


Abstract
Iron is required for key metabolic processes but is toxic in excess. This circumstance forces organisms across the tree of life to tightly regulate iron homeostasis. In hypersaline lakes dominated by archaeal species, iron levels are extremely low and subject to environmental change; however, mechanisms regulating iron homeostasis in archaea remain unclear. In previous work, we demonstrated that two transcription factors (TFs), Idr1 and Idr2, collaboratively regulate aspects of iron homeostasis in the model species Halobacterium salinarum. Here we show that Idr1 and Idr2 are part of an extended regulatory network of four TFs of the bacterial DtxR family that maintains intracellular iron balance. We demonstrate that each TF directly regulates at least one of the other DtxR TFs at the level of transcription. Dynamical modeling revealed interlocking positive feedback loop architecture, which exhibits bistable or oscillatory network dynamics depending on iron availability. TF knockout mutant phenotypes are consistent with model predictions. Together, our results support that this network regulates iron homeostasis despite variation in extracellular iron levels, consistent with dynamical properties of interlocking feedback architecture in eukaryotes. These results suggest that archaea use bacterial-type TFs in a eukaryotic regulatory network topology to adapt to harsh environments.
INTRODUCTION
Iron is an essential element for almost all living organisms as a prosthetic component of enzymes, acting as a biocatalyst or as an electron carrier in metabolic processes such as respiration (1). When present in excess, iron undergoes oxidation-reduction Fenton and/or Haber-Weiss reactions, catalyzing the production of toxic reactive oxygen species that disrupt cell membranes, hinder cellular functions and damage macromolecules such as proteins, lipids and DNA (2–6). Mechanisms of uptake, transport and storage are therefore tightly controlled to maintain iron in the homeostatic range, e.g. low μM in bacteria (7).
To adapt and survive in the face of stress, cells use gene regulatory networks (GRNs) in which transcription factors (TFs) work together, regulating each other and stress response systems. These networks allow nuanced control of the transcriptional response, dynamically adjusting physiology to adapt (8–10). Organisms across the tree of life have evolved a diversity of regulatory network strategies to regulate iron homeostasis. During iron starvation in eukaryotes such as yeast, conserved TFs such as Aft1 and Aft2 coordinately regulate genes encoding the iron regulon (11–13). Post-transcriptional regulators such as Cth1 and 2 promote the degradation of transcripts for iron use (14,15). In bacteria, different families of TFs each regulate specific functions (16) such as iron homeostasis (DtxR (17)), manganese uptake (Mur (18,19)) and cofactor synthesis (IscR (20)). Fur exerts global control over iron homeostasis genes as well as connects the iron response regulatory network to related networks such as those responsive to oxidative stress (16,21).
In general, transcriptional regulation in response to environmental cues in archaea requires bacterial-type TFs (22) that either block or activate transcription initiation with a eukaryote-like basal apparatus (23–25). In contrast to the wealth of knowledge in bacteria and eukaryotes, however, the mechanisms underlying regulation of transcription, particularly for iron homeostasis, remain unclear in archaea. Fur and DtxR family homologs are widespread in currently available archaeal genome sequences, with the majority encoding one or two DtxR homologs (26). Only four of these homologs have yet been experimentally characterized in various species of Euryarchaea. These DtxR proteins repress iron uptake transport during iron sufficiency (27–29). Fur appears to have been inactivated by mutation or does not appear to function in metal regulation (28,29).
Here, we use Halobacterium salinarum as a model organism to characterize the transcriptional regulatory network regulating iron homeostasis in archaea. This hypersaline-adapted species colonizes environments such as solar salterns and hypersaline lakes where sodium chloride levels reach saturation. Under these conditions, water evaporates and iron precipitates in the form of pyrite, resulting in immobilization and biological inaccessibility of iron and other metals (30). When salinity decreases during rainfall, iron levels rise. In contrast to the bacterial species (31), H. salinarum survives under low iron conditions for months (32), but is also robust to iron overload, experiencing normal growth in iron concentrations up to 50 times higher than mesophilic bacteria can withstand (27,33). In previous work, we demonstrated that DtxR homologs Idr1 and Idr2 activate and repress overlapping sets of target genes throughout the genome as well as independent regulons to maintain iron homeostasis (34). For example, Idr2 indirectly controls idr1 expression and directly activates putative siderophore biosynthesis genes (34,35). A third DtxR homolog, SirR, is required to repress Mn uptake during Mn sufficiency and overload (27). The role of TroR, the fourth DtxR family homolog encoded in the H. salinarum genome, remains unknown. It therefore remains unclear how all four DtxR TFs play a role in the maintenance of such a wide homeostatic range of iron given the intermittent scarcity of iron in hypersaline environments.
Based on our prior knowledge that Idr1 and Idr2 collaboratively regulate iron uptake genes (34), here we tested the hypothesis that all four DtxR family TFs work together to control genes during perturbations in extracellular iron levels. We inferred the network topology based on gene expression in knockout strains using statistical methods (ANOVA, (36)). in vivo TF-DNA binding experiments defined direct regulatory edges in this network. The topology of the resultant network consists of two interlocked feedback loops. One regulatory loop is active under iron starvation, the other under sufficiency, and the two loops repress each other. Using Boolean logic simulations (37), we predict bistable network dynamics under constant iron conditions and oscillatory dynamics under fluctuating iron regimes. The predicted dynamical behavior is supported by knockout mutant phenotypes, including growth sensitivity and intracellular iron concentrations quantified by mass spectrometry during growth under iron starvation or iron sufficiency. We discuss how such TF–TF cross-regulation may enable enhanced buffering against perturbations in external iron levels, reinforcing a stable regulatory state and allowing for physiological tolerance to a wider homeostatic range of iron in hypersaline niches.
MATERIALS AND METHODS
Strains
All strains used in the current work were originally derived from H. salinarum NRC-1 (ATCC700922). Deletion mutants in sirR, idr1 and idr2 were previously constructed (27,34). In the current study, an in-frame deletion in troR was newly constructed in the Δura3 parent strain background, a uracil auxotroph, by homologous replacement as previously described (38,39). For epitope-tagged strains used in chromatin immunoprecipitation-quantitative polymerase chain reaction (ChIP-qPCR) experiments, the complete sequence for each of sirR and troR plus 500 bp upstream of the coding sequence (to include their own promoters) were fused in-frame at the C-termini to the hemagglutinin epitope (HA) in backbone vector pMTF-HA (40). HA epitope-tagged strains and knockout of troR were verified by PCR using primers listed in Supplementary Table S1 and Sanger sequencing as previously described (41). Primers used for cloning of HA vectors are also included in Supplementary Table S1. All strains and plasmids used in this study are listed in Supplementary Table S2.
Growth conditions
For routine culturing, cells were first grown from single colonies to early stationary phase (OD600 ≈ 1) in CM medium (250 g/l NaCl, 20 g/l MgSO4·7H2O, 3 g/l sodium citrate, 2 g/l KCl, 10g/l peptone). Cells were then pelleted and washed with basal salts (CM lacking peptone), and then subcultured at initial OD600 ≈ 0.1 in Complete Defined Medium (CDM) (42). In the case of iron starvation, cells were subcultured a second time, washed, then inoculated at 1:100 dilution into CDM iron starvation medium. Prior to culturing, CDM was treated with 10 g/l Chelex resin (BioRad, Hercules, CA, USA) for at least 1 h to eliminate trace metals, and then supplemented with 50 μg/ml metal-free uracil to complement the Δura3 auxotrophy. For iron-replete medium, trace metals were added to 10 μM MnSO4, 100 μM FeSO4, 0.01 μM ZnSO4 and 0.01 μM CuSO4. In the case of CDM iron starvation medium, CDM treated with Chelex as described above and supplemented with 100 μM BPS (4,7-diphenyl-1, 10-phenanthrolinedisulfonic acid, Sigma–Aldrich). The trace metals described above except iron were added. All salts and trace metals added to every one of these CDM medium formulations were free from contaminating metals (Alfa Aesar, 99.999% purity). In the case of ΔsirR::sirR-HA and ΔtroR::troR-HA strains for ChIP-qPCR experiments, 10 μg/ml of mevinolin (Lovostatin, A.G Scientific, San Diego, CA, USA) was added to the medium to maintain selection for plasmids. To avoid metal contamination, all glassware was soaked with 0.1 N HCl overnight and then washed with 18 mΩ filtered ultrapure water (Picopure® Hydro System 150 II, Hydro Service and Supplies, Durham, NC, USA) before being used. For all experiments, cultures were grown at 225 r.p.m. shaking at 42°C under ambient light. For every experiment except growth curves, cultures were harvested in mid-logarithmic phase (OD600 ∼0.5).
High-throughput growth rate measurements
The Δura3 parental strain and Δidr1, Δidr2, ΔsirR and ΔtroR deletion mutants were pre-cultured in CM and then sub-cultured in CDM with or without iron as described above. Upon reaching saturation, cultures were diluted to 600 nm (OD600) of 0.1 into 200 μl iron-starvation or iron-replete medium. Growth was monitored every 30 min at 42°C for 4 days under continuous shaking (∼225 rpm) in a Bioscreen C microbial growth analyzer (Growth Curves USA, Piscataway, NJ, USA). Growth measurements for each replicate were converted to area under the curve (AUC), also known as growth potential, using the bsd Analysis Function in the R coding environment (43,44). Growth potential captures information about the entirety of the growth curve (lag, log and stationary phases), therefore allowing overall growth comparisons between strains (44). Growth potential from three technical and three biological replicates were averaged and standard error calculated using the three biological replicate values. Significant differences of the growth of each mutant strain compared to that of the isogenic parent control strain were evaluated by two-tailed paired t-test (Supplementary Table S3). Error bars in the figures refer to the standard error of the mean.
Iron starvation survival plate assay
A total of 3 ml cultures of Δura3 parent strain, Δidr1, Δidr2, ΔsirR and ΔtroR mutant were grown in biological triplicate in iron-replete or iron-starvation media as described above. Subsequently, ∼10 μl of each culture was individually spotted in serial dilutions (undiluted to 10−4) on CDM plates without iron. Colony density was scored and compared with the Δura3 parent strain after 4 days of growth in the dark at 42°C. A representative spot plate is shown in the main text Figure 1B and the two other biological replicate plates are given in Supplementary Figure S1.
Figure 1.

DtxR TFs are required for normal growth under iron starvation and replete conditions. (A) Growth potential (area under the log-transformed growth curve, AUC) measurement of DtxR mutant strains compared to the isogenic parent under iron replete (blue bars) compared to iron starvation (orange bars) conditions. Each bar represents the data from the average of three independent biological replicates, each with three technical replicates. Error bars represent standard error of the mean (SEM). Stars represent P-value < 0.05 based on t-tests comparing the growth of the parent strain versus each mutant under the same iron condition (P-values listed in Supplementary Table S3). (B) Viable count survival assay. Each drop represents a 10-fold dilution, starting with undiluted on the left. The assay was performed in triplicate for each strain, with a representative plate shown in the figure, and the other two biological replicates shown in Supplementary Figure S1.
RT-qPCR
A total of 50 ml of H. salinarum parental strain Δura3 and knockout mutant strains were grown in metal-clean CDM without iron as described above. A total of 2 ml of each culture at mid-logarithmic phase (OD600 = 0.3–0.5) were harvested by centrifugation (2 min at 16 000 × g) and frozen immediately in liquid nitrogen, then stored at −80°C. Subsequently, 100 μM FeSO4 was added to the remaining 48 ml of culture, and 2 ml samples from iron-treated cultures of each strain were collected 5, 20 and 60 min after iron re-addition. RNA was isolated using the Absolutely RNA mini-prep kit (Agilent Technologies, USA) and the quantity and quality of RNA extracted was determined by PCR and Agilent Bioanalyzer as previously described (45). Three-hundred nanograms of each extracted RNA sample was retrotranscribed to cDNA and quantified using the iTaq Universal SYBR Green One Step Kit according to manufacturer’s instructions (BioRad, Hercules, CA, USA). To minimize handling errors, all plate loading was performed robotically using a Corbitt Life Sciences system. Real-time qPCR reactions were performed in an Eppendorf Mastercycler ep RealPlex thermocycler for 25 cycles with the following parameters: 95°C for 12 min to activate Taq polymerase, melting for 30 s at 95°C, annealing for 30 s at 60°C and extension for 1 min at 72°C. One final extension step was added for 10 min at 72°C. Cycle threshold (CT) was determined automatically by the Eppendorf RealPlex software. Primers (included in Supplementary Table S1) were designed using the H. salinarum sequences from NCBI GenBank as a DNA template in the Clone Manager software (Sci-Ed Software) with previously described parameters (42). Specificity for the region of interest and lack of off-target annealing was ensured by BLAST. Quality of the primers was checked using Primer Star software (DNASTAR, Inc.). Amplification efficiencies for each pair of primers targeting each gene of interest (idr1, VNG2579G; idr2, VNG0835G; sir, VNG0536G; troR, VNG0530G) as well as the reference control, eif1a2 (VNG1756G) were determined by qRT-PCR on three 10-fold serial dilutions of the same sample. These efficiencies were used to calculate RNA enrichment relative to the reference gene (VNG1756G) as described in (46). A melting curve analysis was performed after each run to ensure the specificity of products. Each experiment consisted of three biological replicate samples, each with three technical replicates. The significance of change in gene expression was assessed by t-test for each gene at each time point compared to that of the Δura3 parent strain (P-values provided in Supplementary Table S3).
ANOVA modeling of network topology
RT-qPCR gene expression data were log2 transformed and measurements for each gene were scaled to mean 0 and standard deviation 1. Gene expression measurements were divided into four groups by time point (t = 0, 5, 20, 60) and ANOVA was performed to construct the network topology at each time point independently using the equation:
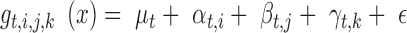 |
where, 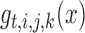 is the expression of gene
is the expression of gene 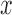 at time point t. Let α, β and γ represent the effects of each of the three other TFs on the fourth gene.
at time point t. Let α, β and γ represent the effects of each of the three other TFs on the fourth gene. 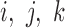 are Boolean variables representing whether that TF is knocked out or if the wild-type gene is present; i.e.
are Boolean variables representing whether that TF is knocked out or if the wild-type gene is present; i.e. 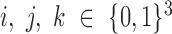 . For example, for expression of gene idr1 in the ΔtroR knockout:
. For example, for expression of gene idr1 in the ΔtroR knockout: 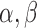 and
and 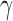 correspond to the influence of Idr2, SirR and TroR, respectively.
correspond to the influence of Idr2, SirR and TroR, respectively. 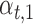 ,
, 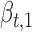 and
and 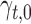 correspond to the specific parameters representing the ΔtroR mutant.
correspond to the specific parameters representing the ΔtroR mutant. 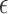 represents independent and identically distributed (i.i.d.) Gaussian noise. The ANOVA model was constructed using the Python package statsmodels (36). Source code is freely available at https://github.com/amyschmid/DtxR. Networks were visualized in Cytoscape (47) with *.sif input file as given in Supplementary Table S4.
represents independent and identically distributed (i.i.d.) Gaussian noise. The ANOVA model was constructed using the Python package statsmodels (36). Source code is freely available at https://github.com/amyschmid/DtxR. Networks were visualized in Cytoscape (47) with *.sif input file as given in Supplementary Table S4.
ChIP-qPCR
At least two 50-ml biological replicate cultures of each of ΔsirR carrying pMTF::sirR-HA and ΔtroR carrying pMTF::troR-HA (AKS111 and AKS112, Supplementary Table S2) were grown in CDM with or without iron as described above. Each plasmid-expressed gene was driven by its native promoter. In mid-logarithmic phase, chromatin crosslinking with formaldehyde was performed according to (34). Immunoprecipitation by virtue of the HA epitope tag was performed according to (40). ChIP samples were analyzed in technical triplicate by qPCR as previously described (34) except that iTaq Universal SYBR Green reagents were used according to manufacturer’s instructions (BioRad, Hercules, CA, USA) in the Eppendorf RealPlex instrument (see RT-qPCR above). Primers (Integrated DNA Technologies, Coralville, IA, USA) amplified multiple regions in each promoter of each gene encoding the DtxR family TFs (two regions for promoters of each of sirR, idr1 and idr2; three regions for troR; primer sequences are detailed in Supplementary Table S1 and a schematic of primer positions relative to TF-coding genes is given in Supplementary Figure S2). Binding enrichment was quantified from resultant data by normalizing CT values of amplicons from each promoter region to the coding sequence of the same gene and to input DNA background (34,46,48). Primer efficiencies were also taken into account as described above for RT-qPCR. Binding enrichment values for multiple amplicons across biological and technical replicates from the same promoter region were averaged and standard error of the mean calculated across biological replicates. Statistical significance was determined by one-tailed t-test comparing the log-transformed binding enrichment distribution for each TF against the log-transformed distribution of all binding data for under the same condition (P-values given in Supplementary Table S3).
Boolean modeling and simulation of gene regulatory network dynamics
The final GRN topology (Figure 5) results from combining topologies for all four time points from ANOVA inference into a consensus network and then pruning edges not supported by ChIP-qPCR binding data (i.e. indirect edges). The network topology of direct regulatory interactions between the four TFs with iron input to each TF was used to construct a synchronously updating Boolean logic model (Table 1) written in Python. To determine the attractor end points, model simulations were conducted for all 32 possible starting states. Simulations reached a steady state at the fourth time step (Supplementary Figure S3). The more stringent initial logic choice of “AND" was assumed for Idr1 and SirR activating inputs to sirR (Table 1, Supplementary Figure S3 and Figure 5). To test the robustness of the attractor states to differences in network logic, simulations with all possible starting states were also run with or logic input to sirR as well as autorepression (Supplementary Figure S3C). “AND" logic between TF input and iron was assumed at all other network nodes (Table 1 and Figure 5). Source code is freely available at https://github.com/amyschmid/DtxR.
Figure 5.

Boolean modeling predicts dynamical properties of the DtxR TF network. (A) Interlocking positive feedback network topology in the absence of regulatory feedback on iron uptake. Blue node outlines and edge colors represent regulatory relationships that are active in the presence of iron, whereas orange depicts activity in the absence of iron. (B) Each graph depicts one of the two attractors (iron sufficiency, top; iron starvation, bottom) for the network at two randomly selected starting states given the network shown in (A) and initial logic functions listed in Table 1. Colored blocks depict Boolean network node activities across the synchronously updating time steps (x-axis), where orange represents 1 (ON) and black represents 0 (OFF). Model simulations for all 32 possible starting states and alternative logic functions are given in Supplementary Figure S3. (C) Network topology including regulatory feedback on iron uptake. Logic functions are as in Table 1, with logic for iron shown at the bottom of the table. Node and edge colors are as in (A). (D) Oscillatory attractor state predicted by Boolean simulation of network topology shown in (C).
Table 1. Boolean logic functions.
| Node | Logic function of node activity |
|---|---|
| Logic functions (without regulated Fe uptake) | |
| SirR |
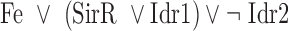
|
| Idr2 |
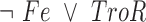
|
| TroR |
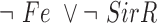
|
| Idr1 |
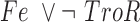
|
| Fe |
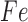
|
| Logic functions (with regulated Fe uptake) | |
| Fe |
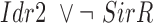
|
Metal quantification by ICP-MS
The metal content of the media before and after cell culturing, as well as the lysate of harvested cells, was determined using inductively coupled plasma mass spectrometry (ICP-MS) as previously described (49). In short, 200 ml of cultures of each of Δura3, Δidr1, Δidr2, ΔsirR and ΔtroR were grown in metal-clean CDM with and without iron until logarithmic phase (OD600 ∼0.5–1.0). Cultures were pelleted by centrifugation (8 min at 4500 × g) in an Eppendorf® 5804R tabletop centrifuge. The 1 ml aliquot of supernatant was reserved to quantify metal amounts in the spent media. Pellets were then washed thrice in metal-free basal salt buffer and resuspended in 800 μl picopure water (H. salinarum is obligately halophilic and therefore lyses spontaneously in hypotonic solutions). The lysates were homogenized by 10 min of sonication with Bioruptor UCD-200 (Diagenode, Liege, Belgium) for 30 s on/off cycles. Cell debris was removed by centrifugation for 5′ at 4500 × g. For ICP-MS analysis, samples were diluted 80-fold to minimize salt crystals. Metal quantities were normalized to protein concentrations from lysates, which were quantified by QubitR fluorimeter (Molecular Probes, Life Technologies). The media and normalized lysate samples were diluted an additional 10–15 times into 2% (vol/vol) nitric acid, incubated at 37°C with shaking at 250 rpm for 1.5 h, and then centrifuged. Samples and media controls were analyzed for cobalt (Co), copper (Cu), manganese (Mn), Nickel (Ni) and Zinc (Zn) content on an octo-pole-based 7500ce ICP-MS using a MicroMist Nebulizer (Agilent Technologies).
RESULTS
Four DtxR family transcription factors are required for ensuring proper growth during iron perturbations in H. salinarum
By amino acid sequence homology, four TFs that belong to the DtxR family of proteins are encoded in the genome of H. salinarum: SirR (VNG0530G; (27)), TroR (VNG0536G), Idr1 (VNG2579G) and Idr2 (VNG0835G) (34). These four paralogs show strong amino acid sequence conservation with one another (minimum 64%, maximum 82% by uncorrelated pairwise distance; Supplementary Figure S4A). Like DtxR family homologs of other archaeal species (26), all four H. salinarum proteins possess two characteristic conserved domains: an N-terminal helix-turn-helix DNA binding domain (PF01325 (50)) and a C-terminal iron binding domain (PF02742; Supplementary Figure S4B). SirR also contains an SH3 (PF04023) dimerization domain and a Mn2+ binding domain at its C-terminus (Supplementary Figure S4B). All four DtxR proteins in H. salinarum have retained the highly conserved, characteristic iron-binding residues that change protein conformation and DNA binding affinity in bacteria (Supplementary Figure S4C; (34,51–55)).
In order to elucidate the importance of these DtxR family TFs in the iron starvation response, we performed growth and survival phenotype analysis in four strains, each deleted of one of the four TFs. According to the AUC metric (also known as growth potential, see ‘Materials and Methods’ section and (44)), the Δura3 parent strain growth was reduced 1.5-fold under iron starvation conditions relative to iron-replete conditions (Figure 1A). Δidr2 and ΔtroR mutants showed a slight sensitive growth phenotype relative to the parent strain in the presence of iron, although this difference was only statistically significant in the case of ΔtroR. However, under iron starvation, both of these mutant strains exhibited significant impairment of growth (Figure 1A) as well as strongly reduced survival (Figure 1B) relative to the Δura3 parent strain. In contrast, ΔsirR exhibited significantly impaired growth under iron replete conditions, but significantly improved growth relative to the parent strain under iron deficiency conditions. Sensitive phenotypes were complemented by in trans expression of a wild-type copy of the gene encoding each TF in the corresponding mutant background (Supplementary Figure S5, (34)). Δidr1 growth phenotype was statistically indistinguishable from that of Δura3 under both conditions tested (Figure 1A and B; Supplementary Figure S1). Together these results suggest that Idr2 and TroR play a critical role in cell growth and survival under iron deficiency conditions, whereas SirR is important under iron replete conditions. Idr1 may have a redundant, minor, or balancing function in iron homeostasis regulation.
Each DtxR TF influences the expression of genes encoding subsets of the other TFs
It was previously demonstrated that Idr1 and Idr2 regulate the expression of the sirR gene and that Idr2 indirectly regulates idr1 (34). Because DtxR TFs play a role during iron stress adaptation (Figure 1), we reasoned that these TFs might work together to regulate the iron starvation response. To test this, we used quantitative RT-PCR (RT-qPCR) to measure the expression of each gene encoding the DtxR TFs in deletion strains of the other TFs (Figure 2). The Δura3, Δidr1, Δidr2, ΔsirR and ΔtroR mutant strains were grown under iron deficiency and expression was measured immediately before and 5, 20 and 60 min after the addition of 100 μM FeSO4. In the Δura3 parent strain during iron starvation, expression of idr2 and troR genes were induced 7- and 4-fold, respectively, and then rapidly repressed upon iron re-addition, reaching a low steady state level within 60 min (Figure 2A and B). In contrast, sirR gene expression was low under iron starvation conditions and was then gradually induced upon iron re-addition, reaching maximal 4-fold induction after 1 h (Figure 2C). idr1 expression was modestly induced by iron re-addition (Figure 2D). These transcriptional changes were reflected at the protein level under steady state conditions in cultures grown with and without iron (Supplementary Figure S6). Taken together, these results are consistent with growth phenotypes (Figure 1), suggesting that Idr2 and TroR are active during iron starvation, whereas sirR is active during iron replete conditions. Idr1 appears to play minor role in variable iron conditions.
Figure 2.

Gene expression analysis by qRT- PCR demonstrates that TF-DNA binding has significant effects on gene expression of TF-coding genes. Expression of the genes (A) idr2 (B) troR (C) sirR and (D) idr1 across the iron re-addition time course. Each line indicates the expression of that gene in each of the knockout backgrounds of the other TFs (see legend for color scheme). Gene expression is relative to the highly expressed housekeeping control gene, VNG1756G. In each graph, the expression of the gene shown is normalized to the minimum value of expression across the genetic backgrounds. Each point on the graph represents the mean of three technical replicates from three independent biological replicates. Error bars represent SEM of biological replicates. Stars represent P < 0.05 from t-tests comparing expression of each gene in each knockout background at each time point to that in the Δura3 parent strain (Supplementary Table S3 for all P-values).
idr2 expression was significantly lower in the ΔtroR knockout background, in contrast to the parent strain under iron starvation conditions (P < 0.05 at 0 min time point by t-test, Supplementary Table S3), but significantly higher in the ΔsirR background (P < 0.05, Supplementary Table S3 and Figure 2A). idr2 expression was not affected by deleting idr1. troR expression was significantly elevated in ΔsirR relative to the parent strain regardless of iron concentration and in Δidr1 under iron starvation (Figure 2B). troR expression was significantly elevated in Δidr2 only at 20 min after iron re-addition. As expected from our previous transcriptomics and genome-wide binding data (34), sirR expression was low in Δidr1 in iron replete conditions, significantly elevated in Δidr2 regardless of condition, and modestly affected by deletion of troR (Figure 2C). idr1 expression was significantly higher under iron starvation in the ΔtroR and Δidr2 knockout backgrounds, and significantly lower in iron replete conditions in the ΔsirR deletion background (Figure 2D). Taken together, these results suggest that each of the four DtxR TFs influences the expression of a specific subset of the others under certain iron conditions.
Inferring network topology for inter-TF regulation between DtxR family homologs in response to different iron environments
To further test the statistical significance of the effect of TF deletions on gene expression and infer the GRN topology among the four DtxR TFs, we constructed an ANOVA statistical model (‘Materials and Methods’ section, (36)) at each time point (Figure 3). In the model output, each edge represents a significant regulatory influence (P < 0.05) of one TF over the expression of the other. These regulatory edges capture effects on gene expression of removing each TF that are both direct (i.e. TF-promoter binding) and indirect (e.g. other TFs, changes in cell state). We observed that the inferred networks exhibit extensive feedback between the four TFs. For example, at the 0 time point under iron starvation, Idr2 is required for significant repression of sirR, activation of troR and repression of idr1 (Figure 3). TroR and Idr1 repress each other, and TroR feeds back to activate idr2. This 0-min time point network and those inferred across the other time points are consistent with t-tests of significance of gene expression in each mutant background versus the parent strain (Figure 2, Supplementary Table S3, see also Supplementary Table S4 for ANOVA network edge values). One exception was SirR-mediated repression of troR at 20 min, which was significant by t-test (Figure 2B) but not in the ANOVA model (Figure 3 and Supplementary Table S4), suggesting that ANOVA is statistically more stringent. In summary, the ANOVA model enabled direct inference of network state at each time point, aiding interpretation of gene expression of the four TFs in the context of extensive and counterintuitive regulatory feedback.
Figure 3.

ANOVA modeling infers the gene regulatory network (GRN) from gene expression data. Each network diagram depicts the union of direct and indirect regulatory influences inferred from gene expression data. Red arrows represent activation relationships, whereas bar-ended blue lines represent repression relationships. Intensity of the colors of the lines represent the magnitude of the regulatory influence predicted by ANOVA (minimum value = −1.15, maximum value = 0.84, Supplementary Table S4). Square nodes represent TFs. As in Figure 2, time points correspond to time relative to iron addition, with 0 immediately prior to addition (i.e. iron starvation).
Each DtxR TF binds promoters of genes encoding other DtxR TFs in a condition-specific and TF-specific manner
ANOVA output topologies were based on the RT-qPCR gene expression data alone and therefore represent the union of direct and indirect regulatory influences (Figure 3). We next sought to determine the GRN topology consisting only of direct regulatory influences. Previous genome-wide transcriptomic and TF-DNA binding data showed that Idr2 directly binds and represses the expression of the sirR gene under iron starvation conditions (34). In contrast, Idr1 binds and activates sirR under iron replete conditions. Here we measured the binding of TroR and SirR to the promoters of genes encoding each DtxR TF under iron starvation versus replete conditions (sequences of primers used are listed in Supplementary Table S1 and schematic of primer positions in Supplementary Figure S2) using chromatin immunoprecipitation coupled with quantitative PCR (ChIP-qPCR).
For SirR in the presence of iron, we observed significant enrichment of binding to its own promoter (∼3.5-fold, Figure 4A; P-values of significance relative to the background controls are listed in Supplementary Table S3) and to the promoter of the troR gene (∼2.5-fold; Figure 4A). SirR binding enrichment to the idr1 and idr2 promoters was not significantly detectable above the background distribution in the presence of iron. SirR binding enrichment was not detected for any promoters in the absence of iron (Figure 4A), suggesting the SirR activity is specific to metal replete conditions. In contrast, TroR bound the idr1 (∼5.5-fold enrichment) and idr2 (∼3-fold) promoters only during iron starvation (Figure 4B). TroR did not bind to its own promoter or the sirR promoter under any conditions tested. These DNA binding results for SirR and TroR, together with those for Idr1 and Idr2 (34), indicate that the binding activity of each DtxR TF to specific subsets of promoters of genes encoding themselves and the other TFs is stimulated under certain iron regimes.
Figure 4.

DtxR TF-DNA binding analysis suggests that SirR and TroR directly regulate genes encoding the other TFs. ChIP-qPCR analysis of binding of (A) SirR and (B) TroR to the promoters of idr1, idr2, sirR and troR genes. Bars represent the mean and SEM of binding enrichment from biological replicate experiments across multiple amplicons for each promoter region (n ranges from 4 to 9). Stars represent P < 0.005 by t-test comparing the binding event of interest to the distribution of all binding data under that condition (n = 41). Primer sequences in Supplementary Table S1, P-values for all comparisons listed in Supplementary Table S3. Positions of the primers used to amplify binding sites of the four promoters are depicted on Supplementary Figure S2.
Boolean prediction of GRN dynamics based on direct TF–TF regulatory influences
To determine the network topology, or architecture, of direct regulatory influences of each TF over the others, we combined edges (connections between TFs) across the four time points of the ANOVA model (Figure 3), then pruned edges from this topology that were not supported by ChIP-qPCR binding data (Figure 4). For example, in the absence of iron, Idr2-mediated repression of idr1 under iron starvation (Figure 2D) is likely indirect through both SirR and TroR because SirR represses troR, and TroR, in turn, directly represses idr1 (Figures 2D and 4). After pruning, the resultant topology of the iron homeostasis network in H. salinarum consists of two interlocking positive feedback loops between the TFs (Figure 5). Specifically, one feedback loop consists of the following edges: TroR activation of Idr2, Idr2 repression of SirR and SirR repression of TroR. The second loop includes SirR repression of TroR, TroR repression of Idr1 and Idr1 activation of SirR.
In other organisms such as yeast and plants, interlocked positive feedback loop architectures are capable of producing oscillatory dynamics in some parameter regimes and bistability in other regimes (56–58). Both of these dynamical properties contribute to the robustness of the system against random fluctuations in external conditions while enabling response flexibility (57,58). We constructed a synchronously updating Boolean model (37) based on the DtxR TF network topology in order to test for these dynamical properties (Table 1 and Figure 5). As a starting point, we assumed autoactivation for SirR autoregulation. Based on the TF activities from the gene expression (Figure 2) and TF-DNA binding data (Figure 4, (34)), we assumed that iron activates SirR and Idr1 but represses Idr2 and TroR regulatory activity (Figure 5A). Based on this regulatory logic (Table 1), we observed two attractors: one in which Idr2 and TroR were active when iron was absent throughout the simulation, the other in which SirR and Idr1 were active in the constant presence of iron (Figure 5B). The attractors were observed in equal proportion for simulations from each of the 32 possible starting states (Supplementary Figure S3A and B). These final attractor states were unaffected by changes in regulatory logic when we simulated: (i) SirR AND Idr1 activate sirR; (ii) SirR OR Idr1 activate sirR; (iii) SirR autoactivation versus autorepression (Supplementary Figure S3). However, with autorepression logic, SirR is active at three intermediate time steps under iron starvation conditions, which is inconsistent with data that SirR is only active under iron replete conditions (Figures 2 and 3, (27); Supplementary Figure S3C). The model is therefore able to predict certain aspects of the complex logic of SirR activation. Taken together, these Boolean simulations resulting in two strong attractor states are consistent with a bistable system under constant iron conditions.
Previous data suggests that Idr2 and SirR are involved in regulating iron uptake: Idr2 induces siderophore biosynthesis genes (34), which we hypothesize enables iron uptake; whereas SirR represses the expression of putative metal uptake genes under iron replete conditions (27). We reasoned that the incorporation of metal uptake regulation as edges in the network topology could enable Boolean model prediction of system responsiveness to fluctuating external iron concentrations (Figure 5C). Indeed, the additional feedback between iron and the GRN produced oscillations (i.e. repeated transitions between opposing network states) between the iron starvation network state (i.e. Idr2 and TroR active) and the iron replete network state (i.e. SirR and Idr1 active; Figure 5D). These oscillations were robust, with the same expression dynamic patterns predicted for all of the 32 possible starting states. Oscillations were slow, taking three time steps for a full transition to the opposite network state. Taken together, the dynamics produced by this Boolean model are consistent with the hypothesis that the four DtxR family TFs in H. salinarum regulate each other in multiple feedback loops, providing the checks and balances needed to maintain intracellular iron concentrations in the homeostatic range during changes in external concentrations.
Transcription network model validation: iron uptake is altered in TF mutants
From the GRN model together with the Boolean predictions (Figure 5), we expect that knockouts of idr2, troR and sirR should have a strong impact on iron uptake. However, we reasoned that idr1 should have a more modest effect on metal uptake given the relatively more extensive regulation of SirR activity. To test this, inductively coupled plasma mass spectrometry (ICP-MS) analysis was conducted to quantify intracellular concentrations of iron and other transition metals. Loss of sirR led to approximately 1.5-fold over-accumulation of intracellular iron in both iron replete and iron-depleted media; these differences were statistically significant in iron replete conditions (Figure 6A; P-values of significance listed in Supplementary Table S3). In contrast, knockout strains Δidr1, Δidr2 and ΔtroR accumulated 1.5- to 5-fold less intracellular iron than the parent strain under both iron-replete and iron starvation conditions (Figure 6). However, these differences were only statistically significant in Δidr2 in both conditions, and in ΔtroR under iron starvation (Figure 6B). These results are strongly concordant with the model predictions that (i) Idr2 and SirR are the major regulators of iron uptake; and (ii) the transcriptional regulatory network plays a major role in the maintenance iron homeostasis under steady state conditions.
Figure 6.

Measurement of cytosolic iron accumulation in DtxR TF mutants by ICP-MS validates the GRN model. Bars represent measurements of ICP-MS iron quantification in the presence (A) and absence (B) of iron. Each bar represents the average and SD of three technical replicates from three independent biological replicates for each strain normalized to protein concentration. P-values of significance of differences between each mutant and the Δura3 parent strain by t-test are shown in Supplementary Table S3.
The quantification of other metals by ICP-MS showed that the parent strain accumulates significantly higher concentrations of manganese (Mn) in iron-deprived conditions than in iron replete medium (Supplementary Figure S7 and Table S3). Δidr2 is significantly impaired for Mn uptake in iron starvation conditions. In contrast, ΔsirR exhibits ∼3-fold higher Mn uptake relative to the parent strain regardless of iron addition. Although not statistically significant due to the high variability in the Mn measurements in ΔsirR, this result is nevertheless consistent with the hypothesized role of SirR in repression of genes encoding Mn uptake transporters (27). Although the overall levels of cobalt (Co) and nickel (Ni) were quite close to the limit of detection (nM range), we observed significantly lower Co, Ni and Mn uptake in ΔtroR, lower Ni uptake in Δidr1, and higher Ni uptake in ΔsirR (Supplementary Figure S7). This suggests that the DtxR network may also regulate the uptake of other transition metals besides iron. Together these metal quantification assays strongly support the transcriptional regulatory network circuitry proposed in our model (Figure 5), which functions to maintain homeostatic intracellular iron concentrations in response to changes in extracellular levels.
DISCUSSION
Here, we report a novel network comprised of four TFs that regulates iron homeostasis and possibly other metals in archaea. Each TF except for Idr1 has an important effect on growth under either iron starvation or iron sufficiency (Figure 1). Such homeostasis is maintained by extensive transcriptional feedback between four DtxR family TFs (Figures 3 and 5), each of which regulates expression of (Figure 2) and directly binds to (Figure 4) at least one other TF. Dynamical modeling predicts that this network exhibits bistability under constant iron conditions and oscillates between iron replete and iron starvation states when iron levels are allowed to fluctuate (Figure 5). Measurements of intracellular iron concentrations in cells deleted of each of the four TFs are consistent with these predictions (Figure 6).
However, because the H. salinarum network regulates the expression of genes involved in iron uptake (34), we cannot yet rule out that changes in internal iron content may also affect the pools of other metals and the activity of the TFs themselves, as has been observed for bacterial DtxR homologs (7). Indeed, our ICP-MS data suggest that manganese uptake is significantly increased during iron starvation (Supplementary Figure S7), consistent with previous reports that halophiles and other stress resistant microbes maintain high intracellular manganese relative to iron, protecting cells against stressors such as oxidative radicals and DNA damage (59,60). Here, we observed that the Mn:Fe ratio is perturbed in the ΔsirR knockout (Figure 6 and Supplementary Figure S7), and previously we showed that the ΔsirR knockout mutant strain is highly sensitive to oxidative stress (61). Together, these data support a regulatory and physiological connection between Mn, Fe and oxidative stress that deserves additional future attention.
Here we observed that the network of DtxR TFs regulate each other in an interlocking positive feedback loop topology (Figure 5). Previous theoretical and experimental studies of exemplar networks across eukaryotes and bacteria have demonstrated that nested feedback topologies enable buffering against random fluctuations in external conditions; for example, maintaining photoperiod length in the fungal circadian clock during seasonal light and temperature variation (62). Our Boolean simulation of network dynamics in H. salinarum also suggest extensive buffering against transient fluctuations in iron concentration endemic to hypersaline environments. Mutual reinforcement of the positive feedback by the two opposing sides of the interlocked loops also enables stable maintenance of a certain physiological state, such as colony morphology in Candida (57) or flowering in plants (56). This property is consistent with our observation of the increased homeostatic range of iron concentration (from ∼3 to 30 μM, Figure 6) relative to that of bacteria (few micromolar (7,31,33)). This feature explains the resistance of H. salinarum to iron starvation (Figure 1, (32)), while also enabling the rapid changes in gene expression (within 5–10 min, Figures 2 and 5). By extension, the DtxR network may remain poised to respond to a sustained alteration in extracellular iron, as supported by our detection of bistability in the model simulations under constant iron starvation or sufficiency (Figure 5A and B). Together, these features of the interlocked feedback loop topology explain our observations and make important predictions for future testing regarding the dynamical properties of the iron homeostasis network in other starvation-resistant organisms.
The results presented here are consistent with the hypothesis that new TF acquisition and subsequent network rewiring is a universally conserved mechanism across the domains of life for increasing resistance to iron starvation stress. For example, certain species of pathogenic fungi have integrated a unique TF called Sef1 to the conserved fungal iron network to enable positive feedback, reinforcing iron uptake during starvation (63). Fur in pathogenic Neisseria bacteria regulates a novel ArsR TF family homolog, which is required for iron starvation resistance and virulence (64). In the model yeast S. cerevisiae, Cth1 and Cth2 auto- and cross-regulate each other, creating a cis/trans regulatory loop that facilitates rapid adaptation to changes in iron availability (14). The results presented here suggest that H. salinarum has also evolved an elaborated network topology, enabling higher resistance to iron starvation (Figures 1 and 6) relative to those conserved across other archaea (∼75% of sequenced genomes encode only 1–3 DtxR TFs (26)). To our knowledge, feedback loop networks composed of purely transcriptional mechanisms have not yet been observed in bacteria (65–68). More specifically, neither TF–TF regulation nor transcriptional feedback has yet been observed for DtxR family TFs in bacteria. The H. salinarum network therefore represents an unusual hybrid with bacterial-like TFs connected through eukaryote-like topology for robust and flexible transcriptional control of iron homeostasis.
AVAILABILITY
All modeling source code, dependencies and instructions for use can be found in the GitHub repository https://github.com/amyschmid/DtxR.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the Schmid Lab for helpful discussions. We thank Paul Magwene for advice on the manuscript and Boolean modeling tutorials.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-1417750, MCB-CAREER-1651117 to A.K.S., Graduate Student Research Fellowship to P.D.T.]; Duke University Energy Initiative grant to A.K.S; ENIGMA-Ecosystems and Networks Integrated with Genes and Molecular Assemblies, U.S. Department of Energy, Office of Science, Office of Biological & Environmental Research (Scientific Focus Area Program at Lawrence Berkeley National Laboratory) [DE-AC02-05CH11231]. Funding for open access charge: National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Andrews S.C., Robinson A.K., Rodriguez-Quinones F.. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003; 27:215–237. [DOI] [PubMed] [Google Scholar]
- 2. de Silva D.M., Askwith C.C., Kaplan J.. Molecular mechanisms of iron uptake in eukaryotes. Physiol. Rev. 1996; 76:31–47. [DOI] [PubMed] [Google Scholar]
- 3. Imlay J.A. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003; 57:395–418. [DOI] [PubMed] [Google Scholar]
- 4. Imlay J.A., Chin S.M., Linn S.. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988; 240:640–642. [DOI] [PubMed] [Google Scholar]
- 5. Imlay J.A., Linn S.. DNA damage and oxygen radical toxicity. Science. 1988; 240:1302–1309. [DOI] [PubMed] [Google Scholar]
- 6. Keyer K., Imlay J.A.. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:13635–13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Helmann J.D. Specificity of metal sensing: iron and manganese homeostasis in Bacillus subtilis. J. Biol. Chem. 2014; 289:28112–28120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonneau R., Facciotti M.T., Reiss D.J., Schmid A.K., Pan M., Kaur A., Thorsson V., Shannon P., Johnson M.H., Bare J.C. et al. . A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007; 131:1354–1365. [DOI] [PubMed] [Google Scholar]
- 9. Brooks A.N., Reiss D.J., Allard A., Wu W.J., Salvanha D.M., Plaisier C.L., Chandrasekaran S., Pan M., Kaur A., Baliga N.S.. A system-level model for the microbial regulatory genome. Mol. Syst. Biol. 2014; 10:740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopez-Maury L., Marguerat S., Bahler J.. Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2008; 9:583–593. [DOI] [PubMed] [Google Scholar]
- 11. Jo W.J., Kim J.H., Oh E., Jaramillo D., Holman P., Loguinov A.V., Arkin A.P., Nislow C., Giaever G., Vulpe C.D.. Novel insights into iron metabolism by integrating deletome and transcriptome analysis in an iron deficiency model of the yeast Saccharomyces cerevisiae. BMC Genomics. 2009; 10:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamaguchi-Iwai Y., Dancis A., Klausner R.D.. AFT1: a mediator of iron regulated transcriptional control in Saccharomyces cerevisiae. EMBO J. 1995; 14:1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rutherford J.C., Jaron S., Winge D.R.. Aft1p and Aft2p mediate iron-responsive gene expression in yeast through related promoter elements. J. Biol. Chem. 2003; 278:27636–27643. [DOI] [PubMed] [Google Scholar]
- 14. Martinez-Pastor M., Vergara S.V., Puig S., Thiele D.J.. Negative feedback regulation of the yeast CTH1 and CTH2 mRNA binding proteins is required for adaptation to iron deficiency and iron supplementation. Mol. Cell. Biol. 2013; 33:2178–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Outten C.E., Albetel A.N.. Iron sensing and regulation in Saccharomyces cerevisiae: Ironing out the mechanistic details. Curr. Opin. Microbiol. 2013; 16:662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodionov D.A., Gelfand M.S., Todd J.D., Curson A.R., Johnston A.W.. Computational reconstruction of iron- and manganese-responsive transcriptional networks in alpha-proteobacteria. PLoS Comput. Biol. 2006; 2:e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hantke K. Iron and metal regulation in bacteria. Curr. Opin. Microbiol. 2001; 4:172–177. [DOI] [PubMed] [Google Scholar]
- 18. Lee J.W., Helmann J.D.. Functional specialization within the Fur family of metalloregulators. Biometals. 2007; 20:485–499. [DOI] [PubMed] [Google Scholar]
- 19. Platero R., de Lorenzo V., Garat B., Fabiano E.. Sinorhizobium meliloti fur-like (Mur) protein binds a fur box-like sequence present in the mntA promoter in a manganese-responsive manner. Appl. Environ. Microbiol. 2007; 73:4832–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giel J.L., Nesbit A.D., Mettert E.L., Fleischhacker A.S., Wanta B.T., Kiley P.J.. Regulation of iron-sulphur cluster homeostasis through transcriptional control of the Isc pathway by [2Fe-2S]-IscR in Escherichia coli. Mol. Microbiol. 2013; 87:478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo S.W., Kim D., Latif H., O’Brien E.J., Szubin R., Palsson B.O.. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 2014; 5:4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perez-Rueda E., Janga S.C.. Identification and genomic analysis of transcription factors in archaeal genomes exemplifies their functional architecture and evolutionary origin. Mol. Biol. Evol. 2010; 27:1449–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gehring A.M., Walker J.E., Santangelo T.J.. Transcription regulation in archaea. J. Bacteriol. 2016; 198:1906–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grohmann D., Werner F.. Recent advances in the understanding of archaeal transcription. Curr. Opin. Microbiol. 2011; 14:328–334. [DOI] [PubMed] [Google Scholar]
- 25. Peeters E., Peixeiro N., Sezonov G.. Cis-regulatory logic in archaeal transcription. Biochem. Soc. Trans. 2013; 41:326–331. [DOI] [PubMed] [Google Scholar]
- 26. Leyn S.A., Rodionov D.A.. Comparative genomics of DtxR family regulons for metal homeostasis in Archaea. J. Bacteriol. 2015; 197:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kaur A., Pan M., Meislin M., Facciotti M.T., El-Gewely R., Baliga N.S.. A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 2006; 16:841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Louvel H., Kanai T., Atomi H., Reeve J.N.. The Fur iron regulator-like protein is cryptic in the hyperthermophilic archaeon Thermococcus kodakaraensis. FEMS Microbiol. Lett. 2009; 295:117–128. [DOI] [PubMed] [Google Scholar]
- 29. Zhu Y., Kumar S., Menon A.L., Scott R.A., Adams M.W.. Regulation of iron metabolism by Pyrococcus furiosus. J. Bacteriol. 2013; 195:2400–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Domagalski J.L., Eugster H.P., Jones B.F.. Spencer RJaCI-M. Trace metal geochemistry of Walker, Mono, and Great Salt Lakes. Fluid-Mineral Interactions: a Tribute to H. P. Eugster. 1990; 2:Washington, D.C.: The Geochemical Society; 315–353. [Google Scholar]
- 31. Hubbard J.A., Lewandowska K.B., Hughes M.N., Poole R.K.. Effects of iron-limitation of Escherichia coli on growth, the respiratory chains and gallium uptake. Arch. Microbiol. 1986; 146:80–86. [DOI] [PubMed] [Google Scholar]
- 32. Hubmacher D., Matzanke B.F., Anemuller S.. Iron-uptake in the Euryarchaeon Halobacterium salinarum. Biometals. 2007; 20:539–547. [DOI] [PubMed] [Google Scholar]
- 33. Ratledge C., Winder F.G.. Effect of iron and zinc on growth patterns of Escherichia coli in iron-deficient medium. J. Bacteriol. 1964; 87:823–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schmid A.K., Pan M., Sharma K., Baliga N.S.. Two transcription factors are necessary for iron homeostasis in a salt-dwelling archaeon. Nucleic Acids Res. 2011; 39:2519–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burrell M., Hanfrey C.C., Kinch L.N., Elliott K.A., Michael A.J.. Evolution of a novel lysine decarboxylase in siderophore biosynthesis. Mol. Microbiol. 2012; 86:485–499. [DOI] [PubMed] [Google Scholar]
- 36. Seabold J.S., Perktold J.. Statsmodels: econometric and statistical modeling with python. Proceedings of the 9th Python in Science Conference. 2010; Austin: SciPy society; 57–61. [Google Scholar]
- 37. Wang R.S., Saadatpour A., Albert R.. Boolean modeling in systems biology: an overview of methodology and applications. Phys. Biol. 2012; 9:055001. [DOI] [PubMed] [Google Scholar]
- 38. Peck R.F., Dassarma S., Krebs M.P.. Homologous gene knockout in the archaeon Halobacterium salinarum with ura3 as a counterselectable marker. Mol. Microbiol. 2000; 35:667–676. [DOI] [PubMed] [Google Scholar]
- 39. Schmid A.K., Reiss D.J., Pan M., Koide T., Baliga N.S.. A single transcription factor regulates evolutionarily diverse but functionally linked metabolic pathways in response to nutrient availability. Mol. Syst. Biol. 2009; 5:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilbanks E.G., Larsen D.J., Neches R.Y., Yao A.I., Wu C.Y., Kjolby R.A., Facciotti M.T.. A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq. Nucleic Acids Res. 2012; 40:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dulmage K.A., Todor H., Schmid A.K.. Growth-phase-specific modulation of cell morphology and gene expression by an archaeal histone protein. Mbio. 2015; 6, doi:10.1128/mBio.00649-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Todor H., Sharma K., Pittman A.M., Schmid A.K.. Protein-DNA binding dynamics predict transcriptional response to nutrients in archaea. Nucleic Acids Res. 2013; 41:8546–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Darnell C.L., Schmid A.K.. Systems biology approaches to defining transcription regulatory networks in halophilic archaea. Methods. 2015; 86:102–114. [DOI] [PubMed] [Google Scholar]
- 44. Todor H., Dulmage K., Gillum N., Bain J.R., Muehlbauer M.J., Schmid A.K.. A transcription factor links growth rate and metabolism in the hypersaline adapted archaeon Halobacterium salinarum. Mol. Microbiol. 2014; 93:1172–1182. [DOI] [PubMed] [Google Scholar]
- 45. Sharma K., Gillum N., Boyd J.L., Schmid A.. The RosR transcription factor is required for gene expression dynamics in response to extreme oxidative stress in a hypersaline-adapted archaeon. BMC Genomics. 2012; 13:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pfaffl M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001; 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T.. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011; 27:431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mukhopadhyay A., Deplancke B., Walhout A.J., Tissenbaum H.A.. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat. Protoc. 2008; 3:698–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vaccaro B.J., Menon A.L., Lancaster A.W., Adams M.W.W.. Metallomics using inductively coupled plasma mass spectrometry. Curr. Protoc. Chem. Biol. 2012; 4:249–274. [Google Scholar]
- 50. Finn R.D., Mistry J., Schuster-Bockler B., Griffiths-Jones S., Hollich V., Lassmann T., Moxon S., Marshall M., Khanna A., Durbin R. et al. . Pfam: clans, web tools and services. Nucleic Acids Res. 2006; 34:D247–D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H., Wu R., Xu G., Fang X., Qiu X., Guo H., Tian B., Hua Y.. DR2539 is a novel DtxR-like regulator of Mn/Fe ion homeostasis and antioxidant enzyme in Deinococcus radiodurans. Biochem. Biophys. Res. Commun. 2010; 396:413–418. [DOI] [PubMed] [Google Scholar]
- 52. Qiu X., Verlinde C.L., Zhang S., Schmitt M.P., Holmes R.K., Hol W.G.. Three-dimensional structure of the diphtheria toxin repressor in complex with divalent cation co-repressors. Structure. 1995; 3:87–100. [DOI] [PubMed] [Google Scholar]
- 53. Schiering N., Tao X., Zeng H., Murphy J.R., Petsko G.A., Ringe D.. Structures of the apo- and the metal ion-activated forms of the diphtheria tox repressor from Corynebacterium diphtheriae. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:9843–9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. D’Aquino J.A., Tetenbaum-Novatt J., White A., Berkovitch F., Ringe D.. Mechanism of metal ion activation of the diphtheria toxin repressor DtxR. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:18408–18413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Feese M.D., Ingason B.P., Goranson-Siekierke J., Holmes R.K., Hol W.G.. Crystal structure of the iron-dependent regulator from Mycobacterium tuberculosis at 2.0-A resolution reveals the Src homology domain 3-like fold and metal binding function of the third domain. J. Biol. Chem. 2001; 276:5959–5966. [DOI] [PubMed] [Google Scholar]
- 56. Jaeger K.E., Pullen N., Lamzin S., Morris R.J., Wigge P.A.. Interlocking feedback loops govern the dynamic behavior of the floral transition in Arabidopsis. Plant Cell. 2013; 25:820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zordan R.E., Miller M.G., Galgoczy D.J., Tuch B.B., Johnson A.D.. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 2007; 5:e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cheng P., Yang Y., Liu Y.. Interlocked feedback loops contribute to the robustness of the Neurospora circadian clock. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:7408–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Robinson C.K., Webb K., Kaur A., Jaruga P., Dizdaroglu M., Baliga N.S., Place A., Diruggiero J.. A major role for nonenzymatic antioxidant processes in the radioresistance of Halobacterium salinarum. J. Bacteriol. 2011; 193:1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Culotta V.C., Daly M.J.. Manganese complexes: diverse metabolic routes to oxidative stress resistance in prokaryotes and yeast. Antioxid. Redox. Signal. 2013; 19:933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tonner P.D., Darnell C.L., Engelhardt B.E., Schmid A.K.. Detecting differential growth of microbial populations with Gaussian process regression. Genome Res. 2017; 27:320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Akman O.E., Rand D.A., Brown P.E., Millar A.J.. Robustness from flexibility in the fungal circadian clock. BMC Syst. Biol. 2010; 4:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gerwien F., Safyan A., Wisgott S., Hille F., Kaemmer P., Linde J., Brunke S., Kasper L., Hube B.. A novel hybrid iron regulation network combines features from pathogenic and nonpathogenic yeasts. Mbio. 2016; 7:e01782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu C., McClure R., Nudel K., Daou N., Genco C.A.. Characterization of the Neisseria gonorrhoeae Iron and Fur Regulatory Network. J. Bacteriol. 2016; 198:2180–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gama-Castro S., Salgado H., Santos-Zavaleta A., Ledezma-Tejeida D., Muniz-Rascado L., Garcia-Sotelo J.S., Alquicira-Hernandez K., Martinez-Flores I., Pannier L., Castro-Mondragon J.A. et al. . RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016; 44:D133–D143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shen-Orr S.S., Milo R., Mangan S., Alon U.. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002; 31:64–68. [DOI] [PubMed] [Google Scholar]
- 67. Thieffry D., Huerta A.M., Perez-Rueda E., Collado-Vides J.. From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in Escherichia coli. Bioessays. 1998; 20:433–440. [DOI] [PubMed] [Google Scholar]
- 68. Milo R., Shen-Orr S., Itzkovitz S., Kashtan N., Chklovskii D., Alon U.. Network motifs: simple building blocks of complex networks. Science. 2002; 298:824–827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.