

Key Points
ATAC-seq provides genome-wide chromatin state in 3 cell types of hematopoietic stem/progenitor cells.
Transcription factor cohorts are associated with dynamic changes of open chromatin during the differentiation of LT/ST-HSCs to MPPs.
Visual Abstract
Introduction
Hematopoietic stem cells (HSCs) are characterized by their self-renewal potential and ability to differentiate into multiple blood lineages.1,2 They are essential for lifelong blood production and represent 1 of the best-studied somatic stem cell systems.2-4 Several decades of successful bone marrow transplants have demonstrated the therapeutic importance of HSCs.5 Much progress has been made to understand the regulatory network of HSC self-renewal and differentiation.6,7 Several studies suggest that epigenetic mechanisms play an important role in controlling HSC renewal and lineage commitment.8-12 Understanding the regulatory mechanisms of HSC self-renewal and differentiation is important for both basic stem cell biology and improving the quality of stem cell transplantation in clinical settings.
HSCs are a heterogeneous population of cells that contain at least 3 populations: long-term HSCs (LT-HSCs), short-term HSCs (ST-HSCs), and multipotent progenitors (MPPs).13 LT-HSCs reside in the bone marrow and can self-renew to maintain the stem cell pool or differentiate into ST-HSCs or lineage-restricted progenitors. The progenitors can further differentiate to produce terminally differentiated, functional hematopoietic cells. LT-HSCs must persist for the lifespan of the organism to constantly replenish the hematopoietic system. ST-HSCs or MPPs are able to sustain hematopoiesis in the short term only. Experimentally, HSCs can be distinguished by surface marker staining, and several different laboratories have developed different antibody combination schemes to identify the different populations of cells within the total population of HSCs. One major scheme is the cell surface phenotype of positive selection for the markers c-Kit and Sca-1 and negative selection for markers of mature hematopoietic cell lineages (typically B220, CD4, CD8, Gr-1, Mac-1, and Ter-119).14 Although this Lin−Sca-1+ c-Kit+ (LSK) phenotype greatly enriches for hematopoietic reconstituting activity, this bone marrow compartment contains progenitor cells in addition to long-term HSCs. Specifically, only approximately 10% of LSK cells are bona fide LT-HSCs. Another staining scheme is based on the signaling lymphocytic activation molecule (SLAM) family of cell surface glycoproteins and used a profile of CD150+; CD244−; CD48− to define LT-HSCs.15 So far, the scheme to combine LSK and SLAM has shown that approximately 50% of single cells with this phenotype are LT-HSC.16-19 It can separate the LSK compartment into LT-HSCs (LSK CD150+CD48−), ST-HSCs (LSK CD150−CD48−), and MPPs (LSK CD150−CD48+). CD34 and Flk2 are also commonly used to separate the LSK population into LT-HSCs (LSK CD34−Flk2−), ST-HSCs (LSK CD34+Flk2−), and MPP cells (LSK CD34+Flk2+).20-23
Chromatin structure seems to play a critical role in the regulation of gene transcription during hematopoiesis. It has been nicely demonstrated that gene expression changes during hematopoietic stem/progenitor cell differentiation, and is programmed by chromatin modifications present at the hematopoietic stem/progenitor cell stage.9 Recently, 4 chromatin modifications were surveyed across 16 stages of hematopoietic differentiation with a high-sensitivity indexing-first chromatin immunoprecipitation technology.10 This study identified transcription factor networks controlling chromatin dynamics and lineage specification in hematopoiesis and provided a comprehensive model of chromatin dynamics during development. However, genome-wide profiles of the open-chromatin landscape of LT-HSCs, ST-HSCs, and MPPs in mice have not been examined to date.
Assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) uses a bacterial (Tn5) transposase, an enzyme that inserts a short fragment of DNA (a transposon) into another molecule of DNA, or, in this assay, inserts 2 short fragments separate from each other.24,25 Because the transposase is unable to access DNA that is either bound by nucleosomes or strongly bound transcription factors, it incorporates its transposons preferentially into open or accessible chromatin. Specifically, the transposase inserts 2 fragments of DNA that serve as tags, and a mechanism to lead to a process known as tagmentation to fragment the accessible DNA. This assay was developed to identify accessible DNA regions, equivalent to DNase I hypersensitive sites. Buenrostro et al convincingly demonstrated that ATAC-seq was able to accurately identify the nucleosome-free regions of a lymphoblastoid cell line. The authors showed that ATAC-seq profiles were comparable to DNaseI-seq in specificity and signal to noise ratio from 100 to 10 000-fold fewer cells than DNaseI-seq.24 Thus, this technique allows us to analyze the chromatin status of hematopoietic stem cells, which is a rare population in the hematopoietic system. In this study, we used this powerful technology to compare chromatin accessibility (“open chromatin”) profiles of LT-HSCs, ST-HSCs, and MPPs. We found that the chromatin is dynamically remodeled at promoters and enhancers of HSCs, thus affecting the accessibility of transcription factors during differentiation of LT-HSCs to ST-HSCs and MPPs.
Methods
Mice
The C57BL/6 wild-type (CD45.2+) mice were from The Jackson Laboratory. Mice were bred in-house in a pathogen-free mouse facility at Temple University. Animal experiments were performed in accordance with guidelines approved by the Institutional Animal Care and Use Committee at Temple University.
Fluorescence-activated cell sorting and isolation of HSCs
Bone marrow cells were flushed from the long bones (tibias and femurs) of mice with phosphate-buffered solution without calcium or magnesium. For detection of LSK cells, whole bone marrow cells were incubated with phycoerythrin anti-mouse Lineage Cocktail antibody, fluorescein isothiocyanate–conjugated antibody to Sca1 (Ly6A/E; D7) and allophycocyanin-conjugated antibody to c-Kit (ACK2). CD150 and CD48 were measured with the following antibodies: phycoerythrin-Cy7 conjugated antibody to CD48 (HM48-1) and Brilliant Violet 421-conjugated antibody to CD150 (TC15-12F12.2). All antibodies were purchased from Biolegend except for antibodies to Sca1 and c-Kit, which were purchased from eBioscience. Antibodies to specific lineages, Sca1, c-Kit, CD150, and CD48 were diluted 1:100. Nonviable cells were excluded using the viability dye 7-AAD (50 μg/mL−1). Cells were sorted with a FACSAria (Becton Dickinson) automated cell sorter. Data were analyzed using FlowJo software (Tree Star). For each experiment, 20 000 cells of LT-HSCs, ST-HSCs, and MPPs were used for ATAC-seq analysis.
Library construction for ATAC-seq
Library construction for ATAC-seq was performed as described.25 In brief, transposed DNA fragments were polymerase chain reaction (PCR) amplified after the transposition reaction and purification. To maintain high library complexity, we used 11 total PCR cycles, which was determined by quantitative PCR method as the optimized cycle number. The deep sequencing was performed at the Fox Chase Cancer Center Genomic Facility. The quality control of libraries was performed using gel electrophoresis. The libraries were quantified using the KAPA Library Quant Kit for Illumina Sequencing Platforms (KAPA Biosystems) as suggested by Buenrostro et al.25 Illumina high-throughput sequencing instrument, Nextera-based sequencing primers, and reagents were used for the deep sequencing. The same amount (molarity) of library DNA of each group was used as the input material for the deep sequencing.
Reads alignment and correlation between replicates
The 50-nucleotide (nt) paired end reads were aligned to the mouse genome, version mm10, using bowtie2 with default parameters. In total, the average mapping reads in LT-HSCs, ST-HSCs, and MPPs were about ∼110, 122, and 114 million, and the average coverage along the mouse genome (2.8 × 109) were 3.92, 4.35, and 4.07, respectively. Mapped reads were normalized to reads per million reads mapped (RPM) and were converted to bigwig format for visualizing using the R package Gviz. After normalizing to RPM, the average coverage is ∼0.035. To assess data reproducibility, normalized read counts in 250 nt bins genome-wide were determined for each set of biological replicates, and principal component analysis (PCA) were performed on the read counts of 6 samples after scaling and centering. The Pearson correlation coefficients of pairwise ATAC-seq samples were calculated using the same data.
Peak calling of open chromatin regions
Peak calling was performed using the findPeaks script in the HOMER software26 package with DNase style. Common peaks between 2 replicates were detected by bedtools and were defined as open chromatin regions for further analysis. The peak distribution along chromosomes with a summarized size of each 1 Mb were plotted using the R package ggbio.
Genome features classification of open chromatin regions
Genome features of open chromatin regions were annotated using the HOMER script annotatePeaks.pl with 8 types: promoter-transcription start site (TSS), 5′ UTR, exon, intron, 3′ UTR, transcription termination site, non-coding RNAs (ncRNAs), and intergenic regions. For determining the enrichment of the open chromatin regions in each genomic feature, 10 000 random genomic regions were sampled as background. The enrichment significance were evaluated using a χ2 test.
Gene ontology enrichment analysis
The list of genes with open chromatin in the promoter-TSS region were ranked by peak score in descending order, and the top 3000 were used for gene ontology (GO) enrichment analysis using DAVID (https://david.ncifcrf.gov) with the default background. The top 3000 highest expressed genes in LT-HSCs (CD34−Flk2−Lin−c-kit+Sca1+) downloaded from Gene Expression Commons (https://gexc.stanford.edu)27 were performed for GO enrichment analysis as previously. The false discovery rates of top enriched GO terms were visualized by heat map or barplot using a customized script.
Heat map of open chromatin around promoter-TSS regions
The flanking 1 kb around TSS, both upstream and downstream, were extracted from all genes or genes with open chromatin in their promoter regions. Heat maps of read density for each gene were plotted along their given TSS as well as the up- and downstream 1 kb flanking regions using the R package “genomation.” The average reads density was visualized as a color image and lines smoothed with loess function.
Identification of enhancer regions in an open chromatin state
Published enhancer regions in blood cells marked by H3K27ac and H3K4me1 were downloaded from the Web site (http://compbio.cs.huji.ac.il/blood-chromatin/Data.html).10 The bed files of open chromatin were converted from mm10 to a mm9 version of the mouse genome using LiftOver tools. Enhancer regions overlapping with open chromatin were defined using bedtools. To define conserved open enhancer regions between mouse and human, the open enhancer regions were further converted from mouse mm9 version to human hg18 version using LiftOver, and were compared with published enhancer regions marked by H3K4me1 in human CD133+ cell.9
Gene expression activity
The gene expression activity in different blood cell types were downloaded from Gene Expression Commons (https://gexc.stanford.edu).27 The gene expression activity was ordered from high to low (100 to −100). Correlation were performed between ATAC-seq signal in 1 kb promoter upstream from TSS and gene expression activity of LT-HSCs (marked by CD34−Flk2−Lin−c-kit+Sca1+), ST-HSCs (CD34+Flk2−Lin−c-kit+Sca1+), and MPPs (CD34+Flk2+Lin−c-kit+Sca1+), respectively.
cis-Element Enrichment and PCA
The DNA sequence motif enrichment in promoter regions and enhancers were identified using the HOMER script findMotifsGenome with length 6, 8, and 10 nt. The known transcription factor binding motifs with significant enrichment (P < .01) were clustered and visualized as a heat map using custom scripts. The motif logos were generated for the given motif matrix using stamp (http://www.benoslab.pitt.edu/stamp/). PCA was applied to the P value matrix of enriched motifs for all 3 cell types.
Results and discussion
ATAC-seq provides open chromatin landscape of HSCs
In this study, we used the combination of Lin−Sca1+c-Kit+ and SLAM surface markers and fluorescence activated cell sorting (FACS) to isolate mouse LT-HSC (LSK CD150+CD48−), ST-HSC (LSK CD150−CD48−), and MPP (LSK CD150−CD48+) cells (Figure 1A). Next, we performed ATAC-seq to survey genome-wide chromatin dynamics for 2 biological replicates of all 3 cell types (LT-HSCs, ST-HSCs, and MPPs). From our ATAC-seq libraries, 85% to 93% of the paired end reads were mapped to the mouse genome. The average mapping reads in LT-HSCs, ST-HSCs, and MPPs are ∼110, 122, and 114 million, respectively. To determine the reproducibility between replicates, we calculated the reads density in 250 nt windows across the genome, and performed PCA and Pearson correlation analysis of pairwise comparisons among ATAC-seq samples from all the 3 cell types. From this analysis, we found that the 2 replicates from the same cell type clustered together, and were separated from other samples (Figure 1B); specifically, the Pearson correlation coefficient of 2 replicates of each cell type were ∼0.9, indicating high reproducibility of the assay (supplemental Figure 1B; Figure 1C).
Figure 1.

ATAC-seq provides the open chromatin landscape of LT-HSCs, ST-HSCs, and MPPs. (A) Representative flow cytometry results to assess the Sca1+c-Kit+ population of cells, then to further distinguish LT-HSCs, ST-HSCs, and MPPs using the CD150 and CD48 combination of markers. (B) PCA for read counts of bins genome-wide among ATAC-seq samples. (C) Scatterplot and Pearson correlation of ATAC-seq data between 2 biological replicates for each of the 3 HSC cell types (LT-HSCs, ST-HSCs, and MPPs). (D-F) Browser track plot of ATAC-seq signal (RPM) in the genomic regions around CD34, Flt3/Flk2, and Hoxb5, respectively. (G) Gene expression activity of CD34, Flt3/Flk2, and Hoxb5 in different types of blood cells. Expression data collected from Gene Expression Commons (https://gexc.stanford.edu). Gene expression activity ranged from −100 (most inactive) to 100 (most active). MEP, megakaryocyte-erythroid progenitor.
Using the peak calling algorithm in the HOMER software package, we globally identified 22 285, 17 249, and 33 585 common open chromatin regions in both replicates for LT-HSCs, ST-HSCs, and MPPs, respectively (supplemental Figure 1A). To begin, we examined the ATAC-seq signal in genes used for FACS-based cell sorting and found ATAC-seq signal is specifically higher at the promoter of CD48 in MPPs, and highest at the promoter of CD150 in LT-HSCs (supplemental Figure 2A-B). Also, there is strong ATAC-seq signal at the promoter of Sca1/Ly6a in all 3 HSC cell types, consistent with HSCs being Sca1 positive (supplemental Figure 2C). Next, we surveyed several well-known HSC marker genes. As mentioned previously, 2 other surface markers CD34 and Flk2 (also known as Flt3) can be used to distinguish LT-HSCs, ST-HSCs, and MPPs by FACS analysis (supplemental Figure 1C). We found that ATAC-seq signal in the promoter region of CD34 displayed a gradual increase from LT-HSCs to ST-HSCs to MPPs (Figure 1D), which is consistent with the expression pattern of CD34 in HSCs (Figure 1G). Similarly, we found that ATAC-seq signal in the promoter region of Flt3 in MPPs is much higher than in LT-HSCs and ST-HSCs (Figure 1E), which is also consistent with the expression pattern of Flt3 in HSCs (Figure 1G). Additionally, HoxB5 has been shown to be specifically expressed in LT-HSCs28 (Figure 1G). We also found that our ATAC-seq data consistently captured a unique peak signal at the HoxB5 promoter region only in LT-HSCs, but not in ST-HSCs and MPPs (Figure 1F). Also, ATAC-seq signal are higher at LT/ST-HSCs compared with MPPs in the promoter of Bmi1, Gfi1, p57, and p21, which are required for maintenance of self-renewing HSCs29-31 (supplemental Figures 2D, and 3A-C), whereas ATAC-seq signal in the promoter of c-Myc is highly maintained in all 3 types of HSCs, which controls the balance between HSC self-renewal and differentiation32 (supplemental Figure 3D). Although open chromatin signals were not always perfectly correlated with gene expression, these examples indicated our ATAC-seq captured high confidence signal in areas of the genome that should be found as open chromatin regions in the 3 HSC cell types. Taken together, our ATAC-seq data provides a global landscape of high confidence open chromatin regions in HSCs.
Genes involved in cell division or cell differentiation are maintained as open chromatin in HSCs
To survey the global distribution of open chromatin regions throughout the whole genome, we categorized the open chromatin regions into 8 types: promoter-TSS, 5′ UTR, exon, intron, 3′ UTR, transcription termination sites, ncRNAs, and intergenic regions. As expected, open chromatin regions are highly enriched in promoter-TSS regions with more than 30 times in these areas compared with a size-matched random genomic background (Figure 2A, P < 10−100), which is corroborated by the observation that many promoter regions display ATAC-seq peak signals (Figures 1D-F). Next, we extracted the genes whose promoter-TSS regions displayed an open chromatin structure (defined as open promoters) for further analysis. From this analysis, we found 5622 genes have open chromatin in their promoters regions in all 3 types of HSCs (LT-HSCs, ST-HSCs, and MPPs). We also found that MPPs have the highest number of specific genes that displayed open promoters (Figure 2B). We were very interested in which populations of genes are most likely to have their promoter regions maintained as open chromatin state in the different categories of HSCs. To answer this question, we performed GO enrichment analysis on the total set of open chromatin promoters for the 3 cell types and found that genes involved in regulating cell cycle, cell division, mitosis, DNA replication, DNA damage, and DNA repair are highly enriched in those with open promoters in all 3 populations; these GO terms are enriched more significantly in MPPs and ST-HSCs than with LT-HSCs, which is a relatively quiescent stem cell population that has a major function in self-renewal. Taken together, these results suggested that more gene-encoding proteins involved in these cellular processes are required for regulating cell differentiation in MPPs and ST-HSCs than controlling self-renewal in LT-HSCs (Figure 2C). In addition, we found enrichment for open chromatin in the promoters of genes encoding proteins involved in ubiquitin conjugation, alternative splicing, and the unfolded protein response in ST-HSCs, suggesting that those genes might play critical roles in the transition from LT-HSCs to MPPs (Figure 2C). Taken together, our results suggest that genes involved in cell division or cell differentiation are frequently maintained in an open chromatin state in HSCs.
Figure 2.

The open chromatin state of protein coding genes and miRNAs changes dynamically in HSCs. (A) Classification of the genomic features in an open chromatin configuration as determined by our ATAC-seq analysis. TTS, transcription termination site. (B) The overlap of genes with an open chromatin configuration in their promoter-TSS region among LT-HSCs, ST-HSCs, and MPPs. (C) Heat map showing enrichment (-log10 [FDR]) of GO terms in gene set with ATAC-seq peaks found in their promoter regions. (D) Heat map of ATAC-seq peak scores ranking by LT-HSCs for the promoter-TSS regions of miRNAs. FDR, false discovery rate.
Next, we examined the relationship between open chromatin in gene promoters and gene expressional level globally in each cell type of HSCs. We found ATAC-seq signal in promoter 1 kb were positively correlated with the gene expression activity (r = 0.44, r = 0.384, and r = 0.379 in LT-HSCs, ST-HSCs, and MPPs, respectively). We then ranked the gene expression activity from high to low, and divided them into 5 groups equally. ATAC-seq signals were increased as a function of gene expression activity, especially in the categories of highly expressed genes but less correlated in low expressed genes (supplemental Figure 4A-C), indicating that open chromatin state in promoter regions were necessary but not sufficient for the gene transcription, which also need the activation by related transcription factors. Furthermore, we performed GO enrichment analysis for top 3000 highest expressed genes in LT-HSCs, and found genes related to cell cycle were also enriched in expression level but not in top. This result was consistent with that as compared with somatic cell, that LT-HSCs can undergo cell division to renew LT-HSCs or produce ST-HSCs, and it also indicated that genes involved in cell cycle were generally maintained in open chromatin state LT-HSCs but some of them need transcription factors to further active their expression level in ST-HSCs and MPPs.
Multiple miRNA encoding regions are found in an open chromatin state in HSCs
Regulatory ncRNAs play important roles in hematopoiesis.33 Accumulating evidence suggests that microRNAs (miRNAs) are critical for HSC maintenance and that many of them display a cell type-specific expression pattern.34 Therefore, we surveyed the open chromatin state of miRNAs in HSCs and found that 40 miRNAs are in an open chromatin state in at least 1 type of HSCs (Figure 2D). Among those 40 miRNAs, miR-7b, miR-7671, and miR-8111 exhibited higher ATAC-seq signal in LT-HSCs, whereas miR-150 and miR-155 exhibited higher ATAC-seq signal in MPPs (Figure 2D). miR-150 and miR-155 function drives MEP differentiation toward megakaryocytes and controls B- and T-cell differentiation.35 Our results suggest that the open chromatin state for these 2 miRNAs is already established in MPPs before it starts to develop into further differentiated lineages.
Chromatin accessibility in promoter regions of many genes changes upon HSC differentiation
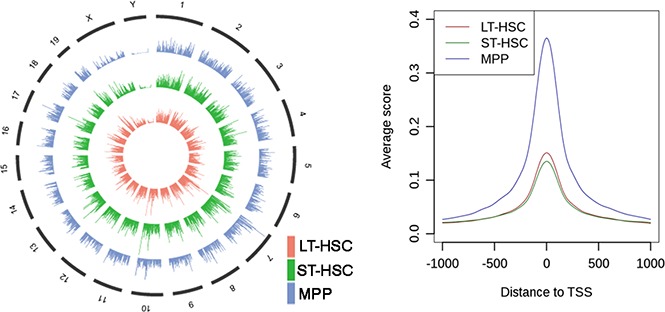
To further understand how chromatin accessibility changes when HSCs differentiate to MPPs, we comprehensively examined the flanking 1 kb regions of gene TSSs. First, we focused on the genes with peaks in the promoter-TSS regions that were common across the 3 cell types; we found peaks were centralized at the TSS in all 3 populations (LT-HSCs, ST-HSCs, MPPs) (Figures 3A-B). From this analysis, we also found that the average ATAC-seq score in these regions in MPPs is significantly higher (χ2 test with P < 10−100) when compared with this signal in LT-HSCs and ST-HSCs, which was also observed for the subset of miRNA regions (described previously) (Figures 2D and 3B). These results indicated that the promoter regions of those genes were occupied less frequently by nucleosomes in the population of MPPs compared with LT-HSCs and ST-HSCs. Next, we surveyed the ATAC-seq signal in promoters of all annotated mouse genes. We found that ATAC-seq read density in flanking TSS regions of all genes again showed that genes in MPPs exhibited globally stronger ATAC-seq signal compared with LT-HSCs and ST-HSCs (supplemental Figure 5A). Interestingly, we found that ATAC-seq signal is focused directly over TSSs in MPPs; instead, periodical peaks and troughs were observed across the upstream regions flanking TSSs in LT-HSCs and ST-HSCs (supplemental Figure 5B-C). The troughs, which are depleted for ATAC-seq reads, likely represent nucleosome-occupied positions, further indicating that more genes have their promoter regions compressed into a more closed chromatin state by nucleosomes in LT-HSCs and ST-HSCs compared with MPPs. This may be a consequence of fewer genes being required to maintain self-renewal of LT-HSCs and ST-HSCs compared with controlling the diverse differentiation processes that MPPs can undergo.
Figure 3.

Chromatin remodeling and enrichment of DNA cis-elements in the promoter regions upon HSC differentiation to MPPs. (A) Heat map of reads density around gene promoters displaying an open chromatin state (from −1000 bp to 1000 bp) in LT-HSCs, ST-HSCs and MPPs, respectively. (B) The average reads density (smoothed) around gene promoters displaying an open chromatin state (from −1000 bp to 1000 bp) in LT-HSCs, ST-HSCs, and MPPs, respectively. (C) Heat map of significant enrichment value (-log [P value]) of transcription factor–binding motifs in gene promoters displaying an open chromatin state. (D) PCA for the enriched DNA motifs in open promoters within the 3 cell types. The first 2 components account for 90.67% and 9.33% of the total variance in the dataset, respectively. Dots represent the PCA loadings for the 166 transcription factors. The different colored dots represent 3 different cohorts. The motif of Krüppel-like factors (Klf9/Klf10) is displayed (top left). (E) Gene expression activity of Klf9 and Klf10 in LT-HSCs, ST-HSCs, and MPPs. Gene Expression Commons (https://gexc.stanford.edu). Gene expression activity ranged from −100 (most inactive) to 100 (most active).
Transcription factor cohorts are associated with cell type-specific open promoters in HSCs
To investigate which transcription factors could potentially access the DNA cis-elements in open promoter regions as determined by our ATAC-seq experiments, we performed DNA sequence motif enrichment analysis using the sequences of all promoter regions from each cell type that our ATAC-seq data found to be in an open confirmation. From this analysis, we found a total of 166 known transcription factor binding motifs significantly enriched in the open promoters as identified by our ATAC-seq analysis in at least 1 cell type of HSCs (Figure 3C; supplemental Figure 6A). Within this total population, 122 of the known transcription factor–interacting motifs were found in open chromatin regions that were common to all 3 HSCs. Furthermore, we did not identify transcription factor binding motifs that were unique within the open chromatin regions identified by our ATAC-seq analysis of ST-HSCs, suggesting that the transcriptional regulatory network of ST-HSCs is in a transitional state between LT-HSCs and MPPs (Figure 3C; supplemental Figure 6A). To further explore the transcription factor cohorts that can distinguish the 3 types of HSCs, we performed PCA using the enriched motifs that we uncovered. From this analysis, we found that along PC1, the cohort of Krüppel-like factors KLF9, KLF10, and KLF14, and ETS transcription factor ETV2, whose binding motifs were specifically enriched in LT-HSCs, were separated from other transcription factors that can access the open promoter regions that are common to the 3 types of HSCs (Figures 3C-D). Interestingly, the gene expression activity of both KLF9 and KLF10, transcription factors that are known to bind C-rich motifs, is higher in LT-HSCs compared with ST-HSCs and MPPs, indicating their potential function in LT-HSCs (Figures 3D-E). Along PC2, we found that another transcription factor cohort that includes SP1, KLF5, ETS1, FLI1, ERG, and GABPA were separated from all others (Figure 3D). Among them, FLI1 is involved in early hematopoietic development,36 whereas GABPA is a critical regulatory module for maintenance and differentiation of HSCs.37 Taken together, our results suggest that these transcription factor cohorts may play important functions during the differentiation of LT-HSCs and ST-HSCs to MPPs.
Chromatin accessibility of enhancer elements changes during hematopoietic differentiation
Next, we focused on identifying the enhancer elements that were in an open chromatin state in the 3 types of HSCs. Enhancer elements are important cis-acting sequences that can mediate long-range activation of transcription of specific target genes. They tend to be located within 1 Mb upstream or downstream of the transcription start site of the gene(s) targeted for regulation.38 Active enhancers are usually marked by histone H3 lysine 27 acetylation (H3K27ac) and H3 lysine 4 monomethylation (H3K4me1), whereas poised enhancers are often marked by H3K4me1 alone.39-41 Therefore, we analyzed the 48 415 putative enhancer regions that have been demonstrated to be marked by high H3K4me1/2 and low H3K4me3.10 We took this list of 48 415 putative enhancer regions and performed an overlap analysis with open chromatin regions identified in our ATAC-seq experiments. From this analysis, we identified 3266, 2424, and 7196 putative enhancer elements that were found in an open chromatin state (defined as open enhancers) in LT-HSCs, ST-HSCs, and MPPs, respectively (Figure 4A). To survey which enhancers were evolutionally conserved between mouse and human, we compared the open enhancers in mouse with enhancers marked by H3K4me1 in human9 through LiftOver tools. A total of 1464, 1094, and 3108 open enhancers in LT-HSCs, ST-HSCs, and MPPs, respectively, in mouse were evolutionally conserved in human; ∼50% of them (775, 533, and 1591 in LT-HSCs, ST-HSCs, and MPPs respectively) were also marked as enhancers in human CD133+ cells, suggesting they may have an important function in HSCs.
Figure 4.

Enrichment of DNA cis-elements in enhancers that display an open chromatin configuration in hematopoietic cells. (A) The overlap of enhancers displaying an open chromatin state in LT-HSCs, ST-HSCs, and MPPs. (B) The overlap of enriched transcription factor–binding motifs found in both promoters and enhancers that display an open chromatin state in our ATAC-seq analysis. (C) PCA for the enriched DNA motifs in open enhancers within the 3 cell types. The first 2 components account for 99.21% and 0.79% of the total variance in the dataset, respectively. Dots represent the PCA loadings for the 153 transcription factors. The different colored dots represent 3 different cohorts. (D-H) Metaplot of ATAC-seq signal around the flanking 1 kb of PU.1, SPIB, ELF5, CCCTC-binding factor (CTCF), and BORIS binding motifs. (I) Venn diagram showing the overlap of poised enhancers in LT-HSCs and ST-HSCs and active enhancers in MPPs.
Through motif discovery analysis, we found 153 DNA sequences that are known to interact with specific transcription factors that were enriched in these putative open enhancer elements (supplemental Figure 4B-C). Similar to the results from this same analysis of promoter regions that demonstrate open chromatin, we did not find any transcription factor interacting motifs that are specifically within the enhancers that show an open chromatin configuration in our ATAC-seq analysis of ST-HSCs (supplemental Figure 6B-C). Interestingly, we uncovered 96 transcription factor interacting motifs that are in both promoter and enhancer regions and that are in an open chromatin configuration within the 3 HSC cell types, suggesting these transcription factors may direct the interaction between specific enhancer elements and their target promoters (Figure 4B). Next, we performed PCA to identify transcription factor cohorts that contribute to the variation of open enhancers within HSCs (Figure 4C). Along PC1, we found a cohort of 9 transcription factors including ERG, GABPA, PU.1, and SPIB were separated from others (Figures 4C-I; supplemental Figure 7). In support of these findings, our ATAC-seq analyses also demonstrated that the enrichment of PU.1, SPIB, and ELF5 binding motifs were decreased in LT-HSCs compared with ST-HSCs and MPPs (Figures 4D-F). Along PC2, we found the motifs for CCCTC-binding factor and BORIS, factors that function in regulating chromatin architecture and transcriptional reprogramming,42,43 were separated from other overrepresented sequence motifs (Figures 4C,G-H). In summary, these 2 cohorts of transcription factors may be associated with dynamic change of open enhancers within the 3 cell types of HSCs.
Poised enhancer elements are established in progenitor cells before they commit to differentiate into specific cell lineages. Then they can become activated to induce expression from specific target genes during subsequent lineage differentiation processes.7 To survey how potentially poised enhancers change during differentiation of LT-HSCs and ST-HSCs to MPPs, we distinguished poised enhancers marked only by H3K4me1 from active enhancers marked by both H3K4me1 and H3K27ac in HSCs. Interestingly, we found 1140 poised enhancers adopt an open chromatin state in LT-HSCs, and 28.2% of these become active enhancers when LT-HSCs differentiate to ST-HSCs (supplemental Figure 6D). Furthermore, we also found that a total of 21.7% of the poised enhancers identified in LT-HSCs and ST-HSCs become active enhancers in MPPs (Figure 4I). Taken together, our results demonstrate that chromatin accessibility can significantly affect the global DNA accessibility for transcription factors at promoter and enhancer regions in different populations of HSCs.
HSCs have the capability to self-renew and differentiate into all blood cell lineages. These 2 opposing forces are finely coordinated by several regulatory mechanisms, consisting of both extrinsic and intrinsic factors. Over the past decade, several studies have discovered the key role of transcription factors, niche factors, and signal transduction pathways in HSC development and homeostasis.1,2 Increasing evidence, however, suggests that a higher level of intrinsic regulation exists (eg, epigenetic regulation, which controls chromatin accessibility can directly regulate HSC development, maintenance of self-renewal, lineage commitment, and aging).8-12 Moreover, aberrant epigenetic marks have been identified in several hematological malignancies, consistent with clinical evidence that mutations targeting epigenetic regulators promote leukemogenesis.44 Our study provides the first chromatin accessibility landscape maps for LT-HSCs, ST-HSCs, and MPPs. It is known that open chromatin is important for transcription factor binding and subsequent regulation of gene expression. Consistent with this notion, we found that open chromatin is highly enriched in promoter regions of coding and noncoding genes in all 3 populations of HSCs profiled here. Furthermore, the ATAC-seq signal in promoter regions of genes is usually correlated with the level of gene expression at that locus. In addition, we identified open chromatin in MPPs for the regions where the transcripts of 40 miRNAs such as mR-7b, miR-150, and miR-155 are found. This is intriguing because the target transcripts of miR-7b are involved in breast cancers,45 and miR-150 and miR-155 are known to play an essential role in hematopoiesis.35 Overall, we identified 7931, 6844, and 9110 genes displaying an open chromatin configuration at their promoter regions in LT-HSCs, ST-HSCs, and MPPs, respectively. The peak number in ST-HSCs is smaller than in LT-HSCs, which may suggest that during the transition state, genes specifically required in LT-HSCs were turned off; many genes have already been maintained in open chromatin state in LT-HSCs but need to be further activated by transcriptional factors in ST-HSCs and MPPs. MPPs, as multipotent stem cells that can differentiate into many types of cells, contain the largest number of open chromatin regions. However, once the MPPs differentiate to more specific cells, the number of open chromatin regions may be decreased. This hypothesis needs further evidence. Furthermore, we discovered that genes with the highest abundance of ATAC-seq signal for open chromatin in their promoters are strongly enriched for those encoding proteins involved in cell division and DNA replication, suggesting that those 2 processes are critical for HSCs playing a role as a stem cell. A recent study using single-cell sequencing suggests the relationship between cell cycle and self-renewal vs differentiation of HSCs is affected by aging.46 We found genes involved in cell cycle were also enriched in highly expressed genes in LT-HSCs (supplemental Figure 4D), consistent with that LT-HSCs can undergo renewal themselves or symmetrically or asymmetrically divide to ST-HSCs or MPPs.
We also found that the promoter regions of many more genes are found in a compressed chromatin state in LT-HSCs and ST-HSCs compared with MPPs, suggesting that only a few genes are involved in regulating self-renewal of HSCs; in contrast, many more genes function in regulation of blood cell differentiation. Enrichment analysis of specific DNA cis-elements in the open chromatin regions identified in our ATAC-seq analyses uncovered a potential transcription regulatory network that contains the known binding sites of at least 166 transcription factors in HSCs. Among them, the binding motifs of GABPA and FLI1 were identified in our analyses, and these 2 proteins are well-known to play important roles in HSC differentiation.4-35 Interestingly, one-quarter of these transcription factor binding sites were found in open chromatin regions that were identified to be cell type–specific, which provides important clues about the differential transcription regulatory networks in promoter regions that may be important in the differentiation of LT-HSCs to ST-HSCs and MPPs. Through PCA, we identified the DNA cis-element known to be bound by the Krüppel-like factor family (KLF9, KLF10, and KLF14) to be enriched specifically in open chromatin regions that were specific to LT-HSCs, which is the cell type that KLF9/KLF10 are known to highly express, suggesting that they may play role in the self-renewal of LT-HSCs.
Finally, we identified 8299 enhancer elements that display an open chromatin configuration in our ATAC-seq analysis of HSCs. Within this group of open enhancer elements, we uncovered the known interaction sites of 153 transcription factors. Importantly, we identified overrepresentation for the known binding motifs of 96 transcription factors, such as GABPA, that are enriched in both enhancer and promoter sequences that our ATAC-seq analysis in HSCs finds to be in an open configuration. These results suggest that it is these transcription factors that drive the enhancer-mediated activation of the genes downstream of the open promoters to regulate gene expression networks during blood cell differentiation. Using PCA, we identified 2 cohorts of transcription factors that were associated with dynamic changes in open chromatin at the enhancers within 3 populations of HSCs. In total, our study reveals the genome-wide landscape of open chromatin in HSCs; these HSC chromatin accessibility data can be used to help facilitate our understanding of self-renewal and differentiation of HSCs.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Peter Klein at University of Pennsylvania and Yuri Persidsky at Temple University School of Medicine for their insightful comments and discussion, all the members of Gregory and Huang laboratories for their help and discussions, and Yuesheng Li from the Genomic Facility of Fox Chase Cancer Center for help with the deep sequencing.
This study is supported by a grant from the National Institutes of Health, National Heart, Lung, and Blood Institute (R00 HL107747-04) (J.H.).
Authorship
Contribution: J.H. designed the study; J.H. and C.W. performed the experiments; D.B., H.W., and B.D.G. provided crucial input for the project; X.Y. performed the bioinformatics analysis; and J.H., X.Y., and B.D.G. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jian Huang, Department of Pathology and Laboratory Medicine, School of Medicine, Temple University, 3500 N. Broad St, MERB 845B, Philadelphia, PA 19075; e-mail: jianh@temple.edu; and Brian D. Gregory, Department of Biology, University of Pennsylvania, 103D Carolyn Lynch Laboratories, Philadelphia, PA 19104; e-mail: bdgregor@sas.upenn.edu.
References
- 1.Giebel B, Bruns I. Self-renewal versus differentiation in hematopoietic stem and progenitor cells: a focus on asymmetric cell divisions. Curr Stem Cell Res Ther. 2008;3(1):9-16. [DOI] [PubMed] [Google Scholar]
- 2.Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2(6):640-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eaves CJ. Hematopoietic stem cells: concepts, definitions, and the new reality. Blood. 2015;125(17):2605-2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354(17):1813-1826. [DOI] [PubMed] [Google Scholar]
- 6.Göttgens B. Regulatory network control of blood stem cells. Blood. 2015;125(17):2614-2620. [DOI] [PubMed] [Google Scholar]
- 7.Qiao W, Wang W, Laurenti E, et al. . Intercellular network structure and regulatory motifs in the human hematopoietic system. Mol Syst Biol. 2014;10(7):741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attema JL, Papathanasiou P, Forsberg EC, Xu J, Smale ST, Weissman IL. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc Natl Acad Sci USA. 2007;104(30):12371-12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui K, Zang C, Roh TY, et al. . Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4(1):80-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, et al. . Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345(6199):943-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng X, Yue S, Chen H, Weber B, Jia J, Zheng Y. Low-cell-number epigenome profiling aids the study of lens aging and hematopoiesis. Cell Reports. 2015;13(7):1505-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corces MR, Buenrostro JD, Wu B, et al. . Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48(10):1193-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci. 2016;1370(1):82-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1(8):661-673. [DOI] [PubMed] [Google Scholar]
- 15.Veillette A, Latour S. The SLAM family of immune-cell receptors. Curr Opin Immunol. 2003;15(3):277-285. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Ellison FM, Keyvanfar K, et al. . Enrichment of hematopoietic stem cells with SLAM and LSK markers for the detection of hematopoietic stem cell function in normal and Trp53 null mice. Exp Hematol. 2008;36(10):1236-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109-1121. [DOI] [PubMed] [Google Scholar]
- 18.Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009;75(1):14-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purton LE, Scadden DT. Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell. 2007;1(3):263-270. [DOI] [PubMed] [Google Scholar]
- 20.Osawa M, Hanada K, Hamada H, Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273(5272):242-245. [DOI] [PubMed] [Google Scholar]
- 21.Christensen JL, Weissman IL. Flk-2 is a marker in hematopoietic stem cell differentiation: a simple method to isolate long-term stem cells. Proc Natl Acad Sci USA. 2001;98(25):14541-14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adolfsson J, Månsson R, Buza-Vidas N, et al. . Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121(2):295-306. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Bryder D, Adolfsson J, et al. . Identification of Lin(-)Sca1(+)kit(+)CD34(+)Flt3- short-term hematopoietic stem cells capable of rapidly reconstituting and rescuing myeloablated transplant recipients. Blood. 2005;105(7):2717-2723. [DOI] [PubMed] [Google Scholar]
- 24.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:29.1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinz S, Benner C, Spann N, et al. . Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seita J, Sahoo D, Rossi DJ, et al. . Gene Expression Commons: an open platform for absolute gene expression profiling. PLoS One. 2012;7(7):e40321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen JY, Miyanishi M, Wang SK, et al. . Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530(7589):223-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park IK, Qian D, Kiel M, et al. . Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423(6937):302-305. [DOI] [PubMed] [Google Scholar]
- 30.Zeng H, Yücel R, Kosan C, Klein-Hitpass L, Möröy T. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 2004;23(20):4116-4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou P, Yoshihara H, Hosokawa K, et al. . p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9(3):247-261. [DOI] [PubMed] [Google Scholar]
- 32.Wilson A, Murphy MJ, Oskarsson T, et al. . c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18(22):2747-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeong M, Goodell MA. Noncoding regulatory RNAs in hematopoiesis. Curr Top Dev Biol. 2016;118:245-270. [DOI] [PubMed] [Google Scholar]
- 34.O’Connell RM, Chaudhuri AA, Rao DS, Gibson WS, Balazs AB, Baltimore D. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proc Natl Acad Sci USA. 2010;107(32):14235-14240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasilatou D, Papageorgiou S, Pappa V, Papageorgiou E, Dervenoulas J. The role of microRNAs in normal and malignant hematopoiesis. Eur J Haematol. 2010;84(1):1-16. [DOI] [PubMed] [Google Scholar]
- 36.Pimanda JE, Ottersbach K, Knezevic K, et al. . Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc Natl Acad Sci USA. 2007;104(45):17692-17697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu S, Cui K, Jothi R, et al. . GABP controls a critical transcription regulatory module that is essential for maintenance and differentiation of hematopoietic stem/progenitor cells. Blood. 2011;117(7):2166-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nat Rev Genet. 2013;14(4):288-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15(4):272-286. [DOI] [PubMed] [Google Scholar]
- 40.Creyghton MP, Cheng AW, Welstead GG, et al. . Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107(50):21931-21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heintzman ND, Stuart RK, Hon G, et al. . Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311-318. [DOI] [PubMed] [Google Scholar]
- 42.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137(7):1194-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pugacheva EM, Rivero-Hinojosa S, Espinoza CA, et al. . Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome Biol. 2015;16(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cullen SM, Mayle A, Rossi L, Goodell MA. Hematopoietic stem cell development: an epigenetic journey. Curr Top Dev Biol. 2014;107:39-75. [DOI] [PubMed] [Google Scholar]
- 45.Foekens JA, Sieuwerts AM, Smid M, et al. . Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci USA. 2008;105(35):13021-13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kowalczyk MS, Tirosh I, Heckl D, et al. . Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25(12):1860-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.