains are sharply separated in the boundaries (Figure 3A and Supplementary Figure S8). Furthermore, the lower level boundaries exhibit stronger insulation effects. As for the controls generated from random shuffling, there are no such sharp separations in the boundaries. These results together indicate that the detected hierarchical boundaries, especially the level 0 and level 1 boundaries, exhibit insulation effects.
matrix was generated, where 20 columns represent the signal strengths of two consecutive domains and rows represent all possible consecutive domains. The correlations of the columns of this matrix reflect how the epigenomic signals in any two bins are correlated. Our calculations show that the signals in the same domain are highly correlated to each other, but the signal correlations between two consecutive domains are sharply separated in the boundaries (Figure 3A and Supplementary Figure S8). Furthermore, the lower level boundaries exhibit stronger insulation effects. As for the controls generated from random shuffling, there are no such sharp separations in the boundaries. These results together indicate that the detected hierarchical boundaries, especially the level 0 and level 1 boundaries, exhibit insulation effects.
Figure 3.

HiTAD evaluation by insulation effects and signal enrichments. (A) Boundary insulation measured by signal correlations. There are 20 bins in each heatmap, in which the first and second 10 bins represent two consecutive domains respectively. The left two heatmaps are calculated from active signal H3K36me3, while the right two heatmaps are calculated from inactive signal H3K27me3. (B) Signal enrichment in different level boundaries. Three representative signals are shown, including H3K4me3, H3K4me1 and CTCF peaks. (C) Enrichment of directional CTCF motifs. The divergent CTCF motifs on boundaries are composed of minus-strand CTCF motifs in upstream regions and plus-strand CTCF motifs in downstream regions.
We next performed enrichment analysis on different level boundaries. Two histone modifications, H3K4me3 and H3K4me1, were selected to represent promoters and enhancers respectively. The key architectural protein CTCF was also included since this protein plays an important role in shaping chromatin domains (40). The calculated results show that different level boundaries are enriched in promoter signal H3K4me3 and CTCF binding sites but are a little depleted in enhancer signal H3K4me1 (Figure 3B and Supplementary Figure S9a), consistent with traditional TAD analysis (9,11). However, higher level boundaries generally show lower signals in both boundaries and near-by background regions. It was reported that the divergent CTCF motifs shaped the domain boundaries (9,36,41–43), so we further analyzed the composition of CTCF motif directions in these binding sites. The result shows that different level boundaries are enriched in divergent CTCF motifs in overall (Figure 3C and Supplementary Figure S9b), but the higher-level boundaries show lower densities on both total and divergent CTCF motifs. The results from histone modifications, CTCF binding sites and motif direction together indicate that almost all level boundaries show similarities with traditional TAD boundaries, but different level boundaries exhibit different signal strengths. These signal differences may partially explain the insulation differences among hierarchical boundaries. However, the limited domain numbers and relatively low reproducibility of higher level domains can influence the reliable analysis on boundary enrichments, and thorough works are needed to further investigate their functional characteristics in the future.
Boundary-level analysis on structural and functional properties of hierarchical TADs
Due to limited domain numbers, level 2 and level 3 domains were excluded from calculations in this and following sections to simplify analysis. The shared boundaries between TADs (level 0 domains) and sub-TADs (level 1 domains) were classified as TAD boundaries. When comparing two cell types, TAD boundaries and sub-TAD boundaries undergo different structural changes, including conserved TAD boundary, conserved sub-TAD boundary, disappeared TAD boundary, disappeared sub-TAD boundary and TAD-to-sub-TAD boundary switch (Supplementary Figure S10). Statistically, TAD boundaries exhibit quite high inter cell-type conservation, whereas sub-TAD boundaries are quite dynamic across cell types (Figure 4A and Supplementary Figure S11). Next, we extended traditional analyses on compartment (44) and replication timing (15) to investigate the similarities and differences between TAD boundaries and sub-TAD boundaries by using both intra cell-type and inter cell-type comparisons. The presented results are mainly calculated from IMR90-I and K562-I since there are replication timing data in human cell types IMR90 and K562.
Figure 4.

Structural and functional analysis on boundary-level changes. (A) The left and right pie charts illustrate the proportion of hierarchical boundary changes by using IMR90-I and K562-I as references respectively. The total boundary number is presented in the bracket for each cell type. (B) The top figure illustrates the enrichment difference between TAD boundaries and sub-TAD boundaries in the same cell type (intra cell-type comparison). The medium and bottom figures illustrate the relationship between hierarchical boundary changes and compartment boundary changes across cell types (inter cell-type comparison). The x-axis denotes the genomic distance to compartment boundaries, and the y-axis denotes the probability density. (C) The enrichments on replication timing boundaries are presented in the same way as compartment boundaries, except that the replication timing transitions are included in each sub-figure. The x-axis denotes the genomic distance to replication timing boundaries. The left y-axis denotes the probability density of domain boundary enrichment, and the right y-axis denotes the replicating timing.
TAD boundaries but not sub-TAD boundaries mainly separate higher-order chromosomal compartments. Lieberman-Aiden et al. proposed A-B (active-inactive) compartments in original Hi-C work (6), so we performed enrichment analysis on these compartment boundaries. Figure 4B illustrates that the A-B compartment boundaries are enriched in TAD boundaries but depleted in sub-TAD boundaries when performing intra cell-type analyses on IMR90-I and K562-I independently. As for inter cell-type comparisons, different change types were analyzed separately. The trends in conserved TAD and sub-TAD boundaries are the same as those from intra cell-type analysis. If TAD boundaries switch to sub-TAD boundaries from one cell type to the other, the enrichment-to-depletion switch is also observed simultaneously. Similarly, if TAD boundaries disappear in the other cell type, the enrichment on compartment boundaries also disappears. In the case of sub-TAD disappearance, the stronger depletion is observed in the corresponding cell type (Figure 4B and Supplementary Figure S12a). The same phenomena are observed in other inter cell-type comparisons (Supplementary Figure S13). These results together suggest that the changes of compartment boundaries are mainly accompanied with the changes of TAD boundaries across cell types. Combining intra cell-type and inter cell-type analyses, we can conclude that the TAD but not sub-TAD boundaries are mainly involved in correlating higher-order compartment.
TAD boundaries but not sub-TAD boundaries mainly separate replication timing domains. Similar to compartment boundaries, replication timing boundaries are enriched in TAD boundaries but depleted a little in sub-TAD boundaries in both IMR90 and K562 independently (Figure 4C). As for inter cell-type comparisons, the conserved TAD boundaries are enriched in replication timing boundaries, and the conserved sub-TAD boundaries in overall are depleted in replication timing boundaries. If TAD boundaries in IMR90-I switch to sub-TAD boundaries or even disappear in K562-I, the enrichments disappear simultaneously. No enrichment is observed in the case of sub-TAD boundary disappearance (Figure 4C and Supplementary Figure S12b). In summary, the changes of replication timing boundaries are also mainly accompanied with the changes of TAD boundaries across cell types, indicating the difference between TAD and sub-TAD boundaries in correlating the replication timing boundaries.
Domain-level analysis on structural and functional properties of hierarchical TADs
We next investigated domain-level changes and corresponding transcriptional associations for hierarchical TADs. Compared to boundary-level change, the domain-level change is more complicated since it covers wider chromatin region and involves simultaneous changes of several boundaries. Through different inter cell-type comparisons, we observed several types of domain-level changes for hierarchical TADs, which are defined as conserved TAD, semi-conserved TAD, merged TAD, split TAD and undefined TAD in this work. Conserved TAD is conserved completely in all hierarchical levels, while semi-conserved TAD is conserved in TAD level but dynamic in sub-TAD level. These two cases together represent traditional TAD conservation across cell types. Merged TAD represents the situation that two or more TADs are merged to form a new TAD from one cell type to another cell type, while split TAD represents the reverse situation. The undefined TAD denotes the other TAD change with no clear definition in this work. Similar to TAD change, the semi-conserved TAD can be further classified as merged sub-TAD, split sub-TAD and undefined sub-TAD (Supplementary Figure S14).
By using IMR90-I as reference, 33.5% TADs are totally conserved and 11.8% TADs are semi-conserved when comparing IMR90-I to K562-I (Figure 5A). Interestingly, the undefined sub-TAD change is not observed in this comparison, and only very few cases are observed in other inter cell-type comparisons in this work (Supplementary Figure S15). These consistent results indicate that most sub-TAD changes in semi-conserved TADs only involve sub-TAD mergence or sub-TAD split without complicated combinations. As for TAD-level change, around 31.7% and 8.6% TADs show TAD mergence and split respectively, and the rest 14.4% TADs undergo complicated domain-level changes in IMR90-I (Figure 5A). Similar trends are observed in K562-I (Figure 5A) and other inter cell-type comparisons (Supplementary Figure S15).
Figure 5.

Structural and functional analysis on domain-level changes. (A) The left and right pie charts illustrate the proportion of hierarchical domain changes by using IMR90-I and K562-I as references respectively. The total TAD number is presented in the bracket for each cell type. (B) The transcriptional fold changes are presented separately for different domain changes when comparing K562 to IMR90. (C) The averaged RNA expressions on merged TADs are presented in the order of inactive to active TADs. Merged TAD represents that the two consecutive TADs in IMR90 are merged to be a new TAD in K562. BPKM: bases per kilobase per million mapped bases. (D) Box plots of average RNA expressions on merged TADs illustrate the transcriptional changes between cell types. The box plots in the left and right figures are calculated from the 100 bins in inactive and active TADs respectively. The Mann–Whitney U test was used to calculate P-values (*P < 0.05, **P < 1e-3 and ***P < 1e-5). (E) The averaged RNA expressions on split TADs are presented in the same order as merged TADs. Split TAD represents that a TAD in IMR90 are split into two consecutive TADs in K562. (F) Box plots of average RNA expressions on split TADs illustrate the transcriptional changes between cell types, in the same way as merged TADs.
We next investigated the relationship between domain changes and gene expressions. The undefined sub-TADs and undefined TADs were excluded from calculation since there are no consistent domain changes in these two cases. Compared to conserved TADs, the sub-TAD mergence and split in semi-conserved TADs exhibit transcriptional upregulation and downregulation respectively. The similar trends are observed in TAD mergence and split (Figure 5B and Supplementary Figure S16). To further reveal the functional roles of TAD mergence and split, we analyzed the gene expression changes on two consecutive TADs jointly by using following procedure. First, each TAD was separated into 100 bins and the total RNA expression in each bin was normalized by corresponding bin size. Second, the RNA expressions on 100 bins were summed and averaged in each TAD of two consecutive TADs. Then the one with lower average RNA expression was classified as inactive TAD, and the other one was classified as active TAD. Third, the average RNA expression in each bin was calculated by using all inactive TADs or active TADs respectively. Figure 5C illustrates the calculated results for TAD mergence. Intuitively, the transcriptional gap between inactive TADs and active TADs is quite large in the original cell type IMR90, but this gap is shrunk after the TAD mergence in cell type K562. To better show the differences, the 100 average RNA expression values in the two kinds of TADs were separately presented by using box plots. Figure 5D clearly illustrates the significant upregulation and downregulation in the originally inactive and active TADs respectively. The reverse effect is observed in TAD split (Figure 5E and F). The same regulatory effects are also observed in other inter cell-type comparisons (Supplementary Figure S17). These converged results suggest the role of TAD mergence in shrinking the transcriptional gap between two consecutive TADs. With respect to sub-TAD mergence and split, we did not observe completely consistent patterns in all inter cell-type comparisons (Supplementary Figure S18). Thorough works are needed to further clarify the transcriptional difference between TAD mergence and sub-TAD mergence in the future.
DISCUSSION
A current question in the field of hierarchical chromatin domain is whether TADs are structurally and functionally distinct from sub-TADs and other domains. Since traditional TAD definition is a little ambiguous, Dixon et al. recently attempted to define TAD to be stable through cell divisions and conserved through cell lineages (45). As discussed in a recent paper (46), this definition is hard to implement computationally. However, by utilizing only chromatin interactions in one cell type, HiTAD detects TADs and corresponding hierarchical domains with pretty high replicate reproducibility and inter cell-type conservation, partially satisfying the TAD definition through biological processes. The detected hierarchical TADs are also evaluated by insulation effects as well as signal enrichments. In summary, our work suggests that the refinement of TAD definition, including hierarchical structure and biological function, can be achieved by only analyzing high-quality chromatin interactions, at least in part.
HiTAD adopts some strategies different from traditional methods. First, except the local insulations used by traditional methods, HiTAD also utilizes global intra-chromosomal interactions by constraining TADs to the best separation on individual chromosomes. In other words, the TAD detection in HiTAD attempts to make use of the advantages of local and global intra-chromosomal interactions in a concerted way. Second, biological replicates are used to generate final hierarchical TADs in our method. The advantage of this strategy is that flexible methods can be used to generate sufficient bottom domains and corresponding hierarchies for every replicate, instead of considering the high variations of chromatin interactions in the initial start. Though the usage of replicate reproducibility can improve the accuracy and reliability from highly variant data, it does not mean that the obtained boundaries and domains are reproducible in all replicates. This is because two replicates cannot represent all cases in the population. In addition, the replicate reproducibility requires the comparable data quality from two replicates. Otherwise, the low quality data will dominate the final results over the high quality data. In this case, we suggest that the results from high quality data or merged data should be used. However, it is not difficult currently to generate Hi-C data with enough quality for HiTAD due to the improvement of sequencing technology and the reduction of sequencing cost. Third, compared to traditional boundary-based alignment, domain-based alignment in our method adaptively matches the domains and boundaries by eliminating the choice of distance threshold between the two aligned boundaries. The domain-based alignment also allows unmatched domains and boundaries automatically.
The change types of hierarchical TADs defined in this work can pave the way for further studies on chromosomal structures and functions. In this work, we defined several inter cell-type changes on boundaries and domains respectively for hierarchical TADs and explored their structural and functional roles for the first time. Compared to boundary-level change, the domain-level change generally covers wider chromatin region and involves several boundaries. For transcriptional regulation, we used a simple method to investigate the domain-level relationship between hierarchical TADs and gene expressions. But for the chromosomal compartment and replication timing domain, their inter cell-type changes also involve several boundaries. In this way, the domain-level analysis will meet the combinatorial problem when simultaneously comparing several boundaries, so the boundary-level enrichments were performed on chromosomal compartment and replication timing by following previous works (15,44) to simplify analysis. In addition, both boundary-level and domain-level analyses were simplified by only considering two levels, TAD and sub-TAD. More complicated structural changes can be observed if taking more hierarchical levels into consideration, but it will be more difficult to depict their biological functions, especially with the limited number of higher level boundaries and domains. In spite of simplified analyses, we still revealed the structural and functional differences between TADs and sub-TADs by using these hierarchical TAD changes. Since these phenomena are quite common in different inter cell-type comparisons, it is quite possible that these analyses can be applied to other mammalian cell types not covered in this work.
TAD and sub-TAD differ in correlating chromosomal compartment and replication timing. In the chromosomal structure, the TAD boundaries but not sub-TAD boundaries mainly correlate the chromosomal compartments. As for the replication timing domain, the same trend is observed. These together argue that TADs are the main structural units in linking higher-order chromosomal organization and replication timing. However, this does not necessarily mean that sub-TADs have no effect. The identification resolution of compartment is relatively low due to sequencing depth, making subtle comparisons difficult. Sub-compartment was recently proposed through extremely deep sequencing, but it is hard to perform reliable analysis on sub-compartment due to the limited data involving only one cell type GM12878 (9). The analysis on replication timing domains also meets the resolution problem. The sub-domains of replication timing, like sub-TADs, may be observed in the future with technology development. The structural and functional differences between TAD and sub-TADs need further investigation with better technologies and algorithms.
TAD and sub-TAD can separate regulatory activity but with different insulation effect. TAD boundaries generally show stronger insulations than sub-TAD boundaries. And the transcriptional association of TAD mergence/split is a little different from that of sub-TAD mergence/split when performing domain-level comparisons. Combining the fact that TAD is more stable than sub-TAD across cell types, these results argue that TAD may insulate the global gene regulation in a relatively stable way and sub-TAD further facilitates local gene regulation in a dynamic way, consistent with previous work (22). With regard to the potential mechanism underlying hierarchical TADs, TAD and sub-TAD boundaries share similar trends in CTCF binding sites and divergent motifs, but with different density. This may partially explain the differences in insulation and stability between TAD boundaries and sub-TAD boundaries. However, recent research showed that transcription could contribute to the formation of chromatin domains by helping position another key architectural protein complex, cohesion (47). Further studies are needed to clarify the detailed relationship among transcription, CTCF binding and hierarchical TAD.
Finally, our method performs well in detecting TADs and sub-TADs, but it is less sensitive to detect higher level domains, especially level ≥3 domains. Figure 2 illustrates that it is currently difficult to sensitively and reproducibly detect higher level domains. This is because the chromatin interactions are highly variable around the boundary regions of these small domains. In HiTAD, the locally high variations and the small window sizes together can make the adaptive DIs fluctuate around candidate boundary regions, which leads to the failure in domain reproducibility. Though this strategy excludes irreproducible domains from two replicates, the percentage of reproducible domains still decreases in higher level domains when more than two replicates are used to measure the reproducibility (Figure 2B). In the future, better methods can be utilized or developed to balance the sensitivity and reproducibility in detecting higher level domains.
CONCLUSION
In this work, we developed a novel method HiTAD to detect hierarchical TADs from Hi-C chromatin interactions by further constraining TAD to optimal chromatin interaction separation in chromosomal level. HiTAD performs well in domain sensitivity, replicate reproducibility and inter-cell-type conservation. We evaluated the detected hierarchical TADs by calculating insulation effects and signal enrichments on different level boundaries. By defining boundary-level and domain-level changes for hierarchical TADs, we systematically investigated the structural and functional differences between TADs and sub-TADs. The intra cell-type and inter cell-type analyses together revealed that TADs and sub-TADs differed in correlating higher-order compartment, replication timing and transcriptional regulation. With better technology and algorithm, the structural and functional characteristics of hierarchical TADs can be further explored in the near future.
AVAILABILITY
HiTAD is integrated to a Python package called TADLib, which is freely available online at https://pypi.python.org/pypi/TADLib.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary data are available at NAR Online.
FUNDING
Fundamental Research Funds for the Central Universities [2662015PY197, 2662016PY034]; National Natural Science Foundation of China [31200951]. Funding for open access charge: Fundamental Research Funds for the Central Universities [2662015PY197, 2662016PY034]; National Natural Science Foundation of China [31200951].
Conflict of interest statement. None declared.
REFERENCES
- 1. Dekker J., Rippe K., Dekker M., Kleckner N.. Capturing chromosome conformation. Science. 2002; 295:1306–1311. [DOI] [PubMed] [Google Scholar]
- 2. Simonis M., Klous P., Splinter E., Moshkin Y., Willemsen R., de Wit E., van Steensel B., de Laat W.. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 2006; 38:1348–1354. [DOI] [PubMed] [Google Scholar]
- 3. Zhao Z., Tavoosidana G., Sjolinder M., Gondor A., Mariano P., Wang S., Kanduri C., Lezcano M., Sandhu K.S., Singh U. et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 2006; 38:1341–1347. [DOI] [PubMed] [Google Scholar]
- 4. Dostie J., Richmond T.A., Arnaout R.A., Selzer R.R., Lee W.L., Honan T.A., Rubio E.D., Krumm A., Lamb J., Nusbaum C. et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006; 16:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fullwood M.J., Liu M.H., Pan Y.F., Liu J., Xu H., Mohamed Y.B., Orlov Y.L., Velkov S., Ho A., Mei P.H. et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009; 462:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kalhor R., Tjong H., Jayathilaka N., Alber F., Chen L.. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 2012; 30:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dryden N.H., Broome L.R., Dudbridge F., Johnson N., Orr N., Schoenfelder S., Nagano T., Andrews S., Wingett S., Kozarewa I. et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014; 24:1854–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G.. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012; 148:458–472. [DOI] [PubMed] [Google Scholar]
- 11. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J. et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanyal A., Lajoie B.R., Jain G., Dekker J.. The long-range interaction landscape of gene promoters. Nature. 2012; 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jin F., Li Y., Dixon J.R., Selvaraj S., Ye Z., Lee A.Y., Yen C.A., Schmitt A.D., Espinoza C.A., Ren B.. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K. et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014; 515:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Franke M., Ibrahim D.M., Andrey G., Schwarzer W., Heinrich V., Schopflin R., Kraft K., Kempfer R., Jerkovic I., Chan W.L. et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016; 538:265–269. [DOI] [PubMed] [Google Scholar]
- 17. Andrey G., Montavon T., Mascrez B., Gonzalez F., Noordermeer D., Leleu M., Trono D., Spitz F., Duboule D.. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science. 2013; 340:1234167. [DOI] [PubMed] [Google Scholar]
- 18. Lupianez D.G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J.M., Laxova R. et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flavahan W.A., Drier Y., Liau B.B., Gillespie S.M., Venteicher A.S., Stemmer-Rachamimov A.O., Suva M.L., Bernstein B.E.. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016; 529:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hnisz D., Weintraub A.S., Day D.S., Valton A.L., Bak R.O., Li C.H., Goldmann J., Lajoie B.R., Fan Z.P., Sigova A.A. et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016; 351:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bouwman B.A., de Laat W.. Getting the genome in shape: the formation of loops, domains and compartments. Genome Biol. 2015; 16:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Phillips-Cremins J.E., Sauria M.E., Sanyal A., Gerasimova T.I., Lajoie B.R., Bell J.S., Ong C.T., Hookway T.A., Guo C., Sun Y. et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang X.T., Dong P.F., Zhang H.Y., Peng C.. Structural heterogeneity and functional diversity of topologically associating domains in mammalian genomes. Nucleic Acids Res. 2015; 43:7237–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weinreb C., Raphael B.J.. Identification of hierarchical chromatin domains. Bioinformatics. 2016; 32:1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhan Y., Mariani L., Barozzi I., Schulz E.G., Bluthgen N., Stadler M., Tiana G., Giorgetti L.. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 2017; 27:479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shavit Y., Walker B.J., Lio P.. Hierarchical block matrices as efficient representations of chromosome topologies and their application for 3C data integration. Bioinformatics. 2016; 32:1121–1129. [DOI] [PubMed] [Google Scholar]
- 27. Filippova D., Patro R., Duggal G., Kingsford C.. Identification of alternative topological domains in chromatin. Algorithms Mol. Biol. 2014; 9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J., Hero A.O. 3rd, Rajapakse I.. Spectral identification of topological domains. Bioinformatics. 2016; 32:2151–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Haddad N., Vaillant C., Jost D.. IC-Finder: inferring robustly the hierarchical organization of chromatin folding. Nucleic Acids Res. 2017; 45:e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Selvaraj S., R Dixon J., Bansal V., Ren B.. Whole-genome haplotype reconstruction using proximity-ligation and shotgun sequencing. Nat. Biotechnol. 2013; 31:1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M.. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A.. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D. et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kheradpour P., Kellis M.. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014; 42:2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gomez-Marin C., Tena J.J., Acemel R.D., Lopez-Mayorga M., Naranjo S., de la Calle-Mustienes E., Maeso I., Beccari L., Aneas I., Vielmas E. et al. Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:7542–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Durand N.C., Shamim M.S., Machol I., Rao S.S., Huntley M.H., Lander E.S., Aiden E.L.. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 2016; 3:95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levy-Leduc C., Delattre M., Mary-Huard T., Robin S.. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics. 2014; 30:i386–i392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shin H., Shi Y., Dai C., Tjong H., Gong K., Alber F., Zhou X.J.. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2016; 44:e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ong C.T., Corces V.G.. CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014; 15:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rudan M.V., Barrington C., Henderson S., Ernst C., Odom D.T., Tanay A., Hadjur S.. Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 2015; 10:1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P., Trzaskoma P., Magalska A., Wlodarczyk J., Ruszczycki B. et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015; 163:1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D.U., Jung I., Wu H., Zhai Y., Tang Y. et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015; 162:900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dixon J.R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J.E., Lee A.Y., Ye Z., Kim A., Rajagopal N., Xie W. et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015; 518:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dixon J.R., Gorkin D.U., Ren B.. Chromatin domains: the unit of chromosome organization. Mol. Cell. 2016; 62:668–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dali R., Blanchette M.. A critical assessment of topologically associating domain prediction tools. Nucleic Acids Res. 2017; 45:2994–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Busslinger G.A., Stocsits R.R., van der Lelij P., Axelsson E., Tedeschi A., Galjart N., Peters J.M.. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature. 2017; 544:503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.