

Abstract
Significance: Pyridine dinucleotides, nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP), were discovered more than 100 years ago as necessary cofactors for fermentation in yeast extracts. Since that time, these molecules have been recognized as fundamental players in a variety of cellular processes, including energy metabolism, redox homeostasis, cellular signaling, and gene transcription, among many others. Given their critical role as mediators of cellular responses to metabolic perturbations, it is unsurprising that dysregulation of NAD and NADP metabolism has been associated with the pathobiology of many chronic human diseases.
Recent Advances: A biochemistry renaissance in biomedical research, with its increasing focus on the metabolic pathobiology of human disease, has reignited interest in pyridine dinucleotides, which has led to new insights into the cell biology of NAD(P) metabolism, including its cellular pharmacokinetics, biosynthesis, subcellular localization, and regulation. This review highlights these advances to illustrate the importance of NAD(P) metabolism in the molecular pathogenesis of disease.
Critical Issues: Perturbations of NAD(H) and NADP(H) are a prominent feature of human disease; however, fundamental questions regarding the regulation of the absolute levels of these cofactors and the key determinants of their redox ratios remain. Moreover, an integrated topological model of NAD(P) biology that combines the metabolic and other roles remains elusive.
Future Directions: As the complex regulatory network of NAD(P) metabolism becomes illuminated, sophisticated new approaches to manipulating these pathways in specific organs, cells, or organelles will be developed to target the underlying pathogenic mechanisms of disease, opening doors for the next generation of redox-based, metabolism-targeted therapies. Antioxid. Redox Signal. 28, 180–212.
Keywords: : NAD, NADP, metabolism, SIRT, PARP
Introduction
In 1929, Sir Arthur Harden and Hans von Euler-Chelpin were awarded the Nobel Prize in Chemistry for “their investigations on the fermentation of sugar and fermentative enzymes.” In 1906, Harden, with his coinvestigator William John Young, demonstrated that fermentation by yeast requires a heat-stable dialyzable substance, which they termed cozymase (81). In 1923, von Euler-Chelpin, with his colleague Karl Myrbäck, determined the structure of cozymase to be a nucleoside sugar phosphate (Fig. 1) (248). Over the subsequent decades, Otto Warburg isolated nicotinamide adenine dinucleotide (NAD) from cozymase and demonstrated its critical role in hydrogen transfer during fermentation (254). During this time, he also discovered a second cozymase, nicotinamide adenine dinucleotide phosphate (NADP) (Fig. 1). Arthur Kornberg, Jack Preiss, and Philip Handler made key discoveries to elucidate the biosynthetic pathways for NAD (128, 191, 192), while others would tie these molecules into familiar bioenergetic pathways, glycolysis, the tricarboxylic acid (TCA) cycle, and electron transport chain.
FIG. 1.

Chemical structures of NAD and NADP. Over one century ago, NAD and NADP were identified as critical factors for fermentation by yeast and named “cozymase” I and II, respectively. These nucleoside sugar phosphate molecules were subsequently found to play a key role in hydride transfer reactions during fermentation.
More recently, new discoveries have demonstrated the fundamental role of these molecules linking metabolism to cellular signaling events to coordinate adaptive responses to environmental changes with marked impacts on many aspects of human health and disease. This review will build from fundamental molecular pathways in NAD(P) biosynthesis and metabolism to the cellular biology of these molecules thereby providing a molecular context for understanding their pathobiological roles in a variety of human diseases. After more than a century of investigation, pyridine dinucleotides continue to offer exciting new biological insights and therapeutic opportunities.
Sources and Sinks of NAD(P)
Biosynthesis of NAD(P)
Mammalian cells synthesize NAD from tryptophan de novo or from preformed precursors (nicotinic acid, nicotinamide, nicotinamide riboside, or nicotinic acid riboside) via salvage pathways (Fig. 2). In the de novo pathway, NAD is synthesized from the tryptophan degradation product, quinolinic acid. Quinolinic acid is converted by quinolinate phosphoribosyltransferase to nicotinic acid mononucleotide (NAMN). NAMN subsequently enters the salvage pathway for NAD synthesis. Notably, tryptophan is a poor precursor for NAD biosynthesis in vivo as its downstream metabolites are typically diverted to other pathways by the enzymatic activity of α-amino-β-carboxymuconate-ɛ-semialdehyde decarboxylase (ACMSD). Only when the enzymatic capacity of ACMSD is overwhelmed does tryptophan metabolism yield quinolinic acid for NAD biosynthesis (103). One might speculate that this represents a mechanism for weakly coupling nutritional status to NAD biosynthesis, as tryptophan is an essential amino acid and, thus, would only be present in sufficient quantities to overcome the capacity of ACMSD when dietary supply is high.
FIG. 2.

Biosynthesis of NAD and NADP. NAD is synthesized de novo from the tryptophan metabolite quinolinic acid or from the preformed precursors nicotinic acid, nicotinic acid riboside, nicotinamide, or nicotinamide riboside. The nicotinamide functional group permits the reversible electron transfer reactions that allow these pyridine dinucleotides to function as electron carriers in a variety of metabolic pathways (inset).
Compared to de novo biosynthesis, salvage pathways play a much more important role in mammalian NAD production. These pathways vary slightly depending on the precursor utilized. In the Preiss-Handler pathway, nicotinic acid from dietary sources is converted to NAMN by nicotinic acid phosphoribosyltransferase (NAPRT). Subsequently, nicotinamide mononucleotide adenylyltransferase (NMNAT) adenylates NAMN to form nicotinic acid adenine dinucleotide (NAAD). NAD synthetase catalyzes the adenosine triphosphate (ATP)-dependent amidation of NAAD to yield NAD.
Nicotinamide recycling is catalyzed by nicotinamide phosphoribosyltransferase (NAMPT) that combines nicotinamide with 5-phosphoribosyl-1-pyrophosphate to form nicotinamide mononucleotide (NMN). NMN, like NAMN in the Preiss-Handler pathway, is also adenylated by NMNAT enzymes to generate NAD.
Nicotinic acid riboside and nicotinamide riboside are phosphorylated by nicotinamide riboside kinases (NRK1 and NRK2) to produce NAMN and NMN, respectively (12, 48, 227).
Cytoplasmic NAD kinase (cNADK) and mitochondrial NAD kinase (mNADK) phosphorylate NAD to form NADP (137, 175). While the human NAD kinases (NADKs) can phosphorylate NADH to produce NADPH, they do so ∼10-fold less efficiently than they phosphorylate NAD to produce NADP (175, 186). These enzymes use ATP as the phosphate donor and preferentially use NAD as the phosphate acceptor with a high degree of specificity.
Mammals have a substantial daily requirement for NAD. In mice, tissue NAD levels range between 200 and 800 μmol/kg depending on the tissue (168), while the half-life of NAD is ∼10 h in mouse liver in vivo (102). Thus, a 75 kg human with an average tissue concentration of 300 μmol NAD/kg requires 50 mmol NAD synthesized per day (equivalent to 6 g of nicotinamide). For every 60 mg tryptophan consumed, ∼1 mg is metabolized to nicotinamide (95) and this rate appears to be independent of nicotinamide consumption (65). Humans consume ∼1 g of tryptophan daily, equivalent to 17 mg nicotinamide (202). The dietary reference intake for vitamin B3 (nicotinic acid, nicotinamide, and nicotinamide riboside) is 16 mg for men and 14 mg for women (1). Nicotinamide riboside is orally bioavailable in humans and increases hepatic NAD more effectively than nicotinic acid or nicotinamide, leading to increasing interest in oral supplementation of nicotinamide riboside (see Section VIII below) (231). Nicotinamide riboside is a major component of milk, where it contributes to 40% of the total NAD precursor pool at a concentration of 5 μM (12, 232). Estimates of the average total dietary intake of nicotinamide riboside are not currently available. In sum, therefore, dietary vitamin B3 provides only about 0.5%–1.0% of the total requirements for NAD synthesis, demonstrating that efficient precursor recycling is the basis for effective NAD maintenance in humans (267). A detailed analysis of NAD pathway metabolite concentrations and synthetic enzyme activities in various tissues indicates that amidated precursors (nicotinamide and nicotinamide riboside) account for nearly all NAD synthesis compared to deamidated precursors (nicotinic acid, quinolinate, nicotinic acid riboside) (168).
Cellular uptake of NAD precursors
Metabolic substrates for cellular NAD production can be obtained from the extracellular environment. Cell culture medium typically contains nicotinamide and tryptophan. Tryptophan alone is insufficient to maintain intracellular NAD (174). NAD may be taken up directly by some human cell lines through connexin 43 hemichannels, which serve as the NAD transporter (21, 286). Evidence also suggests that NADH can be taken up directly from the extracellular medium (280). Other cells rely on uptake of the NAD precursor bases (nicotinamide and nicotinic acid) and nucleosides (nicotinamide riboside and nicotinic acid riboside). Indeed, inhibition of extracellular phosphodiesterases prevents NAD-mediated repletion of mitochondrial NAD pools (174). Likewise, inhibition of extracellular nucleotidase prevented NMN-mediated repletion of mitochondrial NAD pools (174). These observations suggest that NAD must be metabolized to nicotinamide riboside before efficient cellular uptake.
Consumption of NAD(P)
NAD(P) is catabolized by hydrolysis to yield nicotinamide and a variety of adenosine diphosphate (ADP)-ribosyl products.
Sirtuin family deacetylases cleave NAD in the process of removing the acetyl group from acetylated lysines. Similarly, mono- and poly-(ADP-ribose) polymerases cleave NAD to transfer the ADP-ribose moiety to target proteins. NADP does not appear to participate in similar reactions.
CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1) is a multifunctional enzyme that preferentially hydrolyzes NADP to nicotinamide and ADP-ribose(2′-phosphate) (ADPRP) (Fig. 3), although it can use NAD as a substrate as well. CD38 can also catalyze the cyclization of NAD(P) to generate the signaling molecules cyclic ADP-ribose and cyclic ADP-ribose(2′-phosphate) (cADPRP) as well as the hydrolysis of these cyclic molecules to ADP-ribose and ADPRP. NADP can also be converted to nicotinic acid adenine dinucleotide phosphate (NAADP) by CD38 where the nicotinamide base is replaced by nicotinic acid.
FIG. 3.

Multifunctional catalysis of NADP by CD38. CD38 catalyzes several different types of nicotinamide hydrolysis reactions using NADP as substrate. It can catalyze the cyclization of NADP to form cADPRP as well as the hydrolysis of NADP or cADPRP to form ADPRP. It can also catalyze a base exchange reaction through which nicotinamide is substituted by nicotinic acid to form NAADP. ADPRP, ADP-ribose(2′-phosphate); cADPRP, cyclic ADP-ribose(2′-phosphate); NAADP, nicotinic acid adenine dinucleotide phosphate.
The reliance of these enzymes on NAD(P) as a substrate provides a key mechanism by which metabolic effects on cellular NAD(P) are translated into a variety of signaling and regulatory events that will be discussed in additional detail below.
NAD and NADP as electron carriers
In addition to anabolic and catabolic pathways, interconversion between oxidized and reduced forms enables NAD and NADP to play crucial roles as intracellular electron carriers. Oxidized NAD and NADP undergo two-electron reduction to yield NADH and NADPH, respectively (Figs. 2 and 4). While often overlooked, pH can also significantly impact the relative balance of NAD(P) and NAD(P)H, particularly in compartments where proton concentrations are markedly different from standard cytosolic conditions (e.g., mitochondrial matrix or intermembrane space). In addition, proton production from NADH oxidation like contributes to the acidosis of ischemic tissues. Approximately 100 human enzymes utilize NAD as a cofactor for oxidation/reduction reactions catalyzed by dehydrogenases or oxidoreductases and a similar number utilize NADP (185). Classically, NADH accepts electrons in catabolic reactions to supply the electron transport chain, while NADPH provides a reservoir of electrons for anabolic reactions, chemical detoxification, and antioxidant defense. These roles will be discussed in detail below.
FIG. 4.

NAD(P) half reaction. NAD(P) participates in two electron redox reactions. Note that these oxidation/reduction reactions can have consequences on cellular pH due to proton gain/loss.
Quantification of Pyridine Dinucleotides
A variety of analytical approaches have been developed to measure absolute concentrations of NAD, NADP, NADH, NADPH, as well as the redox ratios NAD/NADH and NADP/NADPH. Early, indirect estimates of free cytoplasmic NAD/NADH and NADP/NADPH were based on the principle of chemical equilibrium (242, 258). For example, if one assumes that the reversible reduction of pyruvate to lactate is at equilibrium, one can calculate the NAD/NADH ratio using measurements of lactate and pyruvate, which are easier to obtain, and the chemical equilibrium constant (Keq) of lactate dehydrogenase (LDH) as follows:
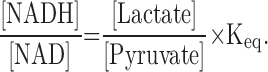 |
An advantage of this method is that it measures free, rather than protein-bound, nucleotides. The primary disadvantage is that these measurements are often not performed under equilibrium conditions, which can lead to substantial errors in the estimation of the redox ratio (223). Subsequently, enzymatic cycling methods were developed to measure pyridine dinucleotides more directly (147). These assays utilize a series of electron transfer reactions to generate a product for fluorescent, colorimetric, or luminescent analysis that reflects the pyridine dinucleotide concentration in the sample (Fig. 5A). Several variations of this approach form the basis of commercially available kits for pyridine dinucleotide and ratiometric quantification. While these kits do not require specialized equipment or expertise, in our experience, it is critically important to validate these methods using appropriate controls, including samples spiked with known concentrations NAD(P)(H). Additional analytical approaches, including ultraviolet or fluorescence spectroscopy and mass spectrometry coupled to capillary electrophoresis or high-performance liquid chromatography, do require specialized equipment and expertise but offer the advantage of truly direct measurement of NAD(P)(H) in a given sample (112, 149, 261).
FIG. 5.

Example of an enzymatic cycling reaction for NAD(H) quantification. (A) An example of an enzymatic cycling reaction to measure NAD(H). Electrons are transferred from ethanol via NAD and PMS to reduce resazurin to yield the fluorescent molecule resorufin at a rate dependent on the overall concentration of NAD(H). Multiple variations are possible, including the substitution of G6P and G6PD from Saccharomyces cerevisiae to measure NADP(H). Diaphorase can replace PMS, and tetrazolium salts can replace resazurin. (B) Selective degradation of NAD or NADH using buffers of different pH facilitates determination of NAD, NADH, and the NAD/NADH redox ratio from the same sample using the enzymatic cycling assay. G6P, glucose-6-phosphate; G6PD, G6P dehydrogenase; PMS, phenazine methiosulfate.
The techniques mentioned above typically rely on cell lysis during sample preparation, thereby providing estimates of whole cell concentrations, but no information on subcellular localization. Recently, rapid antibody-mediated isolation of mitochondria followed by metabolite extraction, liquid chromatography, and mass spectrometry has provided one method for subcellular analysis of pyridine dinucleotides (35). Overcoming the limitations of cell lysis entirely leverages the power of novel genetically encoded fluorescent biosensors to perform live-cell imaging, several of which can be targeted to specific intracellular compartments (13, 23, 281). Monitoring the relatively weak intrinsic fluorescence of the reduced pyridine dinucleotides, NADH and NADPH, using single- or dual-photon spectroscopy is an alternative to biosensors for live-cell imaging, but these techniques are unable to distinguish NADH from NADPH and lack the spatial resolution for subcellular localization (29, 30, 148).
Mulitple factors weigh on the careful assessment of cellular concentrations of pyridine dinucleotides and the ratio of oxidized and reduced forms. First, different methodologies access different pools of these molecules. For example, total cell lysates are unable to distinguish between mitochondrial and cytoplasmic pools. Different lysis conditions may also liberate different amounts of protein-bound pyridine dinucleotides. Second, the relative stabilities of reduced and oxidized forms are different. NADH and NADPH are stable in basic pH solutions, while NAD and NADP are stable in acidic pH solutions. Indeed, most enzymatic cycling assay kits rely on the selective degradation of either the oxidized or reduced forms to obtain the redox ratio (Fig. 5B). Finally, oxidation or reduction during sample preparation can have a dramatic impact on absolute and relative concentrations of these molecules (149).
Cellular Topology of NAD(P)H
Total cellular NAD content is ∼0.5–5 nmol/106 cells (277), 5–15 nmol/mg protein (2, 270), 500 nmol/g tissue (47, 285), or 250–500 μM (35). This pool is asymmetrically distributed between cytoplasmic and mitochondrial compartments where mitochondrial concentrations up to 800 μM have been reported (35, 170, 265). Cytoplasmic concentrations have proven more difficult to estimate. Mouse erythrocyte NAD concentration is 370 μM (263). HeLa cell cytoplasmic concentrations are ∼460 μM, assuming a relative mitochondrial matrix volume of 10% (35, 190).
Intracellular ratios of NAD/NADH also vary by compartment. Cytosolic NAD/NADH ranges between 200 and 800 depending on cell type as determined by the fluorescent ratiometric biosensor, SoNar (279). This finding is consistent with previously reported cytoplasmic ratios based on other techniques (221, 242, 269, 275). Mitochondrial NAD/NADH ratios are ∼100-fold lower, consistent with a more reduced environment (242, 243, 258).
Compared to NAD, cells contain less NADP. Whole cell NADP concentrations are ∼80 μM with a mitochondrial concentration of 20 μM (35). Moreover, the ratio of NADP/NADPH in rat liver tissue lysates is ∼100,000-fold lower than the NAD/NADH ratio in the same sample (0.01 NADP/NADPH vs. 1000 NAD/NADH) (i.e., the majority of the NADP pool is reduced) (242).
A significant portion of intracellular NAD(P) is protein bound, although precise estimates of free/bound ratios for these dinucleotides are difficult to obtain. In erythrocytes, ∼50% of NAD and 10% of NADH are unbound, while 90% of NADP and NADPH are bound (24).
Subcellular distribution of NAD(P) biosynthesis
Nearly all NAD(P) biosynthetic enzymes localize to the cytoplasm or nucleus, except NMNAT3, mNADK, and nicotinamide nucleotide transhydrogenase (NNT) (43, 174, 175). NAMPT has also been associated with mitochondria from HEK293 cells and rat livers (265), although this has not been confirmed in other studies (174, 184). NAMPT appears to be the rate-limiting enzyme in the synthesis of NAD from nicotinamide (200), while NAPRT and NRK appear to limit production of NAD from nicotinic acid and riboside precursors, respectively (174). Overexpression of NMNAT or NAD synthetase fails to increase intracellular NAD, while overexpression of the enzymes that produce the mononucleotides NAMN and NMN leads to marked increases in mitochondrial NAD (174). Thus, adding nicotinic acid to medium containing nicotinamide will cause further increases in cellular NAD as the shared metabolic pathways are not rate limiting (80). Leveraging these nonoverlapping biosynthetic pathways may have therapeutic advantages for increasing cellular NAD.
While the mitochondrial pool of NAD is thought to be substantially protected from fluctuations in the cytoplasmic NAD concentrations, direct evidence in support of this observation is limited. FK866, a potent inhibitor of NAMPT, decreases cytoplasmic NAD to 50% of control levels in HeLa cells treated for 24 h without affecting mitochondrial levels (184). By contrast, activated poly-ADP-ribose polymerase (PARP)-1 markedly depletes cytoplasmic and mitochondrial NAD, although depletion of the mitochondrial pool requires mitochondrial permeability transition pore opening (2, 270). Conversely, HeLa cells treated with 1 mM extracellular NAD increase cytoplasmic and mitochondrial NAD levels twofold (183). NAMPT overexpression will increase mitochondrial NAD when cells are supplemented with nicotinamide (265). Similarly, nicotinamide riboside will increase mitochondrial NAD in vitro and in vivo (26) as will overexpression of NRK1 in the cytoplasm (174).
Without NAD synthetase, mitochondria cannot produce NAD from deamidated precursors, such as NAMN. Assuming the absence of a mitochondrial NAD transporter in mammalian cells and given that some evidence suggests NMNAT3 is the only NAD biosynthetic enzyme localized to the mitochondria, NMN seems to be the primary cytosolic precursor for mitochondrial NAD biosynthesis. However, the gene for NMNAT3 encodes two splice variants, NMNAT3v1 and FKSG76, neither of which appear to be expressed outside of erythrocytes (58). Indeed, siRNA-mediated silencing of these transcripts does not affect NMNAT activity observed in mitochondria and overexpression of FKSG76 depletes the mitochondrial NAD pool, while overexpression of NMNAT3v1 has no effect (58). These data suggest an alternative explanation for the NMNAT activity of mitochondria, perhaps due to the activity of a nucleotidyltransferase/phosphodiesterase (67). Notably, only exogenous NAD can rescue FKSG76-mediated depletion of mitochondrial NAD, the precursors NMN, nicotinamide riboside, nicotinic acid, and nicotinamide, cannot (58). Consistent with this in vitro finding, the NMNAT3 knockout mouse demonstrates preserved mitochondrial NAD content, providing further evidence against an important biosynthetic role for this enzyme in maintaining the mitochondrial NAD pool (264). Based on the observations that NAD is necessary and sufficient to restore mitochondrial NAD pools (58, 183), additional studies are warranted to identify the mechanisms of intracellular NAD transport. Although a mammalian mitochondrial NAD transporter has not yet been identified, NAD transporters have been identified in yeast mitochondria (230) and in intracellular vesicle membranes of sea urchin eggs (44).
Like NAD, the cytoplasmic and mitochondrial pools of NADP are thought to be relatively autonomous. The inner mitochondrial membrane is thought to be impermeable to NADP. Unlike NAD biosynthetic enzymes, both cytoplasmic and mitochondrial isoforms of NADK have been identified (96, 137, 175, 276). Silencing of cNADK with shRNA in HEK293 cells led to a threefold decrease of NADPH, while overexpression increased NADPH four- to fivefold (187). Human deficiency of mNADK causes failure to thrive, developmental delay, lactic acidosis, and severe encephalopathy due to dienoyl-CoA reductase deficiency and hyperlisinemia, as dienoyl-CoA reductase and lysine metabolism both require NADPH as an electron donor (96). Fibroblast mitochondria from one patient with mNADK deficiency contained half of the NADP(H) of control subjects, while the cytoplasmic content was unaffected (96). Mitochondrial NADPH depletion was accompanied by a significant reduction in mitochondrial oxygen consumption capacity compared to that of fibroblasts from normal controls, although whether this was attributable to any specific deficiency in substrate handling or to the oxidant injury that would be expected to accompany mitochondrial NADPH depletion was not investigated. Thus far, this study has been the only one to report the effects of mNADK deficiency on mitochondrial NADP content. Additional studies regarding the relative importance of cytoplasmic and mNADK isoforms in maintaining compartmental NADP levels, the metabolic and bioenergetic effects of NADP deficiency, and the role of NADK in broader oxidant defense remain to be explored.
NADH and Energy Metabolism
NADH wires cells for ATP production
NAD collects electrons from glycolysis, fatty acid oxidation, and the TCA cycle to supply reducing equivalents for the electron transport chain in support of oxidative phosphorylation to generate ATP (Fig. 6). Glycolysis yields two NADH molecules per molecule of glucose. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) utilizes NAD as an electron acceptor in the oxidation of glyceraldehyde 3-phosphate to 1,3-bisphosphoglycerate. These reducing equivalents are subsequently transported into the mitochondria by electron shuttles.
FIG. 6.

NADH wires cells for ATP production. All major catabolic metabolic pathways generate NADH. Glycolysis yields two NADH per glucose molecule through the activity of GAPDH. These NADH are recycled to NAD by LDH. Pyruvate is further oxidized within the mitochondria to acetyl-CoA by PDH, generating an additional molecule of NADH. During fatty acid β oxidation, HADH reduces one NAD molecule per acetyl-CoA liberated. GLUD also generates NADH during glutaminolysis. Three enzymes in the TCA cycle reduce NAD during the oxidation of substrates, IDH, OGDH complex, and MDH. Electrons from NADH then enter the electron transport chain at Complex I. ATP, adenosine triphosphate; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GLUD, glutamate dehydrogenase; HADH, 3-l-hydroxyacyl-CoA dehydrogenase; IDH, isocitrate dehydrogenase; LDH, lactate dehydrogenase; MDH, malate dehydrogenase; OGDH, α-ketoglutarate dehydrogenase; PDH, pyruvate dehydrogenase; TCA, tricarboxylic acid.
NADH is impermeable to the inner mitochondrial membrane. Two shuttle systems, the malate–aspartate shuttle and the glycerol-3-phosphate (G3P) shuttle, conduct electrons from cytoplasmic NADH to reduce mitochondrial NAD (Fig. 7). In the malate–aspartate shuttle, cytoplasmic NADH reduces oxaloacetate to malate in a reaction catalyzed by cytoplasmic malate dehydrogenase (cMDH) (Fig. 7A). Malate is transported into the mitochondria by an α-ketoglutarate/malate antiporter encoded by SLC25A11. A mitochondrial malate dehydrogenase oxidizes malate to oxaloacetate to produce NADH. Mitochondrial aspartate aminotransferase transfers an amino group from glutamate to oxaloacetate, yielding aspartate and α-ketoglutarate. Aspartate is exported from the mitochondria through an aspartate/glutamate antiporter (SLC25A12, Aralar; SLC25A13, Citrin; SLC1A3, excitatory amino acid transporter 1). These calcium-binding transporters facilitate the only irreversible step in the malate–aspartate shuttle, driving the net transit of reducing equivalents into the mitochondria (130, 131). Cytosolic aspartate aminotransferase transfers an amino group from aspartate to α-ketoglutarate to produce glutamate and oxaloacetate to complete the cycle.
FIG. 7.

Shuttling NADH into the mitochondria. (A) The malate–aspartate shuttle is the main NADH translocating apparatus in mammalian cells. cMDH first transfers electrons from NADH to malate, which can enter the mitochondria via the α-ketoglutarate/malate antiporter (SLC25A11). Once inside, mMDH oxidizes malate to regenerate NADH, which can then supply electrons to Complex I. The cycle is completed by the coupled transamination of OAA to form ASP by mAST (GOT2). Aspartate leaves the mitochondria by a calcium-dependent aspartate/glutamate antiporter and is converted back to OAA by GOT1. These reactions are supported by GLU and αKG cycling. (B) NADH can also transfer electrons directly into the electron transport chain through the glycerophosphate shuttle. In this cycle, cytoplasmic GPDH oxidizes NADH to generate G3P and DHAP. G3P is then oxidized by mitochondrial GPDH thereby transferring electrons to ubiquinone via and FAD cofactor. αKG, α-ketoglutarate; ASP, aspartate; cMDH, cytoplasmic malate dehydrogenase; DHAP, dihydroxyacetone phosphate; FAD, flavin adenine dinucleotide; G3P, glycerol-3-phosphate; GLU, glutamate; GPDH, glycerol-3-phosphate dehydrogenase; mAST, mitochondrial aspartate aminotransferase; mMDH, mitochondrial malate dehydrogenase; OAA, oxaloacetate.
NADH also transfers electrons directly to the electron transport chain through the glycerophosphate shuttle (Fig. 7B). In this mechanism, cytosolic glycerol-3-phosphate dehydrogenase (GPDH) oxidizes NADH to generate G3P from dihydroxyacetone phosphate (DHAP). G3P is then oxidized by a mitochondrial isoform of GPDH bound to the inner mitochondrial membrane that subsequently transfers electrons to ubiquinone via a flavin adenine dinucleotide (FAD) cofactor. Since GPDH does not pump protons across the mitochondrial membrane, this shuttling system is a less efficient means of energy transfer for ATP synthesis than the malate–aspartate shuttle, which makes NADH available to drive proton pumping via Complex I as well as through electron transfer between ubiquinone and Complex III.
Within mitochondria, pyruvate is oxidized by pyruvate dehydrogenase (PDH) complex, which reduces NAD to NADH (Fig. 6). Acetyl-CoA produced by PDH provides carbon substrates for a series of oxidative reactions in the TCA cycle. Three enzymes, isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase (OGDH), and malate dehydrogenase (MDH), transfer electrons from their substrates to NAD.
Also, in the mitochondria, β oxidation of acyl-CoA fatty acids yields one NADH molecule per acetyl-CoA generated through the actions of 3-l-hydroxyacyl-CoA dehydrogenase. For example, oxidation of palmitoyl-CoA with its 16-carbon fatty acyl chain produces seven NADH through β oxidation in addition to eight acetyl-CoA. These acetyl-CoA enter the TCA cycle to generate an additional 24 NADH.
Mitochondrial NADH transfers two electrons into the electron transport chain (Fig. 8). Complex I, NADH:ubiquinone oxidoreductase, oxidizes NADH back to NAD. From Complex I, electrons travel through ubiquinone, Complex III, cytochrome c, and Complex IV where they reduce molecular oxygen to water. Electron transport drives protons from the matrix into the inner membrane space at Complexes I, III, and IV to generate a chemiosmotic gradient. Fo F1 ATP synthase (Complex V) uses the proton motive force generated by the electron transport chain to phosphorylate ADP to form ATP. In sum, two electrons from NADH pump 10 protons across the inner mitochondrial membrane and drive the production of 2.5 ATP molecules (87).
FIG. 8.

NADH supplies the electron transport chain. Mitochondrial NADH transfers its electrons to the electron transport chain through Complex I. From there, electrons travel through ubiquinone, Complex III, cytochrome c, and Complex IV where they eventually reduce molecular oxygen to water. Electron transport drives protons from the matrix to the inner membrane space to generate the chemiosmotic gradient that is coupled to ATP production by Complex V. On average, 2 electrons per NADH pumps 10 protons and yields 2.5 ATP.
Regulation of intermediary metabolism by NAD/NADH
Given the ubiquitous requirements for NAD/NADH interconversions in bioenergetic metabolic pathways, this ratio plays a substantial regulatory role in addition to its role as electron carrier (Fig. 9). Moreover, the global NAD/NADH ratio itself affects the relative flux through oxidoreductase and dehydrogenase enzymes. A high NAD/NADH favors substrate oxidation, while a low ratio favors substrate reduction.
FIG. 9.

NADH couples energy supply and demand. When energy demand is high (left panel), ATP hydrolysis and dissipation of the chemiosmotic gradient drive NADH oxidation by Complex I. When energy demand is low (right panel), accumulation of cytoplasmic and mitochondrial NADH inhibits catabolic metabolic pathways to slow bioenergetic metabolism.
NADH can accumulate in the cytosol as a product of GAPDH and can decrease the rate of glycolysis. This phenomenon is perhaps most easily appreciated in erythrocytes, as they lack mitochondria and thus all glycolytic reactions are cytosolic by definition. Accumulation of cytoplasmic NADH in erythrocytes has been shown to slow glycolysis at GAPDH (228). Moreover, GAPDH relies on the availability of NAD to support forward flow of carbon through glycolysis. Two primary mechanisms serve to resupply the cytoplasmic NAD pool and maintain an NAD/NADH ratio favorable for continued glycolytic flux. First, in cells with functional mitochondria, cMDH in the malate–aspartate shuttle oxidizes NADH to facilitate electron transfer into the mitochondria. Second, the cytoplasmic enzyme LDH reduces pyruvate to lactate, thereby regenerating NAD.
After entry into the mitochondria, pyruvate undergoes oxidative decarboxylation by PDH, which transfers electrons from pyruvate to generate NADH. Similar to GAPDH, decreases in mitochondrial NAD/NADH will inhibit pyruvate metabolism by PDH through feedback inhibition (234). NADH can also inhibit PDH through NADH-mediated stimulation of pyruvate dehydrogenase kinase (PDK) (119, 120). PDK is a member of the PDH multiprotein complex that inhibits the activity of PDH by phosphorylation (143).
Enzymes of the TCA cycle are also sensitive to changes in the NAD/NADH ratio. As ATP consumption increases, the NAD/NADH ratio increases, leading to activation of IDH, OGDH, and MDH to provide reducing equivalents to meet the demands of the electron transport chain. Conversely, as NADH accumulates, it exerts strong feedback inhibition on these enzymes to slow flux through the cycle (247). In addition to the TCA cycle dehydrogenases, NADH also inhibits citrate synthase, which catalyzes the condensation of oxaloacetate with acetyl-CoA (129).
As with the TCA cycle, mitochondrial NADH accumulation will inhibit fatty acid β oxidation by inhibition of 3-l-hydroxyacyl-CoA dehydrogenase (213). This leads to accumulation of 3-hydroxy fatty acids and inhibition of upstream enzymes in the β oxidation cycle.
Overall, bioenergetic metabolism relies on the cytoplasmic and mitochondrial NAD/NADH ratios to match the rates of oxidative catabolism to energy demand. Additional study is required to dissect the relative contributions of the cytoplasmic and mitochondrial NAD/NADH ratios to energy metabolism. Interestingly, supplementation of HeLa cells with exogenous NAD stimulated oxygen consumption and ATP production (183); however, bulk transit of NAD from the cytoplasm to the mitochondria by heterologous overexpression of the mitochondrial NAD transporter NDT2 from Arabidopsis thaliana in HEK293 cells markedly decreased oxygen consumption while increasing glycolysis, which was associated with severe growth retardation (241). Additional studies are warranted to investigate the different mechanisms by which these approaches impact cellular bioenergetics.
NAD/NADH couples intermediary metabolism and redox homeostasis
In addition to coordinating energy supply and demand, the NAD/NADH ratio coordinates the metabolic response to changes in cellular redox state. For example, low oxygen availability leads to electron transport chain dysfunction, mitochondrial membrane hyperpolarization, uncoupled electron leak, and the generation of reactive oxygen species (ROS) (32, 121, 273). In response, cells activate a transcriptional program that increases the expression of glycolytic genes, including LDHA, while inhibiting carbon entry into the mitochondria. This gene expression program is mediated by stabilization of the hypoxia inducible factor 1α (HIF1α). Under aerobic conditions, HIF1α is post-translationally modified by prolyl hydroxylase (PHD) in a reaction requiring O2, α-ketoglutarate, and Fe2+ (Fig. 10A). Von Hippel Lindau (VHL), an E3 ubiquitin ligase, recognizes the hydroxylated proline residue on HIF1α, targeting it for proteasomal degradation (Fig. 10B).
FIG. 10.

The HIF1α transcriptional program protects against reducing stress. (A) PHD catalyzes proline hydroxylation on HIF1α in a reaction requiring O2, α-ketoglutarate, and Fe2+. (B) Hydroxylated HIF1α is recognized by the E3-ubiquitin ligase, VHL, which targets HIF1α for proteasomal degradation. As oxygen tension falls, PHD activity decreases, resulting in accumulation of unmodified HIF1α and activation of the HIF1α transcriptional program. HIF1α, hypoxia inducible factor 1α; PHD, prolyl hydroxylase; VHL, von Hippel Lindau.
In the setting of hypoxia or redox stress, the activity of PHD is inhibited, leading to HIF1α stabilization and activation of a hypoxia-like metabolic program (Fig. 10B). While increasing flux through glycolysis may be a way to augment ATP production in the setting of mitochondrial dysfunction, studies of cells derived from Hif1a−/− mice suggest that ATP production is actually greater at 1% oxygen than at 21%, but that ROS production is markedly increased (121, 273). This observation suggests that the primary objective of the HIF1α metabolic program is to mitigate reductive stress rather than supply ATP (215). In this context, LDH serves an important role to maintain continued flow of glucose through glycolysis by renewing cytoplasmic NAD as well as facilitating the clearance of excess reducing equivalents through the cellular export of lactate.
A hypoxia-like metabolic program, characterized by increased glycolysis and lactate fermentation despite sufficient oxygen availability, was associated with cancer cell metabolism by Otto Warburg in 1924 (252, 253). This so-called Warburg effect has since been attributed to stabilization of HIF1α in malignant cells and has been recognized as a normal finding in other proliferating cells (240). Inhibition of LDH in the setting of Warburg metabolism increases ROS generation and inhibits cell proliferation (134). Presumably, LDH inhibition in hypoxia would have similar consequences, although this has not been tested directly.
In addition to NAD recycling in the cytosol by LDH, NAD recycling is also a critically important function of the electron transport chain. Mammalian cells with dysfunctional electron transport require exogenous uridine and pyruvate to support cell proliferation (83, 123, 167). Endogenous uridine biosynthesis depends on a functional electron transport chain as dihydroorotate dehydrogenase is a mitochondrial flavoprotein that transfers electrons to ubiquinone and is required for pyrimidine biosynthesis. Pyruvate seems to be required to support NAD recycling as several other electron-accepting α-keto acids can also rescue pyruvate-dependent cells (163, 222, 229). A direct role for NAD recycling has recently been shown through the heterologous expression of a water-forming NADH oxidase from Lactobacillis brevis, LbNOX, in HeLa cells (229). Whether targeted to the cytoplasm or mitochondria, LbNOX rescues HeLa cell proliferation deficits induced by the electron transport chain inhibitors piercidin (Complex I inhibitor), antimycin (Complex III inhibitor), ethidium bromide (mitochondrial DNA replication inhibitor), and chloramphenicol (mitochondrial translation inhibitor) (229). These findings indicate a critical role for NAD regeneration and restoration of the NAD/NADH ratio in the setting of electron transport chain dysfunction to support cell proliferation.
NADPH Is the Battery of the Cell
Major sources of NADPH
NADPH is required for the de novo biosynthesis of nucleic acids, fatty acid acyl chains, cholesterol, and steroid hormones. It is also a central player in cellular redox balance, being required for the regeneration of unconjugated free glutathione and thus playing a key role in the control of ROS and defense against oxidant injury and also being a critical driving force for the production of ROS by way of NADPH oxidases and of reactive nitrogen species (RNS) by way of nitric oxide synthases (NOSs). In each of these roles, NADPH functions as the electron-donating species in an enzyme-catalyzed redox reaction and can thus be thought of as a stable storage form of electronic reducing potential. Before discussing these central roles for NADPH in detail, however, it is important to understand the major sources of NADPH production.
In most cells, the major source of NADPH is the oxidative portion of the pentose phosphate pathway (PPP), also called the hexose monophosphate shunt (Fig. 11). In this pathway, glucose-6-phosphate (G6P) produced by the action of hexokinase in the first step of glycolysis is “shunted” to G6P dehydrogenase (G6PD), which produces NADPH from NADP and produces 6-phosphoglucono-δ-lactone. This reaction exhibits anywhere from 10- to 500-fold or greater selectivity for NADP over NAD (11, 176). The 6-phosphoglucono-δ-lactone produced is then hydrated by glucolactonase to produce 6-phosphogluconate, and this is metabolized by 6-phosphogluconate dehydrogenase to produce ribulose 5-phosphate and a second molecule of NADPH. Ribulose 5-phosphate can be isomerized to ribose 5-phosphate as an endpoint to be utilized for nucleotide biosynthesis or it can be further metabolized by transaldolase and transketolase to regenerate glyceraldehyde 3-phosphate and fructose 6-phosphate, which can then be “returned” to the glycolytic pathway.
FIG. 11.

NADPH generation by the oxidative portion of the PPP. In most cells, the PPP is the major source of NADPH. Here, G6PD consumes G6P and generates NADPH. Subsequently, 6-phosphogluconate is also oxidized to generate a second molecule of NADPH. Ribulose-5-phosphate generated from this process can then return to the glycolysis pathway as G3P and fructose-6-phosphate through the actions of transketolase and transaldolase. Alternatively, ribulose-5-phosphate can be isomerized to ribose-5-phosphate for nucleotide biosynthesis. PPP, pentose phosphate pathway.
Under normal physiologic conditions, the NADP/NADPH ratio is the major determinant of how much G6P enters the PPP (90, 126). This represents the “committed step” of the oxidative portion of the PPP, while the eventual destination of G6P-derived carbon, either glycolysis or the PPP, is determined primarily at or beyond the level of ribulose 5-phosphate (92, 93). Indeed, in hepatocytes engaged in rapid fatty acid synthesis (an NADPH-requiring process), the glyceraldehyde 3-phosphate and fructose 6-phosphate produced later in the PPP are shunted back into the gluconeogenesis pathway to produce G6P (10, 208). This “regenerated” G6P can be funneled back into the PPP for further NADPH generation, thereby completely oxidizing G6P to carbon dioxide. In cell types incapable of gluconeogenesis, the fate of the downstream PPP intermediates under similar situations of high NADPH demand is to become pyruvate through glycolysis, generating ATP directly and subsequently through further oxidative metabolism of pyruvate.
Although the PPP is a predominant source of NADPH for most cells, it is certainly not the only source. The production of pyruvate from malate by NADP-dependent malic enzyme (ME) results in the production of NADPH and can occur either in the cytosol through the action of ME1 or in the mitochondrion through the action of ME3. Although complete data on relative expression of the various isoforms are somewhat lacking, generally the cytosolic isoform is the more abundantly expressed, and the mitochondrial isoform exhibits lower expression (with ME2 being the mitochondrial NAD-dependent isoform that classically participates in the TCA cycle) (188). Similarly, IDH has two NADP-requiring isoforms, a cytosolic (IDH1) and a mitochondrial (IDH2) isoform, each of which functions as a homodimer. The IDH1 and IDH2 isoforms have received considerable attention recently due to the identification of mutations in these enzymes that contribute to oncogenesis in a variety of tumor types by shifting the enzyme's function from the oxidative decarboxylation of isocitrate to produce α-ketoglutarate (with NADP as the electron acceptor) to a neofunctional enzyme that produces R-2-hydroxyglutarate and consumes NADPH through reductive metabolism of α-ketoglutarate (145).
The mitochondrial isoforms of glutamate dehydrogenase (GLUD) are also sources of NADPH, using NADP as the electron acceptor in a reaction that converts glutamate to α-ketoglutarate and ammonia. GLUD1, the more ubiquitous isoform, can use either NAD or NADP, exhibiting preference for NAD. Interestingly, GLUD1 preferentially binds the reduced forms of both pyridine dinucleotides, suggesting structurally mediated kinetics that favor inhibitory allosteric tone (14, 15). GLUD2, the neural/testicular isoform, is also capable of using either NAD or NADP as electron acceptors. Finally, the mitochondrial NAD(P) transhydrogenase, or NNT, catalyzes a direct hydride transfer from NADH to NADP. The driving force catalyzing this transfer is the proton motive force across the mitochondrial inner membrane. NNT sustains mitochondrial NADPH levels not only for the purposes of antioxidant defense but likely also to support steroid hormone biosynthesis, as evidenced by mutations in the enzyme leading to severe cortisol deficiency (although this may also be secondary to oxidant injury resulting in the destruction of adrenocortical cells) (165).
Macromolecular biosynthesis
NADPH is a required cofactor for the synthesis of deoxyribonucleotide diphosphates (dNDPs) from ribonucleotide diphosphates (NDPs). The removal of the 2′-hydroxyl group on the ribose moiety of NDPs to form dNDPs is accomplished by a rather complex series of redox coupled reactions that are ultimately catalyzed by ribonucleotide reductase (RNR) and that rely on NADPH as the primary, although indirect, electron donor (Fig. 12). NADPH is oxidized to NADP, either by glutathione reductase or by thioredoxin reductase, with the electrons transferred to the flavin prosthetic group of these reductase enzymes (216). For glutathione reductase, the flavin group transfers its electrons directly to reduce a disulfide bond in the active site of the enzyme. Then, glutathione reductase will pass electrons to oxidized glutathione to generate glutathione, which will then reduce free sulfhydryl residues in the active site of glutaredoxin. Glutaredoxin then transfers electrons to reduce a disulfide bond in the active site of RNR to restore its catalytic activity.
FIG. 12.

NADPH provides electrons for nucleotide biosynthesis. RNR, which catalyzes dehydroxylation of NDP to dNDP, forms an active site disulfide during catalysis that must be reduced for continued enzymatic activity. Electrons are transferred from NADPH through two redox pathways. In one, GR oxidizes NADPH to reduce GSSG to GSH. Glutathione reduces disulfides in Grx that subsequently catalyzes the reduction of disulfides in RNR. In the other pathway, NADPH is utilized by TrxR to reduce disulfides in Trx, which, in turn, reduces disulfides in RNR. dNDP, deoxyribonucleotide diphosphates; GR, glutathione reductase; Grx, glutaredoxin; GSH, glutathione; GSSG, glutathione disulfide; NDP, ribonucleotide diphosphates; RNR, ribonucleotide reductase; Trx, thioredoxin; TrxR, thioredoxin reductase.
A similar series of electron transfer steps occur via the thioredoxin pathway (282). First, NADPH donates electrons to reduce an intramolecular selenylsulfide bond via FAD in thioredoxin reductase. Then, a second NADPH molecule reduces an intramolecular disulfide bond in the active site of the thioredoxin reductase. This permits thioredoxin binding through a new selenylsulfide bond that is reduced by the active site disulfide in thioredoxin reductase. Reduced thioredoxin can then transfer electrons to reduce the active site disulfide in RNR.
Fatty acid biosynthesis also requires NADPH as an electron donor to permit elongation of acyl chains (157). The fatty acid synthase enzyme complex catalyzes the formation of saturated fatty acids through the combination of acetyl-CoA (a 2-carbon unit) and malonyl-CoA (a 3-carbon unit) to form palmitate (a 16-carbon acyl chain) in a series of reactions requiring the consumption of ATP and an input of reducing potential from NADPH. In de novo acyl chain synthesis, two steps involve an NADPH-requiring reduction. The first is the reduction of the β-keto group in acetoacetyl-acyl carrier protein (ACP) to produce D-3-hydroxybutyryl-ACP. The second is the reductive saturation of the trans-Δ2 double bond in crotonyl-ACP to form butyryl-ACP. Stoichiometrically, the reactions required to produce one molecule of palmitate de novo will consume 14 molecules of NADPH, equating to 2 NADPH per molecule of malonyl-CoA (each of which is synthesized from acetyl-CoA). Interestingly, while NADPH is the required source of reducing power for fatty acid synthesis, the oxidative breakdown of fatty acids generates NADH and reduced flavin moieties.
The biosynthesis of cholesterol also requires the availability of NADPH as a reducing entity at several steps (189). Two molecules of NADPH are required for the conversion of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) to mevalonate by HMG-CoA reductase, the rate-controlling enzyme in the cholesterol biosynthetic pathway and the primary molecular target of statins. HMG-CoA reductase catalyzes a two-electron reduction at the CoA thioester of HMG to yield mevalonate. The mevalonate is then further metabolized to isopentenyl units, which are then assembled into farnesyl pyrophosphate. At this step, NADPH is again the reducing agent for the final step in squalene synthesis, in which two molecules of farnesyl pyrophosphate are condensed to a single squalene molecule by squalene synthase. It is worth noting that, by extension, the other biosynthetic pathways that stem from the mevalonate/squalene/cholesterol pathway are also dependent upon NADPH as an electron donor. These include the biosynthetic pathways for ubiquinol (coenzyme Q10), dolichol, heme, steroid hormones, bile acids, vitamin D, and others (142). The breakdown of heme to bilirubin also requires the participation of NADPH as a reducing agent.
Finally, although not specifically a macromolecular biosynthesis pathway, it is worth noting here that the vast array of cytochrome P450 oxidoreductase enzymes commonly use NADPH as the electron donor to accomplish the variety of reactions catalyzed by this family of heme-containing enzymes (79, 107). This subject is discussed in greater detail below (under the Cellular Detoxification section), although it bears mention here, as the addition of a hydroxyl group via the monooxygenase activity of some members of the P450 system is a common theme in many biosynthetic reactions. As one example, most of the hydroxylation reactions required for steroid hormone biosynthesis are accomplished by P450-catalyzed monooxygenation reactions driven by NADPH serving as the electron donor to a reductase enzyme (e.g., P450 reductase, cytochrome b5 reductase, adrenodoxin reductase) that serves as the enzymatic partner of the cytochrome P450 monooxygenase.
Generation of ROS/RNS by NADPH oxidases and NOSs
Although cells devote considerable resources to the containment of ROS (including NADPH-dependent systems), there are also complex mechanisms for the deliberate generation of ROS to serve as both cellular signals and as a mechanism for cellular defense from pathogens. As a general scheme, the generation of ROS typically involves the transfer of a single electron to molecular oxygen, which generates a superoxide anion radical. This one-electron reduction of molecular oxygen is directly accomplished by the NADPH oxidase holoenzymes, which, as the name implies, utilize NADPH as the electron donor and molecular oxygen as the electron acceptor.
Seven identified isoforms comprise the NADPH oxidase family, designated NOX1 through NOX5 and DUOX1 and DUOX2 (49). Each isoform, except NOX5, functions as a holoenzyme complex. NOX1 through NOX3 are typically composed of the catalytic core subunit; a membrane-associated stabilizing subunit termed p22phox; cytosolic organizing subunits, including p40phox, p47phox, and/or NOXO1; cytosolic activating subunits, including p67phox or NOXA1; and regulatory Rho family GTPase member RAC1 or RAC2. NOX4 has the p22phox subunit but appears to associate with a DNA polymerase interacting subunit termed POLDIP2. NOX5 has self-contained EF-hand domains that confer calcium sensitivity. The two DUOX family members DUOX1 and DUOX2 each contain EF-hand domains and also associate with stabilizing/activating subunits DUOXA1 and/or DUOXA2. Notably, the DUOX family members also contain a peroxidase-like domain, and these two holoenzymes are generally thought to generate hydrogen peroxide as their ultimate product. NOX4 is also a hydrogen peroxide-generating isoform. Although the precise mechanism by which this occurs is not fully clear, it has been suggested that retention of the first superoxide molecule formed in the active site is accomplished by an extracellular loop of NOX4, thus providing sufficient time for the generation of a second molecule of superoxide, which can then react with the first in a dismutation reaction to form H2O2 (225).
The NADPH oxidases are perhaps best known for their role in host defense. Mutations in various components of the holoenzyme complexes have been identified as causative in most of the multiple clinical subtypes of chronic granulomatous disease (CGD) (37). The CGDs are all characterized clinically by a failure of phagocytes (primarily neutrophils and macrophages) to kill ingested pathogens, due to an inability to generate superoxide via NADPH oxidase in phagosomes. The superoxide generated is directly toxic to pathogens and also serves as a substrate for generating H2O2, which can ultimately be metabolized to hypohalous acid anions (primarily hypochlorite) by myeloperoxidase. The genetic basis of CGD was first attributed to X-linked inheritance of NOX2 mutations (gene name CYBB, protein product also called gp91phox), but subsequent work has identified mutations in CYBA, NCF1, NCF2, and NCF4 (corresponding to p22phox, p47phox, p67phos, and p40phox, respectively) as being causative for autosomal recessive forms of CGD (37). All of these mutations result in a dysfunctional or nonfunctional NADPH oxidase holoenzyme. Consistent with the known biochemistry, there are also case reports of severe G6PD deficiency causing a clinical syndrome very similar to CGD (217), thought due to the relative insufficiency of NADPH availability as an electron donor, which lies at the heart of the hemolytic anemia more classically associated with G6PD deficiency (see “Cellular Detoxification” below).
Apart from the classical association of NADPH oxidase with host defense, this family of ROS-generating enzyme complexes has been ascribed a role in a wide variety of other complex disease phenotypes, including prominent pathogenic roles for NADPH oxidase in atherosclerosis, cardiovascular disease, neurodegeneration, cancer, and fibrotic remodeling of multiple organs (20, 49, 153).
In addition to the controlled generation of superoxide anion by NADPH oxidases, NADPH also serves as the key electron donor for the generation of nitric oxide (NO, a so-called RNS) by NOSs. Three isoforms of NOS are recognized in mammals: neuronal NOS (NOS1), inducible NOS (NOS2), and endothelial NOS (NOS3). Each of these enzymes functions as a homodimer to catalyze the conversion of l-arginine to l-citrulline and NO. The topics of the biology and biochemistry of NO are far too broad to attempt to summarize in this review. However, from the standpoint of redox biochemistry requiring NADPH as an electron donor, the NOS isoforms each contain a reductase domain in the carboxy terminal portion of the monomer that functions in a manner that is biochemically similar to cytochrome P450 reductase (see “Cellular Detoxification” below). Tightly coupled NOSs will consume 1.5 moles of NADPH for each mole of NO produced. When NOS isoforms become uncoupled, the electrons flowing into the enzymatic system from NADPH can allow the one electron reduction of oxygen to superoxide anion (139).
Cellular detoxification
As one of the cell's most flexible electron carriers, NADPH not only participates in ROS generation but it is also a critical requirement for normal antioxidant defenses. This is perhaps best understood and exemplified through a discussion of the biochemistry of G6PD deficiency, the most common human enzymatic defect and the most common genetic disorder affecting red blood cells (84, 151). G6PD deficiency is inherited as an X-linked recessive condition and affects ∼400 million individuals worldwide, including up to 10% of African American men in the United States (84). The primary clinical manifestation is hemolytic anemia that occurs in response to infection, certain medications, and a variety of other stimuli (most notably broad beans, a condition termed favism) that all have as a common feature increased oxidative stress. Normally, oxidant stressors are buffered by glutathione. As discussed above, glutathione, once oxidized, is restored to the reduced state via a series of redox-coupled, enzyme-catalyzed reactions in which NADPH donates electrons to the active site of glutathione reductase (Fig. 12). Glutathione reductase then reduces oxidized glutathione disulfide dimers back to the monomeric form with a free sulfhydryl group that can then act as a direct antioxidant as well as serve as the substrate for glutathione-S-transferase, glutathione peroxidase, and other enzymes involved in antioxidant defense (Fig. 13).
FIG. 13.

NADPH provides electrons to enzymes involved in antioxidant defense. A variety of redox transfer reactions utilize NADPH as the initial electron donor to reduce and detoxify ROS. GSH generated by glutathione reductase is consumed by Gpx enzymes. Thioredoxin also participates in a variety of chemical reductions designed to mitigate oxidant stress through reduction of Prx, H2O2, and other proteins. Gpx, glutathione peroxidase; Prx, peroxiredoxin; ROS, reactive oxygen species.
In erythrocytes, the NADPH needed to maintain an intact free glutathione pool can only be generated by the PPP. In the setting of G6PD deficiency, flux through the PPP cannot be maintained to a degree sufficient to meet the NADPH needs in the face of increased oxidative stress, and erythrocyte lysis occurs. Although illustrative for G6PD deficiency, this same biochemistry is a major contributor to redox homeostasis for all cell types. Erythrocytes are particularly vulnerable to the lack of NADPH due to the absence of other salvage pathways for glutathione as well as the high concentrations of pro-oxidant heme iron and attendant ROS production.
In addition to its central role in the maintenance of free glutathione pools, as mentioned above, NADPH is the major electron donor for cytochrome P450 oxidoreductase-catalyzed reactions. The P450-catalyzed monooxygenation of a huge array of substrates, resulting in the addition of one or more hydroxyl groups, is a cornerstone of the Phase I detoxification and elimination of most xenobiotics as well as a wide variety of endogenous compounds (164). These reactions typically proceed by transfer of electrons from NADPH to the flavin moiety of cytochrome P450 reductase, and then on to the catalytic iron in the heme prosthetic group in the active site of the P450 enzyme. A complex interaction between the heme iron, molecular oxygen, and the porphyrin ring forms to act as the final electron acceptor, and hydroxylation of the target substrate is accomplished through the formation of a highly reactive iron (IV) oxo intermediate, the so-called P450 Compound I (203). Other redox-coupled electron transfer systems can also participate in reactions similar to those catalyzed by the cytochrome P450 oxidoreductases. Notably, the cytochrome b5 class of enzymes and the associated reductase can catalyze a variety of oxygenation reactions (220). These reactions are less selective with regard to the electron donor, frequently using NADH as well as NADPH.
NAD(P) Regulates Cellular Responses to Metabolic Perturbations
Sirtuins
The observation that caloric restriction was associated with extension of natural life span was recognized as early as the 1940s (7). Many theories have been offered to explain the possible mechanism underlying this observation. Two key discoveries focused attention on NAD metabolism as a critical link between caloric restriction and life span. First was the observation that the silent information regulator (SIR) genes in Saccharomyces cerevisiae could regulate aging and longevity (118) in addition to gene expression, chromosomal integrity, and telomere length and function. Second was the finding that the sirtuin family of proteins possessed NAD-dependent enzymatic functions, including ADP-ribosylase (64) and histone/lysine deacetylase (104, 122, 226) activities. Together, these led to the mechanistic hypothesis that caloric restriction works to extend natural life span, in part, through modulation of sirtuin enzymatic activities by way of the availability of the required NAD cofactor. Specifically, caloric restriction increases the NAD/NADH ratio, which activates sirtuins, which works to extend life span by way of chromatin regulation, histone regulation, and regulation of lysine acetylation status. By this hypothesis, metabolism, aging, and gene regulation were all coordinately linked (73).
Subsequent investigations have shown that sirtuins are the key players in a fundamental cellular regulatory network that exerts influence on gene expression, aging, metabolism, redox homeostasis, inflammation, cell motility, proliferation, and many other basic cell functions (4). In humans, seven sirtuin isoforms have been identified, designated SIRT1 through SIRT7. SIRT3, SIRT4, and SIRT5 are largely mitochondrially localized; SIRT7 is localized to the nucleus and nucleolus; and the remaining isoforms are variably distributed in the cytosol and nucleus. The majority of the sirtuins are NAD-dependent lysine deacetylases. SIRT4 has been characterized as having greater activity as an ADP-ribosyltransferase than as a lysine deacetylase, although the ADP-ribosyltransferase activity is still weak, and it has been suggested that the major in vivo enzymatic activity of SIRT4 is yet to be identified (50). SIRT5 is unique in the sirtuin family, as it functions as a desuccinylase, demalonylase, and deglutarylase with a biological function that has thus far been restricted to regulation of the urea cycle and nitrogen balance (170, 249). SIRT6, in addition to its deacetylase activity, has also been shown to possess a deacylase activity with particular affinity for removing long-chain acyl modifications from proteins and modulating cellular secretory activities (57, 110, 278).
All of the sirtuin isoforms utilize NAD as a required cofactor. Nicotinamide and NADH have been suggested as endogenous sirtuin inhibitors through both competitive and noncompetitive mechanisms that nonetheless seem to converge on the single catalytic site within each sirtuin isoform in vitro (5, 156). This reasoning has contributed to the idea that sirtuins are direct sensors of the NAD/NADH ratio. However, more recent investigations have suggested that this view may be overly simplistic, as several sirtuin isoforms' deacylase activities in vitro are inhibited by NADH concentrations that are likely never achieved in normal conditions in vivo (156). Nonetheless, the weight of evidence positions the sirtuins as critical to linking changes in the metabolic and redox state of the cell to regulation of a wide array of cellular functions.
Mono- and poly-ADP-ribosyltransferases/ribose polymerases
It has long been recognized that NAD can serve as the substrate for post-translational covalent modification of proteins. In the late 1960s and early 1970s, this post-translational modification was identified as NAD-dependent ADP-ribosylation by several different groups working in a variety of different cellular and tissue contexts, with most studies being done on poly-ADP-ribosylation modifications. Around the same time, mono-ADP-ribosylation was identified as the mechanism of action of diphtheria toxin and of Pseudomonas aeruginosa exotoxin A, with inhibitory mono-ADP-ribosylation of critical components of the protein synthesis machinery (e.g., eEF-2) being the key molecular event leading to cell death (72, 91, 136). Since these early studies, ADP-ribosylation has emerged as a post-translational modification that is nearly as central to basic cellular processes as phosphorylation or acylation.
For both mono- and poly-ADP-ribosylation, NAD is the required cofactor/donor of the ADP-ribose moiety. ADP-ribosyltransferases, in general, work by catalyzing cleavage of the N-glycosidic bond in NAD, using the target for ADP-ribosylation as the nucleophile. For mono-ADP-ribosyltransferases, common amino acid targets include the functional groups on the side chains of arginine, cysteine, asparagine, threonine, glutamine, lysine, and glutamate (235). For PARPs, the 2′-hydroxyl group of the ADP-ribose moiety already conjugated to a protein target is used as the nucleophile in the active site of PARP (108). In both cases, cleavage of the N-glycosidic bond releases nicotinamide and leaves ADP-ribose covalently linked to the nucleophilic “acceptor” site of the target.
In mammalian cells, the majority of research efforts have focused on the biochemistry and cell biology of PARPs. There are at least seven isoforms of enzymes thought to broadly comprise the PARP family: PARP1, PARP2, PARP3, VPARP, TANK1, TANK2, and TANK3 (173). PARP1 is by far the best studied member of the PARP family and is fairly representative of the cellular processes regulated by PARPs. PARP1 maintains genomic integrity by participating in base excision repair, repair of DNA strand breaks, and by regulating the accessibility of portions of the genome by regulating topoisomerase I. PARPs regulate gene expression through poly-ADP-ribosylation of histones, which serves to directly regulate histone interactions with chromatin thereby “loosening” chromatin structure by providing increased local negative charge density. PARPs also provide a scaffold consisting of poly-ADP-ribose oligomers that can facilitate the assembly of multiprotein complexes. PARP1 is widely recognized as being a direct substrate of caspases, with PARP cleavage being a canonical event in caspase-mediated apoptosis (18). It is thought that PARP cleavage serves to facilitate DNA fragmentation, to drive independent signaling functions mediated by PARP fragments, and to prevent PARP from depleting cellular NAD and energy reserves and thus maintaining these resources for other steps of the apoptosis cascade.
The majority of research regarding mono-ADP-ribosyltransferases has focused on bacterial enzymes such as diphtheria toxin and endotoxin A discussed above. These bacterial toxins tend to inhibit mammalian host enzymes via mono-ADP-ribosylation. Much less is known about endogenous mono-ADP-ribosyltransferases (22). A number of potential endogenous mono-ADP-ribosyltransferases have been identified, designated ARTD7 through ARTD15, although not all of these putative enzymes are thought to be catalytically active. Of the proposed mono-ADP-ribosyltransferases, ARTD10 is probably the best characterized (115). ARTD10 has been shown to shuttle between the cytoplasm and the nucleus, and it has been shown to regulate GSK3β, NFκB (via NEMO), and possibly MYC. All of these targets are thought to be inhibited by mono-ADP-ribosylation catalyzed by ARTD10. Finally, as briefly mentioned above, more than one of the sirtuin isoforms have had mono-ADP-ribosyltransferase activity attributed to them. However, these are often described as “weak” activities, and it has been suggested that any ADP-ribosylation activity associated with sirtuin isoforms should probably be considered more of a side reaction.
The above mechanisms linking metabolic changes within the cell to a variety of response pathways are, generally speaking, reasonably straightforward. Changes in cellular substrate-level metabolism and/or flux through one or more metabolic pathways result directly in changes in the availability of the NAD(P)/NAD(P)H that are the required cofactors for activation of the relevant response enzymes. However, there are examples of more complex interactions between metabolism and redox responses mediated through nicotine adenine dinucleotides. One particularly elegant example involving a redox circuit linked directly to cellular metabolism has been elucidated by P. Darrell Neufer's group (61). When the NAD/NADH ratio is very low (i.e., the NAD pool is in a highly reduced state), the rate of H2O2 production from the PDH complex increases significantly. Containment of the H2O2 produced requires the availability of reduced glutathione to feed into the thioredoxin/peroxidredoxin system as described above (Fig. 13). This, of course, requires the availability of NADPH. In this state of high NADH supply and high NADPH demand, NNT becomes a key link in this redox circuit. Recall that NNT catalyzes a hydride transfer from NADH to NADP to regenerate NADPH, using the proton motive force as the driving force for the reaction. Although other NADPH-generating enzymes (e.g., mitochondrial IDH isoforms discussed above) could partially mitigate H2O2 production, NNT was the key participant in the circuit needed to maintain NADPH levels. Since NNT utilizes the proton gradient across the inner mitochondrial membrane, the redox circuit described by PDH and NNT must be energy-consuming and should thus drive mitochondrial oxygen consumption.
In keeping with this, C57BL/6J mice harboring a known spontaneous loss-of-function deletion in NNT exhibit lower total body oxygen consumption for a given food intake and activity level than that measured in C57BL/6N mice. This complex circuit coupling metabolism and redox homeostasis, with the energy-consuming interconversion between NADH and NADPH as the linchpin, may be an important contributing factor to the particular susceptibility of C57BL/6J mice to diet-induced obesity and insulin resistance, and suggests that matching mitochondrial redox tone and metabolic rate are significant contributors to resting energy expenditure.
NAD(P) in Health and Disease
Pellagra
Pellagra, a systemic disease of niacin deficiency, is the original disease associated with abnormal metabolism of NAD(P). In 1735, a Spanish court physician, Don Gasper Casal described a condition affecting poor peasants of Asturias characterized by a glossy reddish rash on the dorsum of hands and feet, which he called “mal de la rosa.” Italian physician Francesco Frapoli coined the term “pellagra” from the Italian “pelle,” meaning “skin,” and “agra” denoting “rough,” highlighting the thickened rough skin on affected patients. In addition to dermatitis, pellagra is characterized by diarrhea, dementia, and death. The first case of pellagra in the United States was documented in 1902 where the disease quickly developed into an epidemic impacting farming communities in the southern states for over four decades. In this time, estimates suggest there were 3 million cases and 100,000 deaths (86, 193).
Casal also noted that affected patients were poor farmers whose diet consisted mainly of maize with little fresh meat. In 1914, Joseph Goldberger noted that pellagra occurred in inmates of mental institutions whose diet consistent of vegetables and cereals while the staff members, whose diet was more varied and included more meat and dairy products, were unaffected. In a series of human experiments, Goldberger showed that dietary manipulations could both cure and induce pellagra (193). Based on prior investigations, Conrad Elvehjem et al. demonstrated that commercial preparations of nicotinic acid and nicotinamide were highly effective at curing dogs of “black tongue,” a canine manifestation of pellagra (55). As a result of these and other studies, public awareness campaigns, agricultural diversification, and food fortification with nicotinic acid, pellagra was eradicated in the South by 1945 (193). Currently, sporadic cases of pellagra occur in developed countries among people dependent on drugs or alcohol, food faddists, or patients with malabsorption states, while cases continue to occur in developing areas where corn products are the major food source (86).
Aging
NAD metabolism is among many factors that have been implicated in biological aging (113, 244). NAD decreases with age in a variety of mammalian tissues, including the liver, pancreas, kidney, skeletal muscle, heart, white adipose, and skin (19, 69, 158, 169, 271). By contrast, NAD increases with fasting, glucose deprivation, calorie restriction, and exercise, which have been associated with longer life span (25, 33, 41, 66, 198, 205). These two groups of observations suggest that NAD deficiency contributes to the aging process and that supplementation with NAD may offer benefits to slow the aging process.
Two hypotheses address the age-related decrease in NAD. First, NAMPT expression decreases with age (127, 152, 171), which would hamper nicotinamide recovery by the salvage pathway for NAD synthesis. Decreased NAMPT may be a consequence of dysregulated circadian rhythm (171) or redox stress and chronic inflammation associated with aging (105). Second, PARP1 activation due to age-related DNA damage may consume NAD (19, 169). Inhibition of PARP1 activity restored NAD levels in aged worms (169).
NAD deficiency appears to compromise mitochondrial function due to impaired mitochondrial unfolded protein response (UPR). Mitochondrial UPR is triggered by an imbalance between the translation of electron transport chain subunits encoded by nuclear and mitochondrial genes (97). Activation of the UPR promotes longevity in worms (51, 97). Augmenting NAD by PARP1 inhibition or supplementation with nicotinamide or nicotinamide riboside activated the mitochondrial UPR and increased expression of superoxide dismutase, thereby enhancing mitochondrial oxygen consumption, ATP production, and worm life span (169). These effects depended on SIRT1 activation. In a parallel pathway, NAD and SIRT1 deficiency was associated with stabilization of HIF1α resulting in inhibition of oxidative phosphorylation and decreased mitochondrial gene expression (69). SIRT1 seems to be required for VHL transcription and its deficiency leads to HIF1α accumulation. HIF1α sequesters the transcription factor MYC from the mitochondrial transcription factor A (TFAM) promoter. These deleterious effects were reversed by supplementation with NMN (69).
Very recently published findings suggest that NAD deficiency may also be a driving mechanism for molecular aging through interactions with a number of proteins possessing a Nudix homology domain (NHD) (140). The NHD is believed to be an NAD binding pocket and NAD is likely a required cofactor that mediates protein–protein interactions for at least a significant subset of the NHD-containing proteins. Deleted-in-breast-cancer-1 is a specific NHD-containing protein that requires NAD availability to mediate an activating interaction with PARP1 to maintain DNA integrity. Little is known about the mammalian NHD protein family, although these proteins may yet emerge as a key link between NAD and other molecular processes that have been previously associated with aging.
Neurologic disease
Multiple studies have demonstrated a neuroprotective role for NAD, and NAD loss may be a common pathobiologic feature of neurologic disease. For example, toxic prion-treated neuronal cells demonstrate dramatic NAD loss, while NAD repletion by intracerebral or intranasal routes decreases hippocampal neural degeneration and motor activity, respectively (284). Early insights into the mechanism of NAD loss were gained from study of an inbred mouse strain where axonal degeneration after an injury (i.e., Wallerian degeneration) is slow (150). The protective mutation in the WldS mice links the N-terminal fragment of ubiquitination factor E4B to NMNAT1 (154). Wallerian-like degeneration is also a feature of many neurologic diseases, including Parkinson's and Alzheimer's diseases (39). Indeed, expression of WLDS protects animals from deficits associated with models of peripheral nerve injury, toxic neuropathy, glaucoma, hypoxia, ischemia, and Parkinson's disease, among others (39). This protein prevents early NAD loss in injured axons (251). Loss of endogenous NMNAT2 after axonal injury appears to be the key WLDS-reversible event (39).
In addition to NMNAT, an allosteric activator of NAMPT has been identified in a screen for compounds that enhance hippocampal neuron formation in mice (182, 250). This compound, P7C3, and its derivatives protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)- and 6-hydroxydopamine-induced parkinsonism in mice as assessed by preservation of dopamine levels, dopaminergic neuron viability, and normal motor control (45, 46). The SOD1G93A mutant mouse, a model of amyotrophic lateral sclerosis, demonstrated improved walking gait and rotarod stability when treated with a P7C3 analog at symptom onset (46). P7C3 also protects mice from developing learning, memory, and coordination deficits as well as chronic visual system dysfunction after blast-induced traumatic brain injury (52, 268). Overexpression of NAMPT in astrocytes derived from the SOD1 mutant mouse prevented astrocyte-mediated motor neuron cell death in a coculture model. Treating astrocytes with the NAD precursors NMN and nicotinamide riboside also had this protective effect (82). Indeed, supplementation of neurons with NAD, nicotinamide riboside, nicotinamide, or NMN protects against a variety of neurologic insults in rodent models, including axonal injury, ischemia, fetal alcohol toxicity, and Alzheimer's disease, among others (27, 70, 101, 124, 210, 236, 251).
It is worth mentioning that the roles of NAD/NADH in regulating cytoskeletal dynamics and mitophagy may play a larger part in the pathogenesis of neurodegenerative diseases than in some other disease states. Neurons, perhaps more than other cell types, rely on a highly ordered and very specific cellular morphology to maintain normal function. Cytoskeletal organization and maintenance are critical to this specialized morphology, and NAD has a direct impact on cytoskeletal organization by way of SIRT2's activity to modulate the acetylation state of the microtubule network, although whether enhancing or inhibiting SIRT2 activity is beneficial is an open question and may be context specific (56, 68, 178). The importance of dysregulated mitophagy in the pathogenesis of a variety of neurodegenerative diseases is fairly clear, most prominently in the pathogenesis of Parkinson's disease, where exploration of disease pathogenesis has led to much of our most current understanding of mitophagy more generally (133, 181). Although clearly a very complex process, mitophagy is influenced, in part, by cytoskeletal organization and also through NAD-responsive processes in the mitochondrion itself (e.g., regulation of the mitochondrial acetylome) (144, 146, 260).
Metabolic syndrome
Taking a view of metabolic syndrome that uses chronic glucose excess as its starting point, glucose flux through a variety of downstream metabolic pathways increases, including glycolysis and the TCA cycle (89, 106). Under conditions of significant glucose excess (e.g., uncontrolled diabetes mellitus), hexokinase, the first enzyme in the glycolysis cascade, becomes saturated. Glucose will then be metabolized through the polyol pathway, in which glucose is metabolized to sorbitol by aldose reductase, and sorbitol is subsequently metabolized to fructose by sorbitol dehydrogenase (Fig. 14). Functionally, the net redox result is the trading of one molecule of NADPH for one molecule of NADH. The NADH thus formed can then feed into the electron transport machinery in the mitochondrion, although at the expense of NADPH, which is necessary for a variety of other critical reaction pathways including antioxidant defenses. In addition, the continuation of this series of reactions requires the channeling of fructose back into the glycolytic pathway, either via the formation of fructose-6-phosphate or, in the liver, via the formation of fructose-1-phosphate by fructokinase and subsequent enzymatic transformations to DHAP. These reactions tend to proceed relatively less efficiently in situations of high glucose load, however, as the glycolytic pathway is already under high-flux stress. Thus, sorbitol tends to accumulate, and the net effect is the consumption of NADPH.
FIG. 14.
The polyol pathway. Under conditions of significant glucose excess, hexokinase, the initial enzyme in glycolysis, becomes saturated. The accumulating glucose is diverted into the polyol pathway where glucose is metabolized to sorbitol by aldose reductase and sorbitol is metabolized to fructose by sorbitol dehydrogenase. The first reaction requires NADPH and the second reaction yields NADH. Thus, these reactions yield a net exchange of NADPH for NADH, which is likely deleterious under conditions of reductive stress (e.g., diabetes-induced increases in glycolytic flux lowering the NAD/NADH ratio leading to ROS production in the setting of impaired antioxidant defense due to NADPH deficiency).
While polyol flux is low in euglycemia (3% of glucose uptake), estimates suggest that at least 30% of glucose flows through this pathway in chronic hyperglycemia (71). As fuel supply outstrips metabolic demand, mitochondrial and cytoplasmic NAD/NADH ratios fall (63, 259). The ensuing mitochondrial membrane hyperpolarization perpetuates electron leak and ROS formation which, in combination with the accumulation of TCA cycle intermediates fumarate and succinate, lead to PHD inhibition and HIF1α stabilization (63, 109). Thus, the metabolic pathobiology of diabetes has been characterized as “pseudohypoxia” (259).
This finding suggests that restoration of the cellular NAD/NADH ratio may have therapeutic benefit in the treatment of diabetes. Indeed, impaired glucose-stimulated insulin secretion and glucose tolerance in Nampt+/− mice were corrected by supplementation with NMN (201). Similar salutary effects were found in age- and diet-related models of insulin resistance (197, 271). NAM has long been known to protect against the development of diabetes in streptozotocin-treated mice (212). The Otsuka Long-Evans Tokushima Fatty (OLETF) rat model of obesity and diabetes also exhibited improved glucose homeostasis with NAM treatment (266). Nicotinamide riboside protected against diet-induced obesity associated with increased mitochondrial content in brown adipose and skeletal muscle, increased energy expenditure, and improved insulin sensitivity (26).
Normalization of the NAD/NADH ratio activates sirtuin expression and activity. Indeed, substantial evidence suggests that sirtuin deficiency contributes to disease pathogenesis. Nicotinamide riboside supplementation boosted SIRT1 and SIRT3 activities in the liver (26). SIRT1 transgenic mice have normal insulin sensitivity at baseline, but decreased energy expenditure. When challenged, they are able to prevent the development of insulin resistance through increased hepatic insulin sensitivity and decreased whole-body energy requirements (8). SIRT3 deletion causes impaired glucose tolerance, hepatic steatosis, and metabolic syndrome (88). Similarly, SIRT3 activity is reduced in several models of obesity and type 2 diabetes (9, 88, 111, 117). SIRT3 plays a critical role in pancreatic β cell function where its deficiency causes increased ROS production, impaired insulin secretion, and increased apoptosis. SIRT3 is also required for normal insulin action in skeletal muscle, and SIRT3 knockout mice exhibit exaggerated fatty acid oxidation and decreased glucose oxidation in skeletal muscle when fed a high-fat diet (132). Moreover, SIRT3 knockdown blocked the protective effects of NMN (28).
In addition to decreased NAD/NADH ratio, diabetes has been associated with a decrease in absolute NAD in animal models (17). Increased PARP activity may account for some of this finding. Leukocytes from pregnant women demonstrate increasing PARP activity over the course of gestation and the high PARP activity correlated with the development of gestational diabetes (94). A variety of PARP inhibitors attenuate hyperglycemia, NAD depletion, and β cell destruction (245). Similarly, Parp1−/− mice are protected against streptozotocin-induced β cell death and hyperglycemia (159). These animals also have increased mitochondrial content, increased energy expenditure, and resistance to metabolic diseases induced by a high-fat diet (6).
Myocardial infarction
Pyridine nucleotides have a marked impact on the pathophysiology of myocardial ischemia (237). Similar to hypoxia, myocardial ischemia prevents NADH oxidation by the electron transport chain resulting in the accumulation of intracellular NADH. Increased carbon flux through glycolysis, the polyol pathway, and fatty acid β oxidation also contribute to NADH accumulation.
Electron transport chain dysfunction early in myocardial ischemia increases cytoplasmic NADH. Too much NADH will inhibit GAPDH, requiring the activity of LDH to oxidize NADH to produce lactate and to facilitate continued ATP production by glycolysis. Lactate can be excreted from the cardiomyocytes and cleared from the tissue. If the ischemic insult is too severe, however, lactate cannot be cleared. Lactate will continue to accumulate until blood flow is restored, which will contribute to inhibition of glycolysis, LDH activity, and lactate export (207). In addition to NAD recycling, lactate plays a critical role buffering intracellular pH as lactate is excreted with a proton by the monocarboxylate transporter (78). For every glucose molecule hydrolyzed to pyruvate, two protons are generated. Moreover, one proton is produced for every ATP hydrolyzed by energy requiring reactions in cells (e.g., solute transport, myosin cycling). Normally, net proton production by glycolysis is offset by proton utilization by the electron transport chain, however, in conditions of myocardial ischemia where LDH activity and lactate efflux cannot compensate for the rate of proton production, intracellular acidosis occurs (204). This accumulation of protons, lactate, and NADH may contribute to myocardial injury in ischemia (42, 172).
Myocardial ischemia also enhances glucose flux through the polyol pathway due to increased activity of aldose reductase and sorbitol dehydrogenase (Fig. 14). Isolated rodent heart perfusion studies show increased aldose reductase activity under conditions of low-flow ischemia and increased sorbitol dehydrogenase activity under global ischemia (98–100). Inhibition of aldose reductase with zopolrestat increased the NAD/NADH ratio, increased ATP levels, and improved functional recovery in perfused rat hearts (195, 196). Zopolrestat enhances glycolysis and glucose oxidation at baseline and during low-flow ischemia (100). By contrast, isolated hearts from mice overexpressing human aldose reductase had impaired functional recovery and increased release of creatine kinase, indicating worse myocardial injury, associated with reduced glucose oxidation, ATP levels, and decreased NAD/NADH (99). These effects were ameliorated by zopolrestat.
In the ischemic myocardium, most of the residual oxidative metabolism utilizes acetyl-CoA derived from fatty acid β oxidation (62, 257). NADH from fatty acid oxidation contributes to uncoupling of glycolysis from glucose oxidation by inhibiting PDH. This uncoupling exacerbates myocardial acidosis by enhancing lactate production. Inhibition of PDK with dichloroacetate partially restores glucose oxidation and improves cardiac efficiency and contractile function in perfused rat hearts (62). Inhibiting fatty acid β oxidation with a malonyl CoA decarboxylase inhibitor using demand-induced ischemia in pigs reduced myocardial lactate accumulation and improved left ventricular work (53). Continued inhibition of fatty acid oxidation during reperfusion has also been shown to improve recovery from ischemic insult (53, 54, 238).
Perturbations of NADPH metabolism under ischemic conditions also likely play a critical role in mediating myocardial injury and dysfunction. As NADPH oxidases are a ready source of ROS, it is relatively straightforward to suppose that increased NADPH oxidase activity (perhaps driven, at least in part, through increased NAD(P)H availability under ischemic conditions via the mechanisms discussed above) drives oxidant injury in myocardial ischemia or ischemia/reperfusion. Indeed, a number of published studies have implicated NADPH oxidase isoforms, particularly NOX2 and NOX4, in the injury that follows ischemia/reperfusion (138, 161, 194). It would seem, then, that inhibition of NADPH oxidases would be a high-yield strategy for mitigating myocardial ischemic injury. This would not only limit ROS production by NADPH oxidases, it would also increase the availability of NADPH for channeling into antioxidant defense mechanisms to offset ROS produced by other routes during ischemia/reperfusion, such as reverse electron transfer being driven by succinate accumulation in mitochondria (38).
Consistent with this thinking, there are published studies showing benefit to NADPH oxidase inhibition in models of myocardial ischemia/reperfusion (20, 125, 218). However, the situation appears to be more complex. Broad-based inhibition of NADPH oxidase isoforms has been reported to worsen ischemia/reperfusion injury, in part, by inadvertent deleterious decrease in downstream HIF1α and increase in downstream PPARα (160). It has also been shown that both overexpression and quantitative suppression of NOX4 activity worsen myocardial ischemia/reperfusion injury, with overexpression driving oxidant injury and expression of a dominant-negative NOX4 driving reductive stress. In particular, the dominant-negative NOX4 mice showed severe mitochondrial injury during the ischemic period, with a paradoxical increase in ROS that was partially ameliorated by restoring redox balance through increasing the NAD/NADH ratio (272).
Heart failure
Failing hearts increase flux through the PPP (76, 77, 114). In dogs with heart failure, G6PD activity increases and was associated with increased NADPH and superoxide (77). These findings were also observed in discarded left ventricular tissue from patients with heart failure undergoing ventricular restoration procedures (e.g., ventricular assist device placement) (76). Inhibition of the PPP with 6-aminonicotinamide in dogs with heart failure normalized hyperglycemia-induced cardiac isoprostane production, a marker of oxidative stress, and increased cardiac oxygen consumption, glucose oxidation, and stroke work (246). By contrast, G6PD-deficient mice have slightly worse left ventricular dysfunction following myocardial infarction or transverse aortic constriction (85). These studies implicate the PPP in the pathogenesis of heart failure. However, additional studies based on pharmacologic inhibition of the pathway in animal models are warranted to clarify its role in the development of heart failure. Moreover, the mechanisms of increased PPP flux and G6PD activity remain to be elucidated.
Mitochondrial dysfunction and decreased NAD/NADH ratio have also been implicated in the pathogenesis of heart failure (135). This was associated with protein hyperacetylation and SIRT3 inhibition. Increasing the NAD/NADH ratio with NMN supplementation improved left ventricular contractility, dilation, and hypertrophy in both mechanical and genetic models of murine heart failure. Similarly, cardiac overexpression of NAMPT was protective in isoproterenol-induced heart failure. A proteomic analysis of protein acetylation revealed hyperacetylation and inhibition of the malate–aspartate shuttle, which would exacerbate heart failure-induced decreases in cytoplasmic NAD/NADH. Shuttle activity normalized with NAMPT overexpression. Furthermore, hyperacetylation increased the sensitivity of mitochondrial permeability transition pore opening, which was partially reversed by NMN supplementation. These findings suggest that restoring the NAD/NADH ratio is a potential therapeutic goal in the treatment of heart failure (135).
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a deadly disease characterized by progressive obliteration of the pulmonary vascular lumen due to excessive proliferation of endothelial, smooth muscle, and fibroblast cells associated with perivascular inflammation and fibrosis. Affected vascular cells exhibit Warburg metabolism associated with a decreased NAD/NADH ratio (60, 141, 224). Presumably, these metabolic changes support the dysregulated cell growth that is characteristic of the disease and targeting these pathways has had some therapeutic benefits. For example, the PDK inhibitor, dichloroacetate, will ameliorate experimental pulmonary hypertension in rodents by increasing glucose oxidation (166, 180). Decreased NAD/NADH has been shown to activate C-terminal binding protein 1 (CTBP1) in adventitial fibroblasts isolated from humans and cows with pulmonary hypertension (141). Inhibition of CTBP1, a transcriptional repressor that regulates genes involved with cell renewal and pluripotency, genome stability, and epithelial differentiation, attenuated pulmonary vascular cell proliferation, inflammation, and remodeling (141). Recently, NAMPT was shown to be increased in patients with PAH and animal models of the disease (34). Nampt+/− mice develop less severe pulmonary hypertension when exposed to chronic hypoxia, and treatment with FK866 ameliorates experimental pulmonary hypertension in rats (34). Unfortunately, the effects of these interventions on the cellular NAD/NADH ratio were not assessed in this study. Thus, the regulation of cellular NAD/NADH is an active and promising area of investigation in the pathobiology and treatment of pulmonary vascular disease.
Several groups have reported activation of the PPP in cell and animal models of PAH. In a chronic hypoxia model of PAH, G6PD was overexpressed in pulmonary artery smooth muscle cells and in lungs from chronically hypoxic rats, with a corresponding increase in G6PD enzymatic activity (36). A snapshot metabolomic analysis of human pulmonary microvascular endothelial cells expressing either wild-type or mutant isoforms of bone morphogenetic protein receptor type 2 known to cause heritable PAH showed significant increases in multiple PPP intermediates, despite a downregulation of G6PD at the gene expression level (59). Pulmonary artery smooth muscle cells exposed to supraphysiologic flow rates in an ovine model of the PAH seen in children with congenital heart disease showed reduced NADPH/NADP ratios, despite a proposed increase in PPP flux. The authors accounted for this apparent discrepancy by hyperactivation of NADPH oxidase isoform NOX2, and they subsequently showed that inhibition of NOX2 would resolve the hyperproliferative phenotype of the pulmonary artery smooth muscle cells exposed to excess flow (16). It seems very likely that changes in NAD(P)/NAD(P)H ratios and availability play some role in the pathogenesis of PAH. However, the details remain to be elucidated and any effective therapies targeting the PPP will need to be tailored to the molecular details.
Immune function
A rapidly expanding body of research implicates metabolic programming, substrate-level metabolism, and redox balance as being key processes in the function and regulation of nearly all parts of the innate and adaptive immune systems (116, 162, 255). NAD(H) and NADP(H) are thus optimally positioned to serve as dynamic regulators of immune function, and accumulating evidence suggests that this is likely the case. Although an exhaustive review of the topic is well beyond the scope of this article, illustrative examples may be useful. These examples will focus on unique aspects of nicotine adenine dinucleotide biology in the immune system, apart from mechanisms previously discussed in more detail elsewhere (e.g., NADPH oxidase, antioxidant defense pathways, and sirtuin activation), many of which have also been shown to regulate innate and adaptive immune function.
A study recently published by Sanman et al. showed that macrophages require a decrease in availability of NADH produced through glycolytic flux for efficient activation of the NLRP3 inflammasome, proinflammatory cytokine responses, and pyroptotic macrophage cell death in response to the presence of an intracellular pathogen (211). They show that infection with the intracellular pathogen Salmonella typhimurium disrupts macrophage glycolysis through competitive consumption of glycolytic intermediates, the net effect of which is to reduce availability of NADH leading to an increase in mitochondrial ROS flux and assembly of the NLRP3 inflammasome. Exogenous replacement of metabolites that enhanced NADH production (e.g., pyruvate, succinate, or l-glutamine) prevented the assembly of the NLRP3 inflammasome and prevented macrophage cell death.
The metabolic profiles of memory and effector T lymphocytes, particularly CD8+ T lymphocytes, have been shown to be intimately linked to function. For example, memory T cells have been shown to engage in both fatty acid synthesis and fatty acid oxidation simultaneously, in what has been described as a futile cycle (179). This futile cycle is required for maintenance of the memory phenotype, and modulation of the overall metabolic program is required for the generation of a T effector memory response (262). A recent study from Ali et al. suggests that changes in NADP pools can have potent effects on naive and effector T cells through calcium mobilization by way of generation of NAADP (3). NAADP is a potent stimulus for release of calcium from intracellular stores, and production of NAADP is thought to be achieved through deamidation of NADP and using nicotinic acid as the amide acceptor (a reaction accomplished by ADP-ribosyl cyclases in vitro, less clearly so in vivo), yielding NAADP and nicotinamide (Fig. 3). Ali and colleagues showed that increased availability of NAADP in naive CD4+ and CD8+ T cells as well as in effector CD8+ T cells resulted in calcium mobilization sufficient to drive physiologically relevant proliferation and cytokine production responses such as would be expected downstream from T cell receptor activation. They also demonstrated that this is not a universal property of T lymphocytes, as natural T regulatory cells did not exhibit similar responses.
Renal function
NAD/NADH metabolism has been implicated as a key mediator of normal renal function and protection/recovery from kidney injury. A recent study from Tran et al. reported that PGC1α is a critical mediator of resistance to/recovery from acute kidney injury via regulation of NAD biosynthesis (233). Loss of PGC1α resulted in decreased nicotinamide levels, fat accumulation, and loss of normal renal function in the face of an ischemic insult. Restoration of NAD levels via supplementation with exogenous nicotinamide in the setting of an ischemic insult to the kidney improved fat accumulation and renal function in Pgc1a−/− mice. PGC1α was shown to act by coordinately upregulating multiple enzymes in the NAD synthetic pathway, and nicotinamide supplementation specifically required metabolism via NAMPT to mediate protection or recovery from acute kidney injury. Downstream, the protective effects of PGC1α and NAD required signaling via β-hydroxybutyrate and prostaglandin E2.
This study and others reflect the fact that particular portions of the nephron exhibit restricted metabolic behavior and thus specific metabolic requirements for maintenance of NAD/NADH balance. In general, the proximal tubule cannot use glycolysis to sustain metabolic and redox needs due to a lack of expression of hexokinases and, instead, must rely on fatty acid oxidation and amino acid metabolism (40). Similarly, the enzymes directly involved in NAD(P)/NAD(P)H synthesis, balance, and interconversion exhibit specific patterns of regional expression. For example, NADK isoforms exhibit particularly high expression in the collecting portions of the nephron, although the specific isoforms exhibit their own patterns of expression, with NADK more evenly distributed, whereas the mitochondrial NADK2 exhibits particularly high expression in the inner medullary collecting duct and the long descending limb of the inner medulla. This type of regionalization of expression implies specific regional vulnerabilities to metabolic and redox stresses that are only beginning to be established and understood.
Pharmacology of NAD(P) Metabolism
There is increasing interest in targeting NAD(P) metabolism directly as a therapeutic goal in a wide variety of diseases. As the sections above illustrate, NAD(P) metabolism plays a part in the pathogenesis of many of the most prevalent chronic diseases. These are the diseases that account for a huge proportion of adult morbidity and mortality, and they are thus precisely the diseases in need of improved therapies. Several strategies have been and are being actively explored to impact on NAD(P) metabolism.
As has been alluded to above, one of the most straightforward strategies for manipulation of NAD(P) balance is exogenous supplementation of precursors to support NAD(P) production. Several recent studies have shown that NAD levels can be supported or restored in different disease states by supplementation with nicotinamide riboside (209, 274). Nicotinamide riboside appears to be effective for increasing intracellular NAD in murine models and likely in humans as well when supplemented in the diet (231). Supplementation with nicotinamide riboside and NMN relies on NRK activity (199). The strategy of supplementing NAD precursors appears to broadly be effective in humans as well, although predicting beneficial effects accurately may prove difficult as these therapies are developed.
These points were demonstrated directly by van de Weijer et al., who conducted a small randomized trial of the nicotinic acid derivative acipimox compared to placebo in 21 patients with type 2 diabetes mellitus (239). Acipimox treatment increased mitochondrial respiration in skeletal muscle, increased expression of nuclear-encoded mitochondrial genes, drove a protein imbalance in the mitochondrion that may have activated the mitochondrial UPR, and increased NAD levels in cultured myotubes. Despite these apparently beneficial molecular effects, the acipimox group showed an increase in circulating nonesterified fatty acid concentrations, an increase in skeletal muscle lipid content, and a decrease in skeletal muscle insulin sensitivity. Parsing and predicting the beneficial and potentially deleterious effects of precursor supplementation strategies will require significantly more investigation, with a focus on details such as changes in NAD(P) metabolism in specific subcellular compartments in specific tissues.
A variety of more classical pharmacologic approaches to modulating NAD metabolism have been described, although many of the published studies were not necessarily framed in this way. Perhaps the best known pharmacologic modulator of NAD metabolism is metformin, the representative drug of the biguanides. Best studied as a treatment for type 2 diabetes mellitus, metformin has been suggested to have beneficial effects in virtually all of the diseases discussed thus far. Although several mechanisms of action have been ascribed to metformin, most or all of them converge on modulation of NAD metabolism. Metformin and other biguanides (e.g., phenformin) have been suggested to exert their beneficial effects through inhibition of mitochondrial Complex I (NADH:ubiquinone oxidoreductase) at concentrations achieved with standard therapeutic doses (31, 256). This, of course, tends to drive a decrease in the NAD/NADH ratio and decreased mitochondrial availability of NAD, although these effects are likely context specific (74).
Metformin's mechanism of action for the inhibition of hepatic gluconeogenesis has been proposed to be inhibition of mitochondrial GPDH (Fig. 7) (155). This interrupts the glycerol phosphate shuttle, a cytosolic-mitochondrial redox couple, which should result in accumulation of NAD in the cytosol (as cytosolic GPD1 converts DHAP to G3P, normally consuming NADH in the process unless inhibited by metformin). When metformin is present, cytosolic NADH accumulates at the expense of NAD, and this has the effect of inhibiting the redox-coupled exchange of malate and oxaloacetate between the mitochondrion and cytosol. Given that cytosolic oxaloacetate is required for gluconeogenesis to proceed, interruption of this redox couple could be sufficient to inhibit gluconeogenesis; indeed, the argument from redox disturbance seems to be sufficient, as changes in expression of key gluconeogenic enzymes were not observed with metformin dosing (155). The redox balance on the mitochondrial side of this redox couple is somewhat more complicated, as whole cell NAD(P)/NAD(P)H ratios did not change with metformin administration. It is possible that metformin's ability to inhibit Complex I allows for NADH accumulation in the mitochondrion that balances the cytosolic accumulation of NAD, although this is conjecture and would need to be rigorously tested. Nonetheless, inhibition of the glycerol phosphate shuttle represents a second mechanism by which metformin can be used to manipulate NAD(P) metabolism. Finally, metformin activates AMPK, which has the effect of increasing cellular NAD levels overall (25, 283).
Fewer options exist for the direct regulation of NADPH levels in specific cellular compartments or tissues. There is a great deal of interest in developing clinically useful NADPH oxidase inhibitors, although none has yet been approved for use in humans. Dehydroepiandrosterone has been reported to inhibit G6PD, which would have the effect of reducing NADPH availability (214). N-Acetylcysteine could theoretically increase NADPH levels by serving as a direct substrate for glutathione synthesis and/or preservation, thus preserving NADPH levels by reducing the activity of glutathione peroxidase. NADPH oxidase expression has also been reported to decrease with N-acetylcysteine treatment, which should also have the effect of preserving NADPH levels (75).
Apart from the strategies to target NAD(P) directly, there has been significant interest in and research devoted to developing novel therapeutics that target many of the pathways directly affected by nicotine nucleotides. Some of these approaches have leveraged existing therapeutics developed for other purposes. As one example, dicholoracetate, as mentioned above, is an inhibitor of PDK, which has the effect of stimulating PDH activity and enhancing pyruvate metabolism through oxidative pathways as opposed to lactate production. Increased PDH activity would directly contribute to NADH production, and the increased flow of pyruvate-derived carbon into the TCA cycle and away from LDH would have an even greater effect to drive NADH production.
Similarly, other metabolic modulators could be predicted to shift the NAD(P) balance, although their direct targets and intended effects, like dichloroacetate, are not specifically geared toward NAD(P). The PARP inhibitors olaparib and rucaparib, developed as cancer therapeutics (particularly for treatment-resistant and/or BRCA-mutated ovarian cancer), would be predicted to raise NAD levels by preventing the consumption of NAD for poly-ADP-ribosylation, although the intended mechanism of action is sensitizing tumor DNA to strand breaks. Other approaches that indirectly touch on NAD(P) metabolism have involved the development of novel therapeutics targeting specific NAD(P) responsive targets. Direct activators of sirtuins, inhibitors of NADPH oxidase isoforms, next-generation inhibitors of PARP family enzymes, and a variety of other classes of compounds have all been taken to various stages of development.
Conclusion
As the reader can appreciate, the metabolism and regulation of nicotine adenine dinucleotides are the central cellular function that touches on and influences many other fundamental processes in every living cell. We have broadly conceived of the intracellular metabolic environment as a complex of multiple interacting “economies” in which resources are allocated in a dynamic way based upon availability and need at any given time and place within the cell. Just as nucleotide triphosphates (primarily ATP) can be thought of as the “energy currency” of the cell, we offer the concept of nicotine adenine dinucleotides as the “redox currency” of the cell.
With all that is known regarding NAD(P) metabolism, there are still many questions to be answered. Compartmentalization of NAD(P) pools is clearly of major importance to the regulation and function of NAD(P)-dependent processes. Mitochondrial pools are not in a free equilibrium with cytosolic pools. Indeed, one of the major tasks accomplished by the malate–aspartate shuttle is the movement of redox currency in the form of NADH from the cytosol (where NADH is oxidized to NAD) into the mitochondrial matrix (where NAD is reduced to NADH). Fundamental to this is how NAD levels are maintained in the mitochondrion to participate in this and many other redox reactions. This is a largely obscure process, although some data suggest that cytosolic synthesis of NMN from more cell-permeable precursors is followed by mitochondrial uptake of NMN and further metabolism by NMNAT3 to NAD (using local ATP as the other required substrate)—in short, suggesting that local NAD synthesis from specific precursors is responsible for maintaining mitochondrial pools (174). However, it is still unclear how NMN, the required precursor, enters into the mitochondrion, so this is a clearly an area in need of further inquiry (219).
Maintenance of NADP pools is also an area with some significant remaining unanswered questions. The action of NADK is primarily responsible for the conversion of NAD to NADP, and just as NAD pools are localized at least to cytosolic and mitochondrial compartments, the two recognized isoforms of NADK localize to the cytosol (NADK) or mitochondria (mNADK or NADK2). NADK2 mutation/deficiency has been suggested as the underlying genetic cause of dienoyl-CoA reductase deficiency with hyperlysinemia, a rare genetic disorder characterized biochemically by increased plasma levels of C10:2-carnitine and of lysine, and characterized clinically by severe encephalopathy and severe neurologic and metabolic dysfunction (96, 206). A case of partial NADK2 deficiency has recently been identified through the NIH Undiagnosed Diseases Network. This patient presented with neurologic and metabolic complaints that are similar in nature but much less severe than what is seen with dienoyl-CoA reductase deficiency with hyperlysinemia, consistent with a partial loss of function mutation (J.A. Phillips, pers. comm.). Interestingly, what is not seen with NADK2 deficiency is any kind of clinical syndrome reminiscent of CGD, perhaps suggesting that NADPH production and availability—and thus function—are heavily compartmentalized. These are just early steps toward understanding the relative importance of cytosolic and mNADK isoforms and the related compartmentalization of NADPH.
Finally, as touched upon in this review, there is a growing recognition of the importance of “redox couples” in the regulation of cellular redox biology and metabolism. Some of these are classically recognized redox couples, such as the malate–aspartate shuttle and the G3P shuttle. With the rapidly growing interest in and focus on research into cellular metabolism, previously unrecognized redox couples and their associated biology have come to light, such as the PDH-NNT circuit described above, and the redox couple between α-ketoglutarate and L-2-hydroxyglutarate recently shown to regulate bioenergetic metabolic pathways particularly during hypoxia via coupling through NAD/NADH and FAD/FADH2 ratios (177). As these kinds of investigations progress, it seems likely that more such redox couples will be identified and will emerge as attractive therapeutic targets. To accomplish this, tools to more readily and specifically identify redox couples will need to be developed. This will likely involve the development of sensitive, dynamic indicators of NAD(P)(H) that can be efficiently targeted to specific subcellular compartments, readily visualized in either an endpoint or a time-resolved manner, and that have demonstrable utility in vivo. Better definition of the localization, functioning, and regulation of redox couples should then permit targeted manipulation for specific effects.
In sum, even after a century of research, new insights into the fundamental and far-reaching roles of pyridine dinucleotides in human metabolism continue to reform our understanding of the sophisticated interplay between energy homeostasis, cellular signaling, and gene regulation. These molecules have been implicated in nearly every chronic human disease and their biochemistry provides multiple promising targets for therapeutic intervention. Doubtless, not another century will pass before this promise is realized.
Abbreviations Used
- ACMSD
α-amino-β-carboxymuconate-ɛ-semialdehyde decarboxylase
- ACP
acyl carrier protein
- ADP
adenosine diphosphate
- ADPRP
ADP-ribose(2′-phosphate)
- ATP
adenosine triphosphate
- cADPRP
cyclic ADP-ribose(2′-phosphate)
- CGD
chronic granulomatous disease
- cMDH
cytoplasmic malate dehydrogenase
- CTBP1
C-terminal binding protein 1
- DHAP
dihydroxyacetone phosphate
- dNDP
deoxyribonucleotide diphosphates
- FAD
flavin adenine dinucleotide
- G3P
glycerol-3-phosphate
- G6P
glucose-6-phosphate
- G6PD
G6P dehydrogenase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GLUD
glutamate dehydrogenase
- GPDH
glycerol-3-phosphate dehydrogenase
- HIF1α
hypoxia inducible factor 1α
- HMG-CoA
3-hydroxy-3-methylglutaryl-CoA
- IDH
isocitrate dehydrogenase
- LDH
lactate dehydrogenase
- MDH
malate dehydrogenase
- ME
malic enzyme
- mNADK
mitochondrial NAD kinase
- NAAD
nicotinic acid adenine dinucleotide
- NAADP
nicotinic acid adenine dinucleotide phosphate
- NAD
nicotinamide adenine dinucleotide
- NADH
nicotinamide adenine dinucleotide, reduced form
- NADK
NAD kinase
- NADP
nicotinamide adenine dinucleotide phosphate
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced form
- NAMN
nicotinic acid mononucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- NAPRT
nicotinic acid phosphoribosyltransferase
- NDP
ribonucleotide diphosphate
- NHD
Nudix homology domain
- NMN
nicotinamide mononucleotide
- NMNAT
nicotinamide mononucleotide adenylyltransferase
- NNT
nicotinamide nucleotide transhydrogenase
- NO
nitric oxide
- NOS
nitric oxide synthase
- NRK
nicotinamide riboside kinase
- OGDH
α-ketoglutarate dehydrogenase
- PAH
pulmonary arterial hypertension
- PARP
poly-ADP-ribose polymerase
- PDH
pyruvate dehydrogenase
- PDK
pyruvate dehydrogenase kinase
- PHD
prolyl hydroxylase
- PPP
pentose phosphate pathway
- RNR
ribonucleotide reductase
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- TCA
tricarboxylic acid
- UPR
unfolded protein response
- VHL
Von Hippel Lindau
References
- 1.Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press; 1998, p. 592. [PubMed] [Google Scholar]
- 2.Alano CC, Tran A, Tao R, Ying W, Karliner JS, and Swanson RA. Differences among cell types in NAD(+) compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res 85: 3378–3385, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Ali RA, Camick C, Wiles K, Walseth TF, Slama JT, Bhattacharya S, Giovannucci DR, and Wall KA. Nicotinic acid adenine dinucleotide phosphate plays a critical role in naive and effector murine T cells but not natural regulatory T cells. J Biol Chem 291: 4503–4522, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson KA, Green MF, Huynh FK, Wagner GR, and Hirschey MD. SnapShot: mammalian sirtuins. Cell 159: 956–956.e1, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avalos JL, Bever KM, and Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell 17: 855–868, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, and Auwerx J. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13: 461–468, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ball ZB, Barnes RH, and Visscher MB. The effects of dietary caloric restriction on maturity and senescence, with particular reference to fertility and longevity. Am J Physiol 150: 511–519, 1947 [DOI] [PubMed] [Google Scholar]
- 8.Banks AS, Kon N, Knight C, Matsumoto M, Gutierrez-Juarez R, Rossetti L, Gu W, and Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab 8: 333–341, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bao J, Scott I, Lu Z, Pang L, Dimond CC, Gius D, and Sack MN. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic Biol Med 49: 1230–1237, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, and Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 56: 952–964, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Ben-Bassat A. and Goldberg I. Purification and properties of glucose-6-phosphate dehydrogenase (NADP+/NAD+) and 6-phosphogluconate dehydrogenase (NADP+/NAD+) from methanol-grown Pseudomonas C. Biochim Biophys Acta 611: 1–10, 1980 [DOI] [PubMed] [Google Scholar]
- 12.Bieganowski P. and Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 117: 495–502, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Bilan DS. and Belousov VV. Genetically encoded probes for NAD+/NADH monitoring. Free Radic Biol Med 100: 32–42, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Blumenthal KM, Moon K, and Smith EL. Nicotinamide adenine dinucleotide phosphate-specific glutamate dehydrogenase of Neurospora. J Biol Chem 250: 3644–3654, 1975 [PubMed] [Google Scholar]
- 15.Blumenthal KM. and Smith EL. Alternative substrates for glutamate dehydrogenases. Biochem Biophys Res Commun 62: 78–84, 1975 [DOI] [PubMed] [Google Scholar]
- 16.Boehme J, Sun X, Tormos KV, Gong W, Kellner M, Datar SA, Kameny RJ, Yuan JX, Raff GW, Fineman JR, Black SM, and Maltepe E. Pulmonary artery smooth muscle cell hyperproliferation and metabolic shift triggered by pulmonary overcirculation. Am J Physiol Heart Circ Physiol 311: H944–H957, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolaffi JL, Rodd GG, Wang J, and Grodsky GM. Interrelationship of changes in islet nicotine adeninedinucleotide, insulin secretion, and cell viability induced by interleukin-1 beta. Endocrinology 134: 537–542, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, and Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 274: 22932–22940, 1999 [DOI] [PubMed] [Google Scholar]
- 19.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, and Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One 6: e19194, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braunersreuther V, Montecucco F, Asrih M, Pelli G, Galan K, Frias M, Burger F, Quindere AL, Montessuit C, Krause KH, Mach F, and Jaquet V. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol 64: 99–107, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Bruzzone S, Guida L, Zocchi E, Franco L, and De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J 15: 10–12, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Butepage M, Eckei L, Verheugd P, and Luscher B. Intracellular mono-ADP-ribosylation in signaling and disease. Cells 4: 569–595, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cameron WD, Bui CV, Hutchinson A, Loppnau P, Graslund S, and Rocheleau JV. Apollo-NADP(+): a spectrally tunable family of genetically encoded sensors for NADP(+). Nat Methods 13: 352–358, 2016 [DOI] [PubMed] [Google Scholar]
- 24.Canepa L, Ferraris AM, Miglino M, and Gaetani GF. Bound and unbound pyridine dinucleotides in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Biochim Biophys Acta 1074: 101–104, 1991 [DOI] [PubMed] [Google Scholar]
- 25.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, and Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, and Auwerx J. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab 15: 838–847, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canto C, Menzies KJ, and Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22: 31–53, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caton PW, Richardson SJ, Kieswich J, Bugliani M, Holland ML, Marchetti P, Morgan NG, Yaqoob MM, Holness MJ, and Sugden MC. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 56: 1068–1077, 2013 [DOI] [PubMed] [Google Scholar]
- 29.Chance B, Cohen P, Jobsis F, and Schoener B. Intracellular oxidation-reduction states in vivo. Science 137: 499–508, 1962 [DOI] [PubMed] [Google Scholar]
- 30.Chance B. and Thorell B. Localization and kinetics of reduced pyridine nucleotide in living cells by microfluorometry. J Biol Chem 234: 3044–3050, 1959 [PubMed] [Google Scholar]
- 31.Chandel NS, Avizonis D, Reczek CR, Weinberg SE, Menz S, Neuhaus R, Christian S, Haegebarth A, Algire C, and Pollak M. Are metformin doses used in murine cancer models clinically relevant? Cell Metab 23: 569–570, 2016 [DOI] [PubMed] [Google Scholar]
- 32.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, and Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A 95: 11715–11720, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, and Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev 22: 1753–1757, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Sysol JR, Singla S, Zhao S, Yamamura A, Valdez-Jasso D, Abbasi T, Shioura KM, Sahni S, Reddy V, Sridhar A, Gao H, Torres J, Camp SM, Tang H, Ye SQ, Comhair S, Dweik R, Hassoun P, Yuan JX, Garcia JG, and Machado RF. Nicotinamide phosphoribosyltransferase promotes pulmonary vascular remodeling and is a therapeutic target in pulmonary arterial hypertension. Circulation 135: 1532–1546, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Walter W, Freinkman E, Wang T, Birsoy K, and Sabatini David M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166: 1324–1337.e11, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, and Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiriaco M, Salfa I, Di Matteo G, Rossi P, and Finocchi A. Chronic granulomatous disease: clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol 27: 242–253, 2016 [DOI] [PubMed] [Google Scholar]
- 38.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, and Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conforti L, Gilley J, and Coleman MP. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat Rev Neurosci 15: 394–409, 2014 [DOI] [PubMed] [Google Scholar]
- 40.Corcoran CC, Grady CR, Pisitkun T, Parulekar J, and Knepper MA. From 20th century metabolic wall charts to 21st century systems biology: database of mammalian metabolic enzymes. Am J Physiol Renal Physiol 312: F533–F542, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, Church TS, Jubrias SA, Conley KE, and Smith SR. Skeletal muscle NAMPT is induced by exercise in humans. Am J Physiol Endocrinol Metab 298: E117–E126, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cross HR, Clarke K, Opie LH, and Radda GK. Is lactate-induced myocardial ischaemic injury mediated by decreased pH or increased intracellular lactate? J Mol Cell Cardiol 27: 1369–1381, 1995 [DOI] [PubMed] [Google Scholar]
- 43.Danielson L. and Ernster L. Demonstration of a mitochondrial energy-dependent pyridine nucleotide transhydrogenase reaction. Biochem Biophys Res Commun 10: 91–96, 1963 [DOI] [PubMed] [Google Scholar]
- 44.Davis LC, Morgan AJ, Ruas M, Wong JL, Graeff RM, Poustka AJ, Lee HC, Wessel GM, Parrington J, and Galione A. Ca(2+) signaling occurs via second messenger release from intraorganelle synthesis sites. Curr Biol 18: 1612–1618, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Jesus-Cortes H, Miller AD, Britt JK, DeMarco AJ, De Jesus-Cortes M, Stuebing E, Naidoo J, Vazquez-Rosa E, Morlock L, Williams NS, Ready JM, Narayanan NS, and Pieper AA. Protective efficacy of P7C3-S243 in the 6-hydroxydopamine model of Parkinson's disease. NPJ Parkinsons Dis 1: pii: , 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Jesus-Cortes H, Xu P, Drawbridge J, Estill SJ, Huntington P, Tran S, Britt J, Tesla R, Morlock L, Naidoo J, Melito LM, Wang G, Williams NS, Ready JM, McKnight SL, and Pieper AA. Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc Natl Acad Sci U S A 109: 17010–17015, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Lisa F, Menabo R, Canton M, Barile M, and Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276: 2571–2575, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Dolle C. and Ziegler M. Application of a coupled enzyme assay to characterize nicotinamide riboside kinases. Anal Biochem 385: 377–379, 2009 [DOI] [PubMed] [Google Scholar]
- 49.Drummond GR, Selemidis S, Griendling KK, and Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 10: 453–471, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du J, Jiang H, and Lin H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry 48: 2878–2890, 2009 [DOI] [PubMed] [Google Scholar]
- 51.Durieux J, Wolff S, and Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144: 79–91, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dutca LM, Stasheff SF, Hedberg-Buenz A, Rudd DS, Batra N, Blodi FR, Yorek MS, Yin T, Shankar M, Herlein JA, Naidoo J, Morlock L, Williams N, Kardon RH, Anderson MG, Pieper AA, and Harper MM. Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Invest Ophthalmol Vis Sci 55: 8330–8341, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dyck JR, Cheng JF, Stanley WC, Barr R, Chandler MP, Brown S, Wallace D, Arrhenius T, Harmon C, Yang G, Nadzan AM, and Lopaschuk GD. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res 94: e78–e84, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, Kawase Y, Jishage K, and Lopaschuk GD. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 114: 1721–1728, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Elvehjem CA, Madden RJ, Strong FM, and Woolley DW. Relation of nicotinic acid and nicotinic acid amide to canine black tongue. J Am Chem Soc 59: 1767–1768, 1937 [Google Scholar]
- 56.Esteves AR, Arduino DM, Silva DF, Viana SD, Pereira FC, and Cardoso SM. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson's Disease. Mol Neurobiol, 2017. [Epub ahead of print]; DOI: 10.1007/s12035-017-0420-y [DOI] [PubMed]
- 57.Feldman JL, Baeza J, and Denu JM. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J Biol Chem 288: 31350–31356, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Felici R, Lapucci A, Ramazzotti M, and Chiarugi A. Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PLoS One 8: e76938, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, and West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2: 201–213, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fessel JP. and Oldham WM. Pulmonary hypertension as a metabolic disease. In: Pulmonary Hypertension: Basic Science to Clinical Medicine, edited by Maron BA, Zamanian RT, and Waxman AB. Cham: Springer International Publishing, 2016, pp. 135–145. [Google Scholar]
- 61.Fisher-Wellman KH, Lin CT, Ryan TE, Reese LR, Gilliam LA, Cathey BL, Lark DS, Smith CD, Muoio DM, and Neufer PD. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy-consuming redox circuit. Biochem J 467: 271–280, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Folmes CD, Sowah D, Clanachan AS, and Lopaschuk GD. High rates of residual fatty acid oxidation during mild ischemia decrease cardiac work and efficiency. J Mol Cell Cardiol 47: 142–148, 2009 [DOI] [PubMed] [Google Scholar]
- 63.Frizzell N, Thomas SA, Carson JA, and Baynes JW. Mitochondrial stress causes increased succination of proteins in adipocytes in response to glucotoxicity. Biochem J 445: 247–254, 2012 [DOI] [PubMed] [Google Scholar]
- 64.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun 260: 273–279, 1999 [DOI] [PubMed] [Google Scholar]
- 65.Fukuwatari T. and Shibata K. Effect of nicotinamide administration on the tryptophan-nicotinamide pathway in humans. Int J Vitam Nutr Res 77: 255–262, 2007 [DOI] [PubMed] [Google Scholar]
- 66.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, and Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell 14: 661–673, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garavaglia S, D'Angelo I, Emanuelli M, Carnevali F, Pierella F, Magni G, and Rizzi M. Structure of human NMN adenylyltransferase. A key nuclear enzyme for NAD homeostasis. J Biol Chem 277: 8524–8530, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Godena VK, Brookes-Hocking N, Moller A, Shaw G, Oswald M, Sancho RM, Miller CC, Whitworth AJ, and De Vos KJ. Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat Commun 5: 5245, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, and Sinclair DA. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155: 1624–1638, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gong B, Pan Y, Vempati P, Zhao W, Knable L, Ho L, Wang J, Sastre M, Ono K, Sauve AA, and Pasinetti GM. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol Aging 34: 1581–1588, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalez RG, Barnett P, Aguayo J, Cheng HM, Chylack LT., Jr Direct measurement of polyol pathway activity in the ocular lens. Diabetes 33: 196–199, 1984 [DOI] [PubMed] [Google Scholar]
- 72.Goor RS. and Maxwell ES. A proposed mechanism for ADP ribosylation of aminoacyl transferase II by diphtheria toxin. Cold Spring Harb Symp Quant Biol 34: 609–610, 1969 [DOI] [PubMed] [Google Scholar]
- 73.Guarente L. Sir2 links chromatin silencing, metabolism, and aging. Genes Dev 14: 1021–1026, 2000 [PubMed] [Google Scholar]
- 74.Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, Davidson SM, Freinkman E, Thomas CJ, and Vander Heiden MG. Environment dictates dependence on mitochondrial complex I for NAD+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab 24: 716–727, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guo Z, Xia Z, Jiang J, and McNeill JH. Downregulation of NADPH oxidase, antioxidant enzymes, and inflammatory markers in the heart of streptozotocin-induced diabetic rats by N-acetyl-L-cysteine. Am J Physiol Heart Circ Physiol 292: H1728–H1736, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, and Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail 13: 497–506, 2007 [DOI] [PubMed] [Google Scholar]
- 77.Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, and Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol 41: 340–349, 2006 [DOI] [PubMed] [Google Scholar]
- 78.Halestrap AP. The SLC16 gene family—structure, role and regulation in health and disease. Mol Aspects Med 34: 337–349, 2013 [DOI] [PubMed] [Google Scholar]
- 79.Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, and Waskell L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem 284: 11374–11384, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hara N, Yamada K, Shibata T, Osago H, Hashimoto T, and Tsuchiya M. Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J Biol Chem 282: 24574–24582, 2007 [DOI] [PubMed] [Google Scholar]
- 81.Harden A. and Young WJ. The alcoholic ferment of yeast-juice. Proc R Soc London Ser B 77: 405–420, 1906 [Google Scholar]
- 82.Harlan BA, Pehar M, Sharma DR, Beeson G, Beeson CC, and Vargas MR. Enhancing NAD+ salvage pathway reverts the toxicity of primary astrocytes expressing amyotrophic lateral sclerosis-linked mutant superoxide dismutase 1 (SOD1). J Biol Chem 291: 10836–10846, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harris M. Pyruvate blocks expression of sensitivity to antimycin A and chloramphenicol. Somatic Cell Genet 6: 699–708, 1980 [DOI] [PubMed] [Google Scholar]
- 84.Hecker PA, Leopold JA, Gupte SA, Recchia FA, and Stanley WC. Impact of glucose-6-phosphate dehydrogenase deficiency on the pathophysiology of cardiovascular disease. Am J Physiol Heart Circ Physiol 304: H491–H500, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hecker PA, Lionetti V, Ribeiro RF, Jr, Rastogi S, Brown BH, O'Connell KA, Cox JW, Shekar KC, Gamble DM, Sabbah HN, Leopold JA, Gupte SA, Recchia FA, and Stanley WC. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ Heart Fail 6: 118–126, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hegyi J, Schwartz RA, and Hegyi V. Pellagra: dermatitis, dementia, and diarrhea. Int J Dermatol 43: 1–5, 2004 [DOI] [PubMed] [Google Scholar]
- 87.Hinkle PC. P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta 1706: 1–11, 2005 [DOI] [PubMed] [Google Scholar]
- 88.Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV, Jr, Kahn CR, and Verdin E. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell 44: 177–190, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hodgkinson AD, Sondergaard KL, Yang B, Cross DF, Millward BA, and Demaine AG. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int 60: 211–218, 2001 [DOI] [PubMed] [Google Scholar]
- 90.Holten D, Procsal D, and Chang HL. Regulation of pentose phosphate pathway dehydrogenases by NADP+/NADPH ratios. Biochem Biophys Res Commun 68: 436–441, 1976 [DOI] [PubMed] [Google Scholar]
- 91.Honjo T, Nishizuka Y, and Hayaishi O. Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J Biol Chem 243: 3553–3555, 1968 [PubMed] [Google Scholar]
- 92.Horecker BL. Transketolase and transaldolase. In: Comprehensive Biochemistry, Vol 15, edited by Florkin M, and Stotz H. Amsterdam: Elsevier, 1964, pp. 48–70. [Google Scholar]
- 93.Horecker BL. The pentose phosphate pathway. J Biol Chem 277: 47965–47971, 2002 [DOI] [PubMed] [Google Scholar]
- 94.Horvath EM, Magenheim R, Kugler E, Vacz G, Szigethy A, Levardi F, Kollai M, Szabo C, and Lacza Z. Nitrative stress and poly(ADP-ribose) polymerase activation in healthy and gestational diabetic pregnancies. Diabetologia 52: 1935–1943, 2009 [DOI] [PubMed] [Google Scholar]
- 95.Horwitt MK, Harper AE, and Henderson LM. Niacin-tryptophan relationships for evaluating niacin equivalents. Am J Clin Nutr 34: 423–427, 1981 [DOI] [PubMed] [Google Scholar]
- 96.Houten SM, Denis S, Te Brinke H, Jongejan A, van Kampen AH, Bradley EJ, Baas F, Hennekam RC, Millington DS, Young SP, Frazier DM, Gucsavas-Calikoglu M, and Wanders RJ. Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia. Hum Mol Genet 23: 5009–5016, 2014 [DOI] [PubMed] [Google Scholar]
- 97.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, and Auwerx J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497: 451–457, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hwang YC, Bakr S, Ellery CA, Oates PJ, and Ramasamy R. Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. FASEB J 17: 2331–2333, 2003 [DOI] [PubMed] [Google Scholar]
- 99.Hwang YC, Kaneko M, Bakr S, Liao H, Lu Y, Lewis ER, Yan S, Ii S, Itakura M, Rui L, Skopicki H, Homma S, Schmidt AM, Oates PJ, Szabolcs M, and Ramasamy R. Central role for aldose reductase pathway in myocardial ischemic injury. FASEB J 18: 1192–1199, 2004 [DOI] [PubMed] [Google Scholar]
- 100.Hwang YC, Sato S, Tsai JY, Yan S, Bakr S, Zhang H, Oates PJ, and Ramasamy R. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J 16: 243–245, 2002 [DOI] [PubMed] [Google Scholar]
- 101.Ieraci A. and Herrera DG. Nicotinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med 3: e101, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ijichi H, Ichiyama A, and Hayaishi O. Studies on the biosynthesis of nicotinamide adenine dinucleotide. 3. Comparative in vivo studies on nicotinic acid, nicotinamide, and quinolinic acid as precursors of nicotinamide adenine dinucleotide. J Biol Chem 241: 3701–3707, 1966 [PubMed] [Google Scholar]
- 103.Ikeda M, Tsuji H, Nakamura S, Ichiyama A, Nishizuka Y, and Hayaishi O. Studies on the biosynthesis of nicotinamide adenine dinucleotide. Ii. A role of picolinic carboxylase in the biosynthesis of nicotinamide adenine dinucleotide from tryptophan in mammals. J Biol Chem 240: 1395–1401, 1965 [PubMed] [Google Scholar]
- 104.Imai S, Armstrong CM, Kaeberlein M, and Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403: 795–800, 2000 [DOI] [PubMed] [Google Scholar]
- 105.Imai S. and Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab 15 Suppl 3: 26–33, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Iwata K, Nishinaka T, Matsuno K, Kakehi T, Katsuyama M, Ibi M, and Yabe-Nishimura C. The activity of aldose reductase is elevated in diabetic mouse heart. J Pharmacol Sci 103: 408–416, 2007 [DOI] [PubMed] [Google Scholar]
- 107.Iyanagi T. Structure and function of NADPH-cytochrome P450 reductase and nitric oxide synthase reductase domain. Biochem Biophys Res Commun 338: 520–528, 2005 [DOI] [PubMed] [Google Scholar]
- 108.Jagtap P. and Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov 4: 421–440, 2005 [DOI] [PubMed] [Google Scholar]
- 109.Jankovic A, Korac A, Buzadzic B, Otasevic V, Stancic A, Daiber A, and Korac B. Redox implications in adipose tissue (dys)function—a new look at old acquaintances. Redox Biol 6: 19–32, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, and Lin H. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496: 110–113, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, and Kahn CR. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A 108: 14608–14613, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jones DP. Determination of pyridine dinucleotides in cell extracts by high-performance liquid chromatography. J Chromatogr 225: 446–449, 1981 [DOI] [PubMed] [Google Scholar]
- 113.Jones DP. and Sies H. The Redox Code. Antioxid Redox Signal 23: 734–746, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, and Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ Heart Fail 3: 420–430, 2010 [DOI] [PubMed] [Google Scholar]
- 115.Kaufmann M, Feijs KL, and Luscher B. Function and regulation of the mono-ADP-ribosyltransferase ARTD10. Curr Top Microbiol Immunol 384: 167–188, 2015 [DOI] [PubMed] [Google Scholar]
- 116.Kelly B. and O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25: 771–784, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, Watson PA, Birdsey N, Bao J, Gius D, Sack MN, Jing E, Kahn CR, Friedman JE, and Jonscher KR. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 433: 505–514, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kennedy BK, Austriaco NR, Jr., Zhang J, and Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell 80: 485–496, 1995 [DOI] [PubMed] [Google Scholar]
- 119.Kerbey AL, Radcliffe PM, Randle PJ, and Sugden PH. Regulation of kinase reactions in pig heart pyruvate dehydrogenase complex. Biochem J 181: 427–433, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kerbey AL, Randle PJ, Cooper RH, Whitehouse S, Pask HT, and Denton RM. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem J 154: 327–348, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim JW, Tchernyshyov I, Semenza GL, and Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185, 2006 [DOI] [PubMed] [Google Scholar]
- 122.Kim S, Benguria A, Lai CY, and Jazwinski SM. Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell 10: 3125–3136, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.King MP. and Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 246: 500–503, 1989 [DOI] [PubMed] [Google Scholar]
- 124.Klaidman L, Morales M, Kem S, Yang J, Chang ML, Adams JD., Jr Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology 69: 150–157, 2003 [DOI] [PubMed] [Google Scholar]
- 125.Kleikers PW, Wingler K, Hermans JJ, Diebold I, Altenhofer S, Radermacher KA, Janssen B, Gorlach A, and Schmidt HH. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl) 90: 1391–1406, 2012 [DOI] [PubMed] [Google Scholar]
- 126.Kletzien RF, Harris PK, and Foellmi LA. Glucose-6-phosphate dehydrogenase: a “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J 8: 174–181, 1994 [DOI] [PubMed] [Google Scholar]
- 127.Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, Nyakas C, and Radak Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech Ageing Dev 131: 21–28, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kornberg A. The participation of inorganic pyrophosphate in the reversible enzymatic synthesis of diphosphopyridine nucleotide. J Biol Chem 176: 1475, 1948 [PubMed] [Google Scholar]
- 129.Kosicki GW. and Lee LP. Effect of divalent metal ions on nucleotide inhibition of pig heart citrate synthase. J Biol Chem 241: 3571–3574, 1966 [PubMed] [Google Scholar]
- 130.LaNoue KF. and Schoolwerth AC. Metabolite transport in mitochondria. Annu Rev Biochem 48: 871–922, 1979 [DOI] [PubMed] [Google Scholar]
- 131.LaNoue KF. and Tischler ME. Electrogenic characteristics of the mitochondrial glutamate-aspartate antiporter. J Biol Chem 249: 7522–7528, 1974 [PubMed] [Google Scholar]
- 132.Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, James FD, Gius D, and Wasserman DH. SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high-fat-fed mice. Diabetes 64: 3081–3092, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, and Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107: 2037–2042, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, and Tian R. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation 134: 883–894, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Leppla SH. Large-scale purification and characterization of the exotoxin of Pseudomonas aeruginosa. Infect Immun 14: 1077–1086, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lerner F, Niere M, Ludwig A, and Ziegler M. Structural and functional characterization of human NAD kinase. Biochem Biophys Res Commun 288: 69–74, 2001 [DOI] [PubMed] [Google Scholar]
- 138.Li B, Tian J, Sun Y, Xu TR, Chi RF, Zhang XL, Hu XL, Zhang YA, Qin FZ, and Zhang WF. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochim Biophys Acta 1852: 805–815, 2015 [DOI] [PubMed] [Google Scholar]
- 139.Li H. and Poulos TL. Structure-function studies on nitric oxide synthases. J Inorg Biochem 99: 293–305, 2005 [DOI] [PubMed] [Google Scholar]
- 140.Li J, Bonkowski MS, Moniot S, Zhang D, Hubbard BP, Ling AJ, Rajman LA, Qin B, Lou Z, Gorbunova V, Aravind L, Steegborn C, and Sinclair DA. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science 355: 1312–1317, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Li M, Riddle S, Zhang H, D'Alessandro A, Flockton A, Serkova NJ, Hansen KC, Moldvan R, McKeon BA, Frid M, Kumar S, Li H, Liu H, Canovas A, Medrano JF, Thomas MG, Iloska D, Plecita-Hlavata L, Jezek P, Pullamsetti S, Fini MA, El Kasmi KC, Zhang Q, and Stenmark KR. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional co-repressor C-terminal binding protein-1. Circulation 134: 1105–1121, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liao JK. Squalene synthase inhibitor lapaquistat acetate: could anything be better than statins? Circulation 123: 1925–1928, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Linn TC, Pettit FH, and Reed LJ. Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc Natl Acad Sci U S A 62: 234–241, 1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu G, Park SH, Imbesi M, Nathan WJ, Zou X, Zhu Y, Jiang H, Parisiadou L, Gius D. Loss of NAD-dependent protein deacetylase sirtuin-2 alters mitochondrial protein acetylation and dysregulates mitophagy. Antioxid Redox Signal 26: 849–863, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Losman JA, Kaelin WG., Jr What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27: 836–852, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lowery J, Jain N, Kuczmarski ER, Mahammad S, Goldman A, Gelfand VI, Opal P, and Goldman RD. Abnormal intermediate filament organization alters mitochondrial motility in giant axonal neuropathy fibroblasts. Mol Biol Cell 27: 608–616, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lowry OH, Passonneau JV, Schulz DW, and Rock MK. The measurement of pyridine nucleotides by enzymatic cycling. J Biol Chem 236: 2746–2755, 1961 [PubMed] [Google Scholar]
- 148.Lowry OH, Roberts NR, and Kapphahn JI. The fluorometric measurement of pyridine nucleotides. J Biol Chem 224: 1047–1064, 1957 [PubMed] [Google Scholar]
- 149.Lu W, Wang L, Chen L, Hui S, and Rabinowitz JD. Extraction and quantitation of NAD(P)(H). Antioxid Redox Signal, 2017. [Epub ahead of print]; DOI: 10.1089/ars.2017.7014 [DOI] [PMC free article] [PubMed]
- 150.Lunn ER, Perry VH, Brown MC, Rosen H, and Gordon S. Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci 1: 27–33, 1989 [DOI] [PubMed] [Google Scholar]
- 151.Luzzatto L. and Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol 164: 469–480, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ma C, Pi C, Yang Y, Lin L, Shi Y, Li Y, Li Y, and He X. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1. PLoS One 12: e0170930, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK, and Brann DW. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener 12: 7, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, and Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4: 1199–1206, 2001 [DOI] [PubMed] [Google Scholar]
- 155.Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez JP, Lee HY, Cline GW, Samuel VT, Kibbey RG, and Shulman GI. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510: 542–546, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Madsen AS, Andersen C, Daoud M, Anderson KA, Laursen JS, Chakladar S, Huynh FK, Colaco AR, Backos DS, Fristrup P, Hirschey MD, and Olsen CA. Investigating the sensitivity of NAD+-dependent sirtuin deacylation activities to NADH. J Biol Chem 291: 7128–7141, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Martin BR. and Denton RM. The intracellular localization of enzymes in white-adipose-tissue fat-cells and permeability properties of fat-cell mitochondria. Transfer of acetyl units and reducing power between mitochondria and cytoplasm. Biochem J 117: 861–877, 1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, and Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7: e42357, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Masutani M, Suzuki H, Kamada N, Watanabe M, Ueda O, Nozaki T, Jishage K, Watanabe T, Sugimoto T, Nakagama H, Ochiya T, and Sugimura T. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A 96: 2301–2304, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Matsushima S, Kuroda J, Ago T, Zhai P, Ikeda Y, Oka S, Fong GH, Tian R, and Sadoshima J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1alpha and upregulation of peroxisome proliferator-activated receptor-alpha. Circ Res 112: 1135–1149, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Matsushima S, Tsutsui H, and Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc Med 24: 202–205, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McGettrick AF. and O'Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem 288: 22893–22898, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.McKeehan WL. and McKeehan KA. Oxocarboxylic acids, pyridine nucleotide-linked oxidoreductases and serum factors in regulation of cell proliferation. J Cell Physiol 101: 9–16, 1979 [DOI] [PubMed] [Google Scholar]
- 164.McLean KJ, Luciakova D, Belcher J, Tee KL, and Munro AW. Biological diversity of cytochrome P450 redox partner systems. Adv Exp Med Biol 851: 299–317, 2015 [DOI] [PubMed] [Google Scholar]
- 165.Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, Nurnberg P, Mann NP, Banerjee R, Saka HN, Chapple JP, King PJ, Clark AJ, and Metherell LA. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet 44: 740–742, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, and Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 105: 244–250, 2002 [DOI] [PubMed] [Google Scholar]
- 167.Morais R, Gregoire M, Jeannotte L, and Gravel D. Chick embryo cells rendered respiration-deficient by chloramphenicol and ethidium bromide are auxotrophic for pyrimidines. Biochem Biophys Res Commun 94: 71–77, 1980 [DOI] [PubMed] [Google Scholar]
- 168.Mori V, Amici A, Mazzola F, Di Stefano M, Conforti L, Magni G, Ruggieri S, Raffaelli N, and Orsomando G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS One 9: e113939, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, Mottis A, Jo YS, Viswanathan M, Schoonjans K, Guarente L, and Auwerx J. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154: 430–441, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nakagawa T, Lomb DJ, Haigis MC, and Guarente L. SIRT5 Deacetylates carbamoyl phosphate synthetase 1 and regulates the urea cycle. Cell 137: 560–570, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nakahata Y, Sahar S, Astarita G, Kaluzova M, and Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324: 654–657, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Neely JR. and Grotyohann LW. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischemic hearts. Circ Res 55: 816–824, 1984 [DOI] [PubMed] [Google Scholar]
- 173.Nguewa PA, Fuertes MA, Valladares B, Alonso C, and Perez JM. Poly(ADP-ribose) polymerases: homology, structural domains and functions. Novel therapeutical applications. Prog Biophys Mol Biol 88: 143–172, 2005 [DOI] [PubMed] [Google Scholar]
- 174.Nikiforov A, Dolle C, Niere M, and Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J Biol Chem 286: 21767–21778, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ohashi K, Kawai S, and Murata K. Identification and characterization of a human mitochondrial NAD kinase. Nat Commun 3: 1248, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Olavarria K, De Ingeniis J, Zielinski DC, Fuentealba M, Munoz R, McCloskey D, Feist AM, and Cabrera R. Metabolic impact of an NADH-producing glucose-6-phosphate dehydrogenase in Escherichia coli. Microbiology 160: 2780–2793, 2014 [DOI] [PubMed] [Google Scholar]
- 177.Oldham WM, Clish CB, Yang Y, and Loscalzo J. Hypoxia-mediated increases in l–2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab 22: 291–303, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Patel VP. and Chu CT. Decreased SIRT2 activity leads to altered microtubule dynamics in oxidatively-stressed neuronal cells: implications for Parkinson's disease. Exp Neurol 257: 170–181, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, and Choi Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460: 103–107, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, and Archer SL. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate. J Mol Med (Berl) 91: 333–346, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pickrell AM. and Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen CH, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J, De Brabander J, Ready JM, and McKnight SL. Discovery of a proneurogenic, neuroprotective chemical. Cell 142: 39–51, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pittelli M, Felici R, Pitozzi V, Giovannelli L, Bigagli E, Cialdai F, Romano G, Moroni F, and Chiarugi A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol Pharmacol 80: 1136–1146, 2011 [DOI] [PubMed] [Google Scholar]
- 184.Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, and Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem 285: 34106–34114, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Placzek S, Schomburg I, Chang A, Jeske L, Ulbrich M, Tillack J, and Schomburg D. BRENDA in 2017: new perspectives and new tools in BRENDA. Nucleic Acids Res 45: D380–D388, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pollak N, Dolle C, and Ziegler M. The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochem J 402: 205–218, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pollak N, Niere M, and Ziegler M. NAD kinase levels control the NADPH concentration in human cells. J Biol Chem 282: 33562–33571, 2007 [DOI] [PubMed] [Google Scholar]
- 188.Pongratz RL, Kibbey RG, Shulman GI, and Cline GW. Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J Biol Chem 282: 200–207, 2007 [DOI] [PubMed] [Google Scholar]
- 189.Porter TD. Electron transfer pathways in cholesterol synthesis. Lipids 50: 927–936, 2015 [DOI] [PubMed] [Google Scholar]
- 190.Posakony JW, England JM, and Attardi G. Mitochondrial growth and division during the cell cycle in HeLa cells. J Cell Biol 74: 468–491, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Preiss J. and Handler P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J Biol Chem 233: 488–492, 1958 [PubMed] [Google Scholar]
- 192.Preiss J. and Handler P. Biosynthesis of diphosphopyridine nucleotide. II. Enzymatic aspects. J Biol Chem 233: 493–500, 1958 [PubMed] [Google Scholar]
- 193.Rajakumar K. Pellagra in the United States: a historical perspective. South Med J 93: 272–277, 2000 [PubMed] [Google Scholar]
- 194.Rajtik T, Carnicka S, Szobi A, Giricz Z, J OU, Hassova V, Svec P, Ferdinandy P, Ravingerova T, and Adameova A. Oxidative activation of CaMKIIdelta in acute myocardial ischemia/reperfusion injury: a role of angiotensin AT1 receptor-NOX2 signaling axis. Eur J Pharmacol 771: 114–122, 2016 [DOI] [PubMed] [Google Scholar]
- 195.Ramasamy R, Oates PJ, and Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 46: 292–300, 1997 [DOI] [PubMed] [Google Scholar]
- 196.Ramasamy R, Trueblood N, and Schaefer S. Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am J Physiol 275: H195–H1203, 1998 [DOI] [PubMed] [Google Scholar]
- 197.Ramsey KM, Mills KF, Satoh A, and Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in beta cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell 7: 78–88, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, Takahashi JS, Imai S, and Bass J. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324: 651–654, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, Auwerx J, Yanes O, Brenner C, and Canto C. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun 7: 13103, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Revollo JR, Grimm AA, and Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 279: 50754–50763, 2004 [DOI] [PubMed] [Google Scholar]
- 201.Revollo JR, Korner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, and Imai S. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab 6: 363–375, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, and Dougherty DM. L-tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int J Tryptophan Res 2: 45–60, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rittle J. and Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science 330: 933–937, 2010 [DOI] [PubMed] [Google Scholar]
- 204.Robergs RA, Ghiasvand F, and Parker D. Biochemistry of exercise-induced metabolic acidosis. Am J Physiol Regul Integr Comp Physiol 287: R502–R516, 2004 [DOI] [PubMed] [Google Scholar]
- 205.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, and Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118, 2005 [DOI] [PubMed] [Google Scholar]
- 206.Roe CR, Millington DS, Norwood DL, Kodo N, Sprecher H, Mohammed BS, Nada M, Schulz H, and McVie R. 2,4-Dienoyl-coenzyme A reductase deficiency: a possible new disorder of fatty acid oxidation. J Clin Invest 85: 1703–1707, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rovetto MJ, Lamberton WF, and Neely JR. Mechanisms of glycolytic inhibition in ischemic rat hearts. Circ Res 37: 742–751, 1975 [DOI] [PubMed] [Google Scholar]
- 208.Rui L. Energy metabolism in the liver. Compr Physiol 4: 177–197, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mazala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, Bellemin S, Iyer SR, Wang X, Gariani K, Sauve AA, Canto C, Conley KE, Walter L, Lovering RM, Chin ER, Jasmin BJ, Marcinek DJ, Menzies KJ, and Auwerx J. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med 8: 361ra139, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sadanaga-Akiyoshi F, Yao H, Tanuma S, Nakahara T, Hong JS, Ibayashi S, Uchimura H, and Fujishima M. Nicotinamide attenuates focal ischemic brain injury in rats: with special reference to changes in nicotinamide and NAD+ levels in ischemic core and penumbra. Neurochem Res 28: 1227–1234, 2003 [DOI] [PubMed] [Google Scholar]
- 211.Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, Weerapana E, and Bogyo M. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife 5: e13663, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schein PS, Cooney DA, and Vernon ML. The use of nicotinamide to modify the toxicity of streptozotocin diabetes without loss of antitumor activity. Cancer Res 27: 2324–2332, 1967 [PubMed] [Google Scholar]
- 213.Schulz H. Regulation of fatty acid oxidation in heart. J Nutr 124: 165–171, 1994 [DOI] [PubMed] [Google Scholar]
- 214.Schwartz AG. and Pashko LL. Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase, and longevity. Ageing Res Rev 3: 171–187, 2004 [DOI] [PubMed] [Google Scholar]
- 215.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sengupta R. and Holmgren A. Thioredoxin and glutaredoxin-mediated redox regulation of ribonucleotide reductase. World J Biol Chem 5: 68–74, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, Meda Spaccamela V, Belohradsky BH, Speer O, Schmugge M, Kohne E, Hoenig M, Freihorst J, Schulz AS, and Reichenbach J. Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol 139: 212–219 e3, 2017 [DOI] [PubMed] [Google Scholar]
- 218.Somasuntharam I, Boopathy AV, Khan RS, Martinez MD, Brown ME, Murthy N, and Davis ME. Delivery of Nox2-NADPH oxidase siRNA with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 34: 7790–7798, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Stein LR. and Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab 23: 420–428, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Storbeck KH, Swart AC, Fox CL, and Swart P. Cytochrome b5 modulates multiple reactions in steroidogenesis by diverse mechanisms. J Steroid Biochem Mol Biol 151: 66–73, 2015 [DOI] [PubMed] [Google Scholar]
- 221.Stubbs M, Veech RL, and Krebs HA. Control of the redox state of the nicotinamide-adenine dinucleotide couple in rat liver cytoplasm. Biochem J 126: 59–65, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162: 552–563, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sun F, Dai C, Xie J, and Hu X. Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLoS One 7: e34525, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sutendra G. and Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metab 19: 558–573, 2014 [DOI] [PubMed] [Google Scholar]
- 225.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, and Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J Biol Chem 286: 13304–13313, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tanny JC, Dowd GJ, Huang J, Hilz H, and Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell 99: 735–745, 1999 [DOI] [PubMed] [Google Scholar]
- 227.Tempel W, Rabeh WM, Bogan KL, Belenky P, Wojcik M, Seidle HF, Nedyalkova L, Yang T, Sauve AA, Park HW, and Brenner C. Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS Biol 5: e263, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Tilton WM, Seaman C, Carriero D, and Piomelli S. Regulation of glycolysis in the erythrocyte: role of the lactate/pyruvate and NAD/NADH ratios. J Lab Clin Med 118: 146–152, 1991 [PubMed] [Google Scholar]
- 229.Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, and Mootha VK. Complementation of mitochondrial electron transport chain by manipulation of the NAD+/NADH ratio. Science 352: 231–235, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Todisco S, Agrimi G, Castegna A, and Palmieri F. Identification of the mitochondrial NAD+ transporter in Saccharomyces cerevisiae. J Biol Chem 281: 1524–1531, 2006 [DOI] [PubMed] [Google Scholar]
- 231.Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, and Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun 7: 12948, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Trammell SA, Yu L, Redpath P, Migaud ME, and Brenner C. Nicotinamide riboside is a major NAD+ precursor vitamin in cow milk. J Nutr 146: 957–963, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, Clish CB, Stillman IE, Karumanchi SA, Rhee EP, and Parikh SM. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531: 528–532, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Tsai CS, Burgett MW, and Reed LJ. Alpha-keto acid dehydrogenase complexes. XX. A kinetic study of the pyruvate dehydrogenase complex from bovine kidney. J Biol Chem 248: 8348–8352, 1973 [PubMed] [Google Scholar]
- 235.Tsuge H. and Tsurumura T. Reaction mechanism of mono-ADP-ribosyltransferase based on structures of the complex of enzyme and substrate protein. Curr Top Microbiol Immunol 384: 69–87, 2015 [DOI] [PubMed] [Google Scholar]
- 236.Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, and Yalcin A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1–42)-induced rat model of Alzheimer's disease. Free Radic Res 48: 146–158, 2014 [DOI] [PubMed] [Google Scholar]
- 237.Ussher JR, Jaswal JS, and Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res 111: 628–641, 2012 [DOI] [PubMed] [Google Scholar]
- 238.Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, Dyck JR, and Lopaschuk GD. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res 94: 359–369, 2012 [DOI] [PubMed] [Google Scholar]
- 239.van de Weijer T, Phielix E, Bilet L, Williams EG, Ropelle ER, Bierwagen A, Livingstone R, Nowotny P, Sparks LM, Paglialunga S, Szendroedi J, Havekes B, Moullan N, Pirinen E, Hwang JH, Schrauwen-Hinderling VB, Hesselink MK, Auwerx J, Roden M, and Schrauwen P. Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans. Diabetes 64: 1193–1201, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Vander Heiden MG, Cantley LC, and Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.VanLinden MR, Dolle C, Pettersen IK, Kulikova VA, Niere M, Agrimi G, Dyrstad SE, Palmieri F, Nikiforov AA, Tronstad KJ, and Ziegler M. Subcellular distribution of NAD+ between cytosol and mitochondria determines the metabolic profile of human cells. J Biol Chem 290: 27644–27659, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Veech RL, Eggleston LV, and Krebs HA. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem J 115: 609–619, 1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Veech RL, Guynn R, and Veloso D. The time-course of the effects of ethanol on the redox and phosphorylation states of rat liver. Biochem J 127: 387–397, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science 350: 1208–1213, 2015 [DOI] [PubMed] [Google Scholar]
- 245.Vida A, Marton J, Miko E, Bai P. Metabolic roles of poly(ADP-ribose) polymerases. Semin Cell Dev Biol 63: 135–143, 2017 [DOI] [PubMed] [Google Scholar]
- 246.Vimercati C, Qanud K, Mitacchione G, Sosnowska D, Ungvari Z, Sarnari R, Mania D, Patel N, Hintze TH, Gupte SA, Stanley WC, and Recchia FA. Beneficial effects of acute inhibition of the oxidative pentose phosphate pathway in the failing heart. Am J Physiol Heart Circ Physiol 306: H709–H717, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Voet D. and Voet JG. Biochemistry. Hoboken, NJ: John Wiley & Sons, 2011, xxv, 1428, 53 pp [Google Scholar]
- 248.von Euler H. Fermentation of sugars and fermentative enzymes. In: Nobel Lectures in Chemistry, edited by Nobelstiftelsen. New Jersey: World Scientific, 1999, pp. 144–155. [Google Scholar]
- 249.Wagner GR. and Hirschey MD. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol Cell 54: 5–16, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Wang G, Han T, Nijhawan D, Theodoropoulos P, Naidoo J, Yadavalli S, Mirzaei H, Pieper AA, Ready JM, and McKnight SL. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 158: 1324–1334, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, and He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol 170: 349–355, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Warburg O. On respiratory impairment in cancer cells. Science 124: 269–270, 1956 [PubMed] [Google Scholar]
- 253.Warburg O. On the origin of cancer cells. Science 123: 309–314, 1956 [DOI] [PubMed] [Google Scholar]
- 254.Warburg O. and Christian W. Pyridin, der wasserstoffübertragende bestandteil von gärungsfermenten. Helvetica Chimica Acta 19: E79–E88, 1936 [Google Scholar]
- 255.Weinberg SE, Sena LA, and Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42: 406–417, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, and Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3: e02242, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Whitmer JT, Idell-Wenger JA, Rovetto MJ, and Neely JR. Control of fatty acid metabolism in ischemic and hypoxic hearts. J Biol Chem 253: 4305–4309, 1978 [PubMed] [Google Scholar]
- 258.Williamson DH, Lund P, and Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103: 514–527, 1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, van den Enden M, Kilo C, and Tilton RG. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42: 801–813, 1993 [DOI] [PubMed] [Google Scholar]
- 260.Wong YC. and Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A 111: E4439–E4448, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Xie W, Xu A, and Yeung ES. Determination of NAD(+) and NADH in a single cell under hydrogen peroxide stress by capillary electrophoresis. Anal Chem 81: 1280–1284, 2009 [DOI] [PubMed] [Google Scholar]
- 262.Xu Y, Chaudhury A, Zhang M, Savoldo B, Metelitsa LS, Rodgers J, Yustein JT, Neilson JR, and Dotti G. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J Clin Invest 126: 2678–2688, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Yamada K, Hara N, Shibata T, Osago H, and Tsuchiya M. The simultaneous measurement of nicotinamide adenine dinucleotide and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. Anal Biochem 352: 282–285, 2006 [DOI] [PubMed] [Google Scholar]
- 264.Yamamoto M, Hikosaka K, Mahmood A, Tobe K, Shojaku H, Inohara H, and Nakagawa T. Nmnat3 is dispensable in mitochondrial NAD level maintenance in vivo. PLoS One 11: e0147037, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, and Sinclair DA. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130: 1095–1107, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Yang SJ, Choi JM, Kim L, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, and Park CY. Nicotinamide improves glucose metabolism and affects the hepatic NAD-sirtuin pathway in a rodent model of obesity and type 2 diabetes. J Nutr Biochem 25: 66–72, 2014 [DOI] [PubMed] [Google Scholar]
- 267.Yang Y. and Sauve AA. NAD+ metabolism: bioenergetics, signaling and manipulation for therapy. Biochim Biophys Acta 1864: 1787–1800, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yin TC, Britt JK, De Jesus-Cortes H, Lu Y, Genova RM, Khan MZ, Voorhees JR, Shao J, Katzman AC, Huntington PJ, Wassink C, McDaniel L, Newell EA, Dutca LM, Naidoo J, Cui H, Bassuk AG, Harper MM, McKnight SL, Ready JM, and Pieper AA. P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep 8: 1731–1740, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10: 179–206, 2008 [DOI] [PubMed] [Google Scholar]
- 270.Ying W, Alano CC, Garnier P, and Swanson RA. NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res 79: 216–223, 2005 [DOI] [PubMed] [Google Scholar]
- 271.Yoshino J, Mills KF, Yoon MJ, and Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 14: 528–536, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Yu Q, Lee CF, Wang W, Karamanlidis G, Kuroda J, Matsushima S, Sadoshima J, and Tian R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J Am Heart Assoc 3: e000555, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, and Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 274.Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D'Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, and Auwerx J. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352: 1436–1443, 2016 [DOI] [PubMed] [Google Scholar]
- 275.Zhang Q, Piston DW, and Goodman RH. Regulation of corepressor function by nuclear NADH. Science 295: 1895–1897, 2002 [DOI] [PubMed] [Google Scholar]
- 276.Zhang R. MNADK, a novel liver-enriched mitochondrion-localized NAD kinase. Biol Open 2: 432–438, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Zhang T, Berrocal JG, Frizzell KM, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, and Kraus WL. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem 284: 20408–20417, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zhang X, Khan S, Jiang H, Antonyak MA, Chen X, Spiegelman NA, Shrimp JH, Cerione RA, and Lin H. Identifying the functional contribution of the defatty-acylase activity of SIRT6. Nat Chem Biol 12: 614–620, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou HM, Huang X, Yang K, Zhu Q, Wang X, Yi J, Zhu L, Qian X, Chen L, Tang Y, Loscalzo J, and Yang Y. SoNar, a highly responsive NAD(+)/NADH sensor, allows high-throughput metabolic screening of anti-tumor agents. Cell Metab 21: 777–789, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zhao Y, Jin J, Hu Q, Zhou HM, Yi J, Yu Z, Xu L, Wang X, Yang Y, and Loscalzo J. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 14: 555–566, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Zhao Y. and Yang Y. Real-time and high-throughput analysis of mitochondrial metabolic states in living cells using genetically encoded NAD+/NADH sensors. Free Radic Biol Med 100: 43–52, 2016 [DOI] [PubMed] [Google Scholar]
- 282.Zhong L, Arner ES, and Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: the active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc Natl Acad Sci U S A 97: 5854–5859, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, and Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zhou M, Ottenberg G, Sferrazza GF, Hubbs C, Fallahi M, Rumbaugh G, Brantley AF, and Lasmezas CI. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain 138: 992–1008, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Zhu XH, Lu M, Lee BY, Ugurbil K, and Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A 112: 2876–2881, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Zocchi E, Usai C, Guida L, Franco L, Bruzzone S, Passalacqua M, and De Flora A. Ligand-induced internalization of CD38 results in intracellular Ca2+ mobilization: role of NAD+ transport across cell membranes. FASEB J 13: 273–283, 1999 [DOI] [PubMed] [Google Scholar]