


Abstract
The ready availability of vast amounts of genomic sequence data has created the need to rethink comparative genomics algorithms using ‘big data’ approaches. Neptune is an efficient system for rapidly locating differentially abundant genomic content in bacterial populations using an exact k-mer matching strategy, while accommodating k-mer mismatches. Neptune’s loci discovery process identifies sequences that are sufficiently common to a group of target sequences and sufficiently absent from non-targets using probabilistic models. Neptune uses parallel computing to efficiently identify and extract these loci from draft genome assemblies without requiring multiple sequence alignments or other computationally expensive comparative sequence analyses. Tests on simulated and real datasets showed that Neptune rapidly identifies regions that are both sensitive and specific. We demonstrate that this system can identify trait-specific loci from different bacterial lineages. Neptune is broadly applicable for comparative bacterial analyses, yet will particularly benefit pathogenomic applications, owing to efficient and sensitive discovery of differentially abundant genomic loci. The software is available for download at: http://github.com/phac-nml/neptune.
INTRODUCTION
Capacity to cheaply and quickly generate high volumes of sequence reads has made possible the ability to study the genomes of entire populations of organisms, especially those organisms with relatively small genomes such as bacteria. Computational biologists have historically used a wide range of bioinformatics software tools to compare small numbers of bacterial genomes and to perform basic characterizations at the nucleotide, gene and genome scale. However, there now exists a need for bioinformatics software to perform efficient comparative analysis and characterization of entire populations of bacterial genomes. Some tools have emerged. Most of these tools focus on the identification of single nucleotide variants (SNVs) using reference mapping approaches (1,2), or distance estimations based on small exact substrings (k-mers) (3–5) since these approaches scale well using simple parallelization strategies. Microbial genome-wide association studies (GWAS) that analyze bacterial genome populations to correlate genomic features with phenotypic traits are now also possible, thanks to recent methodological developments that address the problems inherent in bacterial genomes that confound conventional GWAS approaches, such as long range linkage disequilibrium and clonal population structure (6). Some software tools for bacterial GWAS have been developed that associate SNVs or k-mers with biological traits (7). However, for bacterial GWAS, it is important to identify all modes of bacterial genomic variation including larger scale genomic gains and losses, particularly for the majority of bacteria that engage in horizontal gene transfer to acquire novel biologic traits. Scalable software that can rapidly extract the large scale genomic loci that differentiate one population from another while tolerating allelic variation within those loci, is valuable to accomplish bacterial GWAS and has utility for many other applications such as developing targeted molecular diagnostics.
To address this challenge, we looked to the field of genomic signature discovery, where a signature is defined as a sequence that is capable of discriminating a group of sequences of interest from a background group of sequences. Signatures may reside in genic or intergenic regions and may correspond to genomic islands, phage regions or entire operons. However, there is no requirement for signatures to contain functionally meaningful content, only that their sequence effectively discriminates the two groups. An effective signature discovery algorithm is both sensitive and specific, while quick to compute. However, in practice, it remains difficult to develop algorithms that possess all three of these attributes. Early algorithmic approaches for signature discovery were developed with the specific aim of generating pathogen detection diagnostic assays (8). In general, these approaches involve exhaustively comparing all sequences using alignment-based methods, such as BLAST (9), to locate signature regions in an inclusion group that are absent in the exclusion group. However, these approaches do not scale efficiently and are focused on generating molecular diagnostic primers of a fixed length.
Other more sophisticated approaches attempt to address efficiency by using computationally optimized string processing approaches that encode fixed size substrings from the genome in rapidly searchable data structures, then analyzing these data structures for unique substrings (10). These approaches are very fast and scale well, but cannot handle variability in the target sequence and are artificially limited to fixed length signatures. Variability in the target can be achieved by grouping similar sequences using multiple sequence alignments (8) or other clustering operations (11). However, these common clustering techniques come at a high computational cost and do not scale well. Some algorithms incorporate a data reduction step prior to clustering to reduce the amount of unnecessary computation. For example, Insignia (10), TOFI (12) and TOPSI (13) use efficient suffix trees to pre-compute exact matches within inclusion targets and an exclusion background. However, depending on the size of the background database, this may remain a computationally expensive operation. One interesting novel implementation is CaSSiS (11), which approaches the problem of signature discovery more thoroughly than other signature discovery pipelines. The software produces signatures simultaneously for all locations in a hierarchically clustered dataset, such as a phylogenetic tree, thereby producing candidate signatures for all possible subgroups. However, this process requires the input data to be provided in a hierarchically clustered format, such as computationally expensive phylogenies. In addition to the efficiency versus sensitivity trade off, most of the programs that have been developed thus far for signature discovery have additional shortcomings that make them unsuitable for identifying common variation between populations of genomes. For example, they may restrict the analysis to a single inclusion genome (12), they might not permit user-supplied genomes for target identification (10), or they might not provide the software to the end user (8).
We designed Neptune as a system for discovering discriminatory bacterial sequence signatures and conducting comparative analyses of arbitrary groups of genome sequences that leverage existing strategies for signature detection, but in a novel way that is both efficient and accurate. Neptune identifies genomic loci uniquely shared among a user-specified interest group but lacking from a background group. Independent of pre-computation, restriction on targets and slow clustering approaches, Neptune applies reference-based, parallelized exact-matching k-mer strategy for speed, while making allowances for inexact matches to enhance sensitivity. Neptune’s signature discovery is guided with probabilistic models that make decisions with a measure of statistical confidence. Neptune is open-source software freely available at github.com/phac-nml/neptune and is broadly applicable for rapid comparative assessments of bacterial populations.
MATERIALS AND METHODS
We define a genomic signature as a string of characters (nucleotides) sufficiently unique to a user-specified set of targets (the ‘inclusion’ group) that discriminates it from a set of user-defined background targets (the ‘exclusion’ group). We define a ‘reference’ sequence as any inclusion target from which to extract signatures. Targets typically comprise draft and closed genome assemblies. Signature discovery aims to locate unique and conserved regions within the inclusion group, but absent or minimally present in the exclusion group.
Neptune uses the distinct k-mers found in each inclusion and exclusion target to identify sequences that are conserved within the inclusion group and absent from the exclusion group. Neptune evaluates all sequence, coding and non-coding, and may therefore produce signatures that correspond to intergenic regions or contain entire operons. The k-mer generation step produces distinct k-mers from all targets and aggregates this information, reporting the number of inclusion and exclusion targets that contain each k-mer. The signature extraction step identifies candidate signatures from one or more references, which are assumed to be drawn from inclusion targets. Candidate signatures are filtered by performing an analysis of signature specificity using pairwise sequence alignments. The remaining signatures are ranked by their Neptune-defined sensitivity and specificity scores, representing a measure of signature confidence.
We provide descriptions of the different stages of signature discovery below and an overview of the signature discovery process is found in Figure 1. The majority of parameters within Neptune are automatically calculated for every reference. However, the user may specify any of these parameters. A full description of the mathematics used in the software is provided in the Supplementary Data. In our probabilistic model, we assume that the probability of observing any single nucleotide base in a sequence is equal to and independent of all other positions and the probability of all SNV events (e.g. mutations, sequencing errors) occurring is equal to and independent of all other SNV events.
Figure 1.

An overview of Neptune’s signature discovery process for a single target reference. The first step involves generating k-mers from all inclusion and exclusion targets. These k-mers are aggregated and provided as input to signature extraction. Signature extraction produces candidate signatures, which are filtered using BLAST (9) and then sorted by their sensitivity and specificity scores.
k-mer generation
Neptune produces the distinct set of k-mers for every inclusion and exclusion target and aggregates these k-mers together before further processing. The software is concerned only with the existence of a k-mer within each target and not with the number of times a k-mer is repeated within a target. Neptune converts all k-mers to the lexicographically smaller of either the forward k-mer or its reverse complement. This avoids maintaining both the forward and reverse complement sequence (14). The number of possible k-mers is bound by the total length of all targets. The k-mers of each target are determined independently and, when possible, in parallel. In order to facilitate parallelizable k-mer aggregation, the k-mers for each target may be organized into several output files. The k-mers in each file are unique to one target (e.g., isolate genome or sequence) and all share the same initial sequence index. This degree of organization may be specified by the user.
The k-mer length is automatically calculated unless provided by the user. A summary of recommended k-mer sizes for various genomes can be found in Supplementary Table S1. We suggest a size of k such that we do not expect to see two arbitrary k-mers within the same target match exactly. This recommendation is motivated by wanting to generate distinct k-mer information, thereby having matching k-mers most often be a consequence of nucleotide homology. Let λ be the most extreme GC-content of all targets and ω be the size of the largest target in bases. The probability of any two arbitrary k-mers, kX and kY, matching exactly, P(kX = kY)A, where x ≠ y, is defined as follows:
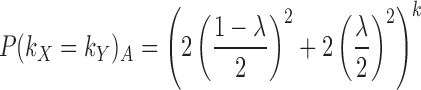 |
(1) |
We use the probability of arbitrary k-mers matching, P(kX = kY)A, to approximate the probability of k-mers matching within a target, P(kX = kY). This is an approximation because the probability of P(kX + 1 = kY + 1) is known to not be independent of P(kX = kY). However, this approximation approaches equality as P(kX = kY)A decreases, which is accomplished by selecting a sufficiently large k such that we do not expect to see any arbitrary k-mer matches. We suggest using a large enough k such that the expected number of intra-target k-mer matches is as follows:
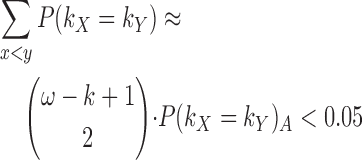 |
(2) |
The distinct sets of k-mers from all targets are aggregated into a single file, which is used to inform signature extraction. This process may be performed in parallel by aggregating k-mers sharing the same initial sequence index and concatenating the aggregated files. Aggregation produces a list of k-mers and two values (the number of inclusion and exclusion targets containing the k-mer, respectively). This information is used in the signature extraction step to categorize some k-mers as inclusion or exclusion k-mers.
Extraction
Signatures are extracted from one or more references, which are drawn from all inclusion targets, unless specified otherwise. However, our probabilistic model assumes all references are included as inclusion targets. In order to identify candidate signatures, Neptune reduces the effective search space of signatures by leveraging the spatial sequencing information inherent within the references. Neptune evaluates all k-mers in each reference, which may be classified as inclusion or exclusion k-mers. An inclusion k-mer is observed in a sufficient number of inclusion targets and not observed in a sufficient number of exclusion targets. The sufficiency requirement is described below. Inclusion and exclusion k-mers are used to infer inclusion and exclusion sequence, with signatures containing primarily inclusion sequence. An inclusion k-mer may contain both inclusion and exclusion sequence because, while they may contain exclusion sequence, k-mers that overlap inclusion and exclusion sequence will often be unique to the inclusion group. An exclusion k-mer is, by default, any k-mer that has been observed at least once in any exclusion target. However, in some applications it may be desirable to relax this stringency. For example, leniency may be appropriate when the inclusion and exclusion groups are not fully understood. This may be the case when metadata is incomplete or unreliable. An exclusion k-mer should, by design, not contain any inclusion sequence. Neptune outputs several ‘candidate signatures’, which begin with the last base position of the first inclusion k-mer, contain an allowable number of k-mer gaps and no exclusion k-mers, and end with the first base position of the last inclusion k-mer (Figure 2). This process is conceptually similar to taking the intersection of inclusion k-mers and allowable k-mer gaps. Furthermore, it avoids generating a candidate containing exclusion sequence found in inclusion k-mers that overlap inclusion and exclusion sequence regions.
Figure 2.

An overview of Neptune’s signature extraction process. The reference is decomposed into its composite k-mers. These k-mers may be classified as either inclusion or exclusion and are used to infer inclusion and exclusion sequence in the reference. A signature is constructed from inclusion k-mers containing sufficiently small k-mer gaps and no exclusion k-mers.
An inclusion k-mer is considered sufficiently represented when it is observed in a number of targets exceeding a minimum threshold. We assume that if there is a signature present in all inclusion targets, then the signature will correspond to homologous sequences in all these targets and these sequences will produce exact matching k-mers with some probability. We start with the probability that two of these homologous bases, X and Y, match is:
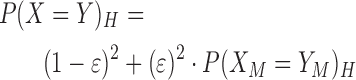 |
(3) |
where ϵ is the probability that two homologous bases do not match exactly, and P(XM = YM)H is the probability that two homologous bases both mutate to the same base. The default probability of ϵ is 0.01. We assume that when the homologous bases do not match, the observed base is dependent on the GC-content of the environment. Let λ be the GC-content of the environment. The probability of P(XM = YM)H is defined as follows:
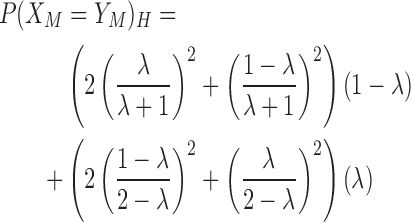 |
(4) |
This probability depends significantly on GC-content of the environment. We assume that the probability of each base matching is independent. Therefore, the probability that two homologous k-mers, kX and kY, match is:
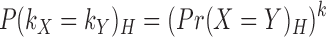 |
(5) |
We model the process of homologous k-mer matches with a binomial distribution. If we are observing a true signature region in a reference, we expect that corresponding homologous k-mers exist in all inclusion targets and infer this homology from aggregated k-mer information. An observed reference k-mer will exactly match a corresponding homologous k-mer in another inclusion target with a probability of p = P(kX = kY)H and not match with a probability of q = 1 − p. The expected number of exact k-mer matches with a reference k-mer will be μ = (n − 1) · p and the variance will be σ2 = (n − 1) · p · q, where n is the number of inclusion targets. We require n − 1 because the reference is an inclusion target and its k-mers will exactly match themselves. However, we compensate for this match in our expectation calculation. We assume the probability of each k-mer match is independent and that k-mer matches are a consequence of homology. When the number of inclusion targets and the probability of homologous k-mers exactly matching are together sufficiently large, the binomial distribution is approximately normal. Let α be our statistical confidence and Φ−1(α) be the probit function. The minimum number of inclusion targets containing a k-mer, ∧in, required for a reference k-mer to be considered an inclusion k-mer is defined as follows:
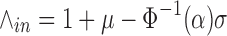 |
(6) |
The ∧in parameter is automatically calculated unless provided by the user and will inform candidate signature extraction. However, there may be mismatches in the reference, which exclude it from the largest homologous k-mer matching group. We accommodate for this possibility by allowing k-mer gaps in our extraction process. We model the problem of maximum k-mer gap size between exact matching inclusion k-mers as recurrence times of success runs in Bernoulli trials. The mean and variance of the distribution of the recurrence times of k successes in Bernoulli trials is described in Feller (1960) (15):
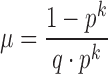 |
(7) |
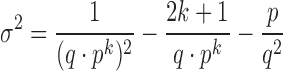 |
(8) |
This distribution captures how many bases we expect to observe before we see another homologous k-mer match. The probability of a success is defined at the base level as p = P(X = Y)H and the probability of failure as q = (1 − p). This distribution may not be normal for a small number of observations. However, we can use Chebyshev’s Inequality to make lower-bound claims about the distribution:
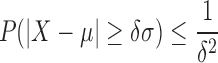 |
(9) |
where δ is the number of standard deviations, σ, from the mean, μ. Let P(|X − μ| ≥ δσ) be our statistical confidence, α. The maximum allowable k-mer gap size, ∨gap, is calculated as follows:
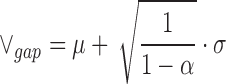 |
(10) |
The ∨gap parameter is automatically calculated unless specified. Candidate signatures are terminated when either no additional inclusion k-mers are located within the maximum gap size, ∨gap, or an exclusion k-mer is identified. In both cases, the candidate signature ends with the last inclusion k-mer match. The consequence of terminating a signature early is that a large, contiguous signature may be reported as multiple smaller signatures. We require the minimum signature size, by default, to be four times the size of k. However, for some applications, such as designing assay targets, it may be desirable to use a smaller or larger minimum signature size. Signatures cannot be shorter than k bases. We found that smaller signatures were more sensitive to the seed size used in filtering alignments. There is no maximum signature size. As a consequence of Neptune’s signature extraction process, signatures extracted from the same target may never overlap each other.
Filtering
The candidate signatures produced will be relatively sensitive, but not necessarily specific, because signature extraction is done using exact k-mer matches. The candidate signatures are guaranteed to contain no more exact matches with any exclusion k-mer than was specified in advance by the user. However, there may exist inexact matches within exclusion targets. Neptune uses BLAST (9) to locate signatures that align with any exclusion target and, by default, removes any signature that shares 50% identity with any exclusion target aligning to at least 50% of the signature, anywhere along the signature. This process is done to avoid investigating signatures that are not highly discriminatory. The remaining signatures are considered filtered signatures and are believed to be sensitive and specific, within the context of the relative uniqueness of the input inclusion and exclusion groups, and the parameters supplied for target identification.
Scoring
Signatures are assigned an overall score corresponding to their highest-scoring BLAST (9) alignments with all inclusion and exclusion targets. This score is the sum of a positive inclusion component and a negative exclusion component, which are analogous to sensitivity and specificity, respectively, with respect to the input data. Let |A(S, Ii)| be the length of the highest-scoring aligned region between a signature, S, and an inclusion target, Ii. Let |S| be the length of signature S, PI(S, Ii) the percent identity (identities divided by the alignment length) between the aligned region of S and Ii, and |I| be the number inclusion targets. The negative exclusion component is similarly defined. The signature score, score(S), is calculated as follows:
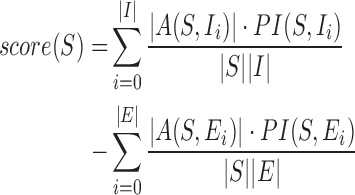 |
(11) |
This score is maximized when all inclusion targets contain a region exactly matching the entire signature and there exists no exclusion targets that match the signature. Signatures are sorted based on their scores with highest-ranking signatures appearing first in the output.
Output
Neptune produces a list of candidate, filtered and sorted signatures for all references. The candidate signatures are guaranteed to contain, by default, no exact matches with any exclusion k-mer. However, there may still remain potential inexact matches within exclusion targets. The filtered signatures contain no signatures with significant sequence similarity to any exclusion target. Sorted signatures are filtered signatures appearing in descending order of their signature scores. A consolidated signature file is additionally provided as part of Neptune’s output. This file contains a consolidated list of the top-scoring signatures produced from all reference targets such that homologous signatures are reported only once. However, because this file is constructed in a greedy manner, it is possible for signatures within this file to overlap each other. To identify redundancy across the reference targets, we recommend evaluating the signatures identified from each individual reference target in combination with this consolidated file when evaluating signatures.
RESULTS
Validation
We applied Neptune to identify differentially abundant genomic loci (genomic signatures) for distinct bacterial datasets from broad phyla. In order to validate methodology and highlight mathematical considerations, we first applied Neptune to a simulated Bacillus anthracis dataset. To demonstrate behavior in populations with genomic variation dominated by gene gain and loss, we applied Neptune to identify signatures within a clinically relevant Listeria monocytogenes dataset. Lastly, we demonstrated Neptune’s capacity to locate genome signatures in a more structurally and compositionally diverse Escherichia coli dataset.
Simulated dataset
In order to show that Neptune identifies signatures as expected, the software was run with an artificially created dataset. We created an initial inclusion genome by interspersing non-overlapping, virulence- and pathogen-associated genes from Vibrio cholerae M66-2 (NC_012578.1) throughout a B. anthracis genome (NC_007530) (Table 1). We selected six signature regions identified with IslandViewer 3 (16), varying from 4 to 50 kb in size, and spaced these signatures evenly throughout the B. anthracis genome with each signature represented only once. The initial exclusion genome consisted of the wild-type B. anthracis genome lacking modification. Lastly, we broadened both the inclusion and exclusion groups to 20 genomes each, by generating copies of the corresponding original inclusion or exclusion genome and incorporating a 1% random nucleotide mutation rate, with all possible mutations being equally probable.
Table 1. Genomic islands naturally found within Vibrio cholerae (NC_012578.1) chromosome I. These islands were used as in silico signatures and artificially inserted within a Bacillus anthracis genome. These islands were identified with IslandViewer 3 (16).
| ID | Length (bp) | Summary |
|---|---|---|
| 1 | 23 338 | O-antigen transport |
| 2 | 50 038 | Toxin pilus |
| 3 | 12 259 | Phage replication |
| 4 | 9652 | Phage integrase |
| 5 | 9652 | N-acetylneuraminate lyase |
| 6 | 10 155 | Neuraminidase |
Neptune was used to identify the inserted pathogenic and virulence regions in our simulated B. anthracis dataset. We specified a k-mer size of 27, derived from Equation (2), and used Neptune’s default SNV rate of 1%. Neptune produced signatures from all 20 inclusion targets and these signatures were consolidated into a single file. We aligned these signatures to the initial inclusion genome and used GView Server (17) to visualize the identified signatures from all references. Neptune identified seven consolidated signatures, corresponding to the six expected V. cholerae regions, with the largest signature region (50 kbp) misreported as two adjacent signatures (10 136 and 39 763 bp) with a gap of 143 bp between them. However, by Equation (9), we expect to see erroneous signature breaks with a frequency inversely proportional to our confidence level (95%) when extending signatures over k-mer gaps. Indeed, upon investigation, the break location contained six mutations almost evenly spaced within the 143 bp region. Importantly, we observed that all Neptune-identified signatures corresponded to the artificially inserted V. cholerae regions and were consistently detected for all references. Neptune reported all of the in silico signatures and reported no false positives. Hence, we conclude that Neptune is able to locate all in silico signatures; although some regions identified are reported as two adjacent signatures.
Listeria monocytogenes
Neptune was used to locate signature regions within two distinct serotypes of Listeria monocytogenes. Listeria monocytogenes is an opportunistic environmental pathogen that causes listeriosis, a serious and life-threatening bacterial disease in humans and animals (18). Listeria monocytogenes is comprised of a group of genetically heterogeneous strains consisting of clonal isolates with very low recombination rates. However, recent L. monocytogenes evolution has been characterized by gene deletion events resulting from horizontally acquired bacteriophage and genomic islands. Hence, we anticipated finding signatures corresponding to these events.
Listeria isolates were serotyped using standard laboratory serotyping procedures (19). Serotypes 1/2a and 4b were selected for evaluation as they represent distinct evolutionary lineages and are clinically relevant (18). Of the 13 L. monocytogenes serotypes, serotype 1/2b and 4b (lineage I) and serotype 1/2a (lineage II) are most commonly associated with human illness globally (18). Listeria monocytogenes lineage I is characterized by low diversity and low recombination rates and strains from this lineage are over-represented among human isolates, as compared to lineage II strains, which exhibit increased levels of genomic diversity, owing to recombination and horizontal gene transfer and have an over representation among food, food-related and natural environments (18). In total, 112 serotype 1/2a and 39 serotype 4b targets were available to be used as inclusion and exclusion groups. These were independently assessed to identify 1/2a signatures as well as the reciprocal 4b signatures, by reversing the inclusion and exclusion groupings. These groups were evenly and randomly subdivided into an experiment set and a validation set.
Neptune was executed on the L. monocytogenes experiment data in order to produce both 1/2a and 4b signatures for validation. Neptune produced 105 1/2a signatures and 75 4b signatures from their respective inclusion targets. We further evaluated the top-scoring (≥ 0.95) 1/2a and 4b signatures. The top-scoring signatures identified for L. monocytogenes serotype 1/2a are listed in Table 2. These signatures included phosphotransferase systems, proteins involved in regulating virulence genes in response to environmental cues and a surface-exposed internalin protein gene, many of which are known to be critical factors for human pathogenesis (20). Furthermore, a lineage II-specific heat shock system (21), constituting an operon with three genes, was present among high scoring signatures. Likewise, the top-scoring signatures identified for L. monocytogenes serotype 4b (Table 3) included proteins related to the cell wall, such as teichoic acid biosynthesis and a cell wall anchor protein, and a variety of other signatures encoding broad functional diversity.
Table 2. A summary of top-scoring (≥ 0.95) Listeria monocytogenes serotype 1/2a signatures generated by Neptune relative to a serotype 4b background. These signatures were mapped against L. monocytogenes 1/2a EGD-e (NC_003210) and 08-5578 (NC_013766) to infer annotations.
| Rank | Score | Length | Locus Information | L. monocytogenes |
|---|---|---|---|---|
| (bp) | Serotype 1/2a str. | |||
| EGD-e coordinates | ||||
| 1 | 0.99 | 4830 | Peptidoglycan-bound protein colossin A | 2 653 185–2 658 013 |
| 2 | 0.99 | 5336 | Phosphotransferase system (PTS), L-ascorbate (L-Asc) family | 2 042 111–2 047 447 |
| 3 | 0.99 | 4059 | bvrABC locus, β-glucoside-specific sensory system | 2 872 894–2 876 952 |
| 4 | 0.99 | 5454 | PTS, glucose–glucoside (Glc) family | 764 364–769 817 |
| 5 | 0.98 | 1938 | Hypothetical | 776 415–778 355 |
| 6 | 0.98 | 4514 | Two-component response regulator and ATP-binding cassette (ABC) transport systems | 1 086 579–1 091 092 |
| 7 | 0.98 | 2839 | Internalin | 169 228–172 066 |
| 8 | 0.98 | 1673 | Glycosyl-transferase | 532 558–534 230 |
| 9 | 0.97 | 967 | Hypothetical | 2 717 382–2 718 348 |
| 10 | 0.96 | 169 | Hypothetical, partial | 270 157–270 325 |
| 11 | 0.96 | 2591 | Lineage II-specific heat shock system | 441 513–444 103 |
| 12 | 0.95 | 548 | Hypothetical | 804 275–804 822 |
Table 3. A summary of top-scoring (≥ 0.95) Listeria monocytogenes serotype 4b signatures generated by Neptune relative to a serotype 1/2a background. The signatures were mapped to L. monocytogenes strain 4b F2365 (NC_002973) to infer annotations.
| Rank | Score | Length | Locus Information | L. monocytogenes |
|---|---|---|---|---|
| (bp) | Serotype 4b str. | |||
| F2365 coordinates | ||||
| 1 | 0.99 | 223 | Hypothetical | 478 246–478 468 |
| 2 | 0.99 | 3081 | gltA–gltB operon | 2 787 943–2 791 023 |
| 3 | 0.99 | 4004 | N-acetylmuramic acid metabolism | 1 685 737–1 689 738 |
| 4 | 0.98 | 1709 | Cell wall anchor | 2 684 246–2 685 954 |
| 5 | 0.97 | 1786 | RHS repeat-containing protein (partial) | 471 882–473 667 |
| 6 | 0.97 | 4912 | RHS repeat-containing protein (partial) | 466 603–471 499 |
| 7 | 0.97 | 5917 | Multiple, including: hypothetical, cell surface membrane anchor, multidrug efflux transporter-like | 428 382–434 298 |
| 8 | 0.97 | 1785 | Pyruvyl-transferase | 117 970–199 754 |
| 9 | 0.95 | 1654 | Teichoic acid biosynthesis | 2 190 231–2 191 883 |
| 10 | 0.95 | 1741 | Serine protease | 1 924 193–1 925 933 |
These experiment-generated signatures were then compared against the wet-lab verified validation datasets to evaluate their in silico sensitivity and specificity. We used BLASTN (9) to independently align the top-scoring signatures against our validation datasets. With a percent identity threshold of 95% and a minimum alignment length of 95%, the size of the signature length, 670 out of 672 (99.7%) 1/2a signature alignments against the 1/2a validation targets met our sensitivity criteria. Likewise, 199 out of 200 (99.5%) 4b signature alignments against 4b validation targets met this strictness. Similarly, when relaxing the percent identity threshold to 50% and the minimum alignment length to 50% the size of the signature length, we found no 1/2a hits against 4b validation targets and no 4b hits against 1/2a validation targets, indicating that the signatures were specific to the inclusion group. These results suggest that our top-scoring Neptune-identified L. monocytogenes serotype 1/2a and 4b signatures have high in silico sensitivity and specificity to their respective serotypes against the other serotype background.
Escherichia coli
We then applied Neptune to locate signatures corresponding to Shiga-toxin producing E. coli (STEC). Specifically, we chose to interrogate E. coli genomes that produce the Stx1 toxin. This toxin requires the expression of both the Stx1a and Stx1b subunits to be functional. Therefore, we expected to locate the genes encoding for these subunits using Neptune. As E. coli exhibits significantly increased genomic diversity over L. monocytogenes, we expect it makes identifying related signatures a more computationally challenging problem.
The inclusion and exclusion datasets were comprised of six STEC (Stx1) and eleven non-STEC draft assemblies, respectively. Neptune identified 371 signatures corresponding to the STEC inclusion group. The top-scoring signature had nearly 100% in silico sensitivity and specificity with respect to the inclusion and exclusion groups. We further investigated the top-scoring (≥ 0.95) consolidated signatures (Table 4) by aligning these signatures against an E. coli O157:H7 str. Sakai reference (NC_002695.1, NC_002127.1, NC_002128.1) to infer sequence annotations. This alignment included the chromosome and both plasmids, pO157 and pSKA1. The Sakai reference was selected because it contains a copy of the Stx1 toxin and is well characterized.
Table 4. A summary of Stx1-containing Escherichia coli signatures generated by Neptune relative to a background of non-toxigenic Escherichia coli. The signatures were mapped to E. coli O157:H7 str. Sakai reference (NC_002695.1, NC_002127.1, NC_002128.1) to infer annotations.
| Rank | Score | Length | Locus Information | E. coli O157:H7 |
|---|---|---|---|---|
| (bp) | Sakai coordinates | |||
| 1 | 1.00 | 1375 | Shiga toxin (A and B subunit) | 2 924 383–2 925 757 |
| 2 | 0.99 | 5433 | Urease gene cluster: ureA-G | 1 390 114–1 395 545 |
| 3 | 0.98 | 3291 | Bacteriophage related, integrase and other | 2 593 022–2 596 313 |
| 4 | 0.98 | 438 | perC, transcriptional activator of EaeA/BfpA, partial | 1 183 201–1 183 639 |
| 5 | 0.98 | 1223 | Phage tail length tape measure protein, partial | 2 170 250–2 171 473 |
| 6 | 0.97 | 7697 | Hemolysin gene cluster: hylC, hylA, hylB, hylD | 15 716–23 412 (pO157) |
| 7 | 0.96 | 1260 | Colonization factor | 1 767 898–1 769 157 |
| 8 | 0.96 | 962 | Hypothetical | 2 200 204–2 201 165 |
| 9 | 0.96 | 495 | Hypothetical | 2 186 120–2 186 614 |
| 10 | 0.96 | 796 | Phage origin, serine/threonine protein phosphatase | 3 488 405–3 489 201 |
| 11 | 0.96 | 1364 | Hypothetical, colicin-like and small toxic polypeptide | 1 397 029–1 398 393 |
| 12 | 0.96 | 987 | Hypothetical, putative membrane protein | 3 486 570–3 487 557 |
| 13 | 0.95 | 916 | Putative serine acetlyltransferase of prophage | 2 605 160–2 606 076 |
| 14 | 0.95 | 300 | Hypothetical, potential T3SS effector | 2 209 466–2 209 765 |
| 15 | 0.95 | 1136 | T3SS effector protein NleH | 1 804 974–1 806 122 |
As expected, Neptune identified the Stx1-encoding region as the highest scoring signature and identified associated phage genes (Table 4). However, due to the polymorphic nature of the stx-associated phage, Neptune’s signature sequence lengths for the stx phage were restricted to the gene level. Interestingly, Neptune identified a 7697 bp hemolysin cluster (Figure 3) that, although not genetically or biologically linked to stx, did segregate with STEC E. coli genomes. This observation underscores Neptune’s strength in identifying large-scale (multigene) features, such as operons, in organisms possessing complex genomic organizations with horizontal gene transfer. In addition, it also serves to demonstrate the value of this type of analysis in identifying unexpected signatures that may provide new insights into the genomic underpinnings of biological traits. Likewise, other salient signatures identified by Neptune included several virulence regions such as the urease gene cluster, intimin transcription regulator (perC) sequences and type 3 secretion system (T3SS)-related regions (Table 4). In the plasmid alignments, the hemolysin-predicted signature was the only top-scoring signature (sixth rank; 0.97 score) located on the pO157 plasmid. Furthermore, using BLASTN (9), we found that many of the Neptune top-scoring signatures aligned to characterized E. coli O157:H7 O-Islands (a set of mobile genetic islands characterized to carry virulence factors). This included signatures 1–3, 5, 7–15; notably Shiga toxin I (as predicted), a urease gene cluster and several phage elements. We conclude that Neptune is effective at locating known pathogenicity-associated regions and horizontally acquired regions within STEC, which have a high in silico sensitivity and specificity with respect to the input genomes for the analysis.
Figure 3.

Genomic synteny for the Neptune signature encoding the hemolysin gene cluster (top) versus the corresponding hlyCABD region of reference plasmid pO157 for Escherichia coli O157:H7 str. Sakai (bottom). GView (17) was used to visualize this 7697 bp signature; gene names and plasmid pO157 coordinates noted according to NC_002128.1.
Comparison
Neptune’s principal distinguishing feature relative to other signature detection software is its ability to accommodate partial representation of the signature sequences in both the inclusion and exclusion groups, and its ability to accommodate variation in the identified signatures. To demonstrate its value, we compared Neptune to mGenomeSubtractor (22) and panSeq (23). mGenomeSubtractor is a web-based tool designed to perform in silico subtractive hybridization. The program accepts as input a single query genome and a set of subtraction genomes, and it reports the regions that are unique to the query genome. panSeq constitutes a suite of genome analysis utilities and is available as a web-based tool or a downloadable application. The novel region finder of panSeq accepts a set of query genomes and a set of reference genomes. The program considers each query genome independently of the others and, like mGenomeSubtractor, reports the sequences that are contained in that query but absent from the reference sequence collection. A crucial difference between Neptune and both mGenomeSubtractor and panSeq is that Neptune simultaneously considers both the entire inclusion and exclusion groups, and reports sequences that differentiate these groups, whereas mGenomeSubtractor and panSeq analyze only a single inclusion genome at a time. This difference is important, since a signature for an individual genome may not be a signature for the group.
We chose to highlight the effect of this difference by comparing these applications’ abilities to detect signatures for 20 Enterococcus hirae isolates against a background of 20 Enterococcus faecium isolates. These genomes were already available on NCBI. This dataset is illustrative due to inter-species variations as well as intra-species diversity attributed to their accessory genomes. In contrast to more clonal organisms, such as L. monocytogenes, the consequence of ignoring signature sequence representation across an entire set of inclusion genomes should become more pronounced for organisms harboring large accessory genomes. An individual genome or a minority population may harbor genomic regions that are specific only to themselves, and thus will not be representative genomic signatures for the broader bacterial population.
We ran all software with default parameters using E. hirae as the inclusion dataset and E. faecium as the exclusion dataset (Supplementary Data). As mGenomeSubtractor analyzes only one inclusion genome, we specified E. hirae ATCC 9790 (NC_018081) from its list of NCBI bacterial genomes as the query genome. Furthermore, mGenomeSubtractor partitions the query genome, either by coding sequence (CDS) or overlapping genome fragments of equal lengths. We chose to have mGenomeSubtractor analyze CDS regions as they generally must remain intact and conserved to perform their biological role, are consequently more likely to harbor stable signature sequences, and offer a more suitable analytical choice for partitioning the genome relative to an arbitrary fixed sequence length. However, we highlight that Neptune and panSeq do not themselves incorporate any information about coding regions for their analysis of signature sequence content. Additionally, these software vary considerably in output, such as the number of signatures reported, the signature sequence length and whether partial or complete CDSs or multi-gene operons are identified as signatures. Importantly, these software differ in the specific analytical application that they were developed to address, which accounted for a large proportion of the observed variation in our parallel analysis. These intrinsic differences impose considerable difficulty for conducting a quantitative side-by-side evaluation; thus, we restricted our comparison to assessing the ability of each respective software to find in silico genome signatures that could differentiate (or represent) a larger bacterial population, while acknowledging that neither panSeq nor mGenomeSubtractor were specifically designed to perform such analyses.
We used BLAST (9) to identify matching signature regions in the input genomes and scored each signature sequence generated by Neptune, mGenomeSubtractor and panSeq, using Equation (11), which assigns a score between −1.0 and +1.0 as a combined measure of signature sensitivity and specificity. These scores represent the in silico discriminatory power of the reported sequences, with positive scores closer to 1.0 being highly discriminatory and scores close to 0.0 being indiscriminate and likely undesirable. A high-valued negative score is a measure of discrimination in the inverse direction (i.e. it indicates that the reported sequence is a strong signature for the exclusion group rather than for the inclusion group). Surprisingly, panSeq and mGenomeSubtractor reported negative scores for some sequences (panSeq: 292 sequences, 61 801 bases, 2.5% of total bases; mGenomeSubtractor: 3 sequences, 515 bases, 0.02% of total bases), implying that they were weakly discriminatory for E. faecium, rather than for E. hirae. As the number and lengths of reported signatures vary considerably between software, as well as the signatures themselves, we constructed a histogram charting the total amount of identified signature sequence (in base pairs) with corresponding combined discriminatory scores falling within a particular interval (Figure 4). Summary statistics of the results are provided in Table 5.
Figure 4.

A comparison of the the population-level discriminatory power, as determined by signature score (Equation 11), for sequences identified by mGenomeSubtractor (pink), panSeq (blue) and Neptune (orange) when operating on a dataset comprised of 20 Enterococcus hirae and 20 Enterococcus faecium genomes. The histogram depicts total signature sequence (y-axis; measured in base pairs) with a combined discrimination score falling within given scoring intervals (x-axis). Each score interval may contain more than one identified signature. Sequences with calculated scores closer to 1 are highly discriminatory, whereas those nearing 0 have no discriminatory power to distinguish between inclusion and exclusion genome groups.
Table 5. Summary statistics of the population-level discriminatory power, as determined by calculated signature score (Equation 11), for sequences identified by Neptune, mGenomeSubtractor and panSeq, and Neptune when operating on a dataset comprised of 20 Enterococcus hirae and 20 Enterococcus faecium genomes.
| Median | Interquartile range | Minimum | Maximum | Average base | |
|---|---|---|---|---|---|
| Neptune | 0.79 | 0.68–0.88 | 0.10 | 1.00 | 0.74 |
| mGenomeSubtractor | 0.71 | 0.35–0.88 | −0.01 | 1.00 | 0.65 |
| panSeq | 0.44 | 0.19–0.72 | −0.73 | 1.00 | 0.43 |
DISCUSSION
Parameters
While many of Neptune’s parameters are automatically calculated, there are a few parameters that deserve special mention. The minimum number of inclusion hits and maximum gap size are sensitive to the SNV rate and the size of k. When estimating these parameters, a slightly higher than expected SNV rate is recommended. This conservative approach will avoid false negatives at the expense of false positives. However, many of these false positives will be removed during the filtering stage at the expense of increased computational time.
Computation time
Neptune is parallelizable and performs well on high-performance computing clusters. In order to show the scalability of Neptune, we created a simulated dataset by generating 100 copies of a L. monocytogenes serotype 1/2a isolate, 100 copies of a serotype 4b isolate and incorporating a 1% random nucleotide mutation rate in each generated copy, with all possible mutations being equally probable. We ran Neptune on a homogeneous computing cluster where there were always more resources available than required by the software. This demonstrates the scalability of Neptune when computing resources are not a limitation (additional information appears in the Supplementary Data). We ran Neptune on 50, 100, 150 and 200 total genomes, with even numbers of inclusion and exclusion genomes, and observed a linear relationship between running time and number of genomes (Supplementary Data). We observed a relationship suggesting each additional genome added as input would require an additional 10.2 s to complete and, more generally, a 53% increase in running time for each additional fold increase in input size.
Neptune may also be run as a parallel process within a single-machine environment. When performing a similar scalability experiment on a smaller real dataset comprised of 112 L. monocytogenes serotype 1/2a isolates and 38 serotype 4b isolates, run on a single compute node with 48 cores and 80 GB of memory, we observed a linear relationship between the number of genomes and completion time (Supplementary Data). We varied the size of the input data such that runs maintained an approximate proportion of 70–75% inclusion genomes and 25–30% exclusion genomes. We observed a relationship suggesting each additional genome added as input would require an additional 7.9 s to complete and, more generally, a 36% increase in running time for each additional fold increase in input size. The observed difference between this experiment and the previous can be partially attributed to the proportionally smaller exclusion group, from which mutations create more work for the algorithm than from within the inclusion group.
Revealing biology
This study demonstrates that Neptune is a very useful tool for the rapid characterization and classification of pathogenic bacteria of public health significance, as it can efficiently discover differential genomic signatures. Although both L. monocytogenes 4b and 1/2a serotypes, belonging to lineages I and II respectively, are associated with human illness, lineage I strains are overrepresented among human cases whereas lineage II isolates are widespread in food-related, natural and farm environments. Among the L. monocytogenes isolates used in our study, 46% of 4b and 17% of 1/2a serotype isolates had a clinical human host origin. Among the signatures for serotype 1/2a, multiple PTS and ABC transport systems were found (Table 2 and Supplementary Data), which may be correlated to the fact that the presence of a variety of transport systems provides L. monocytogenes serotype 1/2a with a competitive advantage to survive under broad environmental conditions due to its ability to utilize a variety of carbon sources. Among the L. monocytogenes 4b serotype signatures found were genes coding for cell wall anchor proteins, rearrangement hotspot (RHS) repeat-containing protein known to be associated with mediating intercellular competition and immunity (24), and cell wall polysaccharides and teichoic acid decoration enzymes (Table 3 and Supplementary Data). In keeping with predilection of lineage I for human clinical disease, such cell surface components play a role in bacterial–host interactions (25). The potential involvement of these genes in the virulence and pathogenesis of serotype 4b should be an interesting area of future inquiry.
Interestingly, two very large, but divergent signature sequences corresponding to the 4b (rank 16; score 0.93; length 12685 nt) and 1/2a (rank 14; score 0.94; length 22937) inclusion groups were found by Neptune (Supplementary Data). These serotype-specific signature regions contained non-homologous teichoic acid biosynthesis and transport system genes at equivalent chromosomal locations in the two serotype subgroupings. In addition, another signature (rank 26; score 0.84; length 6348 nt; Supplementary Data) spanning seven genes corresponding to Listeria pathogenicity island-3 (LIPI-3), or the listeriolysin S cluster (26) was only identified in 4b isolates.
In a recent study by Maury et al., a pattern correlation of gene families with the infection/food ratio of isolates in the Listeria pangenome successfully identified virulence-associated genes such as LIPI-3 and teichoic acid biosynthesis-related gene clusters in serotype 4b strains to be strongly associated with infectious potential at the population level (27). We employed Neptune to analyze the same sequence data for serotypes 1/2a and 4b as was used in Maury et al., and identified signatures that overlapped with our prior, independently generated and distinct genomes for serotype 1/2a and 4b isolates (Supplementary Data).
With the advent of GWAS and their applications in bacteria to rapidly scan genetic markers as the basis of bacterial phenotypes such as host preference, antibiotic resistance and virulence across the complete sets of genomes, Neptune offers to be a promising tool to reveal discriminatory genetic markers and associations with particular phenotypic traits. Hence, by generating such a catalogue of differential loci, Neptune is useful in identifying candidate regions for further investigating the association of identified regions with categorical phenotypes, biological traits or metadata, such as pathogen virulence or persistence in niche environments.
Advancing signature discovery
We compared Neptune against two other genome signature finding programs, mGenomeSubtractor and panSeq, for the ability to identify population level signatures (Figure 4). We observed that all three applications were capable of identifying highly discriminatory sequences (score ≥ 0.95). This result is expected, given that highly discriminatory sequences will be present in virtually all inclusion genomes and thus can be identified by analyzing essentially any arbitrarily selected single genome from the inclusion group. In contrast, we observed significant differences in each software’s ability to report sequences that are present in many, but not necessarily all, inclusion set genomes. This is important in circumstances where no individual locus can serve as a ‘true’ signature for the inclusion group, but there may exist multiple loci that in combination can serve as true signatures. Neptune found many sequences with scores 0.65–0.90 that were present in a majority, but not all, inclusion group genomes, whereas mGenomeSubtractor and panSeq did not identify a comparable amount of sequence with similar scores. While all three applications reported low-scoring sequences indicative of less discriminatory signatures, mGenomeSubtractor reported substantially more low-scoring signatures relative to Neptune, and panSeq’s output was predominantly low-scoring sequences. Overall, Neptune showed the narrowest range of discriminatory scores, followed by mGenomeSubtractor and panSeq (Table 5). Furthermore, Neptune showed the highest average per-base score, followed by mGenomeSubtractor, and panSeq.
Limitations
Neptune’s signature extraction step avoids false negatives at the expense of false positives. The software attempts to locate signatures that may not contain an abundance of exact matches. This approach produces some false positives. However, false positives are removed during signature filtering and requires increased computational time. As signatures are extracted from a reference, repeated regions do not confound signature discovery. However, if a repeated region is a true signature, then Neptune will report each region as a separate signature. In this circumstance, user curation may be required.
Neptune cannot locate isolated SNVs and other small mutations. Any region with a high degree of similarity to the exclusion group will either not produce candidate signatures or be removed during filtering. Neptune is designed to locate general-purpose signatures of arbitrary size (above the k-mer size) and does not consider application-specific physical and chemical properties of signatures. While Neptune is capable of producing signatures as small as the k-mer size, we observed that very short signatures (<100 bases) may not contain any seed matches with targets when performing alignments during the filtering process, thereby preventing the signature from being evaluated correctly. We recommend either using smaller seed sizes during pairwise alignments, at the expense of significantly increased computation time, or discretion when evaluating very short signatures. The largest signatures identified by Neptune in our data were (in bases) 22 937 for L. monocytogenes serotype 1/2a, 12 683 for L. monocytogenes serotype 4b and 14 650 for E. coli. Signature length will be limited by the actual size of the discriminatory sequence and the amount of sequence variation present.
Finally, Neptune makes assumptions about the probabilistic independence of bases and SNV events; while these events do not occur independently in nature, they allow for significant mathematical simplification. Nonetheless, Neptune is capable of producing highly sensitive and specific signatures using these assumptions.
CONCLUSION
Neptune allows one to efficiently and rapidly identify genomic loci that are common to one population and distinguishing them from other populations. When applied to pathogens, top-scoring signatures were specific to known regions encoding mobile islands containing pathogenicity-associated CDSs. By simultaneously considering all of the input data, Neptune is capable of identifying sequences that are representative signatures for bacterial organisms with diverse genome content. While some signatures are reported as smaller, adjacent signatures with intervening gaps, we demonstrated that Neptune can locate signatures in both simulated and biological datasets with high sensitivity and specificity. Neptune provides an array of gene candidates to investigate for their possible role in pathogenesis and functional genomics.
Although Neptune will be useful in broad comparative applications, we anticipate it will be particularly helpful in public health scenarios, where rapid infectious agent screening and characterization is crucial. Neptune may be leveraged to reveal discriminatory signature sequences to uniquely delineate one group of organisms, such as isolates associated with a disease cluster or event, from unrelated sporadic or environmental microbes. Neptune’s computations approach is well suited to comprehensive, ad hoc comparisons. We conclude that Neptune is a powerful and flexible tool for locating signature regions with minimal prior knowledge for wide-ranging applications of bacterial characterization.
AVAILABILITY
Listeria monocytogenes and E. coli data used in the manuscript are stored under NCBI BioProject PRJNA301341. Neptune is developed in Python and the software requires a standard 64-bit Linux environment. The software is available at http://github.com/phac-nml/neptune. Signatures identified in our experiments and additional data files are available at http://github.com/phac-nml/neptune-manuscript.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Franklin Bristow and Eric Enns at The Public Health Agency of Canada for their feedback on various aspects of the software design and implementation. The authors would also like to thank L. Chui, D. Haldane, S. Bekal and J. Wylie for E. coli data.
Author contributions: E.M., R.Z., K.W., M.G., G.V.D. and C.B wrote the manuscript. E.M., G.V.D. and M.G. designed the software and E.M. developed the software. E.M. and M.D. designed the mathematical models. E.M., K.W., M.G. and G.V.D. contributed to all experiment design. C.B. and A.R. designed the L. monocytogenes experiments. L.C. provided the L. monocytogenes data on behalf of L.C., L.P.T., J.Z., F.P., J.F., J.M., K.S., S.B., C.T., J.I.R., N.P., J.C., M.G., G.V.D., N.K., C.B. and P.S. C.B. provided the E. coli data. P.M. and N.K. contributed early work on the problem of signature discovery. E.M. performed all experiments. R.Z., E.M., K.W., C.B., G.V.D. and M.G. interpreted the results of the experiments.
Footnotes
Present addresses:
Rahat Zaheer, Lethbridge Research and Development Centre, Agriculture and Agri-Food Canada, 5403-1st Ave. South, Lethbridge, AB T1J 4B1, Canada.
Kelly A. Weedmark, Bureau of Microbial Hazards, Health Canada, 251 Sir Frederick Banting Driveway, Tunney’s Pasture, Ottawa, ON K1A 0K9, Canada.
Laura Patterson-Fortin, BioLargo Water Inc., 6020–118 Street NW, Edmonton, AB T6H 2V8, Canada.
Jeff Farber, Department of Food Science, University of Guelph, 50 Stone Road East, Guelph, ON N1G 2W1, Canada.
Cécile Tremblay, Centre de recherche du Centre hospitalier de l'Université de Montréal 3840, rue St-Urbain, Bureau 7-355, Montréal, QC H2W 1T8, Canada.
Judy Isaac-Renton, Department of Pathology and Laboratory Medicine, University of British Columbia, 2211 Westbrook Mall, Vancouver, BC V6T 2B5, Canada.
Jessica Chen, Centers for Disease Control and Prevention, 1600 Clifton Rd., Mailstop C0-3, Atlanta, GA 30329, USA.
Peter Slade, Food Safety Consulting, 319 2190 W. 7th Avenue, Vancouver, BC V6K 4K7, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Public Health Agency of Canada; Canadian federal Genomics Research and Development Initiative; Alberta Provincial Laboratory for Public Health; Public Health Laboratory Network of Nova Scotia; Laboratoire de santé publique du Québec; Genome Canada; Alberta Innovates Bio Solutions; Canadian Food Inspection Agency. Funding for open access charge: Public Health Agency of Canada.
Conflict of interest statement. None declared.
REFERENCES
- 1. Davis S., Pettengill J.B., Luo Y., Payne J., Shpuntoff A., Rand H., Strain E.. CFSAN SNP pipeline: an automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput. Sci. 2015; 1:e20. [Google Scholar]
- 2. Sahl J.W., Lemmer D., Travis J., Schupp J.M., Gillece J.D., Aziz M., Driebe E.M., Drees K.P., Hicks N.D., Williamson C.H.D. et al. NASP: an accurate, rapid method for the identification of SNPs in WGS datasets that supports flexible input and output formats. Microb. Genom. 2016; 2:e000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao L., Qi J.. Whole genome molecular phylogeny of large dsDNA viruses using composition vector method. BMC Evol. Biol. 2007; 7:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M.. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016; 17:132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sims G.E., Kim S.-H.. Whole-genome phylogeny of Escherichia coli/Shigella group by feature frequency profiles (FFPs). Proc. Natl. Acad. Sci. U.S.A. 2011; 108:8329–8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Earle S.G., Wu C.-H., Charlesworth J., Stoesser N., Gordon N.C., Walker T.M., Spencer C.C.A., Iqbal Z., Clifton D.A., Hopkins K.L. et al. Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat. Microbiol. 2016; 1:16041–16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lees J.A., Vehkala M., Välimäki N., Harris S.R., Chewapreecha C., Croucher N.J., Marttinen P., Davies M.R., Steer A.C., Tong S.Y.C. et al. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat. Commun. 2016; 7:12797–12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Slezak T., Kuczmarski T., Ott L., Torres C., Medeiros D., Smith J., Truitt B., Mulakken N., Lam M., Vitalis E. et al. Comparative genomics tools applied to bioterrorism defence. Brief. Bioinform. 2003; 4:133–149. [DOI] [PubMed] [Google Scholar]
- 9. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 10. Phillippy A.M., Ayanbule K., Edwards N.J., Salzberg S.L.. Insignia: a DNA signature search web server for diagnostic assay development. Nucleic Acids Res. 2009; 37(Suppl. 2):W229–W234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bader K.C., Grothoff C., Meier H.. Comprehensive and relaxed search for oligonucleotide signatures in hierarchically clustered sequence datasets. Bioinformatics. 2011; 27:1546–1554. [DOI] [PubMed] [Google Scholar]
- 12. Satya R.V., Zavaljevski N., Kumar K., Reifman J.. A high-throughput pipeline for designing microarray-based pathogen diagnostic assays. BMC Bioinform. 2008; 9:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Satya R.V., Kumar K., Zavaljevski N., Reifman J.. A high-throughput pipeline for the design of real-time PCR signatures. BMC Bioinform. 2010; 11:340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Melsted P., Pritchard J.K.. Efficient counting of k-mers in DNA sequences using a bloom filter. BMC Bioinform. 2011; 12:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Feller V. An Introduction to Probability Theory and Its Applications. 1960; 1:NY: J. Wiley & Sons. [Google Scholar]
- 16. Dhillon B.K., Laird M.R., Shay J.A., Winsor G.L., Lo R., Nizam F., Pereira S.K., Waglechner N., McArthur A.G., Langille M.G. et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 2015; 43:W104–W108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petkau A., Stuart-Edwards M., Stothard P., Van Domselaar G.. Interactive microbial genome visualization with GView. Bioinformatics. 2010; 26:3125–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Orsi R.H., den Bakker H.C., Wiedmann M.. Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. Int. J. Med. Microbiol. 2011; 301:79–96. [DOI] [PubMed] [Google Scholar]
- 19. Gilmour M.W., Graham M., Van Domselaar G., Tyler S., Kent H., Trout-Yakel K.M., Larios O., Allen V., Lee B., Nadon C.. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics. 2010; 11:120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bierne H., Sabet C., Personnic N., Cossart P.. Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microb. Infect. 2007; 9:1156–1166. [DOI] [PubMed] [Google Scholar]
- 21. Zhang C., Nietfeldt J., Zhang M., Benson A.K.. Functional consequences of genome evolution in Listeria monocytogenes: the lmo0423 and lmo0422 genes encode σC and LstR, a lineage II-specific heat shock system. J. Bacteriol. 2005; 187:7243–7253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shao Y., He X., Harrison E.M., Tai C., Ou H.-Y., Rajakumar K., Deng Z.. mGenomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Res. 2010; 38(Suppl. 2):W194–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laing C., Buchanan C., Taboada E.N., Zhang Y., Kropinski A., Villegas A., Thomas J.E., Gannon V.P.. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinform. 2010; 11:461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koskiniemi S., Lamoureux J.G., Nikolakakis K.C., de Roodenbeke C.t., Kaplan M.D., Low D.A., Hayes C.S.. RHS proteins from diverse bacteria mediate intercellular competition. Proc. Nati. Acad. Sci. U.S.A. 2013; 110:7032–7037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carvalho F., Sousa S., Cabanes D.. How Listeria monocytogenes organizes its surface for virulence. Front. Cell. Infect. Microbiol. 2014; 4:48–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cotter P.D., Draper L.A., Lawton E.M., Daly K.M., Groeger D.S., Casey P.G., Ross R.P., Hill C.. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008; 4:e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maury M.M., Tsai Y.-H., Charlier C., Touchon M., Chenal-Francisque V., Leclercq A., Criscuolo A., Gaultier C., Roussel S., Brisabois A. et al. Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nat. Genet. 2016; 48:308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.