


Abstract
Non-coding RNAs have critical roles in biological processes, and RNA chaperones can promote their folding into the native shape required for their function. La proteins are a class of highly abundant RNA chaperones that contact pre-tRNAs and other RNA polymerase III transcripts via their common UUU-3′OH ends, as well as through less specific contacts associated with RNA chaperone activity. However, whether La proteins preferentially bind misfolded pre-tRNAs or instead engage all pre-tRNA substrates irrespective of their folding status is not known. La deletion in yeast is synthetically lethal when combined with the loss of tRNA modifications predicted to contribute to the native pre-tRNA fold, such as the N2, N2-dimethylation of G26 by the methyltransferase Trm1p. In this work, we identify G26 containing pre-tRNAs that misfold in the absence of Trm1p and/or La (Sla1p) in Schizosaccharomyces pombe cells, then test whether La preferentially associates with such tRNAs in vitro and in vivo. Our data suggest that La does not discriminate a native from misfolded RNA target, and highlights the potential challenges faced by RNA chaperones in preferentially binding defective substrates.
INTRODUCTION
Nascent RNA transcripts have a significant propensity to become kinetically trapped in non-functional conformations. It has thus been hypothesized that one of the earliest functions of polypeptides would be to help resolve these aberrant structures so that RNAs can better access their functionally relevant, native folds (1). Proteins that promote the native fold have been classified based on their proposed mechanism of action (reviewed in (2)). One such class of factors is the RNA chaperones, which are hypothesized to unwind misfolded structures in the absence of adenosine triphosphate (ATP) hydrolysis via mechanisms that remain ambiguous (3–5). By assisting RNA substrates in attaining their native fold, RNA chaperones can rescue defective or misfolded RNA substrates from RNA quality control systems such as nuclear surveillance (6–8). Though it's been recognized that many non-coding RNAs are susceptible to folding into alternate inactive conformations, mechanisms by which RNA chaperones might discriminate misfolded RNA substrates are not well understood. It has been previously noted that the greater homogeneity of functional groups in RNA relative to amino acids might make for greater challenges in discrimination of fold for RNA chaperones relative to protein chaperones, even if thermodynamic models for misfolded target recognition by RNA chaperones have been proposed (3,5). For RNA chaperones such as StpA and hnRNP A1, it has been proposed that these preferably engage single stranded regions of RNA that are hypothesized to be more prevalent in misfolded conformations (9–11). One significant challenge to accessing insights into target recognition is the identification of physiologically relevant, misfolded RNA conformations that the RNA chaperone in question samples in vivo. Thus, the issue of functional target discrimination by RNA chaperones has not been explored extensively.
The La and La-related proteins are a conserved class of eukaryotic RNA-binding proteins that have been characterized as RNA chaperones (8,12–16). La proteins have important roles in the processing of nascent RNA polymerase (Pol) III transcripts, with pre-tRNAs being their best-characterized substrates (reviewed in (17)). This association relies in part on specific recognition of the uridylate containing trailer sequence (UUU-3′OH) that is found at the end of all Pol III transcripts and some non-coding RNA Pol II processing intermediates. In yeast, La binding to the UUU-3′OH containing trailer results in pre-tRNA stabilization and accumulation, and commits the pre-tRNA to the La-dependent pathway of processing, in which leader processing precedes endonucleolytic trailer processing, rather than trailer removal by exonucleases like Rex1p in the absence of La (18,19).
Structural and biochemical work have indicated that UUU-3′OH dependent La-RNA binding relies on a highly conserved N-terminal domain which comprises a winged-helix like La-motif (LAM) and an adjacent RNA recognition motif (RRM1) that synergize to form the so-called La-module (20,21). Co-crystal structures of the La module interacting with UUU-3′OH containing RNAs have revealed conserved residues in the LAM involved in recognition of the poly-uridylate sequence (22,23), and this binding mode is associated with protecting nascent Pol III transcripts from exonuclease digestion (8). However, other non-UUU-3′OH-dependent RNA contacts also make a major contribution to La/pre-tRNA binding. La binds short UUU-3′OH containing oligonucleotides ∼10× less avidly than the same UUU-3′OH motif presented in the context of a full pre-tRNA (13). This is at least in part due to other regions of the RRM1 domain, as well as disordered amino acids C-terminal to this region, that engage the main body of the tRNA and promote the native fold through RNA chaperone activity (8,13,14,24). Mutation of basic amino acids in RRM1 result in decreased pre-tRNA binding without affecting UUU-3′OH binding (13), and mutation of aromatic amino acids in the RNP motifs of RRM1 normally associated with canonical RRM-associated RNA binding result in defective RNA chaperone activity in vitro and degradation of defective (mutated) pre-tRNAs in vivo (8,14). These mutations also result in decreased levels of the stable pre-tRNA species normally associated with the La-dependent pre-tRNA processing pathway (8). Importantly, decreased pre-tRNA accumulation due to RRM1 mutation only correlates with decreased mature tRNA levels if the tRNA is defective (mutated); conversely, wild-type (WT) pre-tRNAs show normal mature tRNA levels despite decreased pre-tRNA accumulation in the presence of La-RRM1 mutants. These data led to a model in which La engages all pre-tRNAs regardless of folding status for the benefit of rescuing defective substrates and at the expense of processing efficiency for non-defective pre-tRNAs (8). However, the ability of La to differentially bind any folded versus misfolded substrates within the UUU-3′OH containing RNA target cohort has not previously been tested in vitro or in vivo. Since La is hypothesized to be limiting relative to RNA Pol III transcript abundance (25), whether La discriminates native from misfolded substrates could play an important role in determining which pre-tRNAs access the La-dependent versus La-independent processing pathways. Attempting to experimentally test whether the affinity of La for a physiologically relevant, misfolded RNA target differs relative to its natively folded counterpart is a major theme of this work.
Pre-tRNAs are among the most highly post-transcriptionally modified classes of RNA species, with a median of eight modified nucleosides per mature transcript (26). Several cases have been reported in which the absence of an essential modification has resulted in the degradation of select tRNAs. Mature tRNAs lacking particular modifications can be degraded at elevated temperatures via the rapid tRNA decay pathway (27,28), or hypomodified pre-tRNAs can be degraded in the nucleus by nuclear surveillance. One such example is the methylation of A58 (m1A58) by Trm6/61 on pre-tRNAiMet in Saccharomyces cerevisiae; absence of this modification can result in degradation of pre-tRNAiMet via the TRAMP complex and nuclear exosome (7), but this can be rescued by overexpression of the La homolog Lhp1p (6). Similarly, deletion of Lhp1p is synthetically lethal with tRNA mutations shown to affect their folding (29) or with deletion of other tRNA modification enzymes (30,31), consistent with La functioning redundantly with elements that stabilize native pre-tRNA structure.
One tRNA modification associated with La function is the N2,N2-dimethylation of G26 containing tRNAs by the methyltransferase Trm1p (30), a tRNA modification enzyme that localizes to the nuclear membrane (32). N2,N2-dimethylation of G26 has been hypothesized to stabilize tRNA structure in the ‘hinge’ region between the coaxially stacked anticodon- and D-stems (33,34), as well as prioritize the native fold of cytoplasmic tRNA anticodon stem loops over aberrant structures occasionally observed in native mitochondrial tRNAs (35). Deletion of Trm1 is synthetically lethal with deletion of Lhp1 when S. cerevisiae is grown at elevated temperature (30), and N2,N2-dimethylated guanosine containing tRNAs are immunoprecipitated by La protein (36), suggesting that G26 modification occurs during the window of La-pre-tRNA occupancy. Taken together, these data suggest that La binds the nascent pre-tRNA transcript and promotes its stability and/or the native fold until this is reinforced by methylation of G26 by Trm1p. Thus, the study of La binding to Trm1p-modified versus -unmodified pre-tRNAs represents an opportunity to compare the affinity of an RNA chaperone to a natively folded versus misfolded substrate in which the misfolded conformation is physiologically relevant and is sampled by the RNA chaperone in vivo.
Previous work suggests that modification at G26 by Trm1p and La RNA chaperone activity function redundantly in stabilizing tRNA structure (30). However, which tRNAs might misfold in the absence of dimethylation at G26, or whether La can discriminate any folded versus misfolded substrate, has not previously been tested. In this work, we have attempted to directly test whether La differentiates misfolded G26 unmodified versus Trm1p-modified pre-tRNAs both in vitro and in Schizosaccharomyces pombe cells. We demonstrate that similar to S. cerevisiae, deletion of S. pombe La (Sla1p) and Trm1p is synthetically lethal at elevated temperatures. We then further show that Sla1p dependent suppression of lethality relies on amino acids associated with La-dependent RNA chaperone activity, consistent with Trm1p and La functioning redundantly to stabilize pre-tRNA structure. Using lead acetate chemical probing, we identify tRNA-SerUGA as a tRNA whose fold changes on the basis of Trm1 modification, and further demonstrate its impaired charging in a sla1−/trm1− strain, consistent with misfolding in vivo. We also identify cohorts of G26 containing tRNAs whose steady-state abundance is depleted in trm1−, sla1− and sla1−/trm1− cells, consistent with Trm1p functioning in the stabilization of certain tRNA species. Using pulldown of endogenous La–pre-tRNA complexes formed in vivo and gel-shift analysis of La–pre-tRNA complexes in vitro, we then test for the capacity of La to differentially bind pre-tRNAs with respect to their modification status at G26. In sum, our results suggest that La binds natively folded tRNAs and tRNAs that likely misfold due to hypomodification of G26 indiscriminately. Finally, we further demonstrate that human La also binds a fully modified tRNA substrate indiscriminately from its unmodified, T7 transcribed counterpart. Our data suggest that La is poorly capable of differentiating substrates whose folding and accumulation rely on the modification of G26 by Trm1p and the RNA chaperone activity of La, consistent with a model in which alternate tRNA folds may not form a primary binding determinant for La proteins. We propose that these RNA chaperones are directed to their substrates more by features related to the processing stage of their targets and less by their folding status, in an analogous manner to the variant protein molecular chaperones calnexin and calreticulin. Thus, our work adds an interesting new facet with respect to our understanding of how RNA chaperones can engage their targets.
MATERIALS AND METHODS
Northern analyses
Cells were grown in YES at 32°C and then shifted to minimal media for 10 h prior to harvest. Total RNA was extracted and purified from yeast cells and northern analysis was performed as described (13). Values represent the mean fold abundance relative to the respective tRNA in the WT strain over a minimum of three independent growths/total RNA extractions. These were normalized to U5 snRNA as a loading control and presented relative to the respective tRNA abundance in the WT strain. Significant differences in tRNA abundance between mutant strains and the WT strain were assessed by performing a one-way ANOVA to compare means across all treatments, followed by a Tukey posthoc test with α set to 0.05 (*, Figure 4B–D) or 0.01 (**). DNA probes were made for indicated tRNAs by 5′ end labeling of the oligonucleotides using T4 polynucleotide kinase and 32P-γ-ATP. Probes used for northern analysis were all complementary to the TψC-loop of their respective targets. Probe sequences are provided in Supplementary Table S3.
Figure 4.

Variability in relative tRNA abundance in sla1−/trm1− cells. (A) Northern blot of total RNA extracted from indicated strains probed for various tRNAs or the U5 snRNA. (B) Relative abundance of G26-containing and non-G26 containing tRNAs in trm1− cells, normalized to tRNA levels in the WT strain as well as the U5 snRNA as the endogenous loading control. Note that in Schizosaccharomyces pombe, the mature tRNA-SerCGA and tRNA-SerUGA species differ by a single nucleotide in the anticodon loop, and so our probe for northern blot is expected to react with both. White bars: G26-modified tRNAs; black bars: tRNAs lacking a ‘G’ at position 26 or lacking modification at G26 by Trm1p (41). Error bars: SEM. (C) Relative abundance of G26-containing and non-G26 containing tRNAs in sla1− cells. Significant drops in tRNA abundance relative to the WT strain are marked with * (P ≤ 0.05) or ** (P ≤ 0.01). (D) Relative abundance of G26-containing and non-G26 containing tRNAs in sla1−/trm1− cells. (E) Correlation between the product of the tRNA levels in the trm1− and sla1− strains versus the levels of the respective tRNAs in the sla1−/trm1− strain. All tRNAs are represented; some tRNAs are annotated as reference points.
Analysis of aminoacyl-tRNA charging
Cells were grown in YES at 32°C and then shifted to minimal media for 10 h prior to harvest. Total RNA was extracted under acidic conditions using Trizol reagent (Invitrogen). For acid-northern, half of the WT total RNA was deacylated with 0.2 M Tris pH 9.0 for 2 h at 37°C and then ethanol precipitated and stored in 10 mM sodium acetate pH 5.0. All samples were fractionated on a 15% polyacrylamide acid (0.1 M sodium acetate pH 5.0) denaturing gel (vertical Gel Electrophoresis System V16–2, Apogee) and transferred onto a GeneScreen Plus transfer membrane (PerkinElmer) using the iBlot Gel Transfer system (Invitrogen). Periodate oxidation/β-elimination based analysis of charging levels was performed as described (37). Briefly, total RNA was extracted under acidic conditions and half of the total RNA was deacylated as for acid-northern. Both the deacylated and untreated total RNA were subjected to periodate oxidation (5 mM NaIO4, 50 mM NaOAc pH 5.0, 60 min at 37°C). The reaction was stopped with the addition of 50 mM glucose (0°C for 90 min) and ethanol precipitation. The RNA samples were subject to β-elimination by resuspending the total RNA samples in 1 ml of 1M lysine pH 8.0 and incubating 60 min at 45°C. All samples were then treated with 500 μl of 0.4 M Tris pH 9.0 (37°C, 30 min) for deacylation. RNA samples were treated with phenol chloroform and ethanol precipitated. Fraction charged was quantitated as the ratio of upper to lower bands relative to the WT strain.
Electromobility shift assays (EMSAs)
Radiolabeled pre-tRNAs were generated by T7 transcription in the presence of 32P-α-ATP (Ambion Megascript) using T7 promoter containing DNA templates generated by annealing and polymerase chain reaction (PCR) amplification of respective pre-tRNA sequences. Radiolabeled pre-tRNAs were purified using PAGE extraction. Electromobility shift assays (EMSAs) were performed as described (13). Briefly, 3000 cpm (∼0.1 nM) of pre-tRNA was incubated with various concentrations of Sla1p in a 20 μl reaction containing 20 mM Tris pH 7.6, 100 mM or 300 mM KCl and 5 mM β-mercaptoethanol. Pre-tRNAs were initially slow-cooled from 95°C to room temperature and then incubated with protein at 30°C or 37°C for 20 min. Complexes were resolved on 10% (w/v) polyacrylamide non-denaturing gels at 4°C at 100V. Supershifts were treated as supplementary binding events to the primary binding event, and binding curves were fit using a non-linear specific binding curve fitting program (GraphPad, Prism).
In vitro modification of pre-tRNA transcripts by Trm1p
Trm1p was cloned from S. pombe genomic DNA into the KpnI/EcoRI site of pET30a and purified by nickel affinity chromatography using standard methods. Trm1p modification on pre-tRNAs was carried out in a 30 μl reaction containing 33.33 mM Tris–HCl pH 7.6, 0.083 mM ethylenediaminetetraacetic acid (EDTA), 8.3 mM MgCl2, 33.3 mM NH4Cl, 0.83 mM dithiothreitol and 83.3 mM S-Adenosyl methionine (SAM). Reactions were incubated at 32°C for 3 h and purified using phenol chloroform extraction. Charging efficiencies were determined by performing modification reactions in the presence of 14C-SAM (Perkin-Elmer) followed by trichloroacetic acid precipitation of modified RNA, filtration through Whatman GF/C glass microfiber filters and scintillation counting. Reverse transcriptase primer extension for detection of dimethylated Trm1p was performed using SuperScript III (Invitrogen) and standard methods.
In vitro chemical probing assay with lead acetate
Pre-tRNA SerUGA was generated by T7 transcription (Ambion Megascript) and then treated with Calf Intestinal Alkaline Phosphatase (CIP) (NEB) at 37°C for 30 min followed by phenol/chloroform extraction and ethanol precipitation. Pre-tRNA SerUGA was 5′ end labeled using T4 polynucleotide kinase and 32P-γ-ATP and purified using a denaturing gel. In vitro modification of pre-tRNA SerUGA transcript by Trm1p was carried out as above in the presence and absence of SAM. Chemical probing reactions were completed by the addition of 1 μl of 1 mg/ml of yeast RNA and 1× Ambion structure buffer (10 mM Tris pH 7, 0.1 M KCl, 10 mM MgCl2) to heat-renatured pre-tRNA SerUGA in a total volume of 8 μl. Pre-tRNA samples were treated with a final concentration of 5 mM lead acetate with incubation at 37°C for 2 min, compared to mock treated unmodified controls. The reaction was stopped with the addition of loading buffer and incubation on ice for 5 min.
Harvesting yeast cells and immunoprecipitation of Sla1p–PrA pre-tRNA complexes
Integration of the Protein A tag into the sla1+ chromosomal locus was performed and validated by sequencing and western blot as described (38). Yeast strains were grown in YES at 30°C or 37°C to OD600 ∼ 0.8–1.0, harvested and subjected to cryogenic disruption in Retsch PM100 planetary ball mill as described (38). Immunoprecipitation was carried out on 1 g of powder with magnetic Dynabeads (Invitrogen) conjugated with Rabbit IgG (MP-Biomedicals) as described (38). Lysate and wash buffer contained 50 mM NaCl, 0.5% Triton X, 0.1% Tween-20, 20 mM Hepes pH 7.4, 55 mM KOAc, 1 mM β-mercaptoethanol, 0.2 mM PMSF and 1:100 Protease Inhibitor Cocktail (HALT, Pierce). Complexes were treated with proteinase K (Sigma) and RNA was purified using phenol chloroform extraction.
cDNA synthesis and quantitative real time PCR analyses
RNA extracted from immunoprecipitation assays and was quantified by TaqMan qPCR. Primers and probes were from Integrated DNA Technologies (IDT). Reverse transcription on the purified RNA was done using the qScript™ cDNA Synthesis kit (Quanta Biosciences). Elution cDNA was used at 5 ng and total cDNA at 10 ng for pre-tRNA and U5 snRNA analyses. Forward primers and probes used for qPCR provided in Supplementary Table S4. Forward primer (IDT) and PerfeCta® Universal PCR primer (Quanta Biosciences) were used at 0.4 μM and probes (IDT) were used at 0.2 μM. The cycling conditions were as follows: an initial 95°C for 3 min, followed by 40 cycles of 95°C for 3 s, 60°C for 20 s for annealing and extension. qPCR results were evaluated using the ΔΔCt method and pulldown efficiency between inputs and elutions were assessed using U5 snRNA as the endogenous control. Comparison of enrichment within elution samples was performed by measuring the fold enrichment of the query pre-tRNA Ct versus the geometric mean Ct of three Trm1p non-modified pre-tRNAs (IleUAU, AlaCGC and TyrGUA) as described (39). Fold enrichments were calculated from the Ct values for each pre-tRNA performed as triplicate technical replicates; histograms of the means of these triplicate values were assembled from values obtained from two independent pulldowns each obtained from a minimum of two independent growths for each strain and temperature.
RESULTS
La RNA chaperone activity is required to rescue growth of sla1−/trm1− cells at elevated temperature
Mutations to the canonical RNA binding surface of RRM1 impair La RNA chaperone activity in vitro and La dependent rescue of defective (mutated) pre-tRNAs in S. pombe cells (14). Previous work in S. cerevisiae has demonstrated functional overlap of the dimethylation of G26 (N2, N2-dimethylguanosine) by the methyltransferase Trm1p and the La homolog Lhp1p, as a trm1Δ/lhp1Δ strain displays a temperature sensitive lethal phenotype (30). To compare previous insights on La dependent RNA chaperone activity with La function on hypomodified pre-tRNAs in a homologous system, we generated a sla1−/trm1− strain of S. pombe and compared its growth to the respective single deletion mutants at 32°C versus 37°C (Figure 1A and B). We observed that growth of the sla1−/trm1− strain was slightly impaired compared to the sla1−, trm1− or WT strain at 32°C, and that transition to 37°C was lethal in the double mutant, similar to S. cerevisiae. Interestingly, the synthetic lethality at 37°C was seen only when cells were grown in minimal media (EMM), with no synthetic lethality observed when cells were grown in rich media. This is consistent with previous work describing slow growth of sla1− strains under tRNA-associated stresses that accompany growth in media that use NH4 as a source of fixed nitrogen (40). Previous work has indicated that impairing nuclear surveillance by deletion of the nuclear exosome subunit Rrp6p can rescue tRNA-mediated suppression associated with a defective (mutated) pre-tRNA (8). We also tested a sla1−/trm1−/rrp6− triple mutant in case this might suppress the synthetic lethality of the sla1−/trm1− strain. Instead of a rescue, however, we observed that this strain grew worse than the sla1−/trm1− double mutant, as it was unable to grow on minimal media even at 32°C.
Figure 1.

Synthetic lethality of sla1−/trm1− cells relies on La RNA chaperone activity. Indicated yeast strains were grown in liquid EMM media with essential supplements at 32°C (A) and 37°C (B) for 12 h with absorbance collected at OD600. Growth of the sla1−/trm1− was slowed at 32°C and inhibited at 37°C, while sla1−/trm1−/rrp6− did not grow at either temperature. (C) La RNA chaperone mutants are defective in rescuing growth of the sla1−/trm1− cells at 37°C. sla1−/trm1− cells were transformed with empty pRep4 or pRep4 containing hLa, hLa Y114A/F155A, Sla1 or Sla1 Y157A/F201A (8).
To determine whether the RNA chaperone/RRM1 associated binding mode was important for La dependent rescue of the sla1−/trm1− strain, we transformed this strain with plasmids expressing WT human La (hLa) or Sla1p, as well as mutant hLa or Sla1p carrying mutations to the previously characterized RNP aromatic residues of RRM1 important for RNA chaperone function (hLa Y114A/F155A or Sla1 Y157A/F201A), or a vector control (Figure 1C). While all transformants grew at lower temperature, only the WT hLa or Sla1p transformants were able to rescue robust growth at 37°C, and the RRM1 mutants displayed poor rescue more similar to the vector control. We therefore hypothesize that the RNA chaperone activity of La proteins is important for the rescue of pre-tRNAs that misfold due to lack of modification at G26, and that tRNA misfolding due to lack of modification at G26 is sensitive to both temperature and the tRNA-associated stress previously linked to growth in minimal media.
Identification of candidate misfolded tRNAs in trm1− and/or sla1− strains of S. pombe
In most native tRNAs, G26 extends the anticodon stem by making a non-Watson–Crick interaction with N44 (whose identity is biased against ‘C’) in the so-called ‘hinge’ region, generating a 6-bp long (‘Type 6’) anticodon stem (including the non-Watson–Crick 26–44 bp). Dimethylation of G26 has been previously hypothesized to stabilize these non-Watson–Crick G26-N44 interactions over particular folds previously observed for some mitochondrial tRNAs (35). For example, depending on a compatible sequence in the D-stem, lack of dimethylation of G26 could result in an alternate Watson-Crick pairing of G26 with C11 and a resulting non-native 5-bp long anticodon stem (‘Type 5’). Alternatively, an unmodified G26 could form a Watson–Crick pair with a rare C44 and promote a seven base-pair long anticodon stem (‘Type 7’) should nucleotides 25 and 45 be compatible and available for pairing. We examined the sequences of G26 containing tRNAs in S. pombe in an attempt to ascertain which tRNAs may be particularly prone to taking these misfolded forms (Supplementary Table S1). We also took into consideration which G26 containing tRNAs are actually modified at G26 by Trm1, based on published results obtained by next-generation tRNA sequencing (tRNA-HydroSeq, (41)). Surprisingly, we found that contrary to the case in S. cerevisiae (i.e. tRNA-LysCUU), humans (i.e. tRNA-AsnGUU) and other investigated species, there was not a single Trm1 modified tRNA in S. pombe (with the exception of a single allele for tRNA-AsnGUU) that was predicted to be capable of forming the Type 5 fold, due to resultant mismatches in the remodeled D-stem (Supplementary Table S1). Among the G26 containing tRNAs, there was only a single tRNA species (tRNA-GlyGCC) containing a C at position 44, but interestingly, tRNA-GlyGCC is not a Trm1 target in S. pombe (41), consistent with a complete lack of GC base pairs in the Trm1p D-stem recognition element of tRNA-GlyGCC. Together, these data suggest that prevention of the aberrant Type 5 and Type 7 folds may not be as critical a consideration for tRNA misfolding in S. pombe tRNAs compared to other species.
Previous work has identified particular tRNAs that appear to be sensitive to impairment of La function in yeast. In S. cerevisiae, tRNA-ArgCCG has been linked to La associated rescue and is hypothesized to have a fragile anticodon stem due to a UC mismatch (29,30). We therefore also considered the possibility that fragile anticodon stems might sensitize tRNAs to loss of La and/or Trm1. Since La function has also been linked to pseudouridylation of U38 and U39 by Pus3p (30), we also looked for the presence of uridines at these positions in G26 containing tRNAs (Supplementary Table S1). We noted that unlike in S. cerevisiae, the anticodon stem of tRNA-ArgCCG does not have a mismatch in S. pombe. We did note, however, that uniquely among Trm1 targets in S. pombe, tRNA-SerCGA/UGA has only a single G-C base pair in its anticodon stem, with all other Trm1 targets containing between 2 and 4 G-C base-pairs. tRNA-SerCGA and tRNA-SerUGA have also been previously linked to Trm1 function: they have been previously noted to undergo rapid tRNA decay in the context of a trm1Δ/trm4Δ strain in S. cerevisiae (27), and hypomodification of G26 in suppressor tRNA-SerUCA (an anticodon variant of tRNA-SerUGA) results in compromised tRNA-mediated suppression in S. pombe (41).
To experimentally identify candidate tRNAs that might misfold causing functional impairment, we assessed the amino acid charging levels of several G26 containing tRNAs in WT, sla1−, trm1− and sla1−/trm1− strains, as it has been demonstrated that tRNA misfolding can result in lower charging levels by aminoacyl-tRNA synthetases (29,42,43). In order to assess tRNA charging levels we performed acid-Northern (44) (Figure 2A) or β–elimination (45) (Figure 2B) of tRNAs isolated from cells grown in minimal media. As previously noted, we were particularly interested in investigating the charging efficiency of tRNA-SerCGA/UGA, tRNA-ArgCCG, as well as other tRNAs predicted to have less stable anticodon stems. We observed a drop in charging efficiency (∼25%) for tRNA-SerCGA/UGA in the trm1− and sla1−/trm1− strains, but no drop in charging efficiency for tRNA-ArgCCG or for other tested tRNAs containing two GC base pairs in their anticodon stems (tRNA-LeuAAG, tRNA-LysCUU, tRNA-SerAGA, tRNA-IleAAU, tRNA-TrpCCA, tRNA-ArgUCG) or an anticodon stem mismatch (tRNA-LeuUAA). To test the importance of growth conditions on tRNA charging, we also tested for the charging of several tRNAs from cells grown in rich media (Supplementary Figure S1). We found that tRNA-SerUGA/CGA again had the greatest decrease in charging level among the tRNAs tested, with most tRNAs showing no charging defect in the mutant strains, although the degree of impaired charging was slighter than observed in minimal media. These results identify tRNA-SerUGA/CGA as a tRNA whose fold is likely influenced by dimethylation of G26 in vivo, consistent with previous work in S. pombe and S. cerevisiae linking this modification and tRNA-SerUGA/CGA function (27,41).
Figure 2.

Lack of Trm1p modification at G26 results in likely misfolding of tRNA-SerCGA/UGA. Acid-northern (A) and periodate oxidation/β-elimination (B) to assay charging levels of various tRNA species indicates a Sla1p/Trm1p associated tRNA charging defect for tRNA-SerCGA/UGA. (C) Reverse transcriptase primer extension of recombinant Trm1p-modified tRNA-PheGAA confirms specific modification of G26 in vitro. (D) Left: lead acetate chemical probing indicates altered structure in the anticodon stem loop and variable arm of pre-tRNA-SerUGA after in vitro modification with Trm1p. Right: secondary structure of tRNA-SerUGA. Nucleotides with altered chemical reactivity to lead acetate probing +/− Trm1p modification indicated in bold. Bottom: quantitated differential lead acetate reactivity +/− Trm1p modification.
Lack of G26 dimethylation causes conformational changes in tRNA-SerUGAin vitro
Based on our results demonstrating impaired charging of tRNA-SerCGA/UGAin vivo, we attempted to ascertain whether dimethylation of G26 might alter the structure of pre-tRNA-SerUGA as measured by chemical probing in vitro. In order to obtain G26 modified tRNAs, we cloned and purified recombinant Trm1p from S. pombe and used this to modify a T7-transcribed pre-tRNA-SerUGA sequence. We observed between 80 and 100% transcript modification of pre-tRNA-SerUGA as measured by use of 14C S-adenosylmethionine as the methyl donor, followed by TCA precipitation/filter binding and quantitation by liquid scintillation (data not shown). As the in vitro modification of pre-tRNAs by Trm1p from S. pombe has not previously been reported, we confirmed the specificity of our cloned enzyme by taking advantage of the propensity of Trm1p G26 dimethylation to cause an N-1 reverse transcriptase stop (46) and confirmed that a T7 pre-tRNA transcript was modified exclusively at the G26 nucleotide by our recombinant S. pombe Trm1p in vitro (Figure 2C). As measured by primer extension, our Trm1p dimethylated our T7 transcript to ∼60% (ratio of G26 band to run off primer extension product), although this likely an underestimate as reverse transcriptase (Superscript III) can read through modified G26 through misincorporation of non-C nucleotides (41).
We then investigated the structure of pre-tRNA-SerUGA in the absence or presence of dimethylation at G26 by lead acetate chemical probing, as this method has previously been demonstrated to be highly sensitive to changes in secondary and tertiary structures in tRNA substrates (47). Upon Trm1p modification we observed a significant decrease in Pb2+ based cleavage in the A-U rich anticodon stretch (U41, A42 and A43) immediately adjacent to the hinge region of pre-tRNA-SerUGA, and a concomitant slight increase in reactivity at C40 (Figure 2D). Notably, these nucleotides are next to the U44 nucleotide that base pairs with G26 in pre-tRNA-SerUGA, and modification at G26 has been previously hypothesized to stabilize tRNA structure in this region (33–35). Furthermore, the accessibility of this region to chemical probing has been demonstrated to change upon S. cerevisiae La (Lhp1p) binding to pre-tRNA-ArgCCG (29). Interestingly, we also observed a G26 modification dependent decrease in reactivity near the beginning of the variable loop, close to the hinge, around C47:8. It is notable that both the C40/U41/A42/A43 and C47:8 regions lie at or near nucleotides whose mutation (C40U; C47:6U) results in defective tRNA suppression for tRNA-SerUCA (an anticodon mutant derived from tRNA-SerUGA) in S. pombe (48). While the exact nature of structural changes observed cannot be inferred from these results, they are strongly indicative that dimethylation at G26 can influence the stability of the hinge region of tRNA-SerUGA with consequent effects on tRNA function in vivo. Since tRNA-SerUGA/CGA has only a single GC base pair in the anticodon stem, it is thus possible that the stability of the anticodon stem loop may be linked to La and Trm1 function, consistent with other work in S. cerevisiae (29). We did not notice any trend in tRNA fold or stability (see below) correlating with the possibility of pseudouridylation at positions 38 and 39 (Supplementary Table S1).
Trm1 promotes tRNA-mediated suppression of tRNA-SerUCA
In order to further investigate the importance of modification of G26 in the native folding of pre-tRNA-SerUGA, we tested for the capacity of Trm1p dependent dimethylation to promote tRNA-mediated suppression in a red-white assay that relies on the activity of tRNA-SerUCA, a suppressor-tRNA derivative of tRNA-SerUGA (Figure 3). We compared the overexpression of Trm1p (pRep4-Trm1) to a vector alone control (pRep4) in both a sla1−/trm1+ (Figure 3A) and a sla1+/trm1− (Figure 3B) background for the original tRNA-SerUCA as well as two other tRNA alleles (tRNA-SerUCA C40U and tRNA-SerUCA C40U C47:6U) that carry additional mutations that have been previously demonstrated to increasingly rely on Sla1p for their function in the assay (8). We observed that overexpression of Trm1p resulted in enhanced tRNA-mediated suppression relative to the vector control for all three tRNA alleles in the sla1−/trm1+ background, as well as for the tRNA-SerUCA C40U and tRNA-SerUCA C40U C47:6U alleles in the sla1+/trm1− background (the tRNA-SerUCA allele demonstrated no red pigment accumulation in this background and thus could not be assessed in this way), similar to previous results also testing for the function of Trm1p in tRNA-mediated suppression in S. pombe (41). Together, these data further support impaired function for a tRNA-SerUGA derived tRNA in the absence of dimethylation at G26, and are consistent with our other data indicative of a tRNA-SerUGA folding defect that is rescued by Trm1p.
Figure 3.
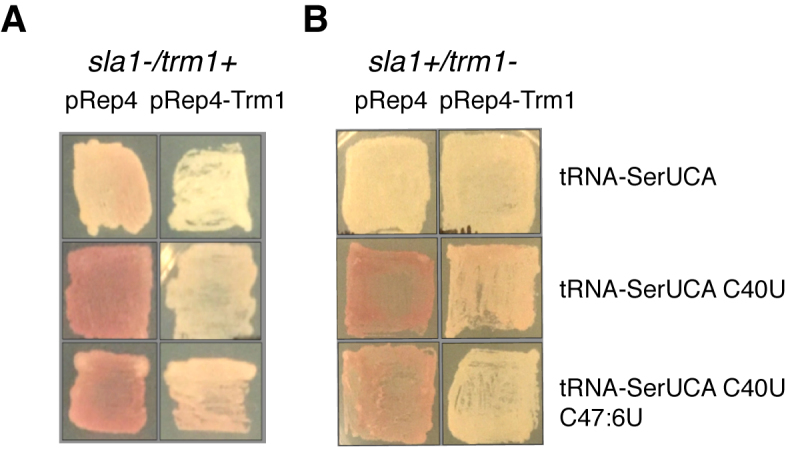
Dependence of tRNA mediated suppression on dimethylation at G26 by Trm1p. (A) tRNA-mediated suppression of tRNA-SerUCA, tRNA-SerUCA C40U and tRNA-SerUCA C40U/C47:6U in a sla1−/trm1+ strain background in the absence (pRep4) or presence (pRep4-Trm1) of overexpressed Trm1p. (B) Same as for A, but in a sla1+/trm1− background.
Sla1 and Trm1 promote the accumulation of selective sets of G26-containing tRNAs
We hypothesized that misfolding of tRNAs in sla1−/trm1− cells may alternatively result in their degradation, analogous to the demonstrated dependence of other tRNAs and pre-tRNAs on their modification status to protect these from rapid tRNA decay and nuclear surveillance. We therefore used Northern blots to quantify steady state levels of G26 containing tRNAs in S. pombe from sla1−, trm1− and sla1−/trm1− strains relative to amounts in the WT parent strain, 10 h after a shift to minimal media (Figure 4). To normalize for differences in loading, we also probed our blots for U5 snRNA. As an internal control, we also measured steady state levels for a subset of tRNAs lacking a guanosine at position 26 (tRNA-IleUAU, tRNA-GlyCCC, tRNA-MetCAU and tRNA-HisGUG) and for which deletion of Trm1p should thus not affect their relative abundance. Some representative northerns are provided in Figure 4A. Certain tRNAs were more sensitive to deletion of Trm1p than others, for example, tRNA-SerAGA & tRNA-ArgACG accumulated to lower levels in the trm1− and sla1−/trm1− strains compared to the WT and sla1− strains (compare lanes 2 and 4 to lanes 1 and 3, Figure 4A). We summarized the levels of all tRNAs sampled in the trm1− strain normalized to a loading control (mature U5 snRNA) and relative to WT levels in Figure 4B. We observed that the levels of some tRNAs dropped slightly in the trm1− strain, and noted that the identities of these seemed to correlate with those tRNAs known to be modified by Trm1p (Trm1p-modified tRNAs represented by white bars, Figure 4B–D; black bars represent tRNAs lacking G26 or known to not be modified at G26 in S. pombe as measured by RNA-seq (41)). Other tRNAs, for example tRNA-PheGAA and tRNA-IleAAU, constituted a non-overlapping set that were more sensitive to deletion of Sla1p (compare lanes 3 and 4 to lanes 1 and 2, Figure 4A; summary data for sla1− strain provided in Figure 4C). Examination of the sla1−/trm1− double mutant strain were indicative of more substantial decreases in several tRNAs, some to the degree to which they became statistically significant (P ≤ 0.05 or 0.01), thus identifying which tRNAs were most dependent on Sla1p and Trm1p for their steady state abundance (Figure 4D). Interestingly, we observed that the fold decrease for tRNAs in the sla1−/trm1− strain correlated positively with the product of the fold decrease of the same tRNAs in the trm1− and sla1− single mutant strains (Figure 4E), suggesting that the degree of depletion of tRNAs in the sla1−/trm1− double mutant is indeed due to the additive effect of the two single mutations impairing a related process. These experiments provided us with a subset of tRNAs for further analysis whose abundance is most sensitive to lack of modification at G26 in the absence of Trm1p.
Sla1p does not differentially engage hypomodified pre-tRNAs in vivo
Our analysis of tRNA charging, tRNA-mediated suppression and tRNA abundance in our mutant strains identified candidate pre-tRNAs that La may preferentially engage should La display affinity for potentially misfolded RNA substrates. In order to sample Sla1p/pre-tRNA complexes formed in vivo, we integrated a C-terminal 4X protein A (PrA) tag into the S. pombe sla1 chromosomal locus in trm1+ and trm1− strains and confirmed its integration by sequencing and western blot (Figure 5A). To confirm the activity of our endogenously tagged Sla1-PrA protein, we compared the ability of our Sla1-PrA strain to promote tRNA-mediated suppression relative to sla1+ and sla1− strains using a previously characterized, Sla1p dependent red/white assay (8) and showed that Sla1-PrA was active in the assay, similar to the WT sla1+ strain (Figure 5B). We pulled down native Sla1p–PrA ribonucleoprotein complexes using a rapid pulldown protocol optimized for retention of endogenous particles (38), and pre-tRNA enrichment was determined by quantitative real-time PCR using tRNA-specific exonuclease hydrolysis probes (i.e. ‘TaqMan’) and mature U5 snRNA as a normalization control. To ensure we sampled nuclear pre-tRNA La targets and not mature tRNAs in our analyses, we took advantage of the order of tRNA processing in yeast. Pre-tRNAs are transcribed by RNA Pol III, are occasionally capped (49) and engage La through both their terminal UUU-3’OH and other contacts to the tRNA, which affects the order of tRNA processing events (18). When tRNAs are bound by La, 5′ processing precedes 3’ trailer processing before tRNAs are exported to the cytoplasm and spliced, should they have an intron (40,50), after which, under some conditions, they can re-enter the nucleus via tRNA import (51–53). We ensured we sampled nascent pre-tRNAs in our work by limiting our qPCR analysis to intron containing pre-tRNAs using intron specific forward primers. This design also ensured that all primers/probes would anneal downstream of G26, so that modification of this residue (if any) would not interfere with cDNA synthesis (see Supplementary Figure S2).
Figure 5.

Association of pre-tRNAs with Sla1p-PrA +/− G26 modification in Schizosaccharomyces pombe cells. Endogenous Sla1p-PrA/pre-tRNA complexes from trm1− and trm1+ cells grown at 30°C versus 37°C were immunoprecipitated and levels of candidate pre-tRNAs were quantitated by ‘TaqMan’ quantitative real-time PCR (qPCR). (A) Western blot confirming the expected size and expression of the Sla1-PrA fusion protein relative to the parent sla1+ strain. (B) Endogenous Sla1p containing an integrated Protein-A tag at the C-terminus (sla1-PrA) is comparably active to WT Sla1p as determined by a tRNA mediated suppression assay using two different defective suppressor tRNA alleles (8). (C) Analysis of fold pulldown levels of candidate misfolded pre-tRNAs (as measured by impaired charging and/or chemical probing, Figure 2) compared to an internal control, the geometric mean pulldown level of three pre-tRNAs not modified by Trm1p (pre-tRNA-IleUAU, pre-tRNA-TyrGUA and pre-tRNA-AlaCGC) in the same elution samples. As a reference for robust pulldown of La targets versus non-targets, fold pulldown of indicated pre-tRNA in elution samples versus total RNA samples (WT strain) is provided relative to the La non-target U5 snRNA (right hand side, histograms). (D): same as for (C) but for candidate pre-tRNAs identified as having decreased accumulation in sla1−/trm1− strains (see Figure 4).
To determine whether Sla1p might discriminate pre-tRNAs that may be misfolded as a result of the absence of G26 N2,N2-dimethylation, we focused on several G26 containing pre-tRNAs that we demonstrated as having impaired charging in vivo/misfolding in vitro (pre-tRNA-SerUGA and SerCGA; Figure 5C) or decreased steady state stability (pre-tRNA-LysCUU, LeuCAA, LeuUAG, LeuCAG, ArgCCG, SerGCU) (Figure 5D; mean differences in pulldowns and associated P-values provided in Supplementary Table S2). In order to be able to compare relative pulldown of candidate misfolded pre-tRNAs across the trm1+ and trm1− strains, we normalized the levels of these candidate G26-containing pre-tRNAs to the mean pulldown values of three pre-tRNAs whose folding and binding to La would not be influenced by Trm1p activity, by virtue of not possessing a G at position 26 (pre-tRNA-IleUAU, pre-tRNA-TyrGUA) or by virtue of being poorly modified by Trm1p at G26 as previously determined by RNA-Seq and Northern blot (pre-tRNA-AlaCGC; (41)). We found that Sla1p association with pre-tRNAs in the presence or absence of G26 modification did not vary significantly for any of the candidate pre-tRNAs tested, with observed fold enrichments in trm1− versus trm1+ strains on the order of 2-fold or less, and with no differences in pulldown efficiency reaching statistical significance (P = 0.05, Supplementary Table S2). This was in contrast to a highly substantial enrichment of each candidate pre-tRNA relative to a non-La target, the U5 snRNA, validating the effectiveness of the pulldown and quantitation (Figure 5C and D). Similarly, pulldown of candidate pre-tRNAs from the trm1− strain grown at 37°C versus 30°C also did not indicate preferential engagement of candidate pre-tRNAs at higher temperature. In order to ensure that our results were not confounded by rearrangement of complexes during the pulldown procedure, we also repeated the analysis but in the presence of formaldehyde crosslink prior to harvest. These experiments were also indicative of a lack of substantial differences in affinity, similar to the minus crosslink cohort (on the order of 2× or less; Supplementary Table S2). Together, these data are consistent with a lack of discrimination of Sla1p binding to pre-tRNAs according to their G26 modification status in vivo.
Sla1p does not differentially engage Trm1p-modified pre-tRNAs in vitro
To test Sla1p binding to Trm1p modified versus unmodified pre-tRNAs more directly, we tested Sla1p affinity for G26-modified versus -unmodified pre-tRNA transcripts in vitro by EMSA (Figure 6A). We performed EMSA analysis for tRNA-SerUGA, whose charging was defective in the sla1−/trm1− strain and whose structure was altered +/− G26 modification in vitro, as well as two pre-tRNA sequences corresponding to tRNAs with decreased abundance in the sla1−/trm1− strain (tRNA-LeuUAG and tRNA-LeuCAG). For these experiments, we first formed complexes at the temperature which should favor misfolding (37°C) and at 100 mM KCl. For each of these substrates, we observed minimal (∼2× or less) differences in affinity, similar to the trends we observed in vivo. Since misfolding of RNAs in vitro has been hypothesized to be temperature and salt dependent, we also compared complex formation between 30°C and 37°C and between 100 and 300 mM KCl and still observed no modification dependent changes in affinity (Supplementary Figures S3–5). Thus, our in vitro and in vivo data are consistent with Sla1p having similar affinity for pre-tRNAs regardless of the presence or absence of N2,N2-modification at G26, despite our biochemical and genetic data confirming the functional overlap of this modification with the importance of La in promoting productive pre-tRNA processing.
Figure 6.

Association of Sla1p and hLa with modified or unmodified pre-tRNAs in vitro. (A): EMSA of Sla1p with T7 transcripts corresponding to candidate pre-tRNAs identified as having impaired charging in vitro (tRNA-SerUGA) or impaired accumulation (tRNA-LeuUAG & tRNA-LeuCAG) in the absence (unmodified) or presence (Trm1p modified) of G26 modification. (B) EMSA of hLa with a T7 transcript corresponding to mature Saccharomyces cerevisiae tRNA-PheGAA or its fully modified counterpart purified from S. cerevisiae cells.
As a final proof of principle, and to compare the conservation of La binding to pre-tRNAs between yeast and humans, we compared the affinity of human La (hLa) for a fully modified, mature tRNA-PheGAA (purified from S. cerevisiae; Sigma-Aldrich) with its completely unmodified, T7 transcribed counterpart (Figure 6B). While this mature tRNA should not normally be a substrate for La proteins in vivo by virtue of nuclear pre-tRNA trailer processing and 3′CCA addition, we reasoned that the UUU-3′OH independent La-tRNA binding mode should still be sensitive to tRNA modification induced structural changes should these be important for La target discrimination. Similar to our earlier results, we observed less than a 2× difference in affinity between the fully modified tRNA and the corresponding S. cerevisiae tRNA-PheGAA T7 transcript, consistent with the specific fold imposed via tRNA modification not being an important determinant for La engagement, at least for the folds assumed by our tRNA substrates under the conditions of our analysis in vitro.
DISCUSSION
In this work, we have attempted to shed light on how La proteins may discriminate targets on the basis of fold by examining pre-tRNAs, their best-characterized substrates. Proteins that enhance the folding of RNAs into their native conformation have been apportioned into a variety of classes (RNA chaperones, RNA cofactors and RNA helicases) based on their proposed mechanisms of action (2,54). Unlike RNA cofactors, RNA chaperones can display activity across a variety of targets and do not need to remain associated with their remodeled RNA targets for these to retain their function. Thus, RNA chaperones are hypothesized to bind RNA relatively non-specifically (2,3,5). Some RNA chaperones have been hypothesized to have a preference for binding single stranded regions of RNA (9–11), and models for RNA chaperone binding have been proposed in which the free energy of binding to unfolded substrates varies from that for folded substrates (3,5). However, it is not clear how generally these principles should be applied across the wide breadth of proteins with RNA chaperone activity, especially ones like La, in which a non-specific RNA chaperone-associated RNA binding mode is juxtaposed with its sequence-specific UUU-3’OH-dependent RNA binding mode (13,14,24).
La proteins engage pre-tRNAs via at least two distinguishable binding modes: the UUU-3’OH-dependent trailer binding mode associated with the La motif, and a UUU-3’OH-independent mode associated with the canonical RNA binding surface of the RRM1 domain and with RNA chaperone function, with both modes contributing additively to maximize pre-tRNA binding affinity (13,14,22,23,55). Disordered regions C-terminal to RRM1 have also been implicated in RNA chaperone-like functions, and have also been proposed to contribute to La-RNA target affinity (24,56–58). While the importance of the UUU-3′OH motif in La recruitment is well-established (55,59), the question of whether other binding determinants influence La selection of targets within the UUU-3′OH containing cohort has not been tested previously. This issue may be of importance in light of the observation that the maturation of defective suppressor tRNAs benefits from additional, plasmid encoded Sla1p in sla1+ cells (25), suggesting that for at least some La targets, endogenous levels of La protein are limiting, and that UUU-3′OH’ containing RNAs could compete for La binding.
Using in vivo pulldown assays of endogenous complexes as well as in vitro EMSAs, we have tested whether La differentially binds hypomodified pre-tRNA substrates. While it is challenging to demonstrate direct evidence for misfolded conformations of precursor tRNAs in vivo, there are several lines of evidence consistent with a lack of modification of G26 causing folding defects for at least some Trm1 targets: (i) we demonstrate that the synthetic lethality associated with the sla1−/trm1− strain is poorly rescued using mutants of La previously demonstrated to be defective in RNA chaperone activity but not UUU-3′OH end binding (8); (ii) we show that tRNA-SerUGA/CGA is charged to a lesser extent in vivo in a sla1−/trm1− strain, similar to other work showing impaired charging by aminoacyl-tRNA synthetases on misfolded tRNAs; (iii) we demonstrate that lack of Trm1p modification at G26 results in defective tRNA mediated suppression in vivo in S. pombe, for a suppressor tRNA (tRNA-SerUCA) derived from tRNA-SerUGA; (iv) we show that the anticodon stem loop hinge region of a T7 transcript of pre-tRNA-SerUGA shows differential reactivity to lead acetate based cleavage in the presence versus absence of Trm1p modification, which to our knowledge is the first experimental result supporting the importance of this modification on the stability of tRNA anticodon stems (35) and (v) we show that several tRNAs accumulate to lower steady state levels in trm1− and sla1−/ trm1−strains relative to WT strains.
We did not observe substantially different binding to modified versus unmodified pre-tRNA substrates in vitro or in vivo, suggesting that Sla1p does not differentiate a potential tRNA misfold associated with G26 hypomodification, despite the apparent link between Sla1p and Trm1p function in cellular growth. This is, to our knowledge, the first demonstrated example of an RNA chaperone being unable to discriminate a native from a physiologically relevant misfolded target. Thus our work is reminiscent of some previously proposed models for RNA chaperones in which the challenges associated with discriminating native from misfolded RNA conformations due to the relative homogeneity of functional groups within nucleotides have been highlighted (2,3,5). A significant caveat in the interpretation of our results is that we cannot account for differential La binding to every possible misfolded pre-tRNA conformation. Studies on the absence of other tRNA modifications or tRNA anticodon stem loop mutants have also been linked to La function (6,29,31,48), and these may present misfolded conformations that are indeed preferentially engaged by La. Nevertheless, for at least the misfolded RNA species used in our study, our data are consistent with previous work suggesting that La engages pre-tRNAs irrespective of their specific fold for the benefit of misfolded species and at the expense of processing efficiency of correctly folded pre-tRNAs (8). Consistent with this, we show that La shows marginal differences in affinity between a fully modified mature tRNA substrate and its modification lacking, in vitro synthesized counterpart.
In the absence of discrimination on the basis of fold, we hypothesize that the specific targeting of RRM1 associated La RNA chaperone activity to nascent transcripts thus relies on the UUU-3’OH-dependent binding mode and the coupled progression of tRNA processing events. In this context, the La motif (which we have previously demonstrated to lack RNA chaperone activity in vitro; (14)) may be viewed as an accessory domain for RNA refolding (similar to analogous domains in some RNA helicases), as we have hypothesized previously (14); (Figure 7). Thus, the time between nascent transcript binding and trailer processing provides La with a fixed window of opportunity to assist the pre-tRNA in reaching the native state, irrespective of whether or not the pre-tRNA has assumed an early misfold that necessitates La-dependent rescue. In this scheme, a pre-tRNA that misfolds will benefit from the window of La engagement; however, this comes at the cost of decreased processing efficiency for pre-tRNAs that may have assumed a native fold even in the absence of La binding. La RNPs contain many post-transcriptionally modified nucleotides found in tRNAs (36), thus one function of this window may be to provide an opportunity for such modifications to reinforce the native fold before La dissociates upon trailer (i.e. UUU-3′OH) removal (13). In this light, the engagement of La proteins to its RNA targets would seem more akin to the ER resident protein chaperones calnexin and calreticulin, which are hypothesized to recognize a specific N-linked glycosylation motif associated with an intermediate stage of lumenal protein maturation (reviewed in (60)), rather than the exposed hydrophobic regions more directly associated with misfolds that are recognized by classical protein chaperones.
Figure 7.

Model for La engagement of folded versus unfolded pre-tRNAs. (A) The RNA chaperone La preferentially engages pre-tRNAs via their UUU-3′OH containing trailers (18,22,23,55) as well as through contacts to the main body of the pre-tRNA (8,13), but without discriminating folded from misfolded pre-tRNA substrates (this work). (B) During the window of La occupancy, misfolded pre-tRNAs acquire the native fold through post-transcriptional modification by Trm1 (30,36) and/or La RNA chaperone activity (8,12–14,16,29). La affinity for the pre-tRNA does not change as the native fold is acquired. Thus this window benefits the misfolded pre-tRNA but not the pre-tRNA that did not benefit from La dependent rescue. (C) La remains with the pre-tRNA until trailer cleavage and La dissociation (13,18).
We observed that the sla1−/trm1− double mutant was synthetically lethal when grown at higher temperature, similar to S. cerevisiae (30). Notably, this synthetic lethality could be fully rescued with overexpression of S. pombe or human La but not with mutants of these previously demonstrated to be defective in RNA chaperone function (8,13,14). Previous work has demonstrated that La RNA chaperone activity associated with the canonical RNA binding surface of RRM1 can rescue defective suppressor tRNAs in a red-white assay. This work builds on this result, pointing specifically to the importance of La-associated RNA chaperone activity in rescuing viability associated with the misfolding of endogenous pre-tRNAs. We hypothesized that this synthetic lethality could be due to degradation of particularly sensitive pre-tRNAs, as previous work has demonstrated that deletion of the nuclear exosome subunit rrp6+ can rescue the degradation and tRNA-mediated suppression associated with suppressor tRNAs that have been mutated such that they are predicted to misfold. We indeed noted decreases in steady state levels of several tRNAs in the context of a sla1−/trm1−strain, as well as a decrease in the charging of tRNA-SerCGA/UGA. Instead of a rescue, we observed an exacerbated lethal phenotype when combining the sla1−/trm1− deletion withrrp6− deletion, as thesla1−/trm1−/rrp6− triple mutant was unable to grow on minimal media even at 32°C. Since the nuclear exosome is predominantly associated with the degradation of defective substrates, this result may be consistent with the synthetic lethality of thesla1−/trm1− strain being due to the persistence of a misfolded Trm1p substrate (or substrates) acting in a dominant negative fashion, although the specific basis for this phenotype is at this point still unresolved. In this sense, La and Trm1p could co-operate with the nuclear surveillance machinery to ensure that defective pre-tRNAs do not enter the pool of mature tRNAs, rather than to ensure such tRNAs are not degraded below unsustainable levels.
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Arimbasseri and R. Maraia for helpful discussion and for sharing their work with us prior to publication. We thank M. Madan Babu and A. Ashok for helpful suggestions upon critical reading of the manuscript. We thank S. Wolin, E. Massé and A. Eyraud for helpful discussion regarding the experimental design.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research’s Institute of Genetics Open Operating Grant (to M.A.B.). Funding for open access charge: CIHR.
Conflict of interest statement. None declared.
REFERENCES
- 1. Poole A.M., Jeffares D.C., Penny D.. The path from the RNA world. J. Mol. Evol. 1998; 46:1–17. [DOI] [PubMed] [Google Scholar]
- 2. Rajkowitsch L., Chen D., Stampfl S., Semrad K., Waldsich C., Mayer O., Jantsch M.F., Konrat R., Bläsi U., Schroeder R.. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 2007; 4:118–130. [DOI] [PubMed] [Google Scholar]
- 3. Herschlag D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995; 270:20871–20874. [DOI] [PubMed] [Google Scholar]
- 4. Tompa P., Csermely P.. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004; 18:1169–1175. [DOI] [PubMed] [Google Scholar]
- 5. Woodson S.A. Taming free energy landscapes with RNA chaperones. RNA Biol. 2010; 7:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson J., Phan L., Cuesta R., Carlson B.A., Pak M., Asano K., Björk G.R., Tamame M., Hinnebusch A.G.. The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev. 1998; 12:3650–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kadaba S., Krueger A., Trice T., Krecic A.M., Hinnebusch A.G., Anderson J.. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004; 18:1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang Y., Bayfield M.A., Intine R.V., Maraia R.J.. Separate RNA-binding surfaces on the multifunctional La protein mediate distinguishable activities in tRNA maturation. Nat. Struct. Mol. Biol. 2006; 13:611–618. [DOI] [PubMed] [Google Scholar]
- 9. Karpel R.L., Miller N.S., Fresco J.R.. Mechanistic studies of ribonucleic acid renaturation by a helix-destabilizing protein. Biochemistry. 1982; 21:2102–2108. [DOI] [PubMed] [Google Scholar]
- 10. Waldsich C., Grossberger R., Schroeder R.. RNA chaperone StpA loosens interactions of the tertiary structure in the td group I intron in vivo. Genes Dev. 2002; 16:2300–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mayer O., Rajkowitsch L., Lorenz C., Konrat R., Schroeder R.. RNA chaperone activity and RNA-binding properties of the E. coli protein StpA. Nucleic Acids Res. 2007; 35:1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Belisova A., Semrad K., Mayer O., Kocian G., Waigmann E., Schroeder R., Steiner G.. RNA chaperone activity of protein components of human Ro RNPs. RNA. 2005; 11:1084–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bayfield M.A., Maraia R.J.. Precursor-product discrimination by La protein during tRNA metabolism. Nat. Struct. Mol. Biol. 2009; 16:430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naeeni A.R., Conte M.R., Bayfield M.A.. RNA chaperone activity of human La protein is mediated by variant RNA recognition motif. J. Biol. Chem. 2012; 287:5472–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hussain R.H., Zawawi M., Bayfield M.A.. Conservation of RNA chaperone activity of the human La-related proteins 4, 6 and 7. Nucleic Acids Res. 2013; 41:8715–8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuehnert J., Sommer G., Zierk A.W., Fedarovich A., Brock A., Fedarovich D., Heise T.. Novel RNA chaperone domain of RNA-binding protein La is regulated by AKT phosphorylation. Nucleic Acids Res. 2015; 43:581–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wolin S.L., Cedervall T.. The La protein. Annu. Rev. Biochem. 2002; 71:375–403. [DOI] [PubMed] [Google Scholar]
- 18. Yoo C.J., Wolin S.L.. The yeast La protein is required for the 3′ endonucleolytic cleavage that matures tRNA precursors. Cell. 1997; 89:393–402. [DOI] [PubMed] [Google Scholar]
- 19. Copela L.A., Fernandez C.F., Sherrer R.L., Wolin S.L.. Competition between the Rex1 exonuclease and the La protein affects both Trf4p-mediated RNA quality control and pre-tRNA maturation. RNA. 2008; 14:1214–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bousquet-Antonelli C., Deragon J.-M.. A comprehensive analysis of the La-motif protein superfamily. RNA. 2009; 15:750–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bayfield M.A., Yang R., Maraia R.J.. Conserved and divergent features of the structure and function of La and La-related proteins (LARPs). Biochim. Biophys. Acta. 2010; 1799:365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Teplova M., Yuan Y.-R., Phan A.T., Malinina L., Ilin S., Teplov A., Patel D.J.. Structural basis for recognition and sequestration of UUU(OH) 3′ temini of nascent RNA polymerase III transcripts by La, a rheumatic disease autoantigen. Mol. Cell. 2006; 21:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kotik-Kogan O., Valentine E.R., Sanfelice D., Conte M.R., Curry S.. Structural analysis reveals conformational plasticity in the recognition of RNA 3′ ends by the human La protein. Structure. 2008; 16:852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kucera N.J., Hodsdon M.E., Wolin S.L.. An intrinsically disordered C terminus allows the La protein to assist the biogenesis of diverse noncoding RNA precursors. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:1308–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang Y., Intine R.V., Mozlin A., Hasson S., Maraia R.J.. Mutations in the RNA polymerase III subunit Rpc11p that decrease RNA 3′ cleavage activity increase 3′-terminal oligo(U) length and La-dependent tRNA processing. Mol. Cell. Biol. 2005; 25:621–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phizicky E.M., Alfonzo J.D.. Do all modifications benefit all tRNAs. FEBS Lett. 2010; 584:265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dewe J.M., Whipple J.M., Chernyakov I., Jaramillo L.N., Phizicky E.M.. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA. 2012; 18:1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chernyakov I., Whipple J.M., Kotelawala L., Grayhack E.J., Phizicky E.M.. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′-3′ exonucleases Rat1 and Xrn1. Genes Dev. 2008; 22:1369–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chakshusmathi G., Kim S.D., Rubinson D.A., Wolin S.L.. A La protein requirement for efficient pre-tRNA folding. EMBO J. 2003; 22:6562–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Copela L.A., Chakshusmathi G., Sherrer R.L., Wolin S.L.. The La protein functions redundantly with tRNA modification enzymes to ensure tRNA structural stability. RNA. 2006; 12:644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johansson M.J.O., Byström A.S.. Dual function of the tRNA(m(5)U54)methyltransferase in tRNA maturation. RNA. 2002; 8:324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rose A.M., Joyce P.B., Hopper A.K., Martin N.C.. Separate information required for nuclear and subnuclear localization: additional complexity in localizing an enzyme shared by mitochondria and nuclei. Mol. Cell. Biol. 1992; 12:5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ladner J.E., Jack A., Robertus J.D., Brown R.S., Rhodes D., Clark B.F., Klug A.. Structure of yeast phenylalanine transfer RNA at 2.5 A resolution. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:4414–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edqvist J., Stråby K.B., Grosjean H.. Enzymatic formation of N2,N2-dimethylguanosine in eukaryotic tRNA: importance of the tRNA architecture. Biochimie. 1995; 77:54–61. [DOI] [PubMed] [Google Scholar]
- 35. Steinberg S., Cedergren R.. A correlation between N2-dimethylguanosine presence and alternate tRNA conformers. RNA. 1995; 1:886–891. [PMC free article] [PubMed] [Google Scholar]
- 36. Hendrick J.P., Wolin S.L., Rinke J., Lerner M.R., Steitz J.A.. Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins: further characterization of the Ro and La small ribonucleoproteins from uninfected mammalian cells. Mol. Cell. Biol. 1981; 1:1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Salazar J.C., Ambrogelly A., Crain P.F., McCloskey J.A., Söll D.. A truncated aminoacyl-tRNA synthetase modifies RNA. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7536–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oeffinger M., Wei K.E., Rogers R., DeGrasse J.A., Chait B.T., Aitchison J.D., Rout M.P.. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Methods. 2007; 4:951–956. [DOI] [PubMed] [Google Scholar]
- 39. Vandesompele J., De Preter K., Pattyn F., Poppe B., Van Roy N., De Paepe A., Speleman F.. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cherkasova V., Maury L.L., Bacikova D., Pridham K., Bähler J., Maraia R.J.. Altered nuclear tRNA metabolism in La-deleted Schizosaccharomyces pombe is accompanied by a nutritional stress response involving Atf1p and Pcr1p that is suppressible by Xpo-t/Los1p. Mol. Biol. Cell. 2012; 23:480–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arimbasseri A.G., Blewett N.H., Iben J.R., Lamichhane T.N., Cherkasova V., Hafner M., Maraia R.J.. RNA polymerase III output is functionally linked to tRNA dimethyl-G26 modification. PLoS Genet. 2015; 11:e1005671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lindahl T., Adams A., Geroch M., Fresco J.R.. Selective recognition of the native conformation of transfer ribonucleic acids by enzymes. Proc. Natl. Acad. Sci. U.S.A. 1967; 57:178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gartland W.J., Sueoka N.. Two interconvertible forms of tryptophanyl sRNA in E. coli. Proc. Natl. Acad. Sci. U.S.A. 1966; 55:948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chernyakov I., Baker M.A., Grayhack E.J., Phizicky E.M.. Chapter 11. Identification and analysis of tRNAs that are degraded in Saccharomyces cerevisiae due to lack of modifications. Meth. Enzymol. 2008; 449:221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Choi H., Gabriel K., Schneider J., Otten S., McClain W.H.. Recognition of acceptor-stem structure of tRNA(Asp) by Escherichia coli aspartyl-tRNA synthetase. RNA. 2003; 9:386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Motorin Y., Muller S., Behm-Ansmant I., Branlant C.. Identification of modified residues in RNAs by reverse transcription-based methods. Meth. Enzymol. 2007; 425:21–53. [DOI] [PubMed] [Google Scholar]
- 47. Brown R.S., Dewan J.C., Klug A.. Crystallographic and biochemical investigation of the lead(II)-catalyzed hydrolysis of yeast phenylalanine tRNA. Biochemistry. 1985; 24:4785–4801. [DOI] [PubMed] [Google Scholar]
- 48. Intine R.V., Sakulich A.L., Koduru S.B., Huang Y., Pierstorff E., Goodier J.L., Phan L., Maraia R.J.. Control of transfer RNA maturation by phosphorylation of the human La antigen on serine 366. Mol. Cell. 2000; 6:339–348. [DOI] [PubMed] [Google Scholar]
- 49. Ohira T., Suzuki T.. Precursors of tRNAs are stabilized by methylguanosine cap structures. Nat. Chem. Biol. 2016; 12:648–655. [DOI] [PubMed] [Google Scholar]
- 50. Yoshihisa T., Yunoki-Esaki K., Ohshima C., Tanaka N., Endo T.. Possibility of cytoplasmic pre-tRNA splicing: the yeast tRNA splicing endonuclease mainly localizes on the mitochondria. Mol. Biol. Cell. 2003; 14:3266–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takano A., Endo T., Yoshihisa T.. tRNA actively shuttles between the nucleus and cytosol in yeast. Science. 2005; 309:140–142. [DOI] [PubMed] [Google Scholar]
- 52. Ohira T., Suzuki T.. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10502–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shaheen H.H., Hopper A.K.. Retrograde movement of tRNAs from the cytoplasm to the nucleus in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:11290–11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weeks K.M. Protein-facilitated RNA folding. Curr. Opin. Struct. Biol. 1997; 7:336–342. [DOI] [PubMed] [Google Scholar]
- 55. Stefano J.E. Purified lupus antigen La recognizes an oligouridylate stretch common to the 3′ termini of RNA polymerase III transcripts. Cell. 1984; 36:145–154. [DOI] [PubMed] [Google Scholar]
- 56. Martino L., Pennell S., Kelly G., Bui T.T.T., Kotik-Kogan O., Smerdon S.J., Drake A.F., Curry S., Conte M.R.. Analysis of the interaction with the hepatitis C virus mRNA reveals an alternative mode of RNA recognition by the human La protein. Nucleic Acids Res. 2012; 40:1381–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fan H., Goodier J.L., Chamberlain J.R., Engelke D.R., Maraia R.J.. 5′ processing of tRNA precursors can be modulated by the human La antigen phosphoprotein. Mol. Cell. Biol. 1998; 18:3201–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brown K.A., Sharifi S., Hussain R., Donaldson L., Bayfield M.A., Wilson D.J.. Distinct dynamic modes enable the engagement of dissimilar ligands in a promiscuous atypical RNA recognition motif. Biochemistry. 2016; 55:7141–7150. [DOI] [PubMed] [Google Scholar]
- 59. Mathews M.B., Francoeur A.M.. La antigen recognizes and binds to the 3′-oligouridylate tail of a small RNA. Mol. Cell. Biol. 1984; 4:1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lamriben L., Graham J.B., Adams B.M., Hebert D.N.. N-glycan based ER molecular chaperone and protein quality control system: the calnexin binding cycle. Traffic. 2015; 17:308–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.