

SUMMARY
Natural killer (NK) cells are innate lymphocytes that possess traits of adaptive immunity, such as clonal expansion, contraction, and generation of long-lived “memory” cells, processes poorly understood at the molecular level. Here, we found that as proliferating NK cells accumulated dysfunctional mitochondria during viral infection, a protective mitophagy pathway was induced during the contraction phase to promote their survival in a reactive oxygen species (ROS)-dependent manner. Inhibition of mechanistic target of rapamycin (mTOR) or activation of AMP-activated protein kinase (AMPK) during the contraction-to-memory phase transition of the antiviral response increased autophagic activity and enhanced memory NK cell numbers through an Atg3-dependent mechanism. Furthermore, we demonstrated a temporally regulated role for mitophagy-inducing proteins BCL2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L) in the generation of robust NK cell memory. Thus, our study reveals the functional importance of mitophagy during the dynamic response of these cytolytic innate lymphocytes.
In Brief
After viral infection, the majority of effector lymphocytes undergo rapid apoptosis. However, it is unclear how a subset of these cells persist to form immunological memory. Sun and colleagues demonstrate that mitophagy promotes the survival of virus-specific NK cells during the contraction phase to promote memory.
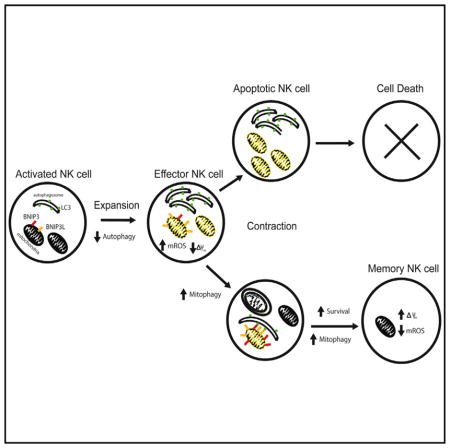
INTRODUCTION
Natural killer (NK) cells play a critical role in immunosurveillance against transformed and virally infected cells (Lanier, 2005). Although traditionally thought to be a cellular component of the innate immune system, NK cells have recently been shown to possess traits of adaptive immunity (Sun and Lanier, 2011; Vivier et al., 2011). During cytomegalovirus infection, virus-specific NK cells undergo robust proliferation (they increase in number by 1,000-fold in mice; Daniels et al., 2001; Dokun et al., 2001; Sun et al., 2009) and induce effector functions to eliminate virally infected cells in both mice and humans. After viral control, most effector NK cells undergo contraction to form a pool of long-lived “memory” NK cells that exhibit enhanced functional capacity upon secondary antigen exposure (Sun et al., 2009). However, the protective pathways that antigen-specific NK cells use to combat apoptosis and mediate survival to form memory cells remain largely unknown.
Induction of apoptosis in cytolytic lymphocytes after viral infection is an essential mechanism to prevent immune-mediated pathology by regulating the numbers of effector cells, and two separate mechanisms control this contraction phase in lymphocytes: extrinsic death receptor signals and cell-intrinsic pathways involving intracellular BH3-only proteins (Marrack and Kappler, 2004). Indeed, it has been shown that the BH3-only family member Bim regulates the contraction of effector T and NK cells by inducing cell-intrinsic death signals (Kurtulus et al., 2010; Min-Oo et al., 2014). During apoptosis, these signals converge at the mitochondria to induce changes in membrane permeability to release pro-apoptotic factors into the cytoplasm and activate degradation of intracellular components via a caspase-mediated cascade (Kroemer and Reed, 2000). This process is accompanied by a decrease in the inner mitochondrial membrane permeability, leading to a loss in the electro-chemical potential (ΔΨm) and dysfunction of the mitochondria (Kroemer and Reed, 2000). Previous work has shown that expanding antigen-specific CD8+ T cells possess decreased mitochondrial cell membrane potential and enhanced mitochondrial-associated reactive oxygen species (ROS) during infection (Grayson et al., 2003), consistent with the increased apoptotic activity in these cells as they enter the contraction phase. Yet how a subset of these effector lymphocytes elude death and persist to generate a long-lived memory pool is not well understood.
Apoptosis and autophagy are evolutionarily conserved pathways that often elicit contrasting cellular outcomes in response to cellular stress (Mariño et al., 2014). Autophagy is a process in which cytosolic contents are engulfed into double-membrane vacuoles, or autophagosomes, and delivered to the lysosome for degradation (Levine et al., 2011; Mariño et al., 2014). Whereas apoptosis executes cell-death programs during periods of metabolic starvation or stress, autophagy can serve as an essential cellular survival mechanism by maintaining energy homeostasis through its self-catabolic activity (Levine et al., 2011; Mariño et al., 2014). Because accumulation of damaged mitochondria in the cell can cause oxidative stress and induce cell death through the production of ROS (Green et al., 2011), they can be selectively sequestered into autophagosomes and undergo lysosomal degradation in a process termed mitophagy to promote cellular homeostasis and survival (Green et al., 2011; Levine et al., 2011; Mariño et al., 2014). However, it has yet to be established whether NK cells accumulate dysfunctional mitochondria as a consequence of virus-driven proliferation and subsequently utilize mitophagy as a pro-survival mechanism to facilitate memory formation.
In this study, we show that the proliferative burst of antigen-specific NK cells during mouse cytomegalovirus (MCMV) infection caused mitochondrial depolarization and accumulation of mitochondrial-associated ROS. We found that dysfunctional mitochondria were rapidly cleared in antigen-specific NK cells during the effector-to-memory phase transition. After MCMV infection, autophagic activity was inhibited in effector NK cells during their rapid expansion but was subsequently induced during the contraction-to-memory phase transition of the antigen-specific NK cell response. We used conditional Atg3 ablation to demonstrate the importance of autophagy during NK cell antiviral response to MCMV. Induction of autophagy through inhibition of mTOR or activation of AMPK at the peak of virus-driven NK cell expansion enhanced memory NK cell numbers in an Atg3-dependent manner. Lastly, we observed that mitochondrial-associated proteins BNIP3 and BNIP3L mediated the removal of ROS and depolarized mitochondria via mitophagy to induce NK cell memory formation after viral infection.
RESULTS
NK Cell Mitochondrial Quality Undergoes Dynamic Changes during Viral Infection
To assess mitochondrial function of antigen-specific NK cells during MCMV infection, we used a well-established adoptive-transfer system (Beaulieu et al., 2014; Karo et al., 2014; Sun et al., 2009; Sun et al., 2012). NK cells expressing the activating Ly49H receptor (which recognizes the MCMV-encoded glycoprotein m157 on infected cells; Arase et al., 2002; Smith et al., 2002) were purified from wild-type (WT) mice and adoptively transferred into Ly49H-deficient hosts, which were infected with MCMV. After a period of rapid MCMV-driven expansion, we found that adoptively transferred effector Ly49H+ NK cells exhibited decreased mitochondrial cell membrane potential (Figure 1A) and a concomitant increase in mitochondrial-associated ROS (Figure 1B), whereas overall mitochondrial content was unchanged at day 7 after infection (Figure 1C). However, during the contraction-to-memory phase transition (days 8–28 after infection), we observed a sequential decrease in mitochondrial-associated ROS and overall mitochondrial content and an increase in mitochondrial membrane potential in the surviving adoptively transferred Ly49H+ NK cells (Figures 1A–1C), indicating that dysfunctional mitochondria are cleared as antigen-specific NK cells enter the memory phase.
Figure 1. NK Cells Undergo Dynamic Changes in Mitochondrial Quality in Response to Viral Infection In Vivo.

WT splenic Ly49H+ NK cells were transferred into Ly49H-deficient mice and infected with MCMV. Shown are representative histograms and quantification of mean fluorescence intensity (MFI) for tetramethylrhodamine ethyl ester (TMRE) staining (A), MitoSox Red staining (B), or MitoTracker Green staining (C) in transferred NK cells from recipient spleens enriched with NK cells at the indicated time points after infection. Data are representative of three independent experiments with n = 3–4 per time point. Samples were compared with an unpaired, two-tailed Student’s t test, and data are presented as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Autophagy Is Induced within NK Cells during Viral Infection
Accumulation of dysfunctional mitochondria in mammalian cells can cause cellular oxidative stress and induce cell death (Green et al., 2011); however, cells can evade apoptosis through sequestration of dysfunctional mitochondria into autophagosomes through a process called mitophagy (Green et al., 2011; Levine et al., 2011; Mariño et al., 2014). We therefore used a specific autophagosome detection reagent (Puleston et al., 2014) to test whether antigen-specific NK cells could induce autophagosome formation in response to viral infection. After MCMV infection, compared to naive NK cells, adoptively transferred effector Ly49H+ NK cells displayed an accumulation of autophagosomes, as revealed by Cyto-ID staining (Figure 2A) and confirmed by transmission electron microscopy (Figures S1A–S1C), suggesting either an induction of autophagic activity or a block in the lysosomal degradation of autophagosomes. In contrast, the relative amount of detected autophagosomes was sequentially decreased during the contraction-to-memory phase transition (Figure 2B), suggestive of an active autophagic process in surviving antigen-specific NK cells.
Figure 2. NK Cells Induce Autophagy in Response to Viral Infection In Vivo.

(A and B) WT splenic Ly49H+ NK cells were transferred into Ly49H-deficient mice and infected with MCMV. Shown are representative histograms and quantification of MFI for Cyto-ID staining of autophagosomes at each indicated time point after infection.
(C–F) LC3-GFP splenic Ly49H+ NK cells were transferred into Ly49H-deficient mice and infected with MCMV. (C) Representative Western blot of LC3b and p62 for adoptively transferred splenic Ly49H+ NK cells at day 8 after infection or for naive NK cells. (D–F) Representative histograms and quantification of LC3-GFP MFI of adoptively transferred Ly49H+ NK cells in the blood on days 0 and 7 after infection (D) and days 7, 14, and 28 after infection (E) and of indicated tissues of recipients on days 0 and 28 after infection (F).
Data are representative of three independent experiments with n = 3–7 per group per time point. Samples were compared with an unpaired, two-tailed Student’s t test, and data are presented as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001). See also Figure S1.
To confirm that NK cells induce autophagy during the contraction phase of MCMV infection, we measured the abundance of microtubule-associated proteins LC3b-I and LC3b-II in effector NK cells. LC3b-II is the lipidated form of LC3b-I and is selectively incorporated into the elongation membrane of early autophagosomes and subsequently degraded after lysosomal fusion with autophagosomes (Klionsky et al., 2012). Whereas naive NK cells expressed low amounts of LC3b-II, we found that LC3b-II was induced in Ly49H+ NK cells at the start of the contraction phase (measured at day 8 after infection) (Figure 2C). Induction of LC3b-II was concurrent with a reduction in the amount of p62 (Figure 2C), another commonly used marker for autophagic activity (Pankiv et al., 2007). To further assess autophagic activity over the course of the contraction phase, we utilized LC3-GFP transgenic mice (Mizushima et al., 2004), where specific autophagic activity can be monitored in lymphocytes by measurement of the degradation of LC3 through reduction in GFP intensity by flow cytometry (Klionsky et al., 2012). We found that adoptively transferred LC3-GFP transgenic Ly49H+ NK cells contained greater GFP intensity at the peak of expansion (day 7 after infection) than did resting NK cells (Figure 2D), indicating either an increase in total LC3 in preparation for autophagy or a block in autophagosome-lysosome fusion in effector NK cells. The latter explanation is more likely because LC3b transcripts remained unchanged in sorted effector Ly49H+ NK cells (Figure S1D), whereas the total amount of LC3b-II and autophagosomes increased (Figures 2A, 2C, and S1C). In contrast to effector NK cells at day 7 after infection, Ly49H+ NK cells showed a sequential decrease in LC3-GFP fluorescence and labeled autophagosomes during the contraction and memory phases (days 14 and 28, respectively, after infection) (Figures 2A and 2E). Memory NK cells isolated from peripheral tissues displayed significant less LC3-GFP fluorescence than did NK cells from uninfected controls (Figure 2F), indicating that virus-specific NK cells display enhanced autophagic activity during the contraction-to-memory phase transition.
Autophagy Is Essential for NK Cell Memory Formation
To study the functional relevance of increased autophagic activity in NK cells during MCMV infection without potential developmental defects, we utilized mice with inducible deletion of the essential autophagosome machinery component Atg3 (Atg3fl/fl Ubc cre-ERT2); these mice have been shown previously to efficiently delete the target Atg3fl allele in splenocytes upon administration of 4-hydroxyta-moxifen (Jia and He, 2011). Equal numbers of congenically distinct WT (CD45.1) and Atg3fl/fl Ubc cre-ERT2 (CD45.2) Ly49H+ NK cells were adoptively co-transferred into Ly49H-deficient mice and infected with MCMV. Recipient mice were then split into two cohorts receiving either tamoxifen (hereafter called NK-Atg3−/−cells) or corn oil control after the initial activation phase of the NK cell response to MCMV (Figure 3A). Whereas early inducible deletion of Atg3 did not affect the expansion of Ly49H+ NK cells on day 7 after infection (Figures S2A and S2B), we observed a significant reduction in the percentage of Ly49H+ NK cells when Atg3 was ablated before the contraction phase of the response (Figures 3B and 3C), such that compared to WT controls, memory NK cells showed a dramatic loss in their percentages (Figure 3D) and absolute numbers (Figure 3E). The recipients treated with control oil contained similar percentages and numbers of functionally and phenotypically similar adoptively transferred NK cells throughout the course of infection (Figures S2C–S2F). Therefore, these data indicate that although autophagy might be dispensable during activated NK cell proliferation, it is essential for the survival of virus-specific NK cells during the transition from effector to long-lived memory cells.
Figure 3. ATG3 Is Required for Generation of NK Cell Memory.

(A) Schematic of experiment. In brief, equal numbers of WT (CD45.1) and Ubc cre-ERT2 × Atg3fl/fl (CD45.2) splenic Ly49H+ NK cells were co-transferred into Ly49H-deficient mice and infected with MCMV. Recipients received 1 mg of tamoxifen daily from day 4 to 8 after infection for the inducible deletion of Atg3 in transferred NK cells (referred to as NK-Atg3−/− cells).
(B and C) Relative percentage of adoptively transferred Ly49H+ NK cells within the total (B) and Ly49H+ (C) NK cell populations in the blood of tamoxifen-treated recipients at the indicated time points after infection.
(D) Plots show percentages of transferred NK cell populations in the spleen at days 7 and 28 after infection.
(E) Absolute number of adoptively transferred Ly49H+ NK cells in the spleen and liver of tamoxifen-treated recipients on day 28 after infection. Data are representative of three independent experiments with n = 4–5 per group per time point and are presented as the mean ± SEM (*p < 0.05; **p < 0.01; ****p < 0.0001). See also Figure S2.
Induction of Autophagy Enhances Memory NK Cell Survival
Previous studies have shown that autophagy can be promoted through inhibition of active mTOR signaling by activation of AMPK (Kim et al., 2011). In order to address whether enhanced autophagic activity could promote the survival of virus-specific NK cells in response to MCMV infection, we adoptively transferred purified LC3-GFP transgenic NK cells, infected with MCMV, and treated recipients with an inhibitor of mTOR (rapamycin) (Araki et al., 2009), an AMPK activator (metformin) (Pearce et al., 2009), or a PBS control during the contraction phase of the response to MCMV (days 8–28 after infection). Interestingly, the percentages (Figure 4A) and absolute numbers (Figure 4B) of memory NK cells in peripheral organs were dramatically higher in recipient mice treated with either rapamycin or metformin than in mice treated with PBS. In order to confirm that metformin and rapamycin increased autophagic activity in NK cells during viral infection, we measured the intensity of GFP expression on adoptively transferred Ly49H+ memory NK cells and found a greater reduction in GFP fluorescence in metformin- or rapamycin-treated mice than in PBS controls (Figure 4C), indicating that inhibition of mTOR, or activation of AMPK, stimulated autophagic activity in NK cells during the transition from effector to memory phase. To test whether increasing autophagic activity with rapamycin or metformin treatment directly enhances survival of NK cells (and to confirm that these compounds are not acting through other pathways), we transferred equal numbers of WT (CD45.1) and Atg3fl/fl × Ubc cre-ERT2 (CD45.2) NK cells, infected recipient mice with MCMV, and administered PBS, metformin, or rapamycin daily from day 8 to 28 after infection (Figure 4D). Indeed, the increase in memory NK cells from metformin- and rapamycin-treated mice was autophagy dependent, given that NK-Atg3−/−NK cells failed to demonstrate enhanced survival after treatment with either compound (Figure 4E). These results support the hypothesis that enhancement of autophagic activity through inhibition of mTOR or activation of AMPK is sufficient to enhance antigen-specific NK cell survival during the effector-to-memory cell transition in response to MCMV infection.
Figure 4. Induction of Autophagy Enhances Virus-Specific NK Cell Survival in an Atg3-Dependent Manner.

LC3-GFP transgenic splenic Ly49H+ NK cells were transferred into Ly49H-deficient mice and infected with MCMV. Recipients received PBS, 200 μg metformin, or 600 μg/kg rapamycin daily from day 8 to 27 after infection.
(A and B) Representative percentages (A) and absolute numbers (B) of splenic Ly49H+ memory NK cells at day 28 after infection are shown for all three groups.
(C) Plots and graph show LC3-GFP intensity.
(D) Schematic of experiment. In brief, equal numbers of WT (CD45.1) and Ubc cre-ERT2 × Atg3fl/fl (CD45.2) splenic Ly49H+ NK cells were co-transferred into Ly49H-deficient mice and infected with MCMV. Recipients received 1 mg of tamoxifen from day 4 to 8 after infection and then PBS, 200 μg of metformin, or 600 μg/kg rapamycin daily from day 8 to 27 after infection.
(E) On day 28 after infection, absolute numbers of transferred Ly49H+ NK cells were quantified in the spleen and liver.
Data are representative of two independent experiment with n = 4–5 per group per time point, and data are presented as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant). See also Figure S3.
Autophagy Regulates NK Cell Survival through Removal of Dysfunctional Mitochondria
Because previous studies have shown that autophagy orchestrates the clearance of dysfunctional mitochondria and ROS in T lymphocytes to promote survival during homeostasis (Jia and He, 2011; Pua et al., 2009), we investigated whether NK cells utilize a similar mechanism during viral infection to promote memory formation. At the peak of expansion, WT and NK-Atg3−/− NK cells displayed similar tetramethylrhodamine ethyl ester (TMRE), MitoSox, and MitoTracker Green staining; however, during the contraction phase (measured at day 10 after infection), compared to NK-Atg3−/− NK cells, WT NK cells demonstrated increased mitochondrial membrane potential but decreased mitochondrial-associated ROS and reduced mitochondrial density (Figure 5A). Induction of autophagy through metformin treatment also decreased mitochondrial mass and mitochondrial-associated ROS while enhancing mitochondrial membrane potential (Figures S3A–S3D), suggesting that autophagy is sufficient and necessary for the removal of dysfunctional mitochondria from antigen-specific NK cells during the contraction phase. Furthermore, the elevated mitochondrial-associated ROS contributed to the survival defect of NK-Atg3−/− NK cells, given that treatment with ROS scavenger N-acetyl-L-cysteine (NAC) during the contraction phase (Figure 5B) selectively enhanced absolute numbers of NK-Atg3−/− memory NK cells in peripheral tissues in comparison to absolute numbers of PBS-treated controls (Figure 5C). Therefore, these data indicate that although autophagy might be dispensable during viral-induced NK cell proliferation, autophagy-mediated clearance of depolarized mitochondria and ROS is essential for the survival of virus-specific NK cells during the transition from effector cells to long-lived memory cells.
Figure 5. Atg3-Deficient NK Cells Display Increased Mitochondrial Depolarization and Production of ROS during Viral Infection.

(A) WT and Ubc cre-ERT2 × Atg3fl/fl (NK-Atg3−/−) splenic Ly49H+ NK cells were co-transferred into Ly49H-deficient mice and infected with MCMV. Recipients received 1 mg of tamoxifen daily from day 4 to 8 after infection, and spleens were harvested at days 7 and 10 after infection. TMRE, MitoSox Red, and MitoTracker Green MFI values of NK-Atg3−/− Ly49H+ NK cells were normalized to those of WT controls within each recipient mouse and plotted as the relative change.
(B) Schematic of experiment. In brief, equal numbers of WT and NK-Atg3−/− Ly49H+ NK cells were co-transferred into Ly49H-deficient mice and infected with MCMV. Recipients received 1 mg of tamoxifen daily from day 4 to 8 after infection and then daily injections of either PBS or 1.25 mg N-acetyl-L-cysteine (NAC) from day 8 to 27 after infection.
(C) Recipient mouse spleens and livers were harvested on day 28 after infection, and the absolute numbers of adoptively transferred WT and NK-Atg3−/− Ly49H+ cells were quantified as mentioned previously.
Data are representative of two independent experiments with n = 4–5 per group per time point and are presented as the mean ± SEM (*p < 0.05; ****p < 0.0001; ns, not significant).
Mitochondrial-Associated Proteins BNIP3 and BNIP3L Are Required for NK Cell Memory Formation
We next investigated the molecular machinery driving mitophagy in virus-specific NK cells during the contraction phase. BH3 domain-containing BCL2 family members BNIP3 and BNIP3-like (BNIP3L, also known as NIX) are mitochondria-associated proteins that can promote mitophagy (Mammucari et al., 2007; Schweers et al., 2007; Zhang and Ney, 2009), and in the hematopoietic system, BNIP3L is required for the survival of developing erythroblasts through the sequestration of mitochondria into autophagosomes for selective clearance (Schweers et al., 2007). However, whether BNIP3L or BNIP3 plays a role in the survival of lymphocytes during viral infection is not known. We found that both Bnip3l and Bnip3 transcripts are dynamically regulated in antigen-specific NK cells during MCMV infection. Compared to cells from uninfected mice, sorted Ly49H+ NK cells from MCMV-infected mice showed strong repression of both Bnip3l and Bnip3 transcripts at day 1.5 after infection. By day 7 after infection, Bnip3l mRNA returned to basal amounts, whereas Bnip3 transcripts did not return to basal amounts until day 14 after infection, revealing a temporally regulated derepression of Bnip3l and Bnip3 (Figure 6A). Modulation of Bnip3l and Bnip3 transcript expression was functionally critical, given that co-adoptive transfer and MCMV infection studies revealed defects in the percentages and absolute numbers of functionally competent memory NK cells deficient in either BNIP3L or BNIP3 during the contraction phase in the blood and peripheral organs (Figures 6B–6F). The impaired survival in BNIP3- or BNIP3L-defi-cient NK cells was not due to inherent developmental defects or altered homeostasis, because BNIP3 and BNIP3L are not required for the development and functionality of mature NK cells or their survival after homeostatic proliferation (Figures S4A–S4H and data not shown). Furthermore, we demonstrated that BNIP3L- and BNIP3-deficient NK cells harbored mitochondria with lower membrane potential (Figures 7A and S5A) and more ROS (Figures 7B and S5B) than those of co-adoptively transferred WT cells. Accumulation of dysfunctional mitochondria in BNIP3-deficient NK cells was due to a loss of mitophagic activity, because we observed greater mitochondrial mass and more autophagosomes in Bnip3−/− and NK-Bnip3l−/− NK cells than in co-transferred WT cells (Figures 7C, 7D, S5C, and S5D). As expected, this defective mitophagic activity coincided with distinct time points during the contraction phase, when BNIP3L and BNIP3 are expressed. The increase in mitochondrial mass observed in BNIP3- or BNIP3L-deficient NK cells was unlikely due to enhanced mitochondrial biogenesis, because sorted Bnip3−/− or NK-Bnip3l−/− Ly49H+ NK cells did not show more expression of transcriptional co-activator peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) or its co-activator, yin-yang 1 (YY1) (Cunningham et al., 2007), than did co-adoptively transferred WT controls during the contraction phase (Figures S5E and S5F). Thus, BNIP3L and BNIP3 play non-redundant roles in the mediation of mitophagy to clear dysfunctional mitochondria and promote virus-specific NK cell survival during the contraction phase of the response to MCMV.
Figure 6. BNIP3 and BNIP3L Are Required for Survival of NK Cells after Viral Infection.

(A) WT Ly49H+ NK cells were adoptively transferred into Ly49H-deficient mice and infected with MCMV. Splenic Ly49H+ NK cells were then sorted at the indicated time points after infection and analyzed for Bnip3 and Bnip3l mRNA by real-time PCR.
(B–F) Equal numbers of WT and Bnip3−/− or Nkp46cre × Bnip3lfl/fl (referred to as NK-Bnip3l−/−) NK cells were co-transferred into Ly49H-deficient mice and infected with MCMV. (B and C) Graphs show relative percentage of transferred Ly49H+ NK cells within the total (B) and Ly49H+ (C) NK cell populations in the blood at various time points after infection. (D) Relative percentage of transferred WT or Bnip3−/− Ly49H+ NK cells within the total and Ly49H+ NK cell populations in the spleen at various time points after infection. (E and F) Absolute numbers of the indicated NK-Bnip3l−/−, Bnip3−/−, or co-transferred WT NK cells in the spleen and liver on either day 21 or 28 after infection are shown.
Data are representative of two or three independent experiments with n = 4–5 per group per time point and are presented as the mean ± SEM (*p < 0.05; **p < 0.01; ***p < 0.0001; ns, not significant). See also Figure S4.
Figure 7. BNIP3 Is Required for the Removal of Dysfunctional Mitochondria in NK Cells after Viral Infection.
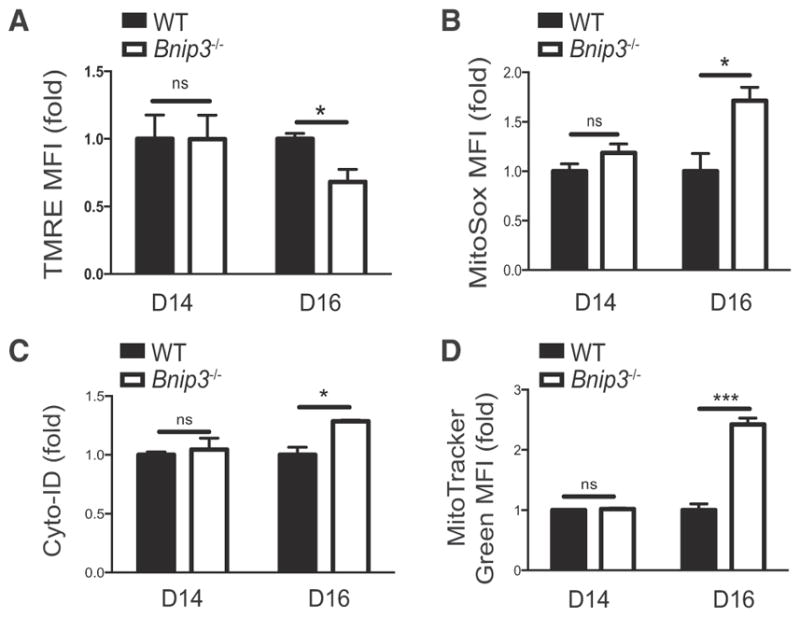
TMRE (A), MitoSox Red (B), Cyto-ID (C), and Mitotracker green (D) MFI values for adoptively transferred Bnip3 −/− Ly49H+ cells were normalized to those of co-transferred WT controls in each recipient mouse and plotted as the relative change in MFI at the indicated time points after infection. Data are representative of two independent experiments with n = 4–5 per group per time point and are presented as the mean ± SEM (*p < 0.05; ***p < 0.0001; ns, not significant). See also Figure S5.
DISCUSSION
Our studies have revealed that mitophagy is dynamically regulated in antigen-specific NK cells during viral infection. Several recent studies have also demonstrated a role for autophagy in antigen-specific B and CD8+ T cell survival during the response to viral infection (Chen et al., 2014; Puleston et al., 2014; Xu et al., 2014); however, these studies did not definitively address whether mitophagy is required for the survival of memory cells. Here, we found that autophagy was dispensable for antigen-specific NK cell proliferation during viral infection but was required for the survival of effector NK cells during the contraction phase, which is consistent with the in vivo kinetics of autophagy in antigen-specific CD8+ T cells (Xu et al., 2014). Proliferating effector CD8+ T cells were found to inhibit autophagy by preventing autophagosome maturation into autolysosomes, but then they induced autophagic flux during the contraction phase to mediate antigen-specific CD8+ T cell survival and memory formation in an Atg5- and Atg7-dependent manner. Given that we also observed an accumulation of autophagosomes and LC3b-II in effector NK cells, it is likely that expanding NK cells utilize a similar mechanism to inhibit autophagy during virus-driven expansion. Although the mechanisms for autophagy-mediated CD8+ T cell survival have yet to be discovered, we provide evidence that NK cells use BNIP3- and BNIP3L-mediated mitophagy to selectively remove dysfunctional mitochondria and mitochondria-associated ROS during the contraction phase after viral infection. Together with the studies in adaptive lymphocytes, our findings suggest that mitophagy might be a hallmark of immunologic memory formation, and it will be of interest to determine whether mitochondria-associated proteins BNIP3 and BNIP3L can also mediate mitophagy in T and B cells responding to pathogens.
Our data represent an essential role for BNIP3 and BNIP3L in the clearance of dysfunctional mitochondria to promote the survival of antigen-specific NK cells. We have shown that expression of Bnip3 and Bnip3l is temporally regulated during viral infection, and their decreased expression is correlated with decreased autophagic activity during activation of antigen-specific NK cells. Although BNIP3 and BNIP3L serve non-redundant roles in NK cell memory formation, the greatest loss of memory cells was observed in BNIP3-deficient NK cells, suggesting that endogenous BNIP3 expression in BNIP3L-deficient NK cells is able to partially restore mitophagic activity at later time points during contraction, whereas endogenous BNIP3L expression is not sufficient to sustain mitophagy in the absence of BNIP3. Indeed, we observed that dysfunctional mitochondria were still present in BNIP3-deficient memory NK cells but were cleared in BNIP3L-deficient memory NK cells (data not shown). In support of our observation, treatment with mitochondrial uncoupling agents has been shown to rescue the mitophagy defects observed in Bnip3l−/− erythroblasts (Sandoval et al., 2008), although it remains unknown whether this process is dependent on BNIP3. Indeed, we also observed that memory NK cell formation was not perturbed in the absence of another known mediator of mitophagy, Parkin (Park2; data not shown), although Parkin’s role in mediating mitophagy in vivo remains controversial (Sterky et al., 2011). Together, these results suggest functional redundancy of mechanisms that can drive mitophagy in NK cells.
Although BNIP3 and BNIP3L have been described to have pro-apoptotic functions in other cell types, various normal tissues can express these proteins at high amounts without inducing cell death (Diwan et al., 2007b; Glick et al., 2012). Thus, additional signals that either modify or disrupt Bnip3 and Bnip3l function are most likely required for these proteins to induce cell death (Webster et al., 2005). Consistent with this hypothesis, we did not observe differences in NK cell development or homeostasis between BNIP3-deficient mice and WT mice. Although we observed that autophagy is critical for the survival of NK cells during development and after homeostatic expansion (T.E.O. and J.C.S., unpublished observations), this process does not require Bnip3, and NK cells do not alter their mitochondrial quality during homeostatic proliferation (data not shown), suggesting a context-specific requirement for mitophagy in NK cell survival or that redundant mechanisms can compensate in the absence of Bnip3. Nevertheless, the precise molecular mechanisms of how BNIP3 and BNIP3L mediate mitophagy in NK cells remain unclear. There is evidence that BNIP3 and BNIP3L can promote mitochondrial depolarization, which is sufficient to induce mitophagy (Elmore et al., 2001; Twig et al., 2008). Other studies have demonstrated that BNIP3 and BNIP3L can serve directly as “mitophagy receptors” through binding their conserved LC3-interacting region (LIR) to LC3b present on autophagosomes (Hanna et al., 2012; Novak et al., 2010; Zhu et al., 2013) to mediate recruitment of autophagosomes to dysfunctional mitochondria. Because we did not observe any defects in mitochondrial depolarization in BNIP3- or BNIP3L-deficient NK cells when their expression was maximal after viral infection, we favor the hypothesis that BNIP3 and BNIP3L directly induce mitophagy by selectively targeting autophagosomes to dysfunctional mitochondria through their LIRs. Understanding the complete molecular mechanisms that regulate BNIP3 and BNIP3L expression will offer valuable insights during antigen-specific lymphocyte responses to viral infections.
Our findings provide further evidence linking mTOR and AMPK as major regulators of autophagy during viral infection. Active mTOR signaling has been shown to repress autophagy, and activation of AMPK can induce autophagy through its ability to actively repress mTOR (Kim et al., 2011); however, whether modulation of these pathways influences autophagic activity in cytolytic lymphocytes during viral infection has remained unanswered. Here, we have demonstrated that inhibition of mTOR by rapamycin treatment or activation of AMPK by metformin treatment during the contraction phase increased NK cell memory formation by enhancing specific autophagic activity. A recent study showing that NK cells induce active mTOR signaling early during MCMV infection (Marçais et al., 2014) corroborates our hypothesis that autophagic activity is inhibited during NK cell expansion. Because autophagic activity is induced during the effector-to-memory NK cell transition, endogenous mTOR signaling is most likely repressed in antigen-specific NK cells during the contraction phase. However, whether AMPK signaling is activated during this process remains unknown. Consistent with our findings, treatment with either rapamycin or metformin increased the generation of memory CD8+ T cells during the contraction phase after LCMV infection (Araki et al., 2009; Pearce et al., 2009).
Furthermore, it has been shown that memory CD8+ T cells use fatty-acid oxidation (FAO) as a primary energy source (van der Windt et al., 2012), and activation of AMPK by metformin stimulates FAO in antigen-specific CD8+ T cells during viral infection (Pearce et al., 2009). Although NK cells stimulated by interleukin-15 show a rapid increase in glucose metabolism in vitro (Marçais et al., 2014), the metabolic profile and use of FAO by virus-specific NK cells during infection have not been studied. Thus, even if activation of AMPK by metformin increases FAO in antigen-specific NK cells during the contraction phase after MCMV infection, autophagy is still required for enhanced survival. Because autophagy-defective effector CD8+ T cells have dysregulated mitochondrial FAO (Xu et al., 2014), it could be possible that mitophagy and FAO are fundamentally linked processes to promote the survival of both memory CD8+ T cells and memory NK cells. Future studies will focus on whether mitophagy is required for the generation of lipid substrates for FAO, consistent with previous studies showing that autophagy can regulate lipid metabolism through the degradation of cellular lipid stores (Singh et al., 2009).
In summary, our study demonstrates the importance of mitophagy during NK cell responses to viral infection. Future work will also determine whether mitophagy is essential for the survival of other innate lymphoid cell lineages in response to pathogen infection. Because of the essential nature of this mechanism in regulating NK cell survival, it will be of interest to test the requirement of autophagy in additional settings of lymphocyte expansion and survival, such as after homeostatic proliferation during conditions of radiation-induced lymphopenia. Recent studies have described the expansion and persistence of memory-like NKG2C+ NK cells in humans infected with human cytomegalovirus (HCMV) (Della Chiesa et al., 2012; Foley et al., 2012a; Foley et al., 2012b; Hendricks et al., 2014; Lopez-Vergès et al., 2011). Thus, it will be important to also determine whether mitophagy is induced in virus-specific human NK cells after rapid proliferation in response to HCMV infection. Given the frequent therapeutic use of mTOR inhibitors and metformin, our results have implications for the development of effective antiviral and anticancer treatments, as well as vaccination strategies targeting the autophagy pathway in NK cells.
EXPERIMENTAL PROCEDURES
Mice
Mice were bred at the Memorial Sloan-Kettering Cancer Center in accordance with the guidelines of the Institutional Animal Care and Use Committee. The following strains were used in this study: C57BL/6 (CD45.2), B6.SJL (CD45.1), Rag2−/− × Il2rg−/−, Klra8−/− (Ly49H deficient) (Fodil-Cornu et al., 2008), Rag2−/− × Klra8−/−, Nkp46iCre (Narni-Mancinelli et al., 2011), Ubc cre-ERT2 × Atg3fl/fl (Jia and He, 2011), LC3-GFP transgenic (Mizushima et al., 2004), Bnip3lfl/fl (Diwan et al., 2007a), and Bnip3−/− (Diwan et al., 2007b). Experiments were conducted with age- and gender-matched mice in accordance with the approved institutional protocols.
Virus Infection
MCMV (Smith strain) was obtained from L. Lanier (University of California, San Francisco). MCMV was passaged serially through BALB/c hosts twice, and then viral stocks were prepared with a dounce homogenizer for dissociating the salivary glands of infected mice 3 weeks after infection. Mice were infected by intraperitoneal injection with 7.5 × 102 plaque-forming units.
NK Cell Enrichment and Adoptive Transfer
T, B, and red blood cells were labeled with 10 μg per spleen of rat monoclonal antibodies against CD4 (GK1.5), CD8 (53.6.72), CD19 (1D3), and Ter119 (Bio-X-cell) and magnetically depleted from total splenocyte suspensions with the use of anti-rat IgG-coupled magnetic beads (QIAGEN). In all experiments, approximately 2 × 105 enriched NK cells were injected intravenously into mice. In adoptive co-transfer experiments, equal numbers of Ly49H+ NK cells from each population (CD45.1+ and CD45.2+) were injected into recipients 1 day before infection. In some experiments, recipient mice were injected intraperitoneally with 1 mg/day of hydroxytamoxifen (4-OHT) dissolved in corn oil or corn oil control (Sigma) for 5 consecutive days starting on day 4 after infection and subsequently injected intraperitoneally with PBS, 200 μg/day metformin (Sigma), 600 μg/kg/day rapamycin (LC Laboratories), or 1.25 mg/day NAC (Sigma) from day 8 to 28 after infection.
Flow Cytometry and Cell Sorting
Cell-surface staining was performed with fluorophore-conjugated antibodies against the following proteins: NK1.1 (PK136, Tonbo), CD11b (M1/70, Tonbo), CD27 (LG.3A10, BioLegend), KLRG1 (2F1, eBioscience), CD69 (H1.2F3, BioLegend), Ly49H (3D10, eBioscience), CD107a (1D4B, BioLegend), CD45.1 (A20, BioLegend), CD45.2 (104, Biolegend), TCRβ (H57–597, BioLegend), IFN-γ (XMG1.2, BioLegend), and Ly49D (4E5, BioLegend). Unless otherwise indicated, NK cells were defined as TCRβ-NK1.1+ cells. Intracellular cytokine staining was performed with the Cytofix/Cytoperm Plus Kit (BD). NK cells were enriched from spleens as mentioned above, stained with cell-surface antibodies, and then incubated with various dyes in Hank’s balanced salt solution plus Mg and Ca as follows: 100 nM Mitotracker Green (Life Technologies) for 30 min at 37°C to measure mitochondrial mass, 100 nM TMRE for 30 min at 37°C to measure mitochondrial membrane potential, 5 μm MitoSOX red (Invitrogen) for 15 min at 37°C to measure mitochondria-associated ROS, or 1:400 Cyto-ID autophagy detection reagent (Enzo Life Sciences) for 30 min at 37°C to measure autophagosomes. Flow cytometry and cell sorting were performed on the LSR II and Aria II cytometers (BD Biosciences), respectively. For experiments involving real-time PCR, cell populations were sorted to >95% purity. Data were analyzed with FlowJo software (Tree Star).
Real-Time PCR
Sorted NK cells were lysed in Tri-Reagent (Ambion). RNA purification and cDNA synthesis were carried out with the QIAGEN RNeasy Kit (with on-column DNase I treatment) and MuLV reverse transcriptase and oligo(dT)16 primers (Applied Biosystems). iQ Sybr Green SuperMix (BioRad) was used for real-time PCR. Data were normalized to that for Actb and expressed as relative target abundance via the ΔΔCt method, where Ct (threshold cycle) is the cycle number at which the amplification curve intersects the threshold value.
Statistical Analyses
For graphs, data are shown as mean ± SEM, and unless otherwise indicated, statistical differences were evaluated with a two-tailed unpaired Student’s t test under the assumption of equal sample variance. p < 0.05 was considered significant. Graphs were produced and statistical analyses were performed with GraphPad Prism.
Supplementary Material
Highlights.
MCMV infection induces dynamic changes in NK cell mitochondrial quality
Autophagy is essential for the generation of NK cell memory
Pharmacologic induction of autophagy enhances NK cell memory formation
Mitophagy is required for anti-viral NK cell survival through removal of ROS
Acknowledgments
We thank members of the J.C.S. lab for technical support and experimental assistance and Sasha Rudensky, Cole Haynes, Michael Overholtzer, Xuejun Jiang, and Aimee Beaulieu for insightful comments and helpful discussions. Gerald Dorn, You-Wen He, Eric Vivier, Liang Deng, and Zhenyu Yu provided mice, reagents, and expertise critical to this study. T.E.O. was supported by a fellowship from the American Cancer Society. J.C.S. was supported by the Searle Scholars Program, the Cancer Research Institute, and NIH grants AI085034 and AI100874.
Footnotes
Supplemental Information includes five figures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.07.012.
AUTHOR CONTRIBUTIONS
T.E.O. and J.C.S. designed the study; T.E.O. and L.R.J. performed cell sorting and EM sample preparation; H.H.K. performed Western blotting; T.E.O. performed all additional experiments; and T.E.O. and J.C.S. wrote the manuscript.
References
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296:1323–1326. doi: 10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- Beaulieu AM, Zawislak CL, Nakayama T, Sun JC. The transcription factor Zbtb32 controls the proliferative burst of virus-specific natural killer cells responding to infection. Nat Immunol. 2014;15:546–553. doi: 10.1038/ni.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Hong MJ, Sun H, Wang L, Shi X, Gilbert BE, Corry DB, Kheradmand F, Wang J. Essential role for autophagy in the maintenance of immunological memory against influenza infection. Nat Med. 2014;20:503–510. doi: 10.1038/nm.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, Welsh RM. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med. 2001;194:29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Chiesa M, Falco M, Podestà M, Locatelli F, Moretta L, Frassoni F, Moretta A. Phenotypic and functional heterogeneity of human NK cells developing after umbilical cord blood transplantation: a role for human cytomegalovirus? Blood. 2012;119:399–410. doi: 10.1182/blood-2011-08-372003. [DOI] [PubMed] [Google Scholar]
- Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, et al. Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA. 2007a;104:6794–6799. doi: 10.1073/pnas.0610666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007b;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. Specific and nonspecific NK cell activation during virus infection. Nat Immunol. 2001;2:951–956. doi: 10.1038/ni714. [DOI] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- Fodil-Cornu N, Lee SH, Belanger S, Makrigiannis AP, Biron CA, Buller RM, Vidal SM. Ly49h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J Immunol. 2008;181:6394–6405. doi: 10.4049/jimmunol.181.9.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, Anasetti C, Weisdorf D, Miller JS. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J Immunol. 2012a;189:5082–5088. doi: 10.4049/jimmunol.1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X, Lopez-Vergès S, Lanier LL, Weisdorf D, Miller JS. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood. 2012b;119:2665–2674. doi: 10.1182/blood-2011-10-386995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW, 2nd, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol. 2012;32:2570–2584. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson JM, Laniewski NG, Lanier JG, Ahmed R. Mitochondrial potential and reactive oxygen intermediates in antigen-specific CD8+ T cells during viral infection. J Immunol. 2003;170:4745–4751. doi: 10.4049/jimmunol.170.9.4745. [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks DW, Balfour HH, Jr, Dunmire SK, Schmeling DO, Hogquist KA, Lanier LL. Cutting edge: NKG2C(hi)CD57+ NK cells respond specifically to acute infection with cytomegalovirus and not Epstein-Barr virus. J Immunol. 2014;192:4492–4496. doi: 10.4049/jimmunol.1303211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. 2011;186:5313–5322. doi: 10.4049/jimmunol.1002404. [DOI] [PubMed] [Google Scholar]
- Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159:94–107. doi: 10.1016/j.cell.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Kurtulus S, Tripathi P, Opferman JT, Hildeman DA. Contracting the ‘mus cells’–does down-sizing suit us for diving into the memory pool? Immunol. Rev. 2010;236:54–67. doi: 10.1111/j.1600-065X.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Vergès S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA, Houchins JP, Miller S, Kang SM, Norris PJ, et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA. 2011;108:14725–14732. doi: 10.1073/pnas.1110900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, Rabilloud J, Mayol K, Tavares A, Bienvenu J, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:749–757. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- Min-Oo G, Bezman NA, Madera S, Sun JC, Lanier LL. Proapoptotic Bim regulates antigen-specific NK cell contraction and the generation of the memory NK cell pool after cytomegalovirus infection. J Exp Med. 2014;211:1289–1296. doi: 10.1084/jem.20132459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc Natl Acad Sci USA. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- Puleston DJ, Zhang H, Powell TJ, Lipina E, Sims S, Panse I, Watson AS, Cerundolo V, Townsend AR, Klenerman P, Simon AK. Autophagy is a critical regulator of memory CD8(+) T cell formation. eLife. 2014;3:3. doi: 10.7554/eLife.03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci USA. 2002;99:8826–8831. doi: 10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, Larsson NG. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci USA. 2011;108:12937–12942. doi: 10.1073/pnas.1103295108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Lanier LL. NK cell development, homeostasis and function: parallels with CD8+ T cells. Nat Rev Immunol. 2011;11:645–657. doi: 10.1038/nri3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Madera S, Bezman NA, Beilke JN, Kaplan MH, Lanier LL. Proinflammatory cytokine signaling required for the generation of natural killer cell memory. J Exp Med. 2012;209:947–954. doi: 10.1084/jem.20111760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster KA, Graham RM, Bishopric NH. BNip3 and signal-specific programmed death in the heart. J Mol Cell Cardiol. 2005;38:35–45. doi: 10.1016/j.yjmcc.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Xu X, Araki K, Li S, Han JH, Ye L, Tan WG, Konieczny BT, Bruinsma MW, Martinez J, Pearce EL, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15:1152–1161. doi: 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem. 2013;288:1099–1113. doi: 10.1074/jbc.M112.399345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.