


Abstract
Gene fusions are known to play critical roles in tumor pathogenesis. Yet, sensitive and specific algorithms to detect gene fusions in cancer do not currently exist. In this paper, we present a new statistical algorithm, MACHETE (Mismatched Alignment CHimEra Tracking Engine), which achieves highly sensitive and specific detection of gene fusions from RNA-Seq data, including the highest Positive Predictive Value (PPV) compared to the current state-of-the-art, as assessed in simulated data. We show that the best performing published algorithms either find large numbers of fusions in negative control data or suffer from low sensitivity detecting known driving fusions in gold standard settings, such as EWSR1-FLI1. As proof of principle that MACHETE discovers novel gene fusions with high accuracy in vivo, we mined public data to discover and subsequently PCR validate novel gene fusions missed by other algorithms in the ovarian cancer cell line OVCAR3. These results highlight the gains in accuracy achieved by introducing statistical models into fusion detection, and pave the way for unbiased discovery of potentially driving and druggable gene fusions in primary tumors.
INTRODUCTION
Identification of cancer biomarkers is a crucial goal of oncology. Gene fusions are excellent tumor-specific biomarkers because they are very rarely present in healthy patients, and thus have the potential to be used for early diagnosis, as drug targets, and as neoantigens. Detection of oncogenic fusions can inform efforts to develop targeted therapy and provide insight into basic cancer biology.
Both the functional importance of gene fusions and how tumor-specific markers can be leveraged for cancer treatment are well-appreciated in leukemias, where fusions were first discovered. In leukemias, recurrent gene fusions and internal tandem duplications are among the most effective therapeutic targets. For example, the discovery of oncogenic gene fusions, including BCR-ABL1 in chronic myelogenous leukemia (CML) and the FLT3 internal tandem duplication in acute myelogenous leukemia (AML), have provided critical insights into pathogenesis and led to important therapeutic advances (1,2). Recurrent gene fusions have also been identified in solid tumors (3,4).
Prior to the advent of next generation sequencing, many fusions, including those above, were found using cytogenetics or clever candidate-based methodologies. Since that time, a variety of gene fusions have been discovered using DNA and RNA-Seq (5–7). Next generation sequencing focused on fusions involving known oncogenes, has enabled discovery of fusions in a variety of other cancers that can be targeted with existing drugs, such as FGFR, ALK and ROS family gene fusions (8–11).
Despite these important discoveries, the unbiased ascertainment of gene fusions from RNA-Seq data remains a difficult and unsolved problem. While discovery of some driver gene fusions described above was aided by algorithms, all required human-guided filtering and heuristic approaches to cull fusions from those likely to be artifactual (12–16).
Recent surveys have concluded that no single RNA-Seq based fusion algorithm has clear dominant performance and consensus between algorithms is needed to attain specificity sufficient for clinical application (17,18). This approach is problematic because systematic false positives present in multiple algorithms will continue to populate consensus lists and lack of sensitivity of a single algorithm can result in rejection of true positives.
For this reason, there is a pressing need for robust algorithms that detect gene fusions with low false positive rates. Fusion detection algorithms with high false positive rates are unfit for use in clinical sequencing applications, as time and resources make it impractical to manually scrutinize a list of gene fusion candidates and/or perform secondary validations (e.g. PCR for the fusion). Further, in clinical samples, it is often impractical or impossible to perform secondary tests of predictions from RNA-Seq because the amount of RNA is limiting or not available.
Another application of a fusion detection algorithm with a low false positive rate is the mining of thousands of publicly available RNA-Seq datasets from tumor samples. These datasets provide an unprecedented opportunity to discover novel oncogenes, drug targets and gene fusions, private or recurrent, that may drive cancers, but algorithms must be trustworthy without secondary validation, because tumor RNA is not available (19–21).
In this paper, we present MACHETE (Mismatched Alignment CHimEra Tracking Engine), a novel, sensitive, and highly specific method to detect fusion RNAs at annotated exonic boundaries from RNA-Seq. This method significantly extends a previous computational framework we introduced to detect circular and linear RNA splicing (22). MACHETE includes key computational and statistical components, leveraging statistical modeling to prioritize fusion transcripts. It weeds out false positives while retaining the ability to identify known true positives in gold standard controls, and in several cases, identify fusions missed by other algorithms.
MACHETE's empirical p value is a key statistical contribution as it provides a measure allowing researchers to prioritize fusions for clinical and research validation. This is an innovation absent in other published algorithms which prioritize potentially oncogenic events by read count, or by scores that lack an underlying null statistical model. We have tested MACHETE against STAR-Fusion, SOAPfuse (23), as well as EricScript, the best performing algorithms as defined by a recent evaluation (18). We have evaluated MACHETE on a simulated negative control dataset (18) and a ‘mixed’ dataset which combines the negative control data-set with simulated reads derived from fusion transcripts. The data used in (18) were not available, so we used published and available simulated data from (24) and the positive control data used in (18). MACHETE reports far fewer false positives in negative controls and provides comparable or better detection of true positives in simulated data and multiple gold standard cell lines.
MATERIALS AND METHODS
Overview of the MACHETE algorithm
The overall workflow of MACHETE is shown in Figure 1. MACHETE begins by running KNIFE (22), which parses sequencing reads into categories that align to the genome, splice junctions, or those that cannot be aligned. Potential gene fusions are nominated by read pairs where R1 and R2 both map to a KNIFE index, but their coordinates are farther away than a user-defined radius (discordant spanning reads), or based on split-read alignment to the genome. Unaligned reads are mapped to the nominated fusions, and MACHETE uses reads that other fusion detection algorithms discard (Figure 2) to model artifactual fusions and assign a statistical score to each fusion candidate (Figure 3). The computational steps that precede assignment of statistical scores are important (see Materials and Methods), but MACHETE's significant advance is due to its use of statistical models described below. Note that the algorithm does not depend on read length.
Figure 1.

MACHETE workflow. MACHETE takes as input the output of KNIFE (21) and generates a list of nominated fusions using spanning reads (red path) or split-reads (blue path). Next, reads that failed to align to any of the reference indices in KNIFE are aligned to the database of nominated fusions. MACHETE statistical models use per-read alignment features and implement an unbiased assessment of the likelihood that a fusion is an artifact due to sequence homology. These models are applied to produce a fusion report prioritized by statistical scores and read counts.
Figure 2.

MACHETE does not censor reads. In the alignment step to the nominated fusion database, MACHETE uses all reads that aligned to a fusion to assign a statistical score. GAPDH-HFM1 is given as an example of an artifactual fusion. Reads aligning to the fusion are aggregated and used in model building and generation of a final statistical score for the fusion in the database even if they have low mapping quality, contain mismatches (depicted with X) or have a small junction overlap. In other algorithms, reads are first censored so that only reads with few mismatches and high overlap are reported, presenting only positive data on the junction even if the junction is an artifact.
Figure 3.

Artifact modeling. (A) Each constructed fusion sequence is aligned to reference indices. Fusions that align to a KNIFE reference index are likely artifacts due to sequence homology (mappable fusions). Fusions are flagged as split mappable fusions and not reported by MACHETE if the first and last 40 bases of each fusion sequence, when treated as pseudo paired end reads, align to the reference genome within a 50 kb window of each other. (B) Unaligned reads are mapped back to putative fusions. If R1 maps across the fusion boundary, the mapping location of R2 can either support that the pair maps consistently (top left) or not (top right). Alternatively, R1 may align to an indel fusion, which MACHETE considers as evidence against the fusion being expressed since this could represent a ligation artifact. Each read is assigned a probability, 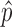 , of being generated from an expressed fusion by the GLM. The reported probabilities for reads providing evidence against the fusion are down-weighted. All junctions are assigned a statistical score based on the aggregated per-read probabilities, termed junction_cdf, reflecting the probability that a fusion is an artifact under a model of simple random sampling of reads. Mappable fusions are used to model the distribution of junction_cdf scores for fusions that are artifacts. All fusions that are not flagged as mappable or split mappable are referred to this distribution and assigned an empirical p value, interpreted as the probability of observing as high a junction_cdf as observed under the null hypothesis that the fusion is an artifact.
, of being generated from an expressed fusion by the GLM. The reported probabilities for reads providing evidence against the fusion are down-weighted. All junctions are assigned a statistical score based on the aggregated per-read probabilities, termed junction_cdf, reflecting the probability that a fusion is an artifact under a model of simple random sampling of reads. Mappable fusions are used to model the distribution of junction_cdf scores for fusions that are artifacts. All fusions that are not flagged as mappable or split mappable are referred to this distribution and assigned an empirical p value, interpreted as the probability of observing as high a junction_cdf as observed under the null hypothesis that the fusion is an artifact.
Splice events and fusions reported by MACHETE
Prior to running MACHETE, data is trimmed and processed by KNIFE (22), an algorithm designed to find mappings to the genome as well as circular and linear splicing or gene fusions between all pairs of annotated exons within a sliding 1 megabase (Mb) window. While KNIFE's main motivation is detection of circular RNA (25), certain types of gene fusions, internal tandem duplications (ITDs) and readthroughs can also be detected by KNIFE (4,21,26). For this reason, MACHETE output includes KNIFE output, reported as separate files, for users interested in detecting local genomic rearrangements or circular RNAs.
In this paper, we focus on the major innovations of MACHETE that enable identification of the following events not detected by KNIFE: (a) splicing between annotated exons on the same chromosome separated by more than a user-defined radius, most likely arising from tandem duplications or large deletions; (b) inversions, which are defined as transcripts containing annotated exons separated by the same user-defined radius or more on the same chromosome in discrepant transcriptional orientations; and (c) translocations, which are defined as transcripts containing annotated exons from two genes on different chromosomes in the reference genome. In this paper we call all of these events ‘fusions’. We have used hg19 and the UCSC GRCh37 gene annotations, but the choice of organism, genome build, annotation, and the user-defined radius for detecting events (a) or (b) are choices which can easily be modified by the user.
A candidate fusion database from discordant spanning reads
MACHETE uses alignment of paired-end RNA seq reads output from KNIFE to generate a database of candidate fusions defined by their diagnostic exon–exon junctions. Reads where mates R1 and R2 were identified by KNIFE as both aligned to the genome index, both aligned to the linear junction index, or one mate aligned to the linear junction index and the other to the genome index, are evaluated and recorded as ‘discordant’ if mates aligned more than the user-defined radius apart or on different chromosomes. These discordant reads signal that there may be a fusion junction nearby. For each discordant read pair, MACHETE defines a pair of 20 kb windows in the genome surrounding R1 and R2 and identifies all exons with a 5΄ or 3΄ boundary within these windows. For each exon in the first window and each in the second, MACHETE adds this exon-exon junction sequence to a nominated fusion database. Similar logic is applied when one or both read mates map to an annotated linear exon-exon junction (see Materials and Methods).
A candidate fusion database from split-reads
MACHETE uses split reads to generate a nominated fusion database supplementing the one generated by discordant spanning reads above. Unlike other algorithms that identify split reads using dynamic programming (27), MACHETE uses a static definition of a split read as a previously unaligned read whose first and last user defined number of nucleotides (nt) map far from one another in the genome index.
The mapping location of the 5΄ and 3΄ piece of each split read are each assigned to the nearest 50 nt bin and the combination of both bins is termed a bin-pair. If multiple reads share the same bin-pair, they are grouped to build a consensus sequence using majority voting to mask potential sequencing errors (see Methods and Supplemental Figure S1). If this consensus sequence is consistent with an annotated exon-exon junction, it is nominated as a fusion junction. If it is not, the consensus is still included in the split-read fusion database, but will not be included in the fusion report. Reads used to construct fusion junction sequences are considered training data and are excluded from the unaligned reads that are subsequently aligned to the nominated fusion database. This leads to a conscientious underestimation of the abundance of fusions nominated by split-reads, but allows a disciplined framework for statistical modeling of the fusions by separating reads used to fit and test expression of junctions, described below.
A junction database to model confounding biochemical artifacts
A critical component of MACHETE is a statistical model of several types of artifacts that can introduce false positives into fusion discovery: ligation artifacts, where two cDNAs from different RNA molecules are ligated (28), and reverse transcriptase (RT) template switching artifacts, where an RT enzyme initiates on one RNA molecule, dissociates and then re-initiates on a second molecule (29). These artifacts appear at non-trivial rates during library preparation and produce what appears to be a sequence compatible with a fusion transcript. An innovation of MACHETE is to model the rate of chimeric artifacts for each candidate fusion junction in our database via a realistic simplifying assumption that a read which is a chimeric artifact due to either of these biochemical events is equally likely to have a spurious fusion breakpoint at any point along the transcript and should not be biased to occurring at an exon–exon boundary. To model this, for each fusion junction, we create indel fusion junctions with symmetric addition and deletion of nucleotides at the exon-exon junction, which we call the indel fusion index.
Classification of read pairs
After building a fusion index from either spanning or split reads, MACHETE maps reads classified as ‘unaligned’ by KNIFE to the original and indel fusion indices. In cases where a read aligns to multiple fusions, the read is determined to align to the fusion with the best alignment score. The alignment of the mate of this read is determined using similar priority rules. Based on relative position of alignments and their orientation, each read is determined to be consistent or inconsistent with being generated from the fusion junction to which it aligns. Consistent and inconsistent reads are used differently in downstream steps of MACHETE.
Prioritizing fusions using statistical scores
The major novelty in MACHETE is its use of statistical scores to prioritize fusions based on their likelihood of being false positives. One source of false positives is biochemical artifact. In addition, sequencing errors convolved with degenerate sequence motifs at exon boundaries can result in identification of false positive junctional sequences (22,30). Bona fide mutations or indels in the cancer genome compound this problem and further contribute to artifacts.
One approach to address this problem is to exclude reads that align to fusions below a certain quality of alignment score or that contain fewer than a certain number of nucleotides overlapping the fusion junction (Figure 2). This procedure is called censoring, and to our knowledge, is used by all fusion detection algorithms. Censoring approaches result in increasing numbers of artifactual fusions being reported as sequencing depth increases, especially among highly expressed genes, due to the convolution of sequencing errors and sequence homology. If enough reads are sequenced, random errors will eventually result in reads that map to a fusion even if the fusion sequence doesn't exist in the input RNA.
MACHETE uses a different approach. Rather than applying hard thresholds to reads that other algorithms would have discarded, MACHETE uses these reads to estimate the probability that each putative fusion is an artifact. The first step in this process uses the information from all reads aligning to a fusion junction, including those with poor alignment scores, reads mapping to the constructed indel indices, and reads where mates map inconsistently (anomalies). MACHETE fits a generalized linear model (GLM) for each read, with predictors being alignment scores, mapping quality, and the amount of junction overlap. The GLM is used to estimate the probability, p̂, that the read's alignment to a putative fusion was due to an artifact.
The per-read probabilities estimated by the GLM are aggregated to generate a cumulative score for each nominated fusion, which is compared to a null distribution. MACHETE constructs a null distribution for each value of N, the number of reads aligning to the fusion junction, by randomly sampling from the empirical distribution of p̂ for all reads in the sequencing library when N is small, and uses the Hoeffding combinatorial central limit theorem to estimate this distribution for large N (31). Comparing the cumulative score for each junction to the null distribution results in assignment of what we deem the junction_cdf value to each fusion junction (see Materials and Methods). Note that all modeling above is done within a single dataset, the same dataset that reports putative fusions. No external ontologies or external control data are needed or used by MACHETE; the model is fit separately and independently each time MACHETE is called using the dataset on which MACHETE is run (see Materials and Methods).
Final statistical scoring: including empirical p value assignment
MACHETE uses a statistical framework to generate an empirical p value for each junction, the algorithm's estimated probability that the junction with a given score is an artifact based on an empirical null. This value is conceptually different from the junction_cdf score because it uses a more realistic null model, accounting for structure in the null distribution lost by simple random sampling of reads that generates the junction_cdf (32). Like other components in MACHETE, the empirical p value is determined by using data that would have otherwise been discarded. In this case, MACHETE models the null distribution of junction_cdf scores using junctions which bioinformatic evidence supports being artifacts because they map to the genome or transcriptome (‘mappable fusions’, see Figure 3A and Methods). The empirical p value for each fusion junction is estimated by referring its junction_cdf to the empirical distribution of junction_cdf scores of mappable fusions. The empirical p value and junction_cdf can be used to determine which fusions are reported by MACHETE and to prioritize fusions for follow-up study. Additionally, standard statistical analysis of these p values could be applied to estimate a false discovery rate (FDR) (33). See Materials and Methods for the statistical thresholds applied in this paper, which were uniform for all samples analyzed. This is a key point because it means that (a) better results could be obtained by manipulating cut-offs on a sample-specific basis; and (b) the thresholds used in this paper are robust and dependable, in the sense that they can be used to obtain similar results on any dataset.
Fetal tissue samples
Ribosomal RNA-depleted (rRNA-) total RNA from fetal tissue samples previously obtained as documented in (22) were analyzed. 43 samples collected within 1 h of pregnancy termination at the following gestational ages were obtained from the following tissues: five adrenal (10.3–20.6 weeks), one brain (20.6 weeks), seven heart (10.4–20.3 weeks), seven intestine (10.3–20.6 weeks), three kidney (10.3–20.0 weeks), six lung (10.2–20.0 weeks), seven stomach (10.3–20.6 weeks) and seven liver (10.3–20.6 weeks).
Normal breast epithelial organoids
Raw fastq files generated from samples of three distinct normal breast epithelial organoids were downloaded on 3 February 2016 from SRA: SRR1027188, SRR1027189, SRR1027190 (34). Samples contained rRNA– RNA.
Human simulated data
Fastq files from two simulated human paired-end RNA-Seq experiments, generated as described in (24), were downloaded on 6 January 2015 from: http://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-1728/files/.
Briefly, transcripts were randomly selected from 11 annotation tracks in the UCSC Genome Browser, the original transcript and novel isoforms generated from the transcript were included, and expression levels were derived from a normal human retina RNA-Seq data set. The second simulated dataset contained more noise than the first. Substitution variants and indel polymorphisms were introduced into both simulated transcriptomes.
Positive control data from fusionmap was downloaded from http://www.arrayserver.com/wiki/index.php?title=FusionMap. This data was combined with “simulation 1" from (24) and is referred to as “mixed" data, following the nomenclature in (18).
CML K562 cell lines
Two replicates containing ribo-depleted and poly-adenylated (rRNA–/polyA+) RNA from Cold Spring Harbor Laboratories (CSHL) were downloaded from ENCODE: https://www.encodeproject.org/experiments/ENCSR000CPS/ on 31 August 2015. ENCODE accession ids: sample 1: library ENCLB555ALY (fastq files ENCFF000HOC and ENCFF000HOE) and sample 2: library ENCLB555ALX (fastq files ENCFF000HOD and ENCFF000HOQ).
A total of nine replicates from two biosamples from UConn were downloaded on 6 April 2016 from SRA under accession number SRP013565. From ENCODE biosample ENCBS087RNA: SRR3192411, SRR3192412, SRR3192417. From ENCODE Biosample ENCBS088RNA: SRR3192409, SRR3192410, SRR3192413, SRR3192415, SRR3192416, SRR3192422. Samples contained rRNA–/polyA+ RNA.
Ewing's sarcoma cell lines
Raw fastq files were downloaded on 12 February 2016 from SRA under accession number SRP048562. Six samples with EWS-FLI1 translocations from 2 different cell lines from (25) were used in the analysis. The EWS-FLI1 fusion was depleted in some samples by transfection with lentiviral shRNA shFLI1 and other samples were treated with a control shGFP which did not deplete the fusion. SRR1594020-SRR1594023 were obtained from Ewing sarcoma cell line SKMNC. SRR1594020 and SRR1594022 were treated with shGFP for 48 and 96 h, respectively. SRR1594021 and SRR1594023 were treated with shFLI1 for 48 and 96 h, respectively. SRR1594024 and SRR1594025 were obtained from Ewing sarcoma cell line A673. The former was treated with shGFP for 48 h. The latter was treated with shFLI1 for 48 h. Samples contained rRNA–/polyA+ RNA.
Ovarian cancer cell lines
Raw fastq files were downloaded on 18 March 2016 from SRA. SRR1772257 (rRNA–/RNase-R+) and SRR1772957 (rRNA–) were obtained from the OVCAR3 cell line.
Data preprocessing
Raw fastq files were processed using default settings of TrimGalore version 0.3.7: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ and Cutadapt version 1.5 (35) and these trimmed files were used as input to SOAPfuse, STAR-Fusion, and KNIFE which yielded MACHETE input files.
SOAPfuse, EricScript and STAR-Fusion
SOAPfuse (v1.27) and STAR-Fusion (version 0.7.0) indices were built using the human reference genome hg19. In addition to the trimmed fastq files, SOAPfuse uses an additional sample file which was generated dynamically with a custom PERL script. Since both STAR-Fusion and SOAPfuse were designed specifically for fusion detection, we ran all samples with the suggested parameters provided by the authors: http://soap.genomics.org.cn/soapfuse.htmlhttps://github.com/STAR-Fusion/STAR-Fusion/wiki
We used the following output files: for STAR-Fusion: *star-fusion.fusion_candidates.final.abridged; for SOAPfuse: *final.Fusion.specific.for.trans
We used filtered results from EricScript 0.5.5 Ensemble version 73 per the user manual from: https://sites.google.com/site/bioericscript/download
Analysis of MACHETE versus SOAPfuse, EricScript and STAR-Fusion
Filtered results for each of the algorithm are available as supplemental tables and scripts. MACHETE source code is available through the Salzman lab website (http://salzmanlab.stanford.edu/software/).
PCR Validation in OVCAR3
RNA was extracted and reverse transcribed using standard protocols. All PCRs were performed for 35 cycles, 30 s extension times using TaqSupermix and the following PCR primers. PCR products were TOPO cloned and Sanger sequencing was performed according to standard protocols.
Benchmarking against SOAPfuse, EricScript and STAR-Fusion
We have benchmarked MACHETE against SOAPfuse (23), EricScript (36) and STAR-Fusion (27). SOAPfuse achieved the best balance between sensitivity and specificity according to recent independent benchmarks (18,32); EricScript was the top performer according to analyses performed in (18). On some samples, STAR-Fusion performed better in our hands than SOAPfuse or EricScript, so we included it in our analysis. We have analyzed these algorithms on publicly available data for three cancer types: (i) two independent datasets from the BCR-ABL1 positive cell line K562, (ii) data from two Ewing's sarcoma cell lines and (iii) the ovarian cancer cell line OVCAR3. We also analyzed these algorithms on three negative control datasets: (i) 43 samples from 8 normal human fetal tissue types, (ii) three normal breast organoids and (iii) two datasets simulated from the human reference transcriptome under two different parameter regimes (24); (iv) a mixed dataset that is comprised of negative control data and simulated positive control fusions as described in (18).
Criteria for benchmarking and algorithm comparison
The vast majority of validated gene fusions, including the BCR-ABL1 and EWS-FLI1 fusions discussed in this paper, occur at exon–exon boundaries (37). MACHETE only detects fusions at exon boundaries, and these stringent criteria improve specificity of other algorithms by eliminating some false positive fusions in normal samples. In order to directly compare the results of fusion algorithms, we imposed a uniform set of filtering criteria on the results of all algorithms that (i) fusions must be reported at annotated exon–exon boundaries, and (ii) candidate fusion junctions on the same chromosome and on the same strand have to occur more than 1 Mb apart or be inversions >100 kb apart. The reason for imposing a different radius on junctions between exons on the same strand is to prevent circular RNA from being reported as a fusion, since it is typically detected between exons within 1 Mb of the genome. MACHETE results are additionally filtered based on statistical scores which are not provided by other algorithms, except EricScript (see Materials and Methods). To assess the effect of imposing a commonly used threshold of more than one read on the false positive rate for all algorithms, we compared results using either all junctions fulfilling the above criteria, or the subset of those junctions with more than one mapping read. For the remainder of the paper we report the number of distinct fusions found by an algorithm in each sample or sample type in the form of ‘Q|R’ where Q and R are the filtered count without and with using the read count threshold of 1, respectively.
RESULTS
MACHETE significantly improves specificity on negative controls
In evaluating specificity, we adopted standards previously applied by third party assessments of fusion detection algorithms (38). Namely, a fusion detection algorithm with high specificity should find no fusions in data simulated from the reference human transcriptome. In normal samples, the algorithm should also rarely, if ever, find interchromosomal fusions, with exceptions occurring only when cryptic translocations have occurred. Because such events are thought to be exceptionally rare, we assume that interchromosomal fusions reported by algorithms in normal samples are artifacts, as other authors have done (39). A subset of individuals harbor local tandem duplications in their genomes, copy number variants (CNVs), that could in principle interrupt protein coding genes and result in gene fusions. However, these events are also considered to be rarely detected in normal cells, especially between exons separated by more than 1 Mb (40,41), motivating our filtering criteria introduced above.
Using the same statistical thresholds that give high true positive rates in the cancer samples, MACHETE has an unprecedented low false positive rate, reporting only one false positive fusion among all of the negative controls. This is a significant advance over SOAPfuse and STAR-Fusion which each report large numbers of distinct fusions in the same samples. Figure 4A depicts the number of fusion isoforms detected by MACHETE and other algorithms in negative control data. In the simulated data, MACHETE and STAR-Fusion reported only three fusions, all of which were TOP3B-PI4KA isoforms that were included in the simulated ground truth (true positives) (24). SOAPfuse found one of the three fusions, but reported an additional 7|5 fusions that were not in the simulated data; similarly, Ericscript had 23|23 fusions that were absent from the simulation, evidence of reporting significant numbers of false positives.
Figure 4.

Performance of MACHETE compared to SOAPfuse, EricScript and STAR-Fusion. (A) Counts of false positive fusions from MACHETE, SOAPfuse and STAR-Fusion are shown for each of the three types of normal samples used as negative controls in this study. Light blue indicates fusions supported by a single read, dark blue indicates fusions supported by at least two reads. (B) Isoforms of the EWSR1-FLI (rows 1–4) and FLI-EWSR1 (rows 5 and 6) detected by MACHETE (yellow), STAR-Fusion (green), SOAPfuse (red) and EricScript (blue) in two distinct Ewing's sarcoma cell lines. Isoforms 1–3 are detected in both cell lines, 4–6 are only detected in A673 cells. Left panel: SKMNC cells under four treatments. Right panel: A673 cells under two treatments. Read counts supporting each isoform are shown in white. shRNA treatments are abbreviated as follows; shGFP (48 h): control treated with shGFP for 48 h; shGFP (96h): control treated with shGFP for 96 h; shFLI1 (48h): cells treated with shFLI1 for 48 h, partially depleting EWSR1-FLI; shFLI1 (96 h): cells treated with shFLI1 for 96 h, with more extreme, but incomplete, depletion of EWSR1-FLI isoforms. Isoform 1: EWSR1-FLI1 chr11:128675261-chr22:29683123; Isoform 2: EWSR1-FLI1 chr11:128675261-chr22:29684775; Isoform 3: EWSR1-FLI1 chr22:29683123-chr11:128677075; Isoform 4: EWSR1-FLI1 chr22:29683123-chr11:128679052; Isoform 5: FLI1-EWSR1 chr11:128651918-chr22:29684595; Isoform 6: FLI1-EWSR1 chr11:128651918-chr22:29688126.
In normal fetal RNA, MACHETE reported no fusions in 43 samples. In the same data, SOAPfuse reported 5|2 fusions, while STAR-Fusion reported 39|32 fusions and Eriscript reported 56|54 In normal breast organoids, SOAPfuse detected 11|4 fusion isoforms and STAR-Fusion detected 45|15 fusions and EricScript reported 37|35. In the same data, MACHETE reported a single potential fusion, MBNL2-GNAS, which was also reported by SOAPfuse.
While STAR-Fusion matched the specificity of MACHETE on simulated data, it reported many more presumed false positives than MACHETE or SOAPfuse in normal samples; EricScript had similar numbers of presumed false positives. These results underline the significant improvement in specificity achieved by MACHETE on both real and simulated data.
MACHETE improves positive predictive value compared to current best performing algorithms
We blindly assessed the sensitivity and Positive Predictive Value (PPV) = TP/(FP + TP) of MACHETE compared to the top performing algorithms from (18) and (42) in a context where the ground truth is known. Because we were not able to obtain the negative control simulated data used in (18), we used third party simulated data from the negative control of Engstrom simulation 1 in (24) that lacked any gene fusions as our negative control data, and concatenated all reads from the positive control dataset containing 50 gene fusions used in (18). In order to have uniform read lengths, the 3΄ most base of the reads from the Engstrom simulation 1 was removed from each read. We refer to this as the ‘mixed’ dataset.
MACHETE reported 33 of the 50 true positive fusions and 0 false positives, a sensitivity of 66% and a PPV of 100%. In contrast, the best performing algorithms from (18), EricScript (resp. SOAPfuse and STAR-Fusion) had sensitivity of 80% (74% and 84%) and PPV of 77% (71% and 91%). STAR-Fusion performs well here, having a PPV close to MACHETE. However, as discussed below, STAR-Fusion suffers from a very high FP rate in other samples, while MACHETE maintains tight control of FPs (see Figure 5).
Figure 5.

Sensitivity analysis and statistical properties of MACHETE versus EricScript, SOAPfuse, and STAR-Fusion. Panel A: Comparison of sensitivity and PPV of top fusion algorithms. MACHETE has highest PPV driven by its use of statistical scores (see B) and lower sensitivity than other algorithms which detect many FP in some or all control data sets. Panel B: MACHETE uses the empirical p value (<0.1) and junction_cdf score (>0.2), thresholds chosen before testing MACHETE on mixed data, and blindly achieves sensitive separation of TP and FP (boxed area). Panel C: Ericscore distributions among FP and TP are similar, showing this score does not discriminate TP and FP.
The use of statistical scores by MACHETE is key to its precision and high PPV. MACHETE detects the sequences of 42 fusions in the ground truth set of fusions, but nine of them are removed based on their poor statistical scores. We display the distribution of empirical p values in Figure 5B stratified by whether the fusion is a TP. Figure 5B shows that all fusions with low empirical p values and high junction_cdf scores are true positives which is why statistical scoring allows MACHETE to achieve a perfect PPV on the blinded, third-party-generated mixed dataset. These features are not true of the EricScore, see Figure 5C: even at a high threshold for the EricScore, the PPV is low: at a threshold for the score of 0.95, 31 TP and 8 FP are reported. MACHETE achieves higher sensitivity (39 fusions detected) and a PPV of 1 (0 FP detected). Finally, we note that MACHETE detects 42 fusions, the same sensitivity of STAR-Fusion, but assigns some TP scores consistent with them being artifacts. This behavior is predicted for any statistic used to test a hypothesis, and reflects the property that no statistical test (or statistical algorithm) can have perfect power (i.e. a power of 1).
MACHETE has comparable detection efficiency of BCR-ABL1 to other algorithms
Increased specificity can always be achieved by shrinking a rejection region, which would correspond to more stringent thresholds imposed to accept a nominated fusion. To address the concern that our low false negative rate could result in decreased statistical power to detect true positives, we performed tests of MACHETE's ability to identify positive control fusions.
We used data from the chronic myelogenous leukemia (CML) cell line K562 to test for one of the best characterized gene fusions, BCR-ABL1, and another validated fusion, NUP214-XKR3 (19). A total of 11 replicates generated in two labs in the ENCODE consortium were analyzed (see Methods). MACHETE, SOAPfuse, EricScript and STAR-Fusion were run on all 11 replicates, although SOAPfuse failed to complete after running for seven days for one sample (SRR3192422) despite repeated attempts.
Across all samples, the algorithms detected only one isoform of each of the validated fusions, BCR-ABL1 and NUP214-XKR3. Within each replicate, these fusions were within the top three results when ranked by read count for each algorithm and were consistently the top two results for MACHETE. Read count filtering did not change detection of these well-documented fusions. NUP214-XKR3 was detected in all replicates and the BCR-ABL1 fusion was identified in all replicates except SRR192416 and SRR192417 by MACHETE, SOAPfuse and STAR-Fusion (Supplementary Tables S2–S5).
Although SRR3192422 was excluded from the above analysis because SOAPfuse failed to complete, we examined the fusions reported by MACHETE and STAR-Fusion in this replicate (Supplementary Tables S2 and S4). Both algorithms reported more fusions than in any of the other 10 replicates: 247|154 for STAR-Fusion and 57|46 for MACHETE. BCR-ABL1 and NUP214-XKR3 remained the fusions with the highest read counts reported by MACHETE in this replicate. NUP214-XKR3 remains the top result for STAR-Fusion, but the algorithm reported 35 other fusions with as many or more reads as BCR-ABL1 in this replicate. These results demonstrate MACHETE's ability to prioritize true positive fusions even in samples that are problematic for other algorithms.
MACHETE improves in vivo sensitivity of fusion detection in Ewing's sarcoma cell lines
Ewing's sarcoma is characterized by a translocation resulting in expression of fusion transcripts between the EWSR1 gene on chromosome 22 and the FLI1 gene on chromosome 11 (11,22). We evaluated the sensitivity of MACHETE's detection of the documented alternative splicing between EWSR1 and FLI1 in the Ewing's sarcoma cell lines SKMNC and A673, as well as the detection of the reciprocal fusion event between FLI1 and EWSR1 in A673 (43) (Figure 4B).
We analyzed RNA-Seq data generated from two experiments using the cell line A673 and four experiments using the cell line SKMNC (44). shRNA against FLI1, targeting the EWSR1-FLI1 fusion, and negative control shRNA against GFP were introduced including four SKMNC samples: treatment of shGFP for 48 h, shGFP for 96 h, shFLI1 for 48 h, or shFLI1 for 96 h. The two A673 samples consisted of the following experiments: treatment of shGFP for 48 h or shFLI1 for 96 h. Because shRNA modulated the expression of the EWSR1-FLI1 transcripts, this dataset allowed us to assess the sensitivity of detection of EWSR1-FLI1.
MACHETE, EricScript and STAR-Fusion detected the expression of EWSR1-FLI1 fusion transcripts in each of the six samples although EricScript failed to detect isoform 2, 3 or 4 (see Figure 4). MACHETE detected more isoforms of EWSR1-FLI1 than other algorithms while reporting fewer overall fusions: STAR-Fusion (resp. EricScript) reports 78 (resp. 60) distinct fusion isoforms across all six samples, from which it would be difficult to select fusions for validation MACHETE reports only 16 distinct expressed fusion isoforms, including one FLI1-EWSR1 and four distinct EWSR1-FLI1 isoforms. SOAPfuse also reports 16 distinct fusion isoforms, although only two of these are EWSR1-FLI isoforms and none are FLI-EWSR1.
The A673 sample is known to express the reciprocal FLI1-EWSR1 transcript (43). SOAPfuse did not detect any fusion transcripts derived from the reciprocal translocation. MACHETE detected one isoform of FLI1-EWSR1 and EricScript had the best sensitivity for this isoform. Together, this data demonstrates that MACHETE is more sensitive and specific than the other algorithms in the analyzed Ewing's sarcoma data.
MACHETE discovers novel fusions in ovarian cancer cell line OVCAR3
All algorithms were used to predict fusions in publicly available RNA-Seq data from the ovarian cancer cell line OVCAR3. We used RT-PCR on RNA we extracted in our own lab to validate the most abundant predicted fusions that passed MACHETE's statistical filters: two splice isoforms of a translocation giving rise to a fusion between the genes SPEN and NEU1, another translocation resulting in a fusion between NUP98 and the gene BEAN1, and a >2 Mb predicted duplication resulting in a fusion between the genes ITSN2 and OTOF. All three fusions, including the predicted splice variant in SPEN-NEU1 were validated (see Materials and Methods).
EricScript predicted NUP98-BEAN1 in both samples from OVCAR3, but it did not detect any of the isoforms of the SPEN-NEU1 fusion, and predicted ITSN2-OTOF in only one of two samples. SOAPfuse predicted only one fusion in each of the two OVCAR3 samples with one read each: in one sample, the fusion LINC00665-HKR1, and in the other, SPEN-NEU1. STAR-Fusion detected 27|17 fusions, but failed to detect either of the two SPEN-NEU1 isoforms that were predicted by MACHETE and validated by PCR underlining in vivo gains in sensitivity contributed by MACHETE.
DISCUSSION
In this paper, we describe a new statistical framework for detecting and quantifying gene fusions. The bioinformatic methodology is transparent and makes no ad hoc or heuristic choice. This allows MACHETE to achieve highly specific and sensitive unsupervised detection of fusions. Statistical models are the workhorse of MACHETE, and allow it to achieve an essentially null background on cytogenetically normal samples, which, to our knowledge, has not been achieved by other published fusion detection algorithms. This feature is an innovation of MACHETE that uniquely situates it for application in cancer diagnosis and discovery of driving and targetable gene fusions.
While achieving specificity, MACHETE is still able to identify true positives at a similar or improved rate compared to current top algorithms; noteworthy cases of improved sensitivity for fusions in the OVCAR3 cell line. In other cases, such as the Ewing's sarcoma data, MACHETE and STAR-Fusion have similarly high sensitivity, but MACHETE is much more specific. The extremely low false positive rate achieved by MACHETE, demonstrated by its performance in normal samples, allows it to discriminate lowly expressed but potentially biologically important gene fusions from false positives regardless of their expression levels. For example, NUP98 is known to be a recurrently fused oncogene (45) and was first identified in AML (46). To our knowledge, this is the first report and validation of a NUP98 fusion in an ovarian cancer cell line.
MACHETE's assignment of a statistical score, including an empirical p value, to each putative fusion, allows for unsupervised detection of fusions. This has important implications for research and for deploying MACHETE to analyze large public datasets. Algorithms that require human guidance and heuristic filtering are too resource intensive for these applications, and introduce biases. High false positives in these algorithms also dilute the statistical signal of fusions that may yield insight into tumor biology.
Furthermore, fusions can be powerful biomarkers, but may have low abundance especially if they are to be detected in bodily fluid containing RNA from normal cells. Thus, for clinical use, comparatively lowly expressed fusions would have to be detected in a background of abundant RNA from normal cells. In addition, clinical applications cannot support secondary validation, as described in the introduction. Thus, the high specificity and sensitivity of MACHETE promises to improve discovery of significant fusions in clinical samples.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Peter Wang and staff from the Stanford University Sherlock computer clusters for technical assistance. Parts of the computing for this project was performed on the Sherlock cluster. We would like to thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that have contributed to these research results. We would like to acknowledge the support of the Stanford Center for Computational, Evolutionary and Human Genomics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NCI [R00 CA168987-03]; NIGMS [R01 GM116847]; JIMB seed grant; NSF CAREER Award; McCormick-Gabilan Fellowship; NIGMS of the NIH [T32GM007276]; Baxter Family Fellowship (to J.S.); J.S. is an Alfred P. Sloan fellow in Computational & Evolutionary Molecular Biology; Stanford Pediatric Clinical Cancer Genomics Program (to E.A.S.C. and A.G.L.). Funding for open access charge: Start-up funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Druker B.J., Talpaz M., Resta D.J., Peng B., Buchdunger E., Ford J.M., Lydon N.B., Kantarjian H., Capdeville R., Ohno-Jones S. et al. . Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001; 344:1031–1037. [DOI] [PubMed] [Google Scholar]
- 2. Weisberg E., Boulton C., Kelly L.M., Manley P., Fabbro D., Meyer T., Gilliland D.G., Griffin J.D.. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002; 1:433–443. [DOI] [PubMed] [Google Scholar]
- 3. Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S., Watanabe H., Kurashina K., Hatanaka H. et al. . Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561–566. [DOI] [PubMed] [Google Scholar]
- 4. Tomlins S.A., Rhodes D.R., Perner S., Dhanasekaran S.M., Mehra R., Sun X.-W., Varambally S., Cao X., Tchinda J., Kuefer R. et al. . Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005; 310:644–648. [DOI] [PubMed] [Google Scholar]
- 5. Ju Y.S., Lee W.-C., Shin J.-Y., Lee S., Bleazard T., Won J.-K., Kim Y.T., Kim J.-I., Kang J.-H., Seo J.-S.. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012; 22:436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carneiro B.A., Elvin J.A., Kamath S.D., Ali S.M., Paintal A.S., Restrepo A., Berry E., Giles F.J., Johnson M.L.. FGFR3-TACC3: a novel gene fusion in cervical cancer. Gynecol. Oncol. Rep. 2015; 13:53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh D., Chan J.M., Zoppoli P., Niola F., Sullivan R., Castano A., Liu E.M., Reichel J., Porrati P., Pellegatta S. et al. . Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012; 337:1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sakamoto H., Tsukaguchi T., Hiroshima S., Kodama T., Kobayashi T., Fukami T.A., Oikawa N., Tsukuda T., Ishii N., Aoki Y.. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011; 19:679–690. [DOI] [PubMed] [Google Scholar]
- 9. Halasi M., Wang M., Chavan T., Gaponenko V., Hay N., Gartel A.. ROS inhibitor N-acetyl-cysteine antagonizes the activity of proteasome inhibitors. Biochemistry. 2013; 454:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gozgit J.M., Wong M.J., Moran L., Wardwell S., Mohemmad Q.K., Narasimhan N.I., Shakespeare W.C., Wang F., Clackson T., Rivera V.M.. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012; 11:690–699. [DOI] [PubMed] [Google Scholar]
- 11. Stransky N., Cerami E., Schalm S., Kim J.L., Lengauer C.. The landscape of kinase fusions in cancer. Nat. Commun. 2014; 5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shugay M., Ortiz de Mendíbil I., Vizmanos J.L., Novo F.J.. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics. 2013; 29:2539–2546. [DOI] [PubMed] [Google Scholar]
- 13. Fernandez-Cuesta L., Sun R., Menon R., George J., Lorenz S., Meza-Zepeda L.A., Peifer M., Plenker D., Heuckmann J.M., Leenders F. et al. . Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biol. 2015; 16:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ortiz de Mendíbil I., Vizmanos J.L., Novo F.J.. Signatures of selection in fusion transcripts resulting from chromosomal translocations in human cancer. PLoS ONE. 2009; 4:e4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Q., Xia J., Jia P., Pao W., Zhao Z.. Application of next generation sequencing to human gene fusion detection: computational tools, features and perspectives. Brief Bioinformatics. 2013; 14:506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M.. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009; 458:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carrara M., Beccuti M., Cavallo F., Donatelli S., Lazzarato F., Cordero F., Calogero R.A.. State of art fusion-finder algorithms are suitable to detect transcription-induced chimeras in normal tissues. BMC Bioinformatics. 2013; 14(Suppl. 7):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumar S., Vo A.D., Qin F., Li H.. Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data. Sci. Rep. 2016; 6:21597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maher C.A., Palanisamy N., Brenner J.C., Cao X., Kalyana-Sundaram S., Luo S., Khrebtukova I., Barrette T.R., Grasso C., Yu J. et al. . Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:12353–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thompson-Wicking K., Francis R.W., Stirnweiss A., Ferrari E., Welch M.D., Baker E., Murch A.R., Gout A.M., Carter K.W., Charles A.K. et al. . Novel BRD4-NUT fusion isoforms increase the pathogenic complexity in NUT midline carcinoma. Oncogene. 2013; 32:4664–4674. [DOI] [PubMed] [Google Scholar]
- 21. Bandopadhayay P., Ramkissoon L.A., Jain P., Bergthold G., Wala J., Zeid R., Schumacher S.E., Urbanski L., O’Rourke R., Gibson W.J. et al. . MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 2016; 48:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szabo L., Morey R., Palpant N.J., Wang P.L., Afari N., Jiang C., Parast M.M., Murry C.E., Laurent L.C., Salzman J.. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015; 16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia W., Qiu K., He M., Song P., Zhou Q., Zhou F., Yu Y., Zhu D., Nickerson M.L., Wan S. et al. . SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013; 14:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engström P.G., Steijger T., Sipos B., Grant G.R., Kahles A., Rätsch G., Goldman N., Hubbard T.J., Harrow J., Guigó R. et al. . Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods. 2013; 10:1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O.. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE. 2012; 7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patch A.-M., Christie E.L., Etemadmoghadam D., Garsed D.W., George J., Fereday S., Nones K., Cowin P., Alsop K., Bailey P.J. et al. . Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015; 521:489–494. [DOI] [PubMed] [Google Scholar]
- 27. Haas B., Dobin A.. STAR-Fusion. STAR-Fusion. 2015. [Google Scholar]
- 28. Quail M.A., Kozarewa I., Smith F., Scally A., Stephens P.J., Durbin R., Swerdlow H., Turner D.J.. A large genome center's improvements to the Illumina sequencing system. Nat. Methods. 2008; 5:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Houseley J., Tollervey D.. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS ONE. 2010; 5:e12271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Salzman J. Datta S, Nettleton D. RNA isoform discovery through goodness of fit diagnostics. Statistical Analysis of Next Generation Sequencing Data. 2014; Cham: Springer International Publishing; 261–276. [Google Scholar]
- 31. Hoeffding W. A combinatorial central limit theorem. Ann. Math. Stat. 1951; 22:558–566. [Google Scholar]
- 32. Barber R.F., Candès E.J.. Controlling the false discovery rate via knockoffs. Ann. Statist. 2015; 43:2055–2085. [Google Scholar]
- 33. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodological). 1995; 57:289–300. [Google Scholar]
- 34. Eswaran J., Horvath A., Godbole S., Reddy S.D., Mudvari P., Ohshiro K., Cyanam D., Nair S., Fuqua S.A.W., Polyak K. et al. . RNA sequencing of cancer reveals novel splicing alterations. Sci. Rep. 2013; 3:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011; 17:10. [Google Scholar]
- 36. Benelli M., Pescucci C., Marseglia G., Severgnini M., Torricelli F., Magi A.. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics. 2012; 28:3232–3239. [DOI] [PubMed] [Google Scholar]
- 37. Wan Q., Dingerdissen H., Fan Y., Gulzar N., Pan Y., Wu T.-J., Yan C., Zhang H., Mazumder R.. BioXpress: an integrated RNA-seq-derived gene expression database for pan-cancer analysis. Database (Oxford). 2015; 2015:bav019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yoshihara K., Wang Q., Torres-Garcia W., Zheng S., Vegesna R., Kim H., Verhaak R.G.W.. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015; 34:4845–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Hsi-Yang Fritz M. et al. . An integrated map of structural variation in 2, 504 human genomes. Nature. 2015; 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kinsella M., Harismendy O., Nakano M., Frazer K.A., Bafna V.. Sensitive gene fusion detection using ambiguously mapping RNA-Seq read pairs. Bioinformatics. 2011; 27:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Greger L., Su J., Rung J., Ferreira P.G., Geuvadis consortium., Lappalainen T., Dermitzakis E.T., Brazma A.. Tandem RNA chimeras contribute to transcriptome diversity in human population and are associated with intronic genetic variants. PLoS ONE. 2014; 9:e104567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu S., Tsai W.-H., Ding Y., Chen R., Fang Z., Huo Z., Kim S., Ma T., Chang T.-Y., Priedigkeit N.M. et al. . Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016; 44:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Elzi D.J., Song M., Houghton P.J., Chen Y., Shiio Y.. The role of FLI-1-EWS, a fusion gene reciprocal to EWS-FLI-1, in Ewing sarcoma. Genes Cancer. 2015; 6:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Riggi N., Knoechel B., Gillespie S., Rheinbay E., Boulay G., Suva M., Rossetti N.. HHS Public Access. Cancer Cell. 2014; 26:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Franks T.M., Hetzer M.W.. The role of Nup98 in transcription regulation in healthy and diseased cells. Trends Cell Biol. 2013; 23:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gervais C., Mauvieux L., Perrusson N., Hélias C., Struski S., Leymarie V., Lioure B., Lessard M.. A new translocation t (9;11) (q34;p15) fuses NUP98 to a novel homeobox partner gene, PRRX2, in a therapy-related acute myeloid leukemia. Leukemia. 2005; 19:145–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.