

Key Points
Novel GM-CSF signaling pathways through IFN-γR/IRF-1 and AKT/mTOR provide monocyte licensing for suppressor function.
Only licensed but not fresh Ly-6Chigh murine or human CD14+ monocytes secrete nitric oxide or IDO for T-cell suppression.
Abstract
Granulocyte-macrophage colony-stimulating factor (GM-CSF) controls proliferation and survival of myeloid cells including monocytes. Here, we describe a time-dependent licensing process driven by GM-CSF in murine Ly6Chigh and human CD14+ monocytes that disables their inflammatory functions and promotes their conversion into suppressor cells. This 2-step licensing of monocytes requires activation of the AKT/mTOR/mTORC1 signaling cascade by GM-CSF followed by signaling through the interferon-γ receptor (IFN-γR)/interferon regulatory factor-1 (IRF-1) pathway. Only licensing-dependent adaptations in Toll-like receptor/inflammasome, IFN-γR, and phosphatidylinositol 3-kinase/AKT/mTOR signaling lead to stabilized expression of inducible nitric oxide synthase by mouse and indoleamine 2,3-dioxygenase (IDO) by human monocytes, which accounts for their suppressor activity. This study suggests various myeloid cells with characteristics similar to those described for monocytic myeloid-derived suppressor cells, Mreg, or suppressor macrophages may arise from licensed monocytes. Markers of GM-CSF–driven monocyte licensing, including p-Akt, p-mTOR, and p-S6, distinguish inflammatory monocytes from potentially suppressive monocytes in peripheral blood of patients with high-grade glioma.
Visual Abstract
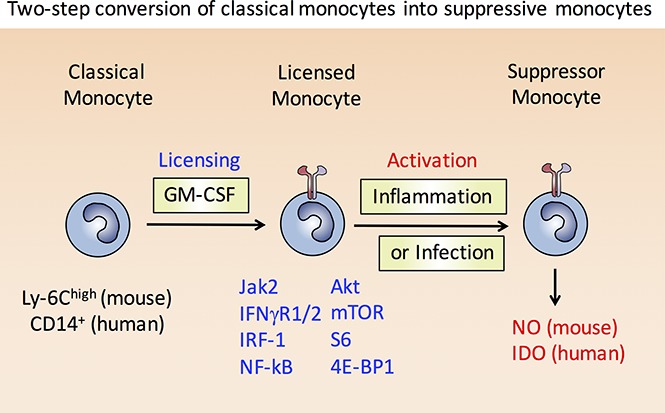
Introduction
It is a puzzle that monocytes should be capable of such opposing immunological activities, especially considering there is no definitive marker profile that allows the reliable and universal demarcation of inflammatory monocytes and monocytic suppressor cells in either mice or humans. Monocytes are essential for protective immunity during acute infections and can augment autoimmune reactions,1 whereas monocytic suppressor cells interfere with immune responses against tumors and chronic infections.2,3 Both inflammatory monocytes and monocytic suppressor cells can generate Nitric oxide (NO) via the inducible isoform of NO synthase (iNOS/NOS2) from the amino acid l-arginine.3-5 Monocytes and macrophages use NO for the killing of pathogens and tumor cells, but in monocytic suppressor cells, iNOS activity causes impaired T-cell function.3,4 Production of NO by mouse monocytes or monocytic suppressor cells can be elicited by interferon-γ (IFN-γ) signaling in combination with a second stimulus, which might be a pathogen-associated factor6 or endogenous inflammatory factors produced by T cells or tumors.7 Confusingly, inflammatory monocytes and monocytic suppressor cells can respond to the same stimuli, often through the same receptor, in opposite ways, as illustrated by the apparently paradoxical effects of IFN-γ8 or ligation of DC-SIGN.9 Thus, despite having cataloged many developmental influences and functional properties of monocytes and monocytic suppressor cells, we lack an integrated understanding of the fundamental differences between inflammatory monocytes and monocytic suppressor cells that accounts for their profoundly different behavior.
Both inflammatory monocytes and monocytic suppressor cells depend on the hematopoietic growth factors granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF), which promote their development in vitro and in vivo.10-14 The ambiguous consequences of GM-CSF, M-CSF, and IFN-γ stimulation in the development of inflammatory monocytes versus monocytic suppressor cells points to the intersection of the GM-CSF/M-CSF signaling cascade with the IFN-γ/interferon regulatory factor-1 (IRF-1) pathway as a key determinant of cell fate. Activation of the phosphatidylinositol 3-kinase (PI3K) and AKT pathways in monocytes after GM-CSF stimulation is now well-documented.15-17 Furthermore, a critical role for PI3K has been described for M-CSF–dependent tumor associated macrophages18 and for mTOR in myeloid-derived suppressor cells (MDSCs).19 However, the mechanisms by which GM-CSF signals control downstream mediators of monocytic suppressor function are unknown.
To clearly dissect the effects of GM-CSF signaling on the IFN-γ/IRF-1 pathway, independently of other growth factor–derived signals, we turned to well-established culture methods for converting GM-CSF–stimulated murine and human monocytes into monocytic suppressor cells.13,20,21 Results from these systems suggest that suppressive monocytes do not arise from a specific lineage-defined precursor cell, but represent a differently polarized state of blood monocytes. Reaching this state involves 2 steps: first, a “licensing” step that requires GM-CSF to activate the PI3K/AKT/mTOR pathway, which results in structural changes to the IFN-γR signaling pathway; second, stimulation of licensed monocytes with IFN-γ then leads to acquisition of suppressor function through posttranscriptionally stabilized expression of iNOS in mice or indoleamine 2,3-dioxygenase (IDO) in humans. Our data indicate that GM-CSF licensed monocytes (L-Mono) serve as a precursor to suppressive monocytes, which some might describe as monocytic-MDSC (M-MDSC). This study has important implications for the therapeutic use of GM-CSF (Sargramostim) and mTOR inhibitors (eg, rapamycin, everolimus) in cancer and transplant patients.
Materials and methods
Mice
C57BL/6, Nos2−/−(purchased from Jackson Laboratories, Bar Harbor, ME), Irf1−/−,22 Ifngr1−/−,23 and Ifng1−/−24 mice were bred in our own animal facilities at Würzburg, Marburg, or Erlangen, kept under specific pathogen-free conditions, and used at an age of 4 to 10 weeks. All animal experiments were performed according to the German animal protection law as well as after approval and under control of the local authorities.
Patient material
In a first analysis, plasma samples from 52 patients with primary or relapsed high-grade gliomas were obtained preoperatively. Plasma from 9 healthy individuals served as a control. GM-CSF was measured using the human Magnetic Cytokine Kit (LHC6003M, Life Technologies) on a MagPix device (Luminex), for M-CSF measurements, an enzyme-linked immunosorbent assay system (Duoset, R&D Systems) was used. For fluorescence-activated cell sorter (FACS) analysis, we collected anonymized EDTA-blood samples from healthy individuals and pediatric patients with advanced solid tumors (untreated primary or progressing refractory). Both analyses have been reviewed by the institutional review board of the University of Würzburg (#135/09 and #102/14), and participants gave their written informed consent.
L-Mono preparation
Murine bone marrow (BM)-derived bulk L-Mono were generated as described before.25 Human L-Mono were generated as described previously.20 Briefly, CD14+ monocytes were isolated from Ficoll-prepared peripheral blood mononuclear cells by positive-selection with anti-CD14 microbeads (Miltenyi, Bergisch-Gladbach, Germany) and were then plated in 6-well Cell+ plates (Sarstedt, Nümbrecht, Germany) at 106 cells/well in RPMI-1640 (Lonza, Cologne, Germany) supplemented with 10% heat-inactivated human AB serum (ZKT, Tübingen, Germany), 2 mM Glutamax (Invitrogen, Karlsruhe, Germany), 100 U/mL penicillin (Lonza), 100 μg/mL streptomycin (Lonza), and recombinant human GM-CSF (rhGM-CSF; R&D Systems, Wiesbaden-Nordenstadt, Germany) at 25 ng/mL carried on 0.1% human albumin (CSL-Behring, Hattersheim-am-Main, Germany). Unless otherwise indicated, cells were stimulated on day 6 of culture for a further 18 to 24 hours with 25 ng/mL rhIFNγ (Chemicon, Billerica, MA).
Reagents
Murine recombinant GM-CSF (200 U/mL), M-CSF (50 U/mL), granulocyte colony-stimulating factor (G-CSF; 50 U/mL), interleukin-6 (IL-6; 50 ng/mL), IL-10 (40 ng/mL), IFN-γ (0.5 μg/mL), and rhFlt3L (100 U/mL) were purchased from Immunotools. Mouse recombinant TNF (500 U/mL) was purchased from Peprotech. Mouse recombinant IL-1β (40 ng/mL) was purchased from eBioscience. The PI3K inhibitors wortmannin (2-10 µM) and Ly294002 (2-10 µM) and lipopolysaccharide (LPS; 100-1000 ng/mL) were purchased from Sigma Aldrich, and rapamycin (1-500 nM, selective mTORC1 inhibitor) from Selleckchem.
Cell sorting and flow cytometry
The murine antibodies used: CD11b-PerCP-Cy5.5 (M1/70), CD11b-fluorescein isothiocyanate (FITC; M1/70), Ly-6G-phycoerythrin (PE; 1A8), Biotin-CD119 (IFN-γR1; clone 2E2), the streptavidin-PE-Cy5 (all BioLegend); Ly-6C-Alexa 647 (ER-MP20, Ab Serotec), FITC-IFN-γR2 (MOB-47, Santa Cruz), CD43-FITC (eBioscience), anti-rabbit DyLight488 (Jackson), IL-6-PE, and iNOS/NOS type II (BD). Human and mouse phosphorylated molecules: p-AKT (S473, SDRNR, eBioscience), p-mTOR (MRRBY, eBioscience), p-STAT1 (pY701, BD), p-STAT3 (pY705, BD), Alexa Fluor647-P-S6 (S235/236, D57.2.2E, Cell Signaling), or PerCP-eFluor710- P-S6 (cupk43k, eBioscience). Staining was performed as described elsewhere.26
Cytospins and confocal microscopy
Sorted cells (5 × 105) were centrifuged onto a glass slide by cytospin at 600g for 5 minutes. Purified anti-IRF-1 (Cell Signaling), IFN-γR1 (CD119) biotin-conjugated, and IFN-γR2-FITC conjugated were diluted 1:100 in phosphate-buffered saline and incubated overnight at 4°C before anti-fluorescein Alexa Fluor 488 (Millipore) or streptavidin-DyLight549 (BioLegend) for 1 hour at room temperature. Nuclei were stained using DRAQ5 (eBioscience). Slides mounted with Fluoromount-G (SouthernBiotech) were analyzed by confocal laser-scanning microscope (LSM 510 Meta, Zeiss).
Western blot
Cells were lysed in ice-cold Triton-X100 lysis buffer and left for 30 minutes on ice. Membrane extraction and preparation was performed using the Mem-PER kit (Thermo Scientific) following the manufacturer's instructions. Proteins were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, followed by semidry western blotting onto a polyvinyl fluoride membrane (Whatman, GE Healthcare). Antibodies against murine Jak1 (#3344), Jak2 (#3230), pY701-STAT1 (#9171), STAT1 (#9172), pY705-STAT3 (#9183), STAT3 (#9139), pS473-AKT (#4060), AKT (#9272), glyceraldehyde-3-phosphate dehydrogenase (#2118), anti-rabbit-horseradish peroxidase (HRP) (#7074), P-4E-BP1 (#2855P), p-mTOR (#5536P), anti-mouse-HRP (#7076), and anti-biotin-HRP (#7727) (all Cell Signaling), Rel A p65 (clone F-6, Santa Cruz), NF-κB p100 (polyclonal rabbit, Cell Signaling), MyD88 (Millipore), anti-iNOS (Calbiochem), human IDO (clone 10.1, Merck Millipore), and human IRF1 (clone d5e4, Cell Signaling Technology). PageRuler plus prestained protein ladder (SM1811) from Fermentas.
Messenger RNA (mRNA) preparation and RT-PCR
After treatment of murine cells total RNA was isolated using the RNeasy kit (Qiagen) according to the manufacturer's instructions. Quantitative real-time reverse transcriptase polymerase chain reaction (RT-PCR) was performed using the FastStart Universal SYBR Green Master (Rox) Kit (Roche Diagnostics) according to the manufacturer's instructions. Primer sequences for quantitative real-time RT-PCR are as follows: murine gp130: forward: 5′-TCCCATGGGCAGGAATATAG-3′, reverse: 5′-CCATTGGCTTCAGAAAGAGG-3′; murine iNOS: forward: 5′-CTTTGCCACGGACGAGAC-3′, reverse: 5′-TCATTGTACTCTGAGGGCTGAC-3′; murine IRF1: forward: 5′- CTCTGCTGTGCGGGTGTA-3′, reverse: 5′- CCACACAGCTTCCTCTTGGT-3′. Quantification of -fold inductions over untreated samples was performed using the mathematical model described by Pfaffl.27
NO measurement
NO was measured as nitrite production using the Griess reaction.28 The evoked color reaction was measured after 10 minutes in the enzyme-linked immunosorbent assay reader (Molecular Devices) at 492 nm.
Proliferation assays
Murine bulk lymph node cells from BALB/c mice, used as a source of responder T cells, were seeded into a 96-well round-bottomed plate (CELLSTAR, Greiner bio-one), activated for proliferation by adding soluble anti-CD3 and anti-CD28 at a final concentration of 2.5 µg/mL each. After 3 days, cell proliferation was detected by 1 μCi/well (3H)-methyl-thymidine (Amersham) pulse for 16 hours. Alternatively, carboxyfluorescein diacetate succinimidyl ester (CFSE)- or eFluor670 (Invitrogen)-labeled T cells were analyzed by flow cytometry.20
Ex vivo suppressor assay
Mice were administered daily (intraperitoneally) with 2 µg of GM-CSF or Flt3L for a total of 10 days. At day 11, mice were euthanized and spleen (SP) and BM collected to isolate CD11b+ cells by MACS beads (Miltenyi Biotec) to be tested in a T-cell suppressor assay for 4 days.
EAE induction and scoring
Experimental autoimmune encephalomyelitis (EAE) induction was performed by a standard protocol.29 GM-CSF (2 μg/mouse) was injected intraperitoneally 10 days before until 5 days after EAE induction. Mice were scored daily for clinical disease symptoms according to the following scale: 0, no disease; 1, limp tail weakness; 2, hind limp weakness; 3, hind limp paralysis; 4, hind and fore limp paralysis; and 5, moribund or death. L-Mono treatment of mice was performed at day −4 of EAE induction by injecting 4 × 106 GM-CSF cultured L-Mono.
Statistics
Comparisons of data were analyzed by the tests indicated in each figure legend for the various types of assays using GraphPad Prism 5.0; in some cases, the Student t test with EXCEL 14.5.3 was used. Data from the experiments are presented as mean values ± standard error of the mean (SEM) or standard deviation (SD), as indicated. Differences of P < .05 were considered significant.
Results
GM-CSF licensing of murine monocyte suppressor function in vitro and in vivo
Earlier work established that GM-CSF acts not only as a growth factor or pro-inflammatory cytokine,30,31 but also conveyed suppressor function on myeloid cells.21,31 However, the relationship between duration of GM-CSF stimulation and acquisition of suppressor function is unclear. Although freshly isolated bone marrow cells did not suppress CD4+ or CD8+ T-cell proliferation in coculture, exposure of the same cells to GM-CSF for 3 days conferred a potent suppressor activity (Figure 1A). Similar results were obtained by isolating CD11b+ cells from BM or SP, and this suppressor function correlated with their capacity to release NO (supplemental Figure 1A-C). Predominantly, Ly-6C+ monocytic cells expressed iNOS, which confirmed that the effect of GM-CSF treatment was primarily mediated by monocytes (supplemental Figure 1D). GM-CSF could be substituted by monocyte-specific M-CSF to confer suppressor cell activity, but granulocyte-specific G-CSF or Flt3L were considerably weaker (Figure 1B). Acquisition of suppressor function required only very low doses of 0.3 ng/mL GM-CSF (equivalent to 5 U/mL) (Figure 1C), but high doses of M-CSF (≥10 ng/mL). Thus, we further will term monocytes that are programmed to become monocytic suppressor cells “licensed” monocytes (L-Mono).
Figure 1.

GM-CSF licensing is a prerequisite to develop suppressor activity in vitro and in vivo. Inhibition of T-cell proliferation was measured using a CFSE dilution assay. (A) Fresh murine BM cells or BM cultured for 1 to 3 days (d) in GM-CSF were added to anti-CD3/CD28–stimulated murine T cells at a ratio 10:1 (T cell/suppressor cell) for 4 days before determining CFSE dilution of T cells by FACS analysis. (B) T-cell suppressor assay where BM cells were used from 3-day cultures with 10 U/mL of GM-CSF or M-CSF, or 100 U/mL of Flt3L or G-CSF. Values correspond to the mean ± SD of 3 independent experiments. Statistics by 1-way analysis of variance (ANOVA) with multiple comparisons and Tukey posttest. *P < .05, ***P < .001. NS, not significant. (C) T-cell suppressor assay where BM cells were used from 3-day cultures with titrated doses GM-CSF (D) C57BL/6 mice were injected daily with GM-CSF or Flt3L with 2 μg/day for 10 days. Then CD11b+ cells from SP and BM were isolated and added to CFSE-labeled, CD3/CD28 antibody-stimulated T cells. Values correspond to the media of 5 independent experiments ± SD. Student t test indicated significance as *P < .05, ***P < .001 when compared with control mice (Ctrl). (E) Clinical autoimmune symptoms in the EAE model with or without preadministration of 4 × 106 L-Mono (day 4 of EAE, IV) in wild-type (WT) mice. Representative of 3 separate experiments. Two-way ANOVA with Bonferroni posttest. *P < .05, **P < .01. (F) L-Mono of WT, Nos2−/−, and Ifngr1−/− mice were tested for suppressive function by titrating them into cultures of CD3/CD28-stimulated T cells. Data represent means ± SD of 2 independent experiments. (G) The T-cell suppressor assay was performed by adding titrated amounts of resting L-Mono alone or L-Mono plus LPS and IFN-γ during the T-cell proliferation assay for 3 days before [3H]-Thymidine was added to the cultures overnight. Extrinsic LPS + IFN-γ activation further boosts suppressor activity of L-Mono by conversion into suppressor monocytes. Statistics by unpaired Student t test by comparing knock-out vs WT controls, ***P < .001.
A similar process of GM-CSF licensing was observed in vivo after 10 days of daily GM-CSF, but not Flt3L injections. GM-CSF treatment massively increased splenic volume and cellularity, as well as the frequency of Ly-6C+ and Ly-6G+ cells in BM and SP, whereas Flt3L was less effective in these respects (supplemental Figure 2A-B). GM-CSF but not Flt3L treatment facilitated NO production by CD11b+ cells isolated from BM or SP (supplemental Figure 2C), which correlated with their capacity to suppress CD4+ and CD8+ T-cell proliferation (Figure 1D). To confirm the in vivo effect of GM-CSF, licensing L-Mono were adoptively transferred into mice immediately before induction of EAE (Figure 1E). Mice receiving GM-CSF–stimulated L-Mono developed less severe disease than untreated controls, demonstrating the conversion of L-Mono into suppressive cells also on a T cell–mediated disease in vivo.
Our previous work showed that IFN-γ–induced iNOS expression was critical for the suppressor function of monocytic suppressor cells in vitro and in vivo. Therefore, we next investigated whether 3 days of exposure to GM-CSF alone was sufficient to drive conversion of CD11b+ monocytes into monocytic suppressor cells or whether IFN-γ–induced signaling and NO generation were also necessary. T cell–derived IFN-γ is critical for activation of the myeloid suppressor function in vitro (supplemental Figure 3A). GM-CSF–stimulated monocytes from IFN-γR1- or iNOS-deficient mice (Nos2−/−) failed to become suppressive (Figure 1F) or to produce NO (supplemental Figure 3B). Production of NO and the potency of monocytic suppressor cells was enhanced by LPS + IFN-γ (Figure 1G). Hence, exposing monocytes to GM-CSF is necessary, but not sufficient, to directly induce monocytic suppressor cells acting through NO. By extension, conversion of monocytes into monocytic suppressor cells can be regarded as a 2-step process: first, monocytes must receive GM-CSF licensing signals for at least 3 days; second, they must then be activated by IFN-γ and perhaps other T cell–derived pro-inflammatory cytokines.6,13,21
GM-CSF licensing does not require cell proliferation and targets only classical Ly-6Chigh monocytes are a source of M-MDSC
Because GM-CSF induces myeloid cell proliferation, GM-CSF could conceivably expand a precursor of suppressor cells that is distinct from inflammatory monocytes. To rule out this possibility, BM cells were eFluor670-labeled before culture with GM-CSF, M-CSF, G-CSF, or Flt3L to assess proliferation and development of CD11b+ Ly-6Clow Ly-6G+ granulocytic and CD11b+ Ly-6Chigh Ly-6G− monocytic suppressor cells.6,32,33 Surprisingly, Ly-6G was downregulated after a number of cell divisions resulting in a Ly-6G− granulocytic population among the dividing monocytes (Figure 2A-B, gate 4). Hence, Ly-6G has limited use as a marker of proliferating granulocytic suppressor cell subsets. When granulocytic Ly-6G+ cells were sorted and subsequently tested for suppressive capacity, they were not suppressive (Figure 2A, gates 1 and 2). In contrast, both nonproliferating (gate 5) and proliferating monocytes (gate 3) as well as the mixed population (gate 4) were suppressive to the same extent (Figure 2C).
Figure 2.

Only classical Ly-6Chighmonocytes acquire suppressor function and NO production independent of proliferation. Fresh murine BM cells were labeled with the cell proliferation dye eFluor670 and grown under the influence of either GM-CSF, M-CSF, G-CSF, or Flt3L at 50 U/mL each for 3 days. (A) Then, cells were harvested and counterstained for Ly-6G or for Ly-6C. Dotted line separates Ly-6Chigh from Ly-6Clow monocytes (1 representative of 4 experiments shown). (B) Murine BM cells cultured in GM-CSF within the gates as indicated in panel A were separated by cell-sorting and individual cytospin preparations were stained with hematoxylin and eosin (n = 2). The sorted cell populations show morphologies of differentiated neutrophils with segmented nuclei (1) that correspond to nondividing cells, dividing preneutrophils with band or ring-shaped nuclei (2) and dividing (3) and nondividing monocytes (5). Population 4 showed a mix composed of preneutrophilic and monocytic cells, indicating that preneutrophilic cells (2) that downregulated the Ly-6G marker during proliferation merge with proliferating monocytic cells. Scale bars (images 1-5), 10 μm. (C) T-cell suppressor assay testing the sorted cell populations as shown in panel A. Populations 1 and 2 were pooled. One representative experiment of 3 is shown. For statistics, granulocytes were compared with L-Mono from pooled data from 3 independent experiments. (D) BM cells of wild-type mice were labeled with eFluor670 and cultured with the indicated doses of GM-CSF for 3 days. Then eFluor670 dilution as a measure for proliferation was tested by FACS analysis (n = 2). (E) Identification of the monocyte subset that acquires T-cell suppressive activity following a 10-day treatment with GM-CSF. (Left) Gating strategy for separating total CD11b+ Ly-6G− myelomonocytic cells (excluding polymorphonuclear granulocytes [PMNs]) into Ly-6Chigh CD43+ classical monocytes, Ly-6Cint CD43+ intermediate monocytes, Ly-6C− CD43+ nonclassical monocytes, and Ly-6C− CD43− double negative cells. (Right) T-cell suppression assay with the sorted cell subsets. One representative experiment is shown. Statistics compare double negative vs classical monocytes from pooled data of 2 independent experiments. (F) Intracellular IL-6 and iNOS expression induced by overnight stimulation with LPS/IFN-γ in Ly-6Chigh classical monocytes freshly isolated from SP, BM, or L-Mono. Values correspond to the median fluorescence intensity (MFI) ± SD of 2 independent experiments. For all panels, unpaired Student t test was used. *P < .05, **P < .01, ***P < .001. Error bars show SD.
It was unclear from these experiments whether GM-CSF–mediated proliferation or GM-CSF-R signaling was required to develop suppressive activity. When using very low doses (0.3 ng/mL = 5 U/mL) of GM-CSF that were sufficient to induce suppressor activity (Figure 1D), myeloid cell proliferation was not further supported (Figure 2D). These data indicate that GM-CSF-R signaling at low doses of GM-CSF is sufficient for CD11b+ Ly-6Chigh monocyte licensing of suppressor monocytes rather than proliferation.34
Next, we sought to identify which monocyte populations from SP acquired suppressor potential after GM-CSF injection. Three different subsets of CD11b+ Ly-6G− monocytes, termed classical (Ly6Chigh CD43+), nonclassical (Ly-6C− CD43+), and intermediate monocytes (Ly6-Cint CD43+),35 were sorted from SP of GM-CSF–treated mice and tested in T-cell suppression assays. Only classical Ly-6Chigh monocytes displayed suppressive capacity (Figure 2E). Notably, Ly-6Chigh cells from L-Mono cultures, but not fresh monocytes, expressed iNOS when activated with IFN-γ/LPS despite producing IL-6 as expected from activated monocytes (Figure 2F). These data indicate that within the pool of CD11b+ myelomonocytic cells, only classical monocytes have the potential to acquire iNOS expression and suppressor function via GM-CSF licensing.
GM-CSF licensing requires AKT and mTOR for suppressor activity
Various signaling molecules have been implicated in monocytic suppressor cell development. Here we tested which of these molecules were induced by GM-CSF licensing in fresh BM cells. The expression level of unphosphorylated STAT1 protein remained largely unchanged, whereas STAT3 protein was upregulated as a result of GM-CSF licensing, as were JAK2, AKT, pAKT, IRF-1, MyD88, RelA p65, and NF-κB p100 (Figure 3A-C; supplemental Figure 4). L-Mono, but not fresh BM cells or Ly6Chigh monocytes, showed that signaling differences were induced by the licensing process downstream of mTORC1 in the S473-pAKT signaling pathway, including mTOR,36 S6 ribosomal protein, and 4E-BP1 translation initiation factor (Figure 3B-C). Similar results were obtained for cells cultured in M-CSF, but not cells cultured in G-CSF (supplemental Figure 5A). Accordingly, Ly-6G+ granulocytic cells were poorly activated by GM-CSF (Figure 3C). The functional relevance of the PI3K/AKT and mTORC1/mTOR signal pathways was confirmed because specific inhibitors abrogated T cell–suppressive potential (Figure 3D-E) and NO release but not mRNA production for IRF-1 or iNOS (supplemental Figure 6). A functional role of STAT3 but not STAT1 or STAT5 described before for MDSC function37,38 was confirmed in L-Mono using specific inhibitors (Figure 3F).
Figure 3.

Functionally relevant signaling intermediates contributing to GM-CSF licensing include PI3K, pAKT, pmTOR, pS6, and p4E-BP1. (A-B) Western blot analyses for the indicated markers of whole cell lysates from fresh BM cells or L-Mono optionally treated with IFN-γ, LPS, or IFN-γ/LPS for 1 hour. p, phosphorylated forms of the indicated marker (for quantification and statistics, see supplemental Figure 3). (C) Expression of the indicated phosphorylated markers in Ly-6G+ granulocytes and Ly-6Chigh monocytes from fresh BM or 3-day GM-CSF cultured BM cells analyzed by FACS. Example of 2 independent experiments each with BM from 2 mice, n = 4 or 5. (D-E) Suppression of T-cell proliferation by murine L-Mono generated from days 0 through 3 in the presence of the PI3K inhibitors Wortmannin or Ly294002, or the mTOR inhibitor rapamycin at the indicated concentrations. One representative of 3 (D) or 2 (E) independent experiments is shown. Statistics by comparison of treated vs L-Mono cells with 10 µM inhibitors. (F) Murine BM cells cultured in GM-CSF in the presence of the different STAT inhibitors (inh.) were titrated into CD3/CD28 stimulated T cells for proliferation. Data represent means ± SD of 2 independent experiments. All statistics by unpaired Student t test by comparing STAT3 treatment to the untreated controls (L-Mono) ***P < .001.
GM-CSF and M-CSF induce IFN-γR platforms to allow IRF-1 control of suppression
Western blot analyses comparing fresh monocytes and L-Mono revealed no differences in IFN-γ signaling with respect to phosphorylation of STAT1 and STAT3; therefore, elevated levels of IFN-γ–dependent transcription factor IRF-1 in L-Mono cannot be explained by STAT1 or STAT3 signaling (Figure 3B; supplemental Figures 4 and 5). An important role for IFN-γ in inducing NOS2/iNOS is well established,5,39 but the observed effect of GM-CSF on IRF-1 is novel and led us to the hypothesis that fresh monocytes and L-Mono were qualitatively different in IFN-γ signaling at the level of its receptors IFN-γR1 and IFN-γR2. Kinetic analysis indicated that GM-CSF licensing upregulated IFN-γR1 and IFN-γR2 protein expression in L-Mono within 3 days (Figure 4A). Confocal microscopy showed that GM-CSF and M-CSF, but not G-CSF and Flt3L, induced IFN-γR1/R2 colocalization and reorganization on the cell surface (Figure 4B) that was associated with a general upregulation of both receptors at the cell surface on Ly6Chigh monocytes (Figure 4C; supplemental Figure 5B). These data indicate that GM-CSF licensing promotes the formation of IFN-γR1/2 signaling platforms on the surface of CD11b+ Ly-6Chigh monocytes to enhance and accelerate T cell–derived IFN-γ–mediated downstream signals.
Figure 4.

GM-CSF licensing induces IFN-γR1/R2 assembly on the cell surface and nuclear translocation of IRF-1 required for MDSC function. (A) Upregulation of IFN-γR1 and IFN-γR2 in western blots of whole cell extracts after 3 days. (B) Colocalization of IFN-γR1 and IFN-γR2 by confocal microscopy in BM cells cultured for 3 days with the indicated cytokines (n = 4). Original magnification ×400. (C) Exemplified cell surface expression of IFN-γR1 and IFN-γR2 of Ly-6Chigh monocytes by FACS analysis after BM cell culture for 3 days with the indicated cytokines; bar graphs for statistical evaluation of MFI values (n = 5). (D) Confocal microscopy analysis of IRF-1 nuclear translocation in fresh or WT or Irf1−/− L-Mono cells stimulated or not with IFN-γ for 1 hour (n = 3). Note that granulocytic cells do not upregulate IRF-1 (lower cell in each panel). (E) Suppressor capacity of L-Mono from WT, Irf-1+/−, or Irf-1−/− mice. Representative of 3 separate experiments. Statistics using 1-way ANOVA, with Tukey posttest. ***P < .001. (F) NO production by L-Mono of the indicated mouse strains after overnight stimulation with LPS/IFN-γ or cytokines (n = 2 from duplicates, unpaired Student t test for each comparison). **P < .01. (G) Clinical autoimmune symptoms in the EAE model with or without 15 daily GM-CSF injections (day 10 to day 5 of EAE) in WT or Irf1−/− mice. Representative of 3 separate experiments. Two-way ANOVA with Bonferroni posttest. *P < .05, **P < .01.
IFN-γ–mediated induction of iNOS in myeloid cells is dependent on the transcription factor IRF-1.40-42 When fresh BM cells were treated with IFN-γ, IRF-1 was undetectable by confocal microscopy in cells with monocytic or granulocytic nuclear morphology. However, IFN-γ treatment of L-Mono, but not granulocytic cells, induced expression and nuclear translocation of IRF-1 (Figure 4D). Functional studies revealed that IRF-1–deficient L-Mono lacked T cell–suppressive potential (Figure 4E) and was incapable of NO production (Figure 4F) in vitro. When mice were injected daily with GM-CSF before and during induction of EAE, the clinical score was strongly reduced, whereas in Irf1−/− mice, this effect was completely abrogated (Figure 4G). Thus, GM-CSF licensing of monocytes modified IFN-γ signaling platforms enabling the synthesis and translocation of IRF-1 to the nucleus, which is required for suppressive NO release in vitro and in vivo.
GM-CSF licensing of human CD14+ monocytes
When human CD14+ monocytes were cultured for 7 days in GM-CSF, the licensing markers pS6 and pAKT were upregulated as compared with day 1 (Figure 5A). pS6 could be further induced by IFN-γ. IRF-1 was only weakly expressed and IDO was not detectable in GM-CSF licensed CD14+ monocytes; however, both were strongly induced after treatment with IFN-γ (Figure 5B). Culture of human L-Mono with the mTORC1 inhibitor rapamycin blocked the upregulation of pS6 and pAKT, whereas IRF-1 and IDO still remained negative in the absence of IFN-γ (Figure 5C). GM-CSF licensing of human monocytes also required 3 days to reach its full suppressive potential (Figure 5D). Suppression could be reverted partially by rapamycin (Figure 5E), indicating that the mTORC1 signaling cascade affected also human monocytic suppressor activity.
Figure 5.

Induction of licensing markers in human monocytes by GM-CSF in vitro and ex vivo from tumor patients. (A) p-S6 and p-AKT expression detected by flow cytometry in fresh (day 1) and GM-CSF–cultured (day 7) human monocytes with or without IFN-γ stimulation. Background-subtracted MFI of p-S6 and p-AKT expression in fresh and GM-CSF–cultured human monocytes with or without IFN-γ stimulation (n = 3; mean ± SD). (B) IRF1 and IDO expression detected by flow cytometry in fresh (day 1) and cultured (day 7) human monocytes with or without IFN-γ stimulation. Background-subtracted MFI of IRF1 and IDO expression in fresh and cultured human monocytes with or without IFN-γ stimulation (n = 3; mean ± SD). (C) Effect of 10 ng/mL rapamycin over 7 days of culture on expression of p-S6, p-AKT, IRF1, and IDO in human monocytes (n = 3; mean ± SD). (D) Freshly isolated human CD14+ monocytes or monocytes cultured for 1 to 3 days in GM-CSF were added to phytohemagglutinin (PHA)-stimulated allogeneic human CD3+ T cells at a ratio 1:1 ratio for 4 days before determining CFSE dilution by FACS analysis and its quantification of T-cell suppression for human CD4+ and CD8+ T cells (human n = 6, statistics by ANOVA with multiple comparisons, *P < .05, ***P < .005). (E) Rapamycin (rapa)-treated human L-Mono were less effective than untreated L-Mono in suppressing PHA-stimulated proliferation of allogeneic human T cells in 1:1 direct cocultures (n = 3; mean ± SD). (F) Example dot plots of peripheral blood mononuclear cells from healthy individuals or tumor patients were stained for HLA-DR and CD14 to identify CD14+ HLA-DRlow M-MDSC (gate) by FACS analysis. (G) Cells gated as in panel F of healthy individuals or tumor patients were further costained for intracellular isotype controls, IDO, p-AKT, p-S6, or p-mTOR. FACS histograms represent examples of the indicated staining. The bar graph summarizes MFI values of the indicated markers with background-subtracted ΔMFI values from healthy (n = 4) or tumor patients (n = 5) and evaluated by unpaired Student t test. (H) Serum samples of (n = 51) tumor patients or (n = 9) healthy donors were analyzed for their GM-CSF (Mann-Whitney U test, mean ± SEM) or (I) for their M-CSF content from (n = 38) tumor patients or (n = 7) healthy donors (Mann-Whitney U test, mean ± SEM). Tumor patients analyzed in panels F and G were different from patients analyzed in panels H and I.
Taken together, our data show that GM-CSF signaling involves the upregulation of important factors for transcription, translation, or signaling molecules that are functionally relevant for T-cell suppression by monocytes. The data further suggest that IDO in human monocytic suppressor cells plays an analogous role to iNOS in their murine counterparts.
Licensing of monocytes from tumor patients
Monocytes from patients with primary or relapsed high-grade gliomas displayed a CD14+ HLA-DRlow phenotype, which is consistent with a human monocytic suppressor cells phenotype (Figure 5F). Analyses of the licensing markers p-AKT, p-mTOR, and p-S6 within this cell population indicated elevated expression of all 3 markers compared with healthy individuals (Figure 5G). The suppressive effector molecule IDO was not detectable in healthy or patient blood samples (Figure 5G), suggesting that these CD14+ HLA-DRlow cells represent L-Mono but not monocytic suppressor cells. Clearly, these data are compatible with longstanding observations with human M-MDSC that required activated T cell–derived factors to achieve their full suppressive potential in vitro and in vivo. However, upregulation of 1 of the serum cytokines GM-CSF (Figure 5H) or M-CSF (Figure 5I) was not universally observed in serum samples of our patient cohort, possibly indicating a redundancy with other cytokines or production within the tissues. Because the elevated expression of “licensing” markers p-AKT, p-mTOR, and p-S6 is consistent, they may be of prognostic value for identifying M-MDSC development in glioma patients to discriminate between p-AKT/mTOR/S6low/neg IDOneg fresh blood monocytes (healthy individuals) from p-AKT/mTOR/S6high IDOneg L-Mono in the blood of cancer patients.
Discussion
There are numerous examples of anti-inflammatory43 or pro-inflammatory activity of GM-CSF.30 The evolutionarily developed role of suppressor monocytes is presumably to limit evolving T-cell responses against pathogenic challenges. Our results suggest a model of suppressor monocyte activity that involves an essential time delay in their development. Early in an immune response, when monocytes encounter activated T cells producing GM-CSF and IFN-γ at inflammatory sites, they differentiate into activated macrophages or dendritic cells and accentuate the response.44,45 Conversely, we found previously that suppressor activity of 3-day GM-CSF myeloid cell cultures was abrogated by adding LPS or IFN-γ or both during the whole 3-day culture period.6 Later in a response, monocytes residing in the BM or SP and exposed to systemically elevated GM-CSF levels for at least 3 days before encountering IFN-γ–producing T cells are licensed to become MDSC, hence suppressing the response. We contend that such a feature of suppressor monocyte function would allow a vigorous immediate T-cell reaction and subsequent containment.
Clearly, time-dependent monocyte licensing has important implications for the balance of immunogenic and suppressive effects of GM-CSF treatment in vivo. Low doses of local GM-CSF exerted immunogenic adjuvant effects, whereas systemic availability of high GM-CSF doses resulted in no or tolerogenic effects in human tumor patients.46 Similarly, early GM-CSF administration has been reported to protect against septic shock in Candida infection.47 Others have shown that GM-CSF production by Th17 and Th1 cells during the effector phase of EAE exacerbates the clinical symptoms by stimulating myeloid cell infiltrates into the central nervous system, which can be reversed by GM-CSF blockade.44,45 Importantly, GM-CSF signaling in monocytes was critical for their development into inflammatory dendritic cells and to promote EAE disease.48 In contrast to these reports, very early it had been observed that tumor cells secreting GM-CSF induced Gr-1+ CD11b+ MDSC that impaired tumor cell killing by CD8+ T cells and promoted CD8+ T-cell apoptosis.14 Recently, blocking of GM-CSF has been shown to effectively inhibit MDSC function in vitro.49 We found that GM-CSF administration before or during EAE induction was suppressive. The protective effect of GM-CSF treatment in our model was dependent upon IRF-1–mediated IFN-γ signaling. Others demonstrated that tumor-induced MDSC suppression was dependent on IFN-γ and its receptor and IRF-1 leading to NO production.50
Together, we interpret these findings as evidence of a time-dependent GM-CSF licensing of monocytes to become suppressive in vivo. Our model suggests that GM-CSF will have suppressive functions if administered to recipients in the absence of inflammation, but will otherwise worsen disease, at least for some period.
Appreciating that monocytes undergo a time-dependent licensing before suppressor functions can be activated, offers a possible resolution to controversies surrounding the generation of M-MDSC, Mreg, or suppressor macrophages. In our model, the L-Mono may serve as the precursor of such suppressor cells, but both ultimately derive from classical monocytes. In our view, this also explains why monocytes and M-MDSC share such similar marker profiles. An important corollary of our work is the identification of L-Mono in peripheral blood using p-AKT, p-mTOR, and p-S6 as biomarkers.
Our data confirm the tolerogenic effects of GM-CSF on monocytes and extend these findings by defining novel proliferation-independent signaling pathways at low doses of GM-CSF through which monocytes transit to L-Mono and then further to suppressive monocytes. Our data identify another low-dose function of GM-CSF that so far has been described only of mediating anti-apoptotic signals.34
In this study, we discovered that murine Ly-6Chigh and human CD14+ classical monocytes can be converted into M-MDSC via a 2-step mechanism. In the first step, classical monocytes treated with GM-CSF undergo “licensing,” consisting of several components that have been unraveled in this study, including: (1) formation of functional IFN-γR1/2 signaling platforms on the cell surface that increases synthesis of IRF-1 and allows its nuclear translocation, which in turn facilitates iNOS or IDO transcription; (2) activation of the PI3K pathway leading to phosphorylation of AKT, mTOR, S6, and 4E-BP1 to potentiate protein translation or posttranslational stabilization of iNOS or IDO; and (3) upregulation of RelA p65, NF-κB p100, and MyD88 required for Toll-like receptor and inflammatory cytokine signaling. In the second step, stimulation of L-Mono with IFN-γ or IFN-γ/LPS drives the IRF-1–dependent expression of the iNOS gene in mice, or IDO gene in humans; critically, however, this stimulation of L-Mono does not lead to induction of a pro-inflammatory program.
In the second step, IFN-γ has long been known as a critical factor for activating suppressor function; however, its dichotomous effects in activating monocytes51 and promoting suppressor function were not mechanistically understood. IFN-γ signaling is initiated by binding of a single IFN-γ molecule to constitutively surface-expressed IFN-γR1.52 This first signal leads to recruitment of IFN-γR2 from intracellular stores or distant surface areas to bind a second IFN-γ molecule, which finally forms the functional IFN-γR1/2 complex.53 We found that GM-CSF or M-CSF, but not G-CSF or Flt3L, increased surface expression of both IFN-γR1 and IFN-γR2 and promoted the formation of IFN-γR1/2 signaling platforms on the surface of CD11b+ Ly-6Chigh monocytes to improve and accelerate IFN-γ–mediated signals.
In freshly isolated human and mouse monocytes, IRF-1 protein synthesis was not altered and nuclear translocation not observed upon IFN-γ stimulation. By contrast, 1 hour’s treatment of L-Mono with IFN-γ led to nuclear relocation of IRF-1. Treatment of IFN-γ–stimulated L-Mono with LPS further induced NO release, similar to a previous report that IFN-γ signals enhanced nuclear transport and MyD88 binding of IRF-1, leading to accelerated cytokine and NO release.42 IRF-1 mRNA could be readily induced by IFN-γ in mouse monocytes and L-Mono, whereas only L-Mono also produced iNOS mRNA after IFN-γ/LPS exposure. In the human, GM-CSF weakly induced IRF-1 protein expression in L-Mono but little in fresh monocytes; in contrast, IFN-γ upregulated IDO protein only in human L-Mono, not in freshly isolated monocytes. These results show that GM-CSF–mediated licensing leads to adaptations of the IFN-γ signaling pathway in L-Mono that result in preferential expression of suppressive molecules (iNOS in mice or IDO in humans). The central importance of the PI3K/mTOR/AKT pathway was functionally confirmed using rapamycin treatment. Recently, PI3K has been found to represent a transcriptional switch in human tumor-associated macrophages toward immune suppression,18 and mTOR plays decisive roles for M-MDSC function in murine allograft and tumor models.19 Expression of mouse iNOS protein-dependent release of NO and suppressor activity of mouse suppressor monocytes, as well as IDO protein expression and suppressor function of human suppressor monocytes, was impaired by rapamycin treatment. This suggests a critical role for mTOR in iNOS and IDO protein translation or stabilization.
Although human cells are able to produce iNOS-dependent NO, major differences to murine cells in their responsiveness on IFN-γ/LPS have been identified, including the promoter structure, epigenetic silencing, histone modifications, and chromatin compaction.54 Overexpression of IDO in human macrophages resulted in a deficit to generate superoxide anions and NO,55 indicating a regulatory effect of IDO on iNOS in human cells. Thus, although the same signaling machinery is used to initiate suppression of human and mouse monocytes, the final effector strategies differ in the use of enzymes catabolizing either arginine or tryptophan to induce metabolic starvation of T cells.4,56
In conclusion, we identified novel signaling pathways of GM-CSF in human and mouse monocytes. These signals induce monocyte licensing, an essential prerequisite to acquire suppressor function by subsequent activation signals. Monocyte licensing includes transcriptional and translational modifications. The licensing process requires only very low doses of GM-CSF, whereas high doses allow cell proliferation functioning as a growth factor. The ability to identify L-Mono in peripheral blood through p-AKT, p-mTOR, and p-S6 upregulation should be valuable in cancer research and diagnostics, and has significant implications for clinical studies with MDSC-based cell therapy and the use of rapamycin.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Thomas Hünig for his continuous support.
This work was supported by the German Research Foundation (grants LU851/6-1 [M.B.L.], FZ82 [H.M.H.], and SFB643 A6 and GRK1660 A5 [C.B. and U.S.]); the European Cooperation in Science and Technology program “Mye-EUNITER”; and by the Interdisciplinary Center for Clinical Research of the University Hospital Erlangen (grant A61) (C.B.).
Authorship
Contribution: E.R., J.A.H., S.H., M. Heuer, J.L., U.S., A-L.J.G., S.J.P., N.M., H.R., M. Huber, and P.R. performed experiments; E.R., J.A.H., S.H., and M.L. analyzed results and made the figures; H.B. provided reagents and permissions; and E.R., J.A.H., E.K.G., I.B., A.B., M.L., C.B., M.E., H.M.H., and M. B. L. designed the research and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Manfred B. Lutz, Institute of Virology and Immunobiology, University of Würzburg, Versbacherstr 7, 97078 Würzburg, Germany; e-mail: m.lutz@vim.uni-wuerzburg.de.
References
- 1.van Furth R, ed. Mononuclear Phagocytes: Biology of Monocytes and Macrophages. Dordrecht, The Netherlands: Kluwer; 1992. [Google Scholar]
- 2.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. 2005;5(8):641-654. [DOI] [PubMed] [Google Scholar]
- 5.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2(10):907-916. [DOI] [PubMed] [Google Scholar]
- 6.Greifenberg V, Ribechini E, Rössner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. 2009;39(10):2865-2876. [DOI] [PubMed] [Google Scholar]
- 7.Gallina G, Dolcetti L, Serafini P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116(10):2777-2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26(7):1641-1646. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Vallejo JJ, van Kooyk Y. DC-SIGN: the strange case of Dr. Jekyll and Mr. Hyde. Immunity. 2015;42(6):983-985. [DOI] [PubMed] [Google Scholar]
- 10.Lacey DC, Achuthan A, Fleetwood AJ, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol. 2012;188(11):5752-5765. [DOI] [PubMed] [Google Scholar]
- 11.al-Ramadi BK, Brodkin MA, Mosser DM, Eisenstein TK. Immunosuppression induced by attenuated Salmonella. Evidence for mediation by macrophage precursors. J Immunol. 1991;146(8):2737-2746. [PubMed] [Google Scholar]
- 12.Hume DA. The mononuclear phagocyte system. Curr Opin Immunol. 2006;18(1):49-53. [DOI] [PubMed] [Google Scholar]
- 13.Rössner S, Voigtländer C, Wiethe C, Hänig J, Seifarth C, Lutz MB. Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol. 2005;35(12):3533-3544. [DOI] [PubMed] [Google Scholar]
- 14.Bronte V, Chappell DB, Apolloni E, et al. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J Immunol. 1999;162(10):5728-5737. [PMC free article] [PubMed] [Google Scholar]
- 15.Hansen G, Hercus TR, McClure BJ, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. 2008;134(3):496-507. [DOI] [PubMed] [Google Scholar]
- 16.Lee AW, States DJ. Colony-stimulating factor-1 requires PI3-kinase-mediated metabolism for proliferation and survival in myeloid cells. Cell Death Differ. 2006;13(11):1900-1914. [DOI] [PubMed] [Google Scholar]
- 17.Hume DA, MacDonald KP. Therapeutic applications of macrophage colony-stimulating factor-1 (CSF-1) and antagonists of CSF-1 receptor (CSF-1R) signaling. Blood. 2012;119(8):1810-1820. [DOI] [PubMed] [Google Scholar]
- 18.Kaneda MM, Messer KS, Ralainirina N, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu T, Zhao Y, Wang H, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep. 2016;6:20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hutchinson JA, Riquelme P, Geissler EK, Fändrich F. Human regulatory macrophages. Methods Mol Biol. 2011;677:181-192. [DOI] [PubMed] [Google Scholar]
- 21.Rössner S, Voigtländer C, Wiethe C, Hänig J, Seifarth C, Lutz MB. Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol. 2005;35(12):3533-3544. [DOI] [PubMed] [Google Scholar]
- 22.Tada Y, Ho A, Matsuyama T, Mak TW. Reduced incidence and severity of antigen-induced autoimmune diseases in mice lacking interferon regulatory factor-1. J Exp Med. 1997;185(2):231-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang S, Hendriks W, Althage A, et al. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259(5102):1742-1745. [DOI] [PubMed] [Google Scholar]
- 24.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259(5102):1739-1742. [DOI] [PubMed] [Google Scholar]
- 25.Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity. 2016;45(5):963-973. [DOI] [PubMed] [Google Scholar]
- 26.Hutchinson JA, Riquelme P, Sawitzki B, et al. Cutting edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. 2011;187(5):2072-2078. [DOI] [PubMed] [Google Scholar]
- 27.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126(1):131-138. [DOI] [PubMed] [Google Scholar]
- 29.Menges M, Rössner S, Voigtländer C, et al. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med. 2002;195(1):15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533-544. [DOI] [PubMed] [Google Scholar]
- 31.Dolcetti L, Peranzoni E, Ugel S, et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. Eur J Immunol. 2010;40(1):22-35. [DOI] [PubMed] [Google Scholar]
- 32.Movahedi K, Guilliams M, Van den Bossche J, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233-4244. [DOI] [PubMed] [Google Scholar]
- 33.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791-5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hercus TR, Thomas D, Guthridge MA, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114(7):1289-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116(16):e74-e80. [DOI] [PubMed] [Google Scholar]
- 36.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7(4):209-219. [DOI] [PubMed] [Google Scholar]
- 37.Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu L, Du H, Li Y, Qu P, Yan C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid-derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am J Pathol. 2011;179(4):2131-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bogdan C. Regulation of lymphocytes by nitric oxide. Methods Mol Biol. 2011;677:375-393. [DOI] [PubMed] [Google Scholar]
- 40.Kamijo R, Harada H, Matsuyama T, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263(5153):1612-1615. [DOI] [PubMed] [Google Scholar]
- 41.Martin E, Nathan C, Xie QW. Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J Exp Med. 1994;180(3):977-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Negishi H, Fujita Y, Yanai H, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci USA. 2006;103(41):15136-15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, Prabhakar BSGM-CSF. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine. 2015;75(2):261-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Codarri L, Gyülvészi G, Tosevski V, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560-567. [DOI] [PubMed] [Google Scholar]
- 45.El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18(2):226-232. [DOI] [PubMed] [Google Scholar]
- 47.Lechner AJ, Lamprech KE, Potthoff LH, Tredway TL, Matuschak GM. Recombinant GM-CSF reduces lung injury and mortality during neutropenic Candida sepsis. Am J Physiol. 1994;266(5 Pt 1):L561-L568. [DOI] [PubMed] [Google Scholar]
- 48.Croxford AL, Lanzinger M, Hartmann FJ, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. 2015;43(3):502-514. [DOI] [PubMed] [Google Scholar]
- 49.Gargett T, Christo SN, Hercus TR, et al. GM-CSF signalling blockade and chemotherapeutic agents act in concert to inhibit the function of myeloid-derived suppressor cells in vitro. Clin Transl Immunology. 2016;5(12):e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schouppe E, Mommer C, Movahedi K, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. 2013;43(11):2930-2942. [DOI] [PubMed] [Google Scholar]
- 51.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623-655. [DOI] [PubMed] [Google Scholar]
- 52.Rosenzweig SD, Schwartz OM, Brown MR, Leto TL, Holland SM. Characterization of a dipeptide motif regulating IFN-gamma receptor 2 plasma membrane accumulation and IFN-gamma responsiveness. J Immunol. 2004;173(6):3991-3999. [DOI] [PubMed] [Google Scholar]
- 53.Dell’Albani P, Santangelo R, Torrisi L, Nicoletti VG, de Vellis J, Giuffrida Stella AM. JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J Neurosci Res. 2001;65(5):417-424. [DOI] [PubMed] [Google Scholar]
- 54.Bogdan C. Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. 2015;36(3):161-178. [DOI] [PubMed] [Google Scholar]
- 55.Keskin DB, Marshall B, Munn D, Mellor AL, Gearhart DA. Decreased protein nitration in macrophages that overexpress indoleamine 2, 3-dioxygenase. Cell Mol Biol Lett. 2007;12(1):82-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.