


Abstract
Toxin-antitoxin (TA) systems are essential for bacterial persistence under stressful conditions. In particular, Mycobacterium tuberculosis express VapBC TA genes that encode the stable VapC toxin and the labile VapB antitoxin. Under normal conditions, these proteins interact to form a non-toxic TA complex, but the toxin is activated by release from the antitoxin in response to unfavorable conditions. Here, we present the crystal structure of the M. tuberculosis VapBC26 complex and show that the VapC26 toxin contains a pilus retraction protein (PilT) N-terminal (PIN) domain that is essential for ribonuclease activity and that, the VapB26 antitoxin folds into a ribbon-helix-helix DNA-binding motif at the N-terminus. The active site of VapC26 is sterically blocked by the flexible C-terminal region of VapB26. The C-terminal region of free VapB26 adopts an unfolded conformation but forms a helix upon binding to VapC26. The results of RNase activity assays show that Mg2+ and Mn2+ are essential for the ribonuclease activity of VapC26. As shown in the nuclear magnetic resonance spectra, several residues of VapB26 participate in the specific binding to the promoter region of the VapBC26 operon. In addition, toxin-mimicking peptides were designed that inhibit TA complex formation and thereby increase toxin activity, providing a novel approach to the development of new antibiotics.
INTRODUCTION
Mycobacterium tuberculosis has co-existed with humans for at least 15,000 years (1). This bacterium is aerobic, non-spore forming, non-motile and may be either gram-negative or gram-positive (2–4). M. tuberculosis causes tuberculosis, which claims ∼2 million lives per year worldwide (5). Notably, multi-drug resistant tuberculosis (MDR-TB) has emerged as a global concern over the past few decades, and ∼350,000 new MDR-TB cases occur annually worldwide (6). Extensively drug-resistant tuberculosis strains (MDR-TB and XDR-TB, which are resistant to fluoroquinolones and second-line injectables) have been reported in 72% of countries studied (7). Therefore, the development of new antibiotics that can be used to eradicate M. tuberculosis by exploiting new therapeutic strategies is urgently needed.
Pathogenic bacteria, such as M. tuberculosis utilize many toxin-antitoxin (TA) systems to survive, but non-pathogenic bacteria, such as Mycobacterium smegmatis, are reported to use only five TA systems (8). M. tuberculosis has a very low growth rate and a long incubation period, whereas M. smegmatis is a free-living bacterium that grows rapidly. Thus, the growth, survival and pathogenicity of these bacterial species are closely related to the number of TA loci. Furthermore, TA loci do not exist in humans but specifically exist in bacteria. Therefore, TA systems represent potential antibiotic targets (9).
There is increasing evidence that TA systems are strongly correlated with bacterial physiology and that they interact with cellular processes involved in gene regulation, growth arrest, survival and apoptosis (10–14). TA loci were first discovered in 1983 on the mini-F plasmid of Escherichia coli, a plasmid addiction module that is responsible for the maintenance of extrachromosomal genetic elements (15). TA systems located on plasmids are transferred to daughter cells and are involved in plasmid stabilization and cell viability (16,17). If the daughter cell primarily inherits plasmids that lack TA genes, it cannot produce antitoxins but still produces residual TA complexes (18). In this situation, a toxin becomes highly activated upon dissociation from the corresponding antitoxin, which is then degraded by cellular proteases, resulting in cell death (19). In contrast, chromosomal TA systems are commonly related to growth arrest, biofilm formation (20) and multidrug tolerance (21,22), all of which facilitate the development of persister or dormant cells (23). These toxins damage host cells by inducing RNA degradation or by binding to topoisomerase or to ribosomes (24–26). In contrast, an antitoxin is an important cellular components for cell maintenance and viability because it neutralizes the detrimental activity of its cognate toxin (27).
TA systems are usually classified into type I, type II and type III TA systems. These three major groups have been subdivided into six groups, each of which has a different mechanism (28,29). The antitoxin in the the type I TA system are antisense RNAs that bind to toxins, which are mRNAs. Type I antitoxins inhibit the translation of toxins and cause gradual toxin degradation. In the type II TA system, the toxins and antitoxins are proteins that interact with each other to form a non-toxic TA complex. In the type III TA system, antitoxins act as RNAs that bind directly to toxin proteins (30,31). In the most widely distributed type II TA systems, the toxin is thermodynamically stable, whereas the antitoxin is unstable and is cleaved by some cellular proteases because its C-terminal region adopts a flexible conformation that is susceptible to proteolysis (32).
Eighty-eight type II TA operons have been identified in M. tuberculosis; of these, 45 belong to the VapBC family (33,34). The VapBC TA family is one of the most widely distributed type II TA families throughout bacterial species. However, crystal structures have been determined for only six VapBC TA complexes to date: four from M. tuberculosis, Protein Data Bank (PDB) codes: 3H87 (VapBC3), 3DBO (VapBC5), 4CHG (VapBC15) and 4XGQ (VapBC30) (35–38); one from Rickettsia felis (PDB code: 3ZVK); and one from Shigella flexneri (PDB code: 3TND) (39,40). However, the physiological roles of these complexes have not yet been clearly elucidated (36).
VapC toxins commonly contain a PilT N-terminal (PIN) domain that exhibits ribonuclease activity toward cellular mRNAs (41,42). The active sites of VapC toxins consist of three conserved acidic residues that coordinate divalent metal ions such as Mg2+ (43–46), suggesting an acid-base catalysis mechanism for the nucleolytic activity of VapC toxins. VapC26 targets the 23S rRNA in the sarcin-ricin-loop, which is crucial for translation and ribosomal activity (47,48). Only two sarcin-ricin-loop endoribonucleases, VapC20 and VapC26, have been reported in M. tuberculosis, (45,47). VapB antitoxins consist of the following two functional motifs: an N-terminal region that binds to the promoter DNA of the TA operon and a C-terminal region that binds to the toxin and thereby abolishes its toxicity (49). The antitoxin alone or in complex with the cognate toxin acts as a repressor that auto-regulates the transcription of the TA operon in a negative feedback loop (50).
Here, we present the crystal structure of the VapBC26 complex from M. tuberculosis at a resolution of 2.65 Å. The structure reveals the important residues involved in binding to the promoter DNA and in the formation of the VapBC26 complex. The VapC26 toxin forms an overall α/β/α structure with four parallel β strands, and VapB26 adopts a ribbon-helix-helix (RHH) DNA-binding motif. The core residues in VapB26 that bind to DNA and the structural changes in VapB26 that result from toxin binding were clarified by nuclear magnetic resonance (NMR). The catalytic site of VapC26 is composed of three conserved acidic residues; two of these, Asp4 and Asp97, interact directly with Mg2+. The ribonuclease activity of VapC26 was confirmed in this study. Several peptides were designed as antibiotic candidates to mimic the binding interface of the VapBC26 complex and thereby suppressing the TA interaction. This approach may contribute to the development of novel, potent antibiotics that can be used to effectively treat antibiotic-resistant M. tuberculosis.
MATERIALS AND METHODS
Cloning and transformation
The VapC26 (Rv0582) and VapB26 (Rv0581) genes were amplified by the polymerase chain reaction (PCR). The primers used in PCR were as follows: for VapB26, forward, 5’-G GAA TTC CAT ATG GAC AAG ACG ACG GTC-3’; reverse, 5’-TTA CCG CTC GAG CCG CTC ACC GAA GCC AGC CAG-3’; for VapC26, forward, 5’-G GAA TTC CAT ATG ATC ATC GAC ACG AGT GCG; and reverse, 5’-CCG CTC GAG TTA CGG AAT GAC GGT GAA CGC CCC. DNA was purchased from Bioneer Innovation (Republic of Korea). The restriction enzymes used for cloning were Nde1 and Xho1; the enzyme cutting sites are underlined above. Restriction enzymes were purchased from New England Biolabs (http://www.neb.com/). The PCR products of VapB26 and VapC26 were double-cut by Nde1 and Xho1 and ligated to vectors that had been cut by the same enzymes. For structure determination and biological assays, VapB26 and VapC26 were ligated to pET21a without a tag and to pET28b, respectively. The pET28b N-terminal tag (MGSSHHHHHHSSGLVPRGSH) was added to VapC26 as an additional residual tag. For NMR experiments, VapB26 was ligated to pET28b resulting in an N-terminal additional residual tag (MGSSHHHHHHSSGLVPRGSH). Each plasmid was then transformed into E. coli DH5α competent cells.
Protein expression and purification
For crystallization, the cloned plasmids of VapB26 and VapC26 were co-transformed into E. coli Rosetta2(DE3) pLysS competent cells. The transformed cells were grown at 37°C in Luria broth until the OD600 of the culture reached 0.8. Protein overexpression was induced by the addition of 0.5 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG), and the culture was further incubated at 37°C for 4 h. The cultured cells were harvested by centrifugation at 11 355 × g at 4°C and stored at −80°C. The harvested cells were then suspended in buffer A (20 mM Tris-HCl, pH 7.9, and 500 mM NaCl) and lysed by ultrasonication. After centrifugation for 1 h at 28 306 × g, the supernatant containing soluble proteins was loaded on a Ni2+ affinity column (Bio-Rad) equilibrated with buffer A and washed with buffer A containing 100 mM imidazole. The protein bound to the Ni2+ column was eluted using an imidazole gradient (150−800 mM), and the TA complex present in the elution fractions was identified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Finally, the buffer in fractions containing the TA complex was exchanged for buffer consisting of 50 mM Tris-HCl, pH 7.9, 500 mM NaCl and 250 mM imidazole using an Amicon Ultra centrifugal filter unit (Millipore), and the sample was concentrated to 5 mg/ml. The purity of the TA complex protein was verified by SDS-PAGE. The VapBC26 complex displaced by selenomethionine (SeMet) (Calbiochem, USA) was expressed and purified by the same procedure except that the cells, containing the complex displaced by SeMet were grown in M9 medium containing extra essential amino acids. Because there are only three methionine residues in the VapB26/VapC26 complex, we mutated Leu50 of VapB26 to Met50 to obtain sufficient phasing power. The primers used for mutation of VapB26 were as follows: forward, 5’-GGCGGGGCCTGGGAGATGGCCAACTGCGGTGCC-3’ and reverse, 5’-GGCACCGCAGTTGGCCATCTCCCAGGCCCCGCC-3’. Mutation was conducted using the EZchange™ Site-Directed Mutagenesis Kit (Enzynomics, Korea) according to the protocol provided by the manufacturer. For NMR experiments, the N-terminal (His)6-tagged VapB26 protein was expressed in E. coli strain BL21(DE3) using M9 medium containing 1.0 g/l [U-13C] glucose and 1.0 g/l [15N] NH4Cl (Cambridge Isotopes Laboratory) as the sole carbon and nitrogen sources, respectively. The procedure for the initial purification of VapB26 was the same as that used for the TA complex; further purification was conducted by size-exclusion chromatography on a HiLoad 16/60 Superdex 75 preparatory-grade column (GE Healthcare) in a buffer consisting of 20 mM MES, pH 6 and 50 mM NaCl. Expression of VapC26 was difficult because of its toxicity to E. coli (51). Therefore, as an alternative method of obtaining the VapC26 protein, limited proteolysis of the VapBC26 complex using pig pepsin protease (Sigma-Aldrich) was performed (52). Pepsin was added to the VapBC26 protein in proportion to its mass, and an adequate number of units which were used in accordance with the manufacturer's instructions. An appropriate amount of pepsin was added to the purified VapBC26, and the mixture was incubated for 3 h at 37°C. The proteolyzed VapBC26 was further purified by size-exclusion chromatography on a HiLoad 16/60 Superdex 75 preparatory-grade column (GE Healthcare) under the same buffer condition as used in VapBC26 purification.
Crystallization, data collection and processing
Initial crystal screening of purified VapBC26 complex protein was conducted using the crystal screening 1, 2 and Index 1, 2 kit (Hamton Research) by mixing 1 μl of protein solution at 5 mg/ml in 50 mM Tris-HCl, pH 7.9, 500 mM NaCl and 250 mM Imidazole with 1 μl of reservoir solution. Crystals of the VapBC26 complex were grown using the sitting-drop vapor diffusion method at 4°C. The crystallization solution for VapBC26 was 25% Tacsimate (pH 7.0). The crystals were frozen immediately in liquid nitrogen because the crystals underwent extensive cracking damage when they came into contact with cryo-protectants containing glycerol. Data were collected using the ADSC Quantum Q270 CCD detector at beamline 7A of the Pohang Light Source, Republic of Korea. Crystals of the VapBC26 complex belong to the tetragonal space group P41, with unit cell parameters of a = 64.35 Å, b = 64.35 Å, c = 216.96 Å, α = β = γ = 90.00° for the native VapBC26 crystal and a = 64.22 Å, b = 64.22 Å, c = 216.13 Å, α = β = γ = 90.00° for the SeMet-labeled VapBC26 crystal. The calculated total mass of the protein complex, including the N-terminal (His)6 tag, was 24116.3 Da. All raw data were scaled and processed using the HKL2000 (53). The structure of the VapBC26 complex from M. tuberculosis was determined at 2.65 Å resolution by single-wavelength anomalous dispersion using SeMet (2.55 Å for the native complex). Despite the slightly better resolution of the native complex, the structure of the SeMet crystal was as well refined as that of the native complex and the crystallization conditions and the space groups were equal. Therefore, further analysis was conducted using the SeMet data. The Met50 mutation did not affect protein folding. The two structures were almost identical, and the protein–protein interaction patterns were also equal. Detailed statistical information is presented in Supplementary Table S1. We determined the positions of 12 selenium sites in the total asymmetric unit. First, PHENIX (54) was used to automatically build the total residues; the remaining residues were built by Coot (55) to provide the final model, which was refined to Rwork and Rfree(56) of 20.8 and 23.9%, respectively, using REFMAC (57). The overall geometry of the structure was validated using Molprobity (58); 96.2% of the residues were in the favored region of the Ramachandran plot, and 99.9% were in the allowed region. PyMOL (59) was used to generate all figures. The electrostatic potential surface shown in Figure 3C and Supplementary Figure S4 was calculated using Adaptive Poisson-Boltzmann solver (APBS) method (60). Secondary structure analysis was conducted using the 2Struc server (61). The interface area was calculated using the PISA server (62). Sequence alignment was performed using Clustal Omega 1.2.2 (63) and visualized using ESPript 3.0 (64).
Figure 3.

(A and B) Hydrophobic interface of the VapC26 homodimer. The residues participating in hydrophobic interactions are shown as stick models. (A) Close-up view of the binding interface between two VapC26 monomers. More than 30 residues participate in cross-contacts between the α5 helix of one toxin monomer and the α3 and α4 helices of a neighboring toxin monomer. (B) Range of hydrophobic forces from Ile78 and Ile98. (C and D) Analysis of the active site. Mg2+ is shown in yellow. (C) Fo-Fc composite omit map of Mg2+ site contours at 3σ [calculated with PHENIX (52)]. Hydrogen bonds are shown as black dotted lines. Asp4, Glu42, Asp97, Asp116 and Mg2+ organize the active site. (D) The electrostatic surface potential of VapC26.
Multi-angle light scattering coupled with size exclusion chromatography
We performed multi-angle light scattering (MALS) to determine the oligomeric states of VapB26 and VapBC26. Size-exclusion chromatography was conducted on a BioSep SEC-s3000 column (Phenomenex) on 1260 Infinity HPLC system (Agilent Technologies). The scattering data were obtained in a miniDAWN-TREOS line for emission at 657.4 nm (Wyatt Technology) and analyzed by ASTRA 6.0.1.10 software (Wyatt Technology). VapB26 and VapBC26 were used at 100 μM in the experiment. VapB26 was run in 20 mM MES pH6, and 50 mM NaCl as the condition of NMR spectroscopy, and VapBC26 was run in 50 mM Tris-HCl, pH 7.9, 500 mM NaCl and 250 mM Imidazole, the same conditions as those used for crystallization. All experiments were performed at room temperature.
Inductively coupled plasma mass spectrometry
Inductively coupled plasma mass spectrometry was conducted to confirm whether Mg2+ was co-purified and co-crystallized with the VapBC26 complex. The crystals of VapBC26 used in this measurement were prepared in the same buffer as was used in crystallization (50 mM Tris-HCl, pH 7.9, 500 mM NaCl and 250 mM imidazole). An aliquot of the same buffer was used as a control. ELAN 6100, NexION 350D (Perkin-Elmer, USA) and argon plasma (6000 K) were used for detection. The radio frequency power was 1100 W and the sample injection flow rate was 1 ml/min.
Isothermal titration calorimetry (ITC) measurements
Isothermal titration calorimetry (ITC) experiments were performed using a MicroCal 200 (GE Healthcare) at 25°C. The proteins and promoter dsDNAs were prepared in a buffer consisting of 20 mM MES, pH 6 and 50 mM NaCl for VapB26 and in a buffer containing 50 mM Tris-HCl, pH 7.9, 500 mM NaCl and 250 mM Imidazole for VapBC26. The promoter region of VapB26 was selected as [forward-TTCGCGGCTACGCCGACAA; reverse-TTGTCGGCGTAGCCGCGAA] and the annealed dsDNA was purchased from Bioneer Innovation. A control experiment was performed with DNA ‘X’ [forward-GATTTTTTTTGATTTTTTT; reverse-AAAAAAATCAAAAAAAATC], purchased from Bioneer Innovation. DNA ‘X’ was annealed to form double-stranded DNA (dsDNA) by heating to 94°C for 5 min and then cooling to 4°C. Affinity experiments were conducted with the protein solution (10 μM, 320 μl) in the cell and the promoter dsDNA solution (600 μM) as the injected titrant. A total of 19 injections made at 150-s intervals were used for data collection. The MicroCal Origin software was used for curve fitting to calculate the binding affinity (Kd), the enthalpy of binding (ΔH), the entropy of binding (ΔS) and the stoichiometry (n). The raw data were fitted using one-site binding. The Gibbs free energies (ΔG) were calculated using the standard equation ΔG = ΔH − TΔS.
Electrophoretic mobility shift assay
To distinguish the binding uniqueness of VapB26 and its promoter DNA, electrophoretic mobility shift assays (EMSA) were conducted for the own palindrome promoter sequence of the VapB26 [forward-TTCGCGGCTACGCCGACAA; reverse-TTGTCGGCGTAGCCGCGAA], and two other palindromic sequences: ‘a’ [forward-TATTTTAATAACTTAAAAGT; reverse-ACTTTTAAGTTATTAAAATA], and ‘b’ [forward-TAATAGAATAATAAGTATCACTCCTTTA; reverse-TAAAGGAGTGATACTTATTATTCTATTA]. The DNAs were annealed to form double-stranded DNA (dsDNA) by heating to 94°C for 5 min and then cooling to 4°C. dsDNAs and protein were prepared in a binding buffer pH 6 consisting of 20 mM MES and 50 mM NaCl. Varying amounts of VapB26 protein was mixed with each DNA to give a final volume of 10 μl and incubated for 20 min at 4°C. The samples were loaded onto 0.8% agarose gels in 0.5 × TBE (45 mM Tris-borate, 1 mM ethylenediaminetetraacetic acid (EDTA)) buffer. The results were visualized using a Printgraph 2M (ATTO).
In vitro ribonuclease assay for the addition of metals or peptides mimicking the binding region
We confirmed the ribonuclease activity of VapC26 using an RNase Alert kit (IDT). We conducted the fluorescence quenching assay according to the protocol provided by the manufacturer. In this system, a fluorophore is covalently attached to one end of a synthetic RNA strand and is quenched by a quencher group at the other end of the RNA strand. If synthetic RNA containing a fluorophore-quencher pair interacts with ribonuclease, the synthetic RNA is digested and the quencher is released. The released fluorophore emits fluorescence at 520 nm upon excitation at 490 nm. The relative fluorescence units (RFU) were observed on a SPECTRAmax GEMINI XS spectrofluorometer. Using concentrations of VapC26 and VapBC26 of 2.5 μM, several metals, including Mg2+, Mn2+ and Zn2+ as well as peptides mimicking the binding region, were examined to determine the influence of metal and peptide binding on the ribonuclease activity of VapC26. We first investigated the metal-dependent ribonuclease activity of VapC26 using Mg2+, Mn2+ and Zn2+. VapC26 was pretreated with 50 mM EDTA to remove metals that may have been bound during purification. Its metal binding ability was then regenerated by serial dialysis against a buffer consisting of 50 mM Tris-HCl, pH 7.9, 500 mM NaCl and 250 mM imidazole. The concentration of metal ions in the buffer was gradually increased to 0.5 μM, 5 μM, 50 μM, 500 μM, 5 mM and 50 mM. We also designed seven short peptides that mimic the binding regions of VapB26 and VapC26; two of these mimicked the binding region of VapB26, and the other five peptides mimicked the binding region of VapC26. Theoretically, these peptides should compete with the original protein for binding to the complex (65); if the peptides are bound, the toxin is released from the complex, a process that can be monitored by fluorescence quenching. The peptides used in these experiments were purchased from AnyGen (Korea, www.anygen.com).
For the mutational study of the key binding residues of VapB26 and VapC26, Pro46 and Tyr51 of VapB26 and Leu46 of VapC26 were mutated to alanine and glutamate, to diminish or remove the hydrophobic tendency. The resulting mutant proteins were designated P46A, P46E, Y51A, Y51E, L46A and L46E. Mutation was conducted using the EZchange™ Site-Directed Mutagenesis Kit (Enzynomics, Korea) according to the manufacturer's protocol. Mutated VapBC26 complexes were expressed and purified using the same procedures as was used for the native VapBC26 complex and were used in fluorescence quenching assay. The primers used to create these mutations are described in Supplementary Table S2.
NMR study of full-length VapB26 antitoxin and DNA titration
The NMR spectrum of antitoxin VapB26 was measured on a Bruker AVANCE DRX 800 spectrometer. All the experiments were performed at 25°C. The samples were prepared in a buffer containing 20 mM MES, pH 6 and 50 mM NaCl containing 10% (v/v) D2O. The data were processed using NMRPipe/nmrDraw (66) and further analyzed by NMRviewJ (67). The data for carbonyl carbon were obtained through spectrum HNCO and HNCACO, and those of α/β carbon were acquired through the spectrum HNCA, HNCOCA, HNCACB and CBCACONH. Additionally, to understand the structural transition that occurs in the specific RHH domain on VapB26 binding to DNA, a 19-bp DNA fragment from the upstream region (promoter DNA) of VapB26 was added to the VapBC26 complex as a palindromic form. The palindromic region of VapB26 was selected as [forward-TTCGCGGCTACGCCGACAA, reverse-TTGTCGGCGTAGCCGCGAA]; the annealed dsDNA was purchased from Bioneer Innovation. DNA titration was conducted three times during the measurement of the heteronuclear single quantum correlation (HSQC) spectrum. The concentration of VapB26 was 0.4 mM in a total of three 1H, 15N-HSQC spectra and the DNA concentration was varied from 0 to a maximum of 0.08 mM, which was 20% of the protein concentration. Titration of VapB26 and the control DNA ‘X’ was conducted in the same manner. The intensity of the chemical shift perturbation (CSP) was calculated by NMRViewJ. The average CSP values of 15N and 1H were calculated from Equation (1), where ΔδN and ΔδH represent the CSP values of the amide nitrogen and protons.
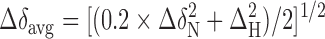 |
(1) |
RESULTS
Overall structure of the VapBC26 complex
The crystal structure of the VapBC26 complex from M. tuberculosis was determined at a resolution of 2.65 Å. The asymmetric unit of the crystal of the VapBC26 complex contains four VapB26 antitoxins and four VapC26 toxins in a hetero-octameric assembly. The calculated molecular weight of VapBC26 was 97.5 ± 1.6 kDa, which matched the theoretical molecular weight of the hetero-octamer model of VapBC26 (97.0 kDa) (Supplementary Figure S1). Four of the heterodimeric VapBC26 complexes are included in the asymmetric unit. The VapB26 dimer binds two VapC26 monomers, and two of the VapB2C2 complexes are related to each other by a dyad axis. The flexible hinge loop of the antitoxin wraps the toxin as a hook, known as a looped arm (Figure 1A–D and Supplementary Figure S2).
Figure 1.

Overall structures of VapBC26 and its subunits. (A) Ribbon representation of the VapBC26 hetero-octamer. Chains A and E are shown in red and chains C and G of VapB26 are shown in salmon. Chains B and F and chains D and H of VapC26 are shown in slate and blue, respectively. (B) Structure of the VapBC26 heterodimer. An arm of VapB26 covers VapC26. (C and D) Structure of (C) the VapC26 and (D) VapB26 dimer detached from the VapBC26 assembly. VapC26 displays cross-contacts between the α5 helix of one toxin monomer and the α3 and α4 helices of another proximal toxin monomer.
In the structure of the VapBC26 heterodimer, VapB26 binds to VapC26 via a deep valley formed by four α-helices (α1–α4) of VapC26 (Figure 2). Two C-terminal segments of VapB26, Arg59–Val61 and Phe68–Glu70, adopt turns at the binding interface, and the Asp62, Glu63 and Leu64 residues form a 310-helix. The long loop between the α2 and α3 helices of VapB26 mainly contributes to the binding to VapC26 through hydrophobic interactions (Figure 2) involving the following VapB26 residues: Pro44, Pro46, Tyr51, Ala52, Pro56, Ile57 and Ala58 in a loop between the α2 and α3 helices; Val61, Leu64 and Leu65 in the α3 helix; and Phe68. The following residues in VapC26 are important for binding: Leu9, Ala10, Tyr11 and Phe12 in the α1 helix; Tyr45, Leu46, Val47, Val51, Ala58 and Val59 in the α3 and α4 helices; and Ala15, Pro17, Ile26, Leu33, Ala41, Ala67, Trp68 and Leu115 in the loop between the α4 and α5 helices. The aromatic residues may be involved in the arrangement of the catalytic site of VapC26 and in the formation of the VapB26 and VapC26 dimer (Figure 2). The interfaces of the VapBC26 complex consist of areas of 1174.1 Å2 (chains A and B), 1246 Å2 (chains C and D), 998.9 Å2 (chains E and F) and 1080.1 Å2 (chains G and H) (62). The interface areas between chains E and F and between chains G and H are relatively underestimated because the electron density of the VapB26 antitoxin (chains E and G) is only shown for Val61.
Figure 2.

Heterodimeric interface between VapB26 and VapC26. The residues participating in hydrophobic interactions are shown as stick models. The same colors as those used in Figure 1 are employed. The aromatic residues that exhibit substantial contributions to the hydrophobic interactions in VapB26 and VapC26 are shown in pink and light green, respectively. The driving force required to generate the heterodimer is derived from hydrophobic interactions.
Structure of the VapC26 toxin
The M. tuberculosis VapC26 toxin contains seven α-helices and five β-strands. The structure of the VapC26 toxin adopts an α/β/α sandwich fold composed of five β-strands and seven α-helices in the following order: β1 (residues 1–4), α1 (residues 5–13), α2 (residues 18–27), β2 (residues 33–36), α3 (residues 37–52), α4 (residues 54–65), β3 (residues 68–71), α5 (residues 74–92), α6 (residues 94–108), β4 (residues 110–114), α7 (residues 116–124) and β5 (residues 129–134) (Supplementary Figure S2). Four-stranded parallel β-sheets (β2-β1-β4-β5) are surrounded by five α-helices, with the two remaining α-helices (α3 and α4) positioned toward the outside of the structure.
VapC26 forms a homodimer such that the α5 helix of one monomer contacts the α3 and α4 helices of the other proximal monomer and the α3 helices partially interact with each other in the middle of the interface. This binding is primarily achieved through hydrophobic interactions. Specifically, the Pro37, Tyr38, Val40 and Ala41 residues participate in the α3–α3 contact, and the Leu57, Leu60 and Ala64 residues in the α4 helix interact with Ala75, Ile78, Ala82 and Val85 in the α5 helix (Figure 3A). Among these residues, Ile78 is a key residue that is required for dimerization of the toxin, as it participates in hydrophobic interactions with Val40, Leu60, Ala64 and Leu70. Ile95 in the α6 helix is also involved in dimerization, participating in hydrophobic interactions with Val40, Ala41 and Leu60 (Figure 3B). Overall, more than 30 residues are involved in the formation of the dimer (Figure 3A). The interface of the VapC26 homodimer has an average area of 1085 Å2 (1065.1 Å2 between chains B and H and 1104.8 Å2 between chains D and F).
The active site of VapC26 is formed by the β1-α1 loop, α3, α6, the β3 loop and the α7 loop. The metal coordination site of VapC26 is surrounded by carboxylate oxygen atoms of the following four well-conserved residues: Asp4 (the N-terminus of the α1 helix), Glu42 (α3 helix), Asp97 (α6 helix) and Asp116 (α7 helix) (Figure 3C). These four acidic residues comprise a negatively charged pocket at the active site (Figure 3D). APBS method (60) was used to calculate the electrostatic surface potential. Among the four VapC26 chains in the asymmetric unit, chain H clearly participates in metal coordination. Heterodimers consisting of chain A-chain B, chain C-chain D and chain A-chain C of VapB26 display an electron density map at Glu70. However, for the chain E-chain H heterodimer, chain E only displays an electron density map at Val61. Therefore, due to the lack of a C-terminus in chain E, a spatial constraint is not present in chain H, thus allowing a definite metal ion site to be identified. The presence of metal ions in the VapBC26 complex was confirmed using inductively coupled plasma mass spectrometry (ICP-MS), which showed that 1.73 μM Mg2+ bound to 5.0 μM of VapBC26 complex. The control buffer contained Mg2+ at <0.1 ppb (μg/kg). In addition, the ribonuclease activity of VapC26 largely depends on the Mg2+ concentration. Thus, Mg2+ is likely coordinated within the active site of VapC26. Mg2+ coordination may be mediated by hydrogen bonding with the Oδ1 atom of Asp4 or Asp97 (Figure 3C). We identified several conserved residues, including Thr5, Ser6, Asn100 and His119, that support the structural integrity of the active site. The Oγ1 atom of Thr5 forms a hydrogen bond with the Oε2 atom of Glu42, and the Oγ atom of Ser6 and the Nδ2 atom of Asn100 form hydrogen bonds with the Oδ1 and Oδ2 atoms of Asp4. The Nε2 atom of His119 forms a hydrogen bond with the Oδ2 atom of Asp97. To illustrate the active site, a figure comparing the active sites of the four toxin chains is included in Supplementary Figure S3.
Structure of the VapB26 antitoxin
The M. tuberculosis VapB26 antitoxin contains three α-helices and one β-strand with a topology of β1-α1-α2-α3. The four secondary structure elements correspond to residues 3–6 (β1), residues 10–23 (α1), residues 27–39 (α2) and residues 60–65 (α3) in chain A and residues 4–7 (β1), residues 10–23 (α1), residues 27–39 (α2) and residues 60–65 (α3) in chain C. The structure features an N-terminal β-sheet, two adjacent α-helices and a small C-terminal α-helix (Supplementary Figure S2) with a long hinge loop between the α2 and α3 helices. Three consecutive prolines (Pro44, Pro45 and Pro46) are located between the α2 helix and the long loop. Gly24, which is located in the short loop region between the α1 and α2 helices, forms a hydrogen bond with nearby residues, thereby creating a turn conformation. The C-terminal α3 helix is visible in chains A and C but not in chains E or G due to the lack of an electron density map.
The structure of the VapB26 antitoxin shows a clear electron density map for all residues in chains A and C. According to the analysis of the structure performed using the 2Struc server (61), chain A of VapB26 exhibits the most ordered structure. VapB26 consists of the following two domains: an N-terminal DNA-binding domain and a C-terminal toxin-binding domain.
The two antitoxins interact with each other through their N-terminal β-strands to form a homodimer. The calculated molecular weight of VapB26 is 19.0 ± 0.4 kDa, consistent with the theoretical molecular weight of the VapB26 dimer (19.2 kDa) (Supplementary Figure S1). The N-terminal domain of the dimer adopts an RHH motif. The average area of the dimeric interfaces is 1372.3 Å2 (1332.2 Å2 between chains E and G; and 1412.4 Å2 between chains A and C). More than 30 residues of each antitoxin participate in dimerization. The most notable difference between the two antitoxin structures was observed in the N-terminal domain. The N-terminal domains of chains A and C are structurally well ordered, whereas the N-terminal domains of chains E and G are not well folded.
The paired residues on the two β1 strands of the VapB26 dimer, including Asp2-Leu8 and Thr4-Val6, form hydrogen bonds through their backbone atoms (Figure 4A), resulting in the formation of antiparallel β-sheets. The main chain O atom of Tyr7 acts as an acceptor for hydrogen bonding with the Nε and NH2 atoms of Arg32 in the α2 helix. Glu11 in the α1 helix and Arg36 in the α2 helix play important roles in the cross stabilization between the α1 and α2 helices by participating in hydrogen bonds via their side chain NH and Oε atoms, which support dimerization (Figure 4B). In addition to the hydrogen bonds, non-polar residues also contribute to formation of the dimerization interface. Residues in the α2 helix, including Val30, Ile31, Ile35 and Val39, cross-interact with equivalent residues in the α2 helix of the proximal antitoxin (Figure 4C). In addition, Ile35 and Val39 play important roles in the interactions with Leu12, Ala15, Val16 and Ala19 in the α1 helix of the adjacent antitoxin monomer. Ala28, Ile31 and Ile35 provide additional hydrophobic interactions to Val6 in β1. Leu8 in the β1-α1 loop also interacts with Ile31 and Ile35 in the antitoxin (Figure 4D).
Figure 4.

Interfaces of the VapB26 dimer. More than 30 residues of each VapB26 N-terminal RHH DNA-binding domain participate in dimerization. Hydrogen bonds are shown as black dotted lines, and residues participating in hydrophobic interactions are shown as stick models. (A) Hydrogen bonding networks of antiparallel β-sheets. (B) Hydrogen bonds in the α1 and α2 helices. (C) Hydrophobic interaction in the α2 helices. (D) Range of hydrophobic forces from Ile35, Val39, Val6 and Leu8.
Unique aspects of the formation of the VapBC26 complex
A search for structural homologs of VapBC26 using the DALI server (68) was conducted to analyze their structural similarities, and obvious structural differences between the VapBC26 complex and its structural homologs were observed.
In the structural homologs, the toxin binding region of the antitoxin consists primarily of one or two α-helices. The antitoxin-binding region of the toxin consists of α-helices and four antiparallel β sheets. The structures of VapB5, VapB15 and VapB30 include only one α-helix (VapB30) or two α-helices (VapB5 and VapB15). VapBs from R. felis and S. flexneri have one α-helix (α2) in the toxin-binding region. VapB3 from M. tuberculosis displays a complete electron density map at the N-terminus due to the presence of a long α-helix (α3). However, VapB26 does not contain a specific helix in the binding region, with the exception of a short α-helix in the C-terminus (α3) of chains A and C. VapB26 forms a flexible hinge loop without any secondary structure at the binding groove, which is formed by the α-helices of VapC26 (Figure 5A).
Figure 5.

Comparison of the structures of the VapBC complexes (A) and VapB antitoxins (B) with their homologs. (A) The complete structures of Mycobacterium tuberculosis VapBC3 and VapBC from Rickettsia felis and Shigella flexneri are shown; other known complexes lack a considerable moiety of the antitoxin. (B) VapB from R. felis and S. flexneri forms a β-barrel hairpin motif. VapB3 from M. tuberculosis forms an RHH N-terminal domain similar to that of VapB26, but the toxin-binding domain contains more α-helices than that of VapB26.
Interestingly, VapB26 does not share significant structural similarity with other VapB proteins, with the exception of VapB3 (35). VapB proteins do not share the same DNA-binding domain and the complete structures have only been resolved for a few VapB proteins, which are similar to VapB26. As mentioned above, VapB26 contains an RHH domain in its N-terminal region. The DNA-binding domains of other VapB proteins are normally located in the N-terminal region. However, VapB5, VapB15 and VapB30 from M. tuberculosis lack an N-terminal structure. Thus, the structures of the DNA-binding regions of these proteins are not available. The VapB proteins from R. felis and S. flexneri show clear electron density maps for the DNA-binding domain region. However, these VapB proteins possess a different motif, namely, a β-barrel hairpin. Among the VapB proteins with determined structures, only VapB3 from M. tuberculosis shares the same RHH DNA-binding motif as VapB26. Nonetheless, VapBC3 from M. tuberculosis does not show the highest structural similarity to VapBC26. Structural similarity is only observed between antitoxin VapB proteins (Figure 5B).
Like VapB26, VapB3 and FitA (69) contain a RHH DNA-binding motif in their N-terminal domains. For a detailed comparison with VapB26, the structural similarity of VapB3 and FitA was analyzed using DALI (68). The results indicated that VapB3 from M. tuberculosis (35) [PDB code: 3H87 (chains C and D) with root mean square (r.m.s.) deviations of 5.4 (chain D) and 12.7 (chain C) Å, Z-scores of 4.1–4.2 and a sequence identity of 16%] and the DNA-bound structure of antitoxin FitA from Neisseria gonorrhoeae (69) [PDB codes: 2BSQ and 2H1O (both show chains E, F, G and H) with r.m.s. deviations of 6.5 (chain G of 2BSQ) and 9.2 (chain F of 2H1O) Å, Z-scores of 4.1–4.2 and sequence identities of 15–17%] had low similarity (Supplementary Table S3A).
Mycobacterium tuberculosis VapC26 shares structural features similar to a PIN domain motif, as described above. Despite the low level of sequence similarity (16−25%), the secondary structures of VapC proteins resemble one another to a considerable extent (Figure 6). The structural homologs include (i) the VapC30 toxin from M. tuberculosis (38) [PDB code: 4XGR (chains A, C, E and G) with a r.m.s. deviation of 2.3 Å, Z-scores of 15.8–16.2 and a sequence identity of 22%], (ii) the VapC toxin from S. flexneri (40) [PDB code: 3TND (chains A, C, E and G) with r.m.s. deviations of 2.4–2.5 Å, Z-scores of 14.2–14.3 and a sequence identity of 16%], (iii) the VapC15 toxin from M. tuberculosis (37) [PDB code: 4CHG (chains A, B, C, D, E and F) with r.m.s. deviations of 2.4–2.5 Å, Z-scores of 13.7–14.1 and sequence identities of 24–25%], (iv) the VapC toxin from R. felis (39) [PDB code: 3ZVK (chains A, B, C and D) with r.m.s. deviations of 2.4–2.5 Å, Z-scores of 13.7–14.1 and sequence identity of 16%], (v) the VapC3 toxin from M. tuberculosis (35) [PDB code: 3H87 (chains A and B) with a r.m.s. deviation of 2.5 Å, Z-scores of 12.1–12.3 and a sequence identity of 18%] and (vi) the VapC5 toxin from M. tuberculosis (36) [PDB code: 3DBO (chain B) with a r.m.s. deviation of 2.7 Å, a Z-score of 11.3 and a sequence identity of 23%]. Among these VapC structures, VapC3, VapC15 and VapC30 contain Mg2+, whereas VapC15 also contains Mn2+ (Supplementary Table S3B).
Figure 6.

Sequence alignment of VapC proteins from Mycobacterium tuberculosis, Shigella flexneri and Rickettsia felis. The known secondary structure elements of the proteins are shown above the alignment. Identical and similar residues are highlighted in red and yellow, respectively. Conserved residues are indicated by red circles, and the residues participating in the stabilization of active sites are indicated by blue circles. VapC proteins are listed in order of their structural homology with VapC26. The alignments were conducted with ESPript (http://espript.ibcp.fr).
VapC contains not only conserved acidic residues but also other conserved residues that support the active site, as shown in Figure 6. Unlike S. flexeneri and R. felis, each M. tuberculosis VapC contains Ser6. However, the Tyr45 of M. tuberculosis VapC26 is only conserved in the VapCs of S. flexneri and R. felis. Other M. tuberculosis VapCs have phenylalanine or another amino acid residue at the same position. Phe120 is conserved in every VapC in the figure except the VapC15 of M. tuberculosis. In addition, the fourth conserved acidic residue, Asp117, shows slight flexibility, and may be glutamate or aspartate. This flexibility can be applied to Glu54, which does not constitute part of the active site.
Characterization of the interaction between VapB26 and promoter DNA
DNA-binding motifs in proteins are largely categorized into the following four groups: helix-turn-helix (70), RHH (71), AbrB-type looped-hinge-helix (72) and the YefM type of three antiparallel β-strands and two helices (73). Structural studies have revealed that VapB26 contains an RHH DNA-binding motif. The functional unit of VapB26 consists of a tight, stable antiparallel β-sheet dimer that binds to DNA. The β-strand and the two subsequent helices (the first and second α-helices) are the basic components of the RHH superfamily. In the structure of DNA bound CopG, which has RHH domain (PDB code: 1B01) (74), the electrostatically positive portion of the two α-helices faces the phosphates of the DNA backbone (Supplementary Figure S4). Antiparallel β-sheet dimer binds to the major groove of the DNA and the two helices anchor the RHH domain to the DNA, allowing the protein to interact optimally with the DNA (71). N-terminus of α2 contacts with DNA phosphate backbone and the loop between α1 and α2 allows the correct positioning of α2. VapB26 is predicted to have dimer functional unit as RHH2 (71).
ITC was used to estimate the binding affinity of VapB26 to promoter DNA. As shown in Figure 7A, the binding titration curve displays a typical hyperbolic form. The promoter DNA-binding reaction of VapB26 is exothermic and enthalpically driven, with thermodynamic parameters of −51.8 ± 6.3 kcal mol−1 (ΔH) and −146 cal mol−1 deg−1 (ΔS). The equilibrium dissociation constant (Kd) for the binding of VapB26 to DNA was measured as 4.69 ± 0.81 μM. The calculated stoichiometry (n) measured in the experiment was 1.1 ± 0.1, indicating that the VapB26 homodimer interacts with DNA.
Figure 7.

Isothermal titration calorimetry assay and electrophoretic mobility shift assays (EMSA) of VapB26 and the promoter DNA. (A) The binding curve of titration indicates that VapB26 is capable of binding to its upstream promoter DNA. (B) Upper: EMSA experiment testing the binding of VapB26 to its own promoter DNA. Middle: EMSA experiment testing the binding of VapB26 to another palindromic DNA ‘a’. Lower: EMSA experiment involving VapB26 and one more different palindromic DNA ‘b’. The concentrations of protein and DNA in each lane are indicated. The results indicate the gradual formation of DNA–protein complexes only between VapB26 and its own promoter DNA as the ratio of protein to DNA is increased. As DNA binds to a large amount of protein, the bands corresponding to the DNA–protein complex move upward.
VapB26 is capable of binding to its upstream promoter DNA. Experimentally obtained values show a hydrophilic effect derived from the formation of hydrogen bonds between VapB26 and the promoter DNA (75). In our study, calorimetric trials were also performed in the absence of VapB26 under the same experimental conditions. No thermal changes were observed in the injections during the entire experiment. In addition, an ITC experiment using control DNA ‘X’ showed no affinity of VapB26 for this DNA (Supplementary Figure S5A).
To confirm the binding specificity of VapB26, an EMSA experiment was conducted using the palindromic DNA of VapB26 and two other palindromic sequences. The DNA concentration was fixed at 0.01 mM, and the protein concentration was increased from 0 to 0.8 mM. As the amount of added protein increased, the shifted band corresponding to the DNA-protein complex increased noticeably in the samples containing VapB26 promoter DNA (Figure 7B). No band shifts were observed in the other samples.
Metal-dependent ribonuclease activity of VapC26
We performed assays of the RNAse activity of VapC26 using fluorescent RNA substrates. When these substrates are cleaved, they emit fluorescence in proportion to the amount of the substrate cleaved by VapC26. The fluorescence data are shown in Figure 8A and B, and the data showing the structural integrity of VapC26 are shown in Supplementary Figure S6A. Mg2+ and Mn2+ significantly increase the enzymatic activity of VapC26 as the metal concentration was increased up to 5 mM, whereas the influence of Zn2+ on the cleavage of the RNA molecules was is negligible. The RNase activity decreased at metal ion concentrations greater than 5 mM. At 50 mM Mg2+ and Mn2+, VapC26 activity was relatively well maintained in the presence of Mg2+, but approximately half of the VapC26 activity was lost in the presence of 50 mM Mn2+. Thus, VapC26 activity is saturated in the presence of ∼5 mM Mg2+, but Mn2+ concentrations greater than 5 mM destroy the VapC26 structure.
Figure 8.

In vitro ribonuclease activity of Mycobacterium tuberculosis VapC26 and comparison of active sites with homologs. When a synthetic RNA containing a fluorophore-quencher pair binds to VapC26, the RNA is digested and the quencher is removed. The released fluorophore generates fluorescence (A and B) Fluorescence measurements, as a function of time, following the addition of Mg2+ and Mn2+. Based on the results, Mg2+ and Mn2+ are essential for catalytic activity. VapC26 was treated with EDTA prior to the assay to remove metal ions. Various concentrations of Mg2+ and Mn2+ were prepared, and 40 units of RiboLock™ (Thermo Scientific) RNase inhibitor was used to prevent contamination. The control contained 50 mM MgCl2 (or 50 mM MnCl2), 50 mM Tris-HCl (pH 7.9), 500 mM NaCl, 250 mM Imidazole and 40 units of RiboLock™ (Thermo Scientific) RNase inhibitor. Each experiment was performed in triplicate. (C) Superposition of conserved residues in active sites of VapC26 and its homologs. Residues and metal ions are marked with different colors depending on the family they belonged to.
The ribonuclease activities of PIN domain proteins, such as VapC toxins, are strongly dependent on the presence of divalent metal ions. VapC proteins commonly bind Mg2+ or Mn2+ as cofactors in their catalytic sites, which consist of three or four conserved acidic residues, including Asp or Glu. However, as shown by structural and experimental studies, only three VapBC complexes obtained from M. tuberculosis exhibit metal-dependent ribonuclease activity. VapC3 and VapC30 from M. tuberculosis contain Mg2+ in their active sites, and VapC15 from M. tuberculosis contains both Mg2+ and Mn2+ in its active site (35,37,38). Based on our results, we concluded that Mg2+ or Mn2+ is required for optimal catalytic activity of VapBC26. Moreover, Mn2+ tends to destroy the structure of the protein, resulting in the preferential presence of Mg2+ in VapBC26 protein.
In order to understand the structural and functional implications of the active site, we superimposed the four conserved active site residues of VapC15 toxin from M. tuberculosis (PDB code: 4CHG), VapC26 toxin from M. tuberculosis (PDB code: 5X3T), VapC30 toxin from M. tuberculosis (PDB code: 4XGR), and VapC toxin from S. flexneri (of PDB code: 3TND), which are indicated by red circles in Figure 6 (Figure 8C). The superposition shows a good fit of three conserved residues, but the location of fourth conserved residue Asp116 from VapBC26 is remote from corresponding residues of other structures. First metal ions are observed in three VapC toxin from M. tuberculosis but second metal ions are only observed in VapC15 toxin from M. tuberculosis.
Influence of peptides on the RNase activity of VapC26
We designed seven peptides as potential inhibitors of the interaction between VapB26 and VapC26. These peptides mimic the binding interface of VapB26 and VapC26. The sequences of peptides two and five correspond to those of portions of the binding regions of VapB26 and VapC26, respectively. Theoretically, these mimicking peptides could compete with their binding partners VapB26 and VapC26 and thereby preventing the formation of the TA complex (65). If the peptides bind with high affinity, free VapC26 would be more predominant, resulting in increased RNase activity. The sequences of the seven peptides are presented in Supplementary Table S4.
As expected, the ribonuclease activity of the M. tuberculosis VapBC26 complex is weak compared to that of VapC26. As a result of competition with the peptide, the ribonuclease activity of VapBC26 at 2.5 μM increases. We did not observe an increase in the fluorescence of VapBC26 following the addition of VapB26-mimicking peptides at peptide-to-protein ratios of 1:1, 10:1 or 100:1 (Supplementary Figure S6B). However, we confirmed that VapC26-mimicking peptides act as inhibitors (Figure 9A and Supplementary Figure S7). VapBC26 showed increased activity following the addition of the α4-mimicking peptide compared to the addition of the α3-mimicking peptide and of peptides corresponding to both helices.
Figure 9.

Ribonuclease activity of Mycobacterium tuberculosis VapC26 measured using a VapC26 α4-mimicking peptide and experiments using mutants. (A and B) The pretreatment was performed as described in Figure 8, and 5 mM Mg2+ was added to each well for optimal activity. (A) The concentration of VapBC26 was fixed at 2.5 μM, and the concentration of peptide was increased from 2.5 μM to 1 mM. The peptide inhibited the binding of VapB26 to VapC26 by ∼50% at 10 μM and by 80% at 200 μM. (B) The concentration of the α4-mimicking peptide was fixed at 2.5 μM, and the concentration of VapBC26 was increased from 0.625 to 20 μM. Based on the data, 2.5 μM peptide interacts with ∼10 μM VapBC26. The experiment was performed in triplicate. (C and D) Comparison of the ribonuclease activity of native VapBC26 and mutated VapBC26. Pretreatment was performed as described in panels A and B. (C) A total of 10 μM of the α4-mimicking peptide was added to the mutated complex (P46A, P46E, Y51A, Y51E, L46A and L46E). Native VapBC26, which shows approximately half the activity of VapC26 in the presence of 10 μM of the α4-mimicking peptide, was used as a reference for comparison. The concentration of each VapBC26 mutant 2.5 μM was the same as that of the native VapBC26 complex. RFU obtained with the reference was taken as 100%. Tyr51 of VapB26 showed the largest effect on the binding of VapB26 and VapC26. Error bars represent the standard deviation of three replicate reactions. (D) Size exclusion chromatography of various proteins. The UV absorption at 280 nm is plotted as a function of the elution volume.
We conducted an additional experiment in which we fixed the concentration of the α4-mimicking peptide at 2.5 μM and increased the concentration of VapBC26 from 0.625 to 20 μM. The plot of RFU for concentrations of VapBC26 greater than 10 μM was not markedly different from the graph for 10 μM VapBC26 (Figure 9B).
Based on our ITC data, VapB26 showed affinity for DNA. In our NMR titration experiments, which are presented in the next section, the promoter DNA of VapB26 caused a chemical shift in the α3 helix residues of VapB26 that are involved in VapC26 binding. Therefore, we titrated VapBC26 with an increasing amount of promoter DNA to examine whether DNA weakens the interaction between VapB26 and VapC26. However, we did not observe increased fluorescence with the addition of the promoter DNA at DNA-to-protein ratio of 1:10, 2:10, 3:10, 5:10 or 20:10 (Supplementary Figure S6C).
To verify that the key binding residues are necessary for the interaction between VapB26 and VapC26, an additional mutational experiment was conducted. Based on the interface, Pro46 and Tyr51 of VapB26 and Leu46 of VapC26, which occupy hydrophobic networks with multiple counterparts, were selected. The selected key residues were mutated to alanine and glutamate to diminish or remove the hydrophobic tendency. Then, 10 μM of the α4-mimicking peptide was added to the mutated complexes. The results show that Tyr51 of VapB26 had the greatest influence on interaction between VapB26 and VapC26, resulting in the emission of more fluorescence (Figure 9C). To confirm the statistical validity of this result, t-test was performed for Y51A and wild-type, Y51E and wild-type. The P-values were calculated as 0.004 for Y51A and wild-type, and 0.003 for Y51E and wild-type. Thus it was confirmed that the distribution of variance of each data were different. To support these results, VapB26 antitoxin, VapC26 toxin, native VapBC26 complex, Y51E complex, which is expected to have the greatest impact on interaction, and α4-mimicking peptide added each complexes were loaded into Superdex 75 10/300 prepacked column (GE Healthcare) and chromatograms were collected. The peptide added VapBC26 complexes showed peaks at different positions from the original peak corresponding VapB26 and VapC26, and the peptide added Y51E complex contained more disassembled proteins than the peptide added native complex (Figure 9D). Calculated area corresponding to VapC26 of α4-mimicking peptide added Y51E complex was 15.42% higher than that of α4-mimicking peptide added native complex, which is well consistent with 15.07% increased fluorescence of Y51E in Figure 9C. The volume of injected protein was 100 μl and the concentration of proteins and α4-mimicking peptide concentration was 200 μM and 1 mM, respectively.
DNA-binding site of VapB26
In type II TA systems, the antitoxin alone or in complex with toxin typically interacts with its corresponding promoter DNA, repressing the transcription of the TA operon. Therefore, the DNA-binding properties of the antitoxin are important for the regulation of TA systems. We characterized the interactions between specific residues in VapB26 and the promoter DNA using NMR titration experiments.
We assigned the peaks to the VapB26 backbone, and 69 out of 71 cross-peaks (97.2%) observed in the 1H-15N HSQC spectra were assigned to individual residues of VapB26, with the exception of Pro44 and Pro45 in the proline bridge. Based on the assigned backbone 1H and 15N resonances, we monitored the residues showing changes in the chemical shifts or resonance broadenings upon binding to DNA. We compared the HSQC spectra of VapB26 (0.4 mM) in the absence and presence of increasing concentrations of promoter DNA (0.04 and 0.08 mM). DNA concentrations >0.08 mM caused few additional changes in the chemical shifts of VapB26, suggesting that the DNA-binding site of the protein is saturated at concentrations above 0.08 mM. These comparative spectra are shown as an overlay in Figure 10C and D (dotted square in the center of the figure).
Figure 10.

NMR titration of VapB26 and its promoter DNA. (A and B) Secondary structure elements based on the TALOS+ analysis are presented above the plots. (A) The chemical shift perturbation of each residue in the presence of 0.08 mM promoter DNA. The residues in the N-terminal RHH DNA-binding domain (Glu11, Leu12, Lys13, Ala14, Ala15, Glu27, Ala28, Arg32, Ile35 and Val39 in the α1 and α2 helices) show CSP values >0.05 (blue dotted line). (B) Relative peak intensity compared to that of the DNA-free form. The peak intensities for Glu27, Ala28, Arg32 and Ile35 in the α2 helix exhibited the largest decreases of <5% compared to the peak intensity of the free form. (C) Overlay of 2D 1H-15N HSQC spectra of 0.4 mM VapB26 with increasing ratios of added DNA. VapB26 with no DNA is shown in light blue, VapB26 with 0.04 mM DNA is shown in medium blue and VapB26 with 0.08 mM DNA is shown in dark blue. The red arrows show the peak shift direction. The Glu27, Ala28, Arg32 and Ile35 peaks disappeared during the interaction with DNA (inverted red triangle). The unassigned residues are not shown. (D) Expanded views of the peak overlap region highlighted with a black dotted square in (C). The spectrum shows DNA/protein ratios of 0 and 0.2.
The shifts in the peaks observed in the spectra indicate that the VapB26–DNA interaction is achieved in a fast exchange on the NMR time scale, which is typically observed when molecules bind with a low to moderate affinity. We calculated the combined 1H and 15N CSP values of VapB26 in the presence of 0.08 mM DNA using Equation (1) shown in the ‘Materials and Methods’ section to quantify the peak shifts. These values are plotted as a function of VapB26 residues (Figure 10A).
The CSP values of the residues belonging to the RHH DNA-binding domain were large. Residues belonging to the α1 helix (Glu11, Leu12, Lys13, Ala14 and Ala15) showed an even distribution of CSP values. Some residues in the α2 helix (Glu27, Ala28, Arg32, Ile35 and Val39) exhibited significant changes in their chemical shifts. Glu27, Ala28, Ile35 and Val39 located in the α2 helix showed CSP values >0.07. The average CSP value of β1 residues (Asp2-Tyr7) was less (0.021) than the CSP values of residues in the α1 (0.038) and α2 helices (0.045). Interestingly, moderate CSP values were observed for the residues corresponding to the α3 helix in the C-terminal domain (average CSP: 0.023) in the structure of the complex, which is involved in binding to the VapC26 toxin.
As shown in our fluorescence assay, the binding of DNA to VapBC26 did not disrupt binding of VapB26 and VapC26, when VapB26 and VapC26 were already bound. Based on the observed changes in the CSP values of the residues in the α3 helix, it is estimated that DNA binds directly to the N-terminal domain of VapB26. However, the binding of DNA to VapB26 affects the conformation of the α3 helix of VapB26, indicated by the moderate CSP values and the large peak intensity change. Because this region is a toxin-binding region, the binding of DNA to VapB26 might also have an effect on the binding of VapC26 to VapB26.
The results of the titration experiment were consistent with the ITC data. According to the Kd determined in the ITC experiment, the binding between VapB26 and DNA is not strong; thus, the binding mode between VapB26 and DNA is expected to show fast exchange. This result is highly consistent with the observed peak movements in the spectra of VapB26 titrated with DNA. Upon titration with DNA, the spectra of VapB26 showed peak broadening and a shift but not a peak split.
The secondary structure of VapB26 in solution was derived from the program TALOS+ (76) using the assigned chemical shift values (Supplementary Table S5). VapB26 adopts a conformation that includes two α-helices (α1 and α2) and one short β-strand; this is partially consistent with the structure of VapB26 in the VapBC26 complex. Val6 and Tyr7 at the N-terminus adopt a β-strand conformation. However, the presence of the α3 helix (residues 60–67) in VapC26-bound VapB26 is not consistent with an unfolded structure of the corresponding region of VapB26 alone. Because this region participates in toxin binding, helix formation may be coupled to toxin binding.
In addition, the overall resonance broadenings of VapB26 residues may have occurred due to the formation of a transient complex between VapB26 and DNA, increasing the transverse relaxation rate of the protein. However, considerable resonance broadenings for specific residues are mainly attributable to their interaction with DNA in an intermediate exchange on the NMR time scale. The residues in the DNA-binding region showed low intensities and peak disappearance due to the decreased transverse relaxation. Changes in peak intensity (I) upon DNA binding also indicate protein recognition (77). The ratios of peak intensities for DNA-bound and free VapB26 (IDNA/I0) are plotted against the VapB26 residues to quantify the resonance broadening effect (Figure 10B). Severe resonance broadening upon binding to DNA is mainly observed for the α1 and α2 helices. In particular, four residues, namely Glu27, Ala28, Arg32 and Ile35, located in the α2 helix show the largest reductions in peak intensities of up to ∼95%, suggesting that these residues are key residues involved in the interaction between VapB26 and the promoter DNA. These results are in highly consistent with the CSP data showing that the α1 and α2 helices of VapB26 are involved in the interaction with DNA. Reduced peak intensities of several residues in the C-terminal region may be explained by changes in the dynamics from fast motion to intermediate motion when VapB26 binds to DNA.
NMR titration was also conducted using control DNA ‘X’. In contrast to the results obtained using promoter DNA, the results showed almost no peak shifts. The ratios of peak intensities were well maintained with the addition of ‘X’ (Supplementary Figure S8).
Based on these data and on the NMR results, we characterized the DNA-binding region of VapB26 and identified the changes in the VapB26 structure upon binding to VapC26. The regions showing relatively high CSP values and peak intensity changes are mapped onto the surface representation of VapB26 in Figure 11.
Figure 11.

Schematic representation and surface representation of VapB26 mapped with CSP values (red) and intensity-related values (blue). The residues are colored with a gradient indicating their CSP values and intensity decrease upon the addition of promoter DNA. As predicted, large CSP values were observed for the residues belonging to the RHH DNA-binding domain, particularly residues in the α1 and α2 helices. Moreover, large decreases in peak intensities were observed for residues in the α1 and α2 helices. The β1 region in the crystal structure showed relatively low values for both parameters.
DISCUSSION
Insights into the unique structure of the VapBC26 complex
Based on the complete analysis of the VapBC26 structure, the binding affinities of VapB26 and VapC26 may be stronger than those of other VapBC structures. Due to their binding affinity for the toxin via the binding domain, the N-terminus-lacking structures of VapBC might have lower steric hindrance than the complete structure. The average binding interface area of VapBC30 (1002.1 Å2) is considerably smaller than the average area of the complete chains of VapBC26 (1255.05 Å2), and the average ΔG (kcal/mol) for VapBC26 (−15.9) is considerably different from the values for VapBC5 (−12.0) and VapBC15 (−12.9), calculated by PISA server (62).
This binding force of VapBC26 is mainly attributed to hydrophobic interactions. The proportion of hydrophobic interactions in the total interactions is higher than that of other VapBC complexes. Ring-type amino acids, such as prolines and aromatic amino acids, including phenylalanine, tryptophan and tyrosine, comprise a large proportion of hydrophobic interactions. The abundant aromatic amino acids in VapBC26 may largely contribute to maintaining the structural integrity and proper function of the protein (78). The strong hydrophobic force derived from the ring results in strong binding (79). Pro44, Pro46, Tyr51, Pro56 and Phe68 of VapB26 and Tyr11, Phe12, Pro17, Tyr45 and Trp68 of VapC26 participate in the interaction between VapB26 and VapC26; together, these residues represent more than 30% (10/29) of the total number of amino acids that participate in the interaction between VapB26 and VapC26.
The proline bridge (Pro44, Pro45 and Pro46) in VapB26 plays a supporting role at the beginning of the toxin binding region. Proline is a strong disruptor of secondary structure in both α-helices and β-strands due to its spatial hindrance, which can prevent the formation of secondary structure. Moreover, the proline bridge of VapB26 serves as a boundary for the DNA-binding and toxin-binding moieties, as it is located in the middle of the disordered region (80–83). Tyr45, Pro37 and Tyr38 of VapC26 participate in binding to the adjacent VapC26 protein. In addition, Tyr45 and Phe120 of VapC26 are involved in maintaining protein functions by stabilizing the active site of the VapC26 protein (84). Peptides, including Tyr45 in the α3 helix, show less activity than other peptides (Supplementary Figure S7).
In the case of Haemophilus influenza VapC1, mutation of 17 amino acid residues in VapC1 showed that these residues do not play essential roles in the interaction of the protein with its cognate antitoxin (85). However, in our study, although single mutations of Pro46 and Tyr51 in VapB26 and of Leu46 in VapC26 did not totally disrupt the interaction required to form VapBC26, the slightly increased ribonuclease activity of the mutated VapBCs and decreased complex formation of Y51E compared to native complex when α4-mimicking peptide was added in size exclusion chromatography supports a role for these hydrophobic residues in forming the VapBC26 interface.
DNA-binding mechanism of VapB26 and the ensuing conformational change
According to studies of FitAB from N. gonorrhoeae, VapBC2 from R. felis and VapBC from S. flexneri (39,40,69), the β1 residues of VapB26 mediate the contact between VapB26 and DNA but do not directly participate in the binding interface. Upon binding to DNA, the conserved residues in β1 recognize DNA, key residues in the two helices directly contacting DNA and residues in the α2 helix bind to the phosphate backbone of DNA at the major groove (71). Alterations in the N-terminal region induced by the interaction with DNA are maintained by two flexible loops between β1 and α1 as well as between α1 and α2.
In our study, the notable CSP and decrease in the intensity of residues in the α1 and α2 helices revealed that the Glu27, Ala28, Arg32, Ile35 and Val39 residues are important for binding to DNA. In this interaction, the promoter site is located in close proximity to RNA polymerase, and transcription is repressed. Therefore, the normal biological functions of bacteria are blocked (86). Promoter DNA is bound by VapBC26 in an extracellular environment, but as M. tuberculosis enters epithelial cells, DNA is released and transcription is initiated (69). Our identification of the residues required for the interaction between VapB26 and promoter DNA will help elucidate the transcriptional repression mechanism and downstream biological actions.
VapB proteins with a β-barrel hairpin domain do not form antiparallel β-sheets until they bind to DNA. Upon interacting with DNA, the β-strands pack tightly to form a complete DNA-binding domain (40). Proteins, such as HU, have flexible β-arms and bend inward when binding to DNA (87). However, VapB26 naturally forms a homodimer in solution, as shown in our MALS data (Supplementary Figure S1) and the RHH domain of VapB26 was confirmed by analyzing the crystal structure of the DNA-free protein. Thus, VapB26 does not undergo dramatic conformational changes in the N-terminal region before and after DNA binding.
In contrast, the TALOS+ (76) secondary structure prediction showed that the binding of DNA to VapB26 generates conformational and dynamic changes in the α3 helix of VapB26, unlike the VapC26-bound structure. VapC26 does not directly participate in the interaction with DNA. However, for FitAB, HipAB and MazEF, the binding affinity of VapB26 to DNA is increased when the antitoxins bind the toxins (69,88,89). VapC26 might thermodynamically contribute to the interaction between VapB26 and DNA by forming a complex with VapB26 and stabilizing the N-terminal-binding domain of VapB26 (90). Similarly, transcription factors undergo conformational changes that increase structural stability or substrate specificity by induced fit or local folding (91–93). Studies of the relationship between the conformational changes that occur in VapB26 and its binding to VapC26 or DNA would be interesting.
Although we were unable to calculate parameters for the interaction between VapBC26 and promoter DNA due to the poor stability of the protein under low salt conditions (Supplementary Figure S5B and C), the C-terminal toxin-binding domain of VapB26 is affected by DNA binding according to our NMR data. Importantly, VapB26, VapC26 and DNA have a mutual relationship (94,95).
Mechanism of the in vitro ribonuclease activity of VapC26
According to our experimental data, the ribonuclease activity of VapC26 was only observed when VapC26 was released from VapB26. EDTA-treated VapC26 did not display ribonuclease activity. Thus, Mg2+ and Mn2+ are essential for VapC26 to block translation by degrading the RNA and inhibiting bacteria growth. Because Mg2+ is smaller than Mn2+, Mg2+ has a strong tendency to mediate water-mediated interactions with the active site. Mg2+ activates the water molecule for nucleophilic attack and stabilizes the active site. This mechanism is shared by other nucleases and ribonucleases (96–98).
The C-terminal α3 helix of VapB26 may affect the position of Mg2+ and Mn2+, which are essential for ribonuclease activity. The α3 helix of VapB26 precisely blocks the putative RNA-binding site of VapC26 and Mg2+ binding by covering the active site. When VapB26 is not bound to VapC26, Mg2+, the RNA substrate and the active site of VapC26 form an active complex (35,37,42,85,99).
Although VapC toxins share structural similarity of active site, they could recognize different targets. VapCs from S. flexneri, Salmonella typhimurium and Leptospira interrogans cleave tRNA in the anticodon stem loop (ASL) (47,100). In M. tuberculosis, VapC4, VapC11, VapC15, VapC25, VapC28, VapC29, VapC30, VapC32, VapC33, VapC37 and VapC39 cleave tRNA (47). VapC20 and VapC26 cut the 23S rRNA in the sarcin-ricin loop (SRL) (45,47). On the basis of previously suggested mechanism of two-metal ion (101), first metal ion in VapBC system can accommodate phosphate hydrolysis, whereas second metal ion seems to be required to stabilize the conformation of active site. In the structural comparison of active sites with reported VapC structures (Figure 8C), second metal ion is coordinated by fourth conserved residue. Corresponding residues of the ASL enzymes, VapC toxin from S. flexneri, and VapC15 and VapC30 toxins from M. tuberculosis showed conservation of geometry. However, the VapC26 toxin from M. tuberculosis, SRL enzyme exhibited different position of the fourth conserved residue. These structural differences of VapC toxins would cause target specificity in spite of their structural similarity, resulting in the different function of ASL enzyme and SRL enzyme.
Importance of the VapBC26 system as an antibiotic target
VapBC systems are of great importance in the study of M. tuberculosis because they are present in quite small numbers in other mycobacteria, compared to M. tuberculosis (33). Furthermore, VapBC26 has great importance as a potential antibiotic target. First, in the hypoxic state, VapB26 expression varies greatly (33), suggesting the possible importance of VapB26 in the stress state. Second, VapC26 is a ribotoxin, cutting the SRL. SRL is the longest conserved sequence in 23s rRNA and is crucial for triggering of GTP hydrolysis and for the anchoring of the elongation factor during mRNA-tRNA translocation (48,102). In particular, sarcin and ricin, which actually targets the SRL, completely inactivate the ribosome. Third, strong growth arrest of E. coli occurs when L. interrogans VapC is expressed (103). Even more crucially, the expression of VapC26 strongly inhibits the growth of M. smegmatis (104,105).
In conclusion, VapC26 can exhibit growth inhibition effect of target cell and act as an effective translation inhibitor. Therefore, the VapBC26 system is expected to bring about new antibiotic target and peptide inhibitor of TA interaction could be a potential antibiotics.
Artificial toxin activation by inhibition of the TA complex
We designed several VapB26 mimetic peptides in an attempt to disrupt the VapBC26 complex. In B. anthracis PemIK, peptide inhibitors that mimic the C-terminal toxin-binding region of PemI influence the TA interaction (106). A peptide designed to mimic the backbone of MoxX also influences the TA interaction (107). The active site of VapC26 interacts with the α3 helix and the C-terminal region of VapB26, but the peptide mimicking the α3 helix of VapB26 does not show any enzymatic activity. With respect to the disruption of the VapB30–VapC30 interaction by the designed peptides, VapB30 lacks the N-terminal residues from 1–46 unlike the complete form of VapB26 (38); this may weaken and prevent complete binding. However, the VapC26-mimicking peptides show great potential as antimicrobial agents. The toxin-mimicking peptides used in the present study interrupted the binding of VapB26 and VapC26 by ∼80% or more at the highest peptide concentration. These data provide evidence suggesting that this peptide may represent novel pharmaceutical compound.
The development of antibiotic drugs based on the TA systems of pathogenic bacteria has attracted interest, and the artificial activation of toxins is a powerful antibacterial strategy. For this purpose, the inhibitor molecule must bind tightly to the TA interaction site to disrupt the TA interaction. The VapC26-mimicking peptide generated in the present study shows possibility as a bactericidal agent based on protein (antitoxin)-protein (toxin) interaction (PPI-approaches) (19,108,109). We may be able to obtain a more optimized inhibitor by applying bio-active techniques, such as stapled peptides. Stapled peptides are stabilized by a hydrocarbon cross-link to form a bioactive conformation and have enhanced membrane permeability (110). Attaching cell-penetrating peptides (CPPs) to the peptide inhibitors might be considered as a way of improving the delivery of antibiotic peptides to appropriate target tissues, and the limited side effects of these peptides in vivo and in vitro and their rapid penetration into bacterial cells of CPPs could enhance their efficiency (111,112). In addition, various modifications can be attached to peptide by conjugation strategies. Combination therapy and surface modulation can increase antibacterial activity and membrane permeability (113). Furthermore, peptide nanoparticles are economical and biocompatible for therapeutic applications (114).
Adequate activation of toxins may induce trivial chronic infections by encouraging the formation of persister or dormant cells. Based on the experimental data obtained in this study, the peptide inhibitors described here may be a new antimicrobial agent.
DATA ACCESS
The structure has been deposited in Protein Data Bank (PDB) under the accession code 5X3T.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the beamline staff at Pohang Light Source, Korea (BL-7A) and Spring-8, Japan (BL44XU).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea (NRF) [2014K1A3A1A19067618, 2015R1A2A1A05001894]; 2016 BK21 Plus Project for Medicine, Dentistry, and Pharmacy. Funding for open access charge: National Research Foundation of Korea [2015R1A2A1A05001894].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hershkovitz I., Donoghue H.D., Minnikin D.E., May H., Lee O.Y., Feldman M., Galili E., Spigelman M., Rothschild B.M., Bar-Gal G.K.. Tuberculosis origin: the neolithic scenario. Tuberculosis (Edinb). 2015; 95(Suppl. 1):S122–S126. [DOI] [PubMed] [Google Scholar]
- 2. Fu L.M., Fu-Liu C.S.. Is Mycobacterium tuberculosis a closer relative to Gram-positive or Gram-negative bacterial pathogens?. Tuberculosis (Edinb). 2002; 82:85–90. [DOI] [PubMed] [Google Scholar]
- 3. Pollock C. Mycobacterial infection in the ferret. Vet. Clin. North Am. Exot. Anim. Pract. 2012; 15:121–129. [DOI] [PubMed] [Google Scholar]
- 4. Lunn J.A., Martin P., Zaki S., Malik R.. Pneumonia due to Mycobacterium abscessus in two domestic ferrets (Mustelo putorius furo). Aust. Vet. J. 2005; 83:542–546. [DOI] [PubMed] [Google Scholar]
- 5. Keri R.S., Chand K., Ramakrishnappa T., Nagaraja B.M.. Recent progress on pyrazole scaffold-based antimycobacterial agents. Arch. Pharm. (Weinheim). 2015; 348:299–314. [DOI] [PubMed] [Google Scholar]
- 6. Comolet T. Multidrug-resistant tuberculosis: challenges of a global emergence. Bull. Soc. Pathol. Exot. 2015; 108:290–298. [DOI] [PubMed] [Google Scholar]
- 7. Pechalrieu D., Lopez M.. Compounds for use in the treatment of mycobacterial infections: a patent evaluation (WO2014049107A1). Expert Opin. Ther. Pat. 2015; 25:729–735. [DOI] [PubMed] [Google Scholar]
- 8. Shao Y., Harrison E.M., Bi D., Tai C., He X., Ou H.Y., Rajakumar K., Deng Z.. TADB: a web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011; 39:D606–D611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pandey D.P., Gerdes K.. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005; 33:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buts L., Lah J., Dao-Thi M.H., Wyns L., Loris R.. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005; 30:672–679. [DOI] [PubMed] [Google Scholar]
- 11. Gerdes K., Christensen S.K., Lobner-Olesen A.. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005; 3:371–382. [DOI] [PubMed] [Google Scholar]
- 12. Van Melderen L. Toxin-antitoxin systems: why so many, what for?. Curr. Opin. Microbiol. 2010; 13:781–785. [DOI] [PubMed] [Google Scholar]
- 13. Hayes F., Van Melderen L.. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011; 46:386–408. [DOI] [PubMed] [Google Scholar]
- 14. Long Q.X., He Y., Xie J.P.. [The molecular physiological and genetic mechanisms underlying the superb efficacy of quinolones]. Yao Xue Xue Bao. 2012; 47:969–977. [PubMed] [Google Scholar]
- 15. Ogura T., Hiraga S.. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. U.S.A. 1983; 80:4784–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Magnuson R.D. Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 2007; 189:6089–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Monti M.C., Hernandez-Arriaga A.M., Kamphuis M.B., Lopez-Villarejo J., Heck A.J., Boelens R., Diaz-Orejas R., van den Heuvel R.H.. Interactions of Kid-Kis toxin-antitoxin complexes with the parD operator-promoter region of plasmid R1 are piloted by the Kis antitoxin and tuned by the stoichiometry of Kid-Kis oligomers. Nucleic Acids Res. 2007; 35:1737–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hayes F. Toxins-antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science. 2003; 301:1496–1499. [DOI] [PubMed] [Google Scholar]
- 19. Chan W.T., Balsa D., Espinosa M.. One cannot rule them all: Are bacterial toxins-antitoxins druggable?. FEMS Microbiol. Rev. 2015; 39:522–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X., Wood T.K.. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011; 77:5577–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vazquez-Laslop N., Lee H., Neyfakh A.A.. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 2006; 188:3494–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korch S.B., Hill T.M.. Ectopic overexpression of wild-type and mutant hipA genes in Escherichia coli: effects on macromolecular synthesis and persister formation. J. Bacteriol. 2006; 188:3826–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yamaguchi Y., Park J.H., Inouye M.. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011; 45:61–79. [DOI] [PubMed] [Google Scholar]
- 24. Zhu L., Zhang Y., Teh J.S., Zhang J., Connell N., Rubin H., Inouye M.. Characterization of mRNA interferases from Mycobacterium tuberculosis. J. Biol. Chem. 2006; 281:18638–18643. [DOI] [PubMed] [Google Scholar]
- 25. Dao-Thi M.H., Van Melderen L., De Genst E., Afif H., Buts L., Wyns L., Loris R.. Molecular basis of gyrase poisoning by the addiction toxin CcdB. J. Mol. Biol. 2005; 348:1091–1102. [DOI] [PubMed] [Google Scholar]
- 26. Christensen S.K., Gerdes K.. RelE toxins from bacteria and Archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003; 48:1389–1400. [DOI] [PubMed] [Google Scholar]
- 27. Ramisetty B.C., Santhosh R.S.. Horizontal gene transfer of chromosomal Type II toxin-antitoxin systems of Escherichia coli. FEMS Microbiol. Lett. 2016; 363:1–7. [DOI] [PubMed] [Google Scholar]
- 28. Fiedoruk K., Daniluk T., Swiecicka I., Sciepuk M., Leszczynska K.. Type II toxin-antitoxin systems are unevenly distributed among Escherichia coli phylogroups. Microbiology. 2015; 161:158–167. [DOI] [PubMed] [Google Scholar]
- 29. Aakre C.D., Phung T.N., Huang D., Laub M.T.. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol. Cell. 2013; 52:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Janssen B.D., Garza-Sanchez F., Hayes C.S.. YoeB toxin is activated during thermal stress. Microbiologyopen. 2015; 4:682–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park S.J., Son W.S., Lee B.J.. Structural overview of toxin-antitoxin systems in infectious bacteria: a target for developing antimicrobial agents. Biochim. Biophys. Acta. 2013; 1834:1155–1167. [DOI] [PubMed] [Google Scholar]
- 32. Brzozowska I., Zielenkiewicz U.. Regulation of toxin-antitoxin systems by proteolysis. Plasmid. 2013; 70:33–41. [DOI] [PubMed] [Google Scholar]
- 33. Ramage H.R., Connolly L.E., Cox J.S.. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009; 5:e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sala A., Bordes P., Genevaux P.. Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins (Basel). 2014; 6:1002–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Min A.B., Miallau L., Sawaya M.R., Habel J., Cascio D., Eisenberg D.. The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg(2)(+) ion in the active site and a putative RNA-binding site. Protein Sci. 2012; 21:1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miallau L., Faller M., Chiang J., Arbing M., Guo F., Cascio D., Eisenberg D.. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 2009; 284:276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Das U., Pogenberg V., Subhramanyam U.K., Wilmanns M., Gourinath S., Srinivasan A.. Crystal structure of the VapBC-15 complex from Mycobacterium tuberculosis reveals a two-metal ion dependent PIN-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J. Struct. Biol. 2014; 188:249–258. [DOI] [PubMed] [Google Scholar]
- 38. Lee I.G., Lee S.J., Chae S., Lee K.Y., Kim J.H., Lee B.J.. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015; 43:7624–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mate M.J., Vincentelli R., Foos N., Raoult D., Cambillau C., Ortiz-Lombardia M.. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res. 2012; 40:3245–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dienemann C., Boggild A., Winther K.S., Gerdes K., Brodersen D.E.. Crystal structure of the VapBC toxin-antitoxin complex from Shigella flexneri reveals a hetero-octameric DNA-binding assembly. J. Mol. Biol. 2011; 414:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arcus V.L., Rainey P.B., Turner S.J.. The PIN-domain toxin-antitoxin array in mycobacteria. Trends Microbiol. 2005; 13:360–365. [DOI] [PubMed] [Google Scholar]
- 42. Arcus V.L., McKenzie J.L., Robson J., Cook G.M.. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 2011; 24:33–40. [DOI] [PubMed] [Google Scholar]
- 43. Winther K.S., Gerdes K.. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:7403–7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ahidjo B.A., Kuhnert D., McKenzie J.L., Machowski E.E., Gordhan B.G., Arcus V., Abrahams G.L., Mizrahi V.. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS One. 2011; 6:e21738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Winther K.S., Brodersen D.E., Brown A.K., Gerdes K.. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat. Commun. 2013; 4:2796. [DOI] [PubMed] [Google Scholar]
- 46. McKenzie J.L., Duyvestyn J.M., Smith T., Bendak K., Mackay J., Cursons R., Cook G.M., Arcus V.L.. Determination of ribonuclease sequence-specificity using Pentaprobes and mass spectrometry. RNA. 2012; 18:1267–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winther K., Tree J.J., Tollervey D., Gerdes K.. VapCs of Mycobacterium tuberculosis cleave RNAs essential for translation. Nucleic Acids Res. 2016; 44:9860–9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi X., Khade P.K., Sanbonmatsu K.Y., Joseph S.. Functional role of the sarcin-ricin loop of the 23S rRNA in the elongation cycle of protein synthesis. J. Mol. Biol. 2012; 419:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Goeders N., Van Melderen L.. Toxin-antitoxin systems as multilevel interaction systems. Toxins (Basel). 2014; 6:304–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bukowski M., Rojowska A., Wladyka B.. Prokaryotic toxin-antitoxin systems–the role in bacterial physiology and application in molecular biology. Acta Biochim. Pol. 2011; 58:1–9. [PubMed] [Google Scholar]
- 51. Lioy V.S., Rey O., Balsa D., Pellicer T., Alonso J.C.. A toxin-antitoxin module as a target for antimicrobial development. Plasmid. 2010; 63:31–39. [DOI] [PubMed] [Google Scholar]
- 52. Gao Y.Z., Xu H.H., Ju T.T., Zhao X.H.. The effect of limited proteolysis by different proteases on the formation of whey protein fibrils. J. Dairy Sci. 2013; 96:7383–7392. [DOI] [PubMed] [Google Scholar]
- 53. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 54. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brunger A.T. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992; 355:472–475. [DOI] [PubMed] [Google Scholar]
- 57. Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. DeLano W.L., Lam J.W.. PyMOL: a communications tool for computational models. Abstr. Pap. Am. Chem. S. 2005; 230:U1371–U1372. [Google Scholar]
- 60. Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A.. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Klose D.P., Wallace B.A., Janes R.W.. 2Struc: the secondary structure server. Bioinformatics. 2010; 26:2624–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 63. McWilliam H., Li W., Uludag M., Squizzato S., Park Y.M., Buso N., Cowley A.P., Lopez R.. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013; 41:W597–W600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Verma S., Kumar S., Gupta V.P., Gourinath S., Bhatnagar S., Bhatnagar R.. Structural basis of Bacillus anthracis MoxXT disruption and the modulation of MoxT ribonuclease activity by rationally designed peptides. J. Biomol. Struct. Dyn. 2015; 33:606–624. [DOI] [PubMed] [Google Scholar]
- 66. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 67. Johnson B.A. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 2004; 278:313–352. [DOI] [PubMed] [Google Scholar]
- 68. Holm L., Rosenstrom P.. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010; 38:W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mattison K., Wilbur J.S., So M., Brennan R.G.. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 2006; 281:37942–37951. [DOI] [PubMed] [Google Scholar]
- 70. Xiong W., Li T., Chen K., Tang K.. Local combinational variables: an approach used in DNA-binding helix-turn-helix motif prediction with sequence information. Nucleic Acids Res. 2009; 37:5632–5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schreiter E.R., Drennan C.L.. Ribbon-helix-helix transcription factors: variations on a theme. Nat. Rev. Microbiol. 2007; 5:710–720. [DOI] [PubMed] [Google Scholar]
- 72. Coles M., Djuranovic S., Soding J., Frickey T., Koretke K., Truffault V., Martin J., Lupas A.N.. AbrB-like transcription factors assume a swapped hairpin fold that is evolutionarily related to double-psi beta barrels. Structure. 2005; 13:919–928. [DOI] [PubMed] [Google Scholar]
- 73. Kumar P., Issac B., Dodson E.J., Turkenburg J.P., Mande S.C.. Crystal structure of Mycobacterium tuberculosis YefM antitoxin reveals that it is not an intrinsically unstructured protein. J. Mol. Biol. 2008; 383:482–493. [DOI] [PubMed] [Google Scholar]
- 74. Gomis-Ruth F.X., Sola M., Acebo P., Parraga A., Guasch A., Eritja R., Gonzalez A., Espinosa M., del Solar G., Coll M.. The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO J. 1998; 17:7404–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Setny P., Baron R., McCammon J.A.. How can hydrophobic association be enthalpy driven?. J. Chem. Theory Comput. 2010; 6:2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shen Y., Delaglio F., Cornilescu G., Bax A.. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 2009; 44:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. de Paula V.S., Razzera G., Barreto-Bergter E., Almeida F.C., Valente A.P.. Portrayal of complex dynamic properties of sugarcane defensin 5 by NMR: multiple motions associated with membrane interaction. Structure. 2011; 19:26–36. [DOI] [PubMed] [Google Scholar]
- 78. Anjana R., Vaishnavi M.K., Sherlin D., Kumar S.P., Naveen K., Kanth P.S., Sekar K.. Aromatic-aromatic interactions in structures of proteins and protein-DNA complexes: a study based on orientation and distance. Bioinformation. 2012; 8:1220–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Meyer E.A., Castellano R.K., Diederich F.. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. Engl. 2003; 42:1210–1250. [DOI] [PubMed] [Google Scholar]
- 80. Chou P.Y., Fasman G.D.. Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins. Biochemistry. 1974; 13:211–222. [DOI] [PubMed] [Google Scholar]
- 81. Chou P.Y., Fasman G.D.. Secondary structural prediction of proteins from their amino-acid sequence. Trends Biochem. Sci. 1977; 2:128–131. [Google Scholar]
- 82. Eisenhaber F., Persson B., Argos P.. Protein-structure prediction—recognition of primary, secondary, and tertiary structural features from amino-acid-sequence. Crit. Rev. Biochem. Mol. Biol. 1995; 30:1–94. [DOI] [PubMed] [Google Scholar]
- 83. Malkov S.N., Zivkovic M.V., Beljanski M.V., Stojanovic S.D., Zaric S.D.. A reexamination of correlations of amino acids with particular secondary structures. Protein J. 2009; 28:74–86. [DOI] [PubMed] [Google Scholar]
- 84. Serrano L., Bycroft M., Fersht A.R.. Aromatic-aromatic interactions and protein stability. Investigation by double-mutant cycles. J. Mol. Biol. 1991; 218:465–475. [DOI] [PubMed] [Google Scholar]
- 85. Hamilton B., Manzella A., Schmidt K., DiMarco V., Butler J.S.. Analysis of non-typeable Haemophilous influenzae VapC1 mutations reveals structural features required for toxicity and flexibility in the active site. PLoS One. 2014; 9:e112921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Christensen S.K., Maenhaut-Michel G., Mine N., Gottesman S., Gerdes K., Van Melderen L.. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 2004; 51:1705–1717. [DOI] [PubMed] [Google Scholar]
- 87. Kim do H., Im H., Jee J.G., Jang S.B., Yoon H.J., Kwon A.R., Kang S.M., Lee B.J.. beta-Arm flexibility of HU from Staphylococcus aureus dictates the DNA-binding and recognition mechanism. Acta Crystallogr. D Biol. Crystallogr. 2014; 70:3273–3289. [DOI] [PubMed] [Google Scholar]
- 88. Black D.S., Kelly A.J., Mardis M.J., Moyed H.S.. Structure and organization of hip, an operon that affects lethality due to inhibition of peptidoglycan or DNA-synthesis. J. Bacteriol. 1991; 173:5732–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Marianovsky I., Aizenman E., Engelberg-Kulka H., Glaser G.. The regulation of the Escherichia coli mazEF promoter involves an unusual alternating palindrome. J. Biol. Chem. 2001; 276:5975–5984. [DOI] [PubMed] [Google Scholar]
- 90. Ahmad S., Keskin O., Sarai A., Nussinov R.. Protein-DNA interactions: structural, thermodynamic and clustering patterns of conserved residues in DNA-binding proteins. Nucleic Acids Res. 2008; 36:5922–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Andrabi M., Mizuguchi K., Ahmad S.. Conformational changes in DNA-binding proteins: relationships with precomplex features and contributions to specificity and stability. Proteins. 2014; 82:841–857. [DOI] [PubMed] [Google Scholar]
- 92. Petty T.J., Emamzadah S., Costantino L., Petkova I., Stavridi E.S., Saven J.G., Vauthey E., Halazonetis T.D.. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011; 30:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Spolar R.S., Record M.T. Jr. Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994; 263:777–784. [DOI] [PubMed] [Google Scholar]
- 94. Johnson E.P., Strom A.R., Helinski D.R.. Plasmid RK2 toxin protein ParE: purification and interaction with the ParD antitoxin protein. J. Bacteriol. 1996; 178:1420–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Afif H., Allali N., Couturier M., Van Melderen L.. The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 2001; 41:73–82. [DOI] [PubMed] [Google Scholar]
- 96. Nowotny M., Yang W.. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. EMBO J. 2006; 25:1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nowotny M., Gaidamakov S.A., Crouch R.J., Yang W.. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005; 121:1005–1016. [DOI] [PubMed] [Google Scholar]
- 98. Zhao H., Lin Z., Lynn A.Y., Varnado B., Beutler J.A., Murelli R.P., Le Grice S.F., Tang L.. Two distinct modes of metal ion binding in the nuclease active site of a viral DNA-packaging terminase: insight into the two-metal-ion catalytic mechanism. Nucleic Acids Res. 2015; 43:11003–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Xu K., Dedic E., Cob-Cantal P., Dienemann C., Boggild A., Winther K.S., Gerdes K., Brodersen D.E.. Protein expression, crystallization and preliminary X-ray crystallographic analysis of the isolated Shigella flexneri VapC toxin. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2013; 69:762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cruz J.W., Sharp J.D., Hoffer E.D., Maehigashi T., Vvedenskaya I.O., Konkimalla A., Husson R.N., Nickels B.E., Dunham C.M., Woychik N.A.. Growth-regulating Mycobacterium tuberculosis VapC-mt4 toxin is an isoacceptor-specific tRNase. Nat. Commun. 2015; 6:7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hwang K.Y., Baek K., Kim H.Y., Cho Y.. The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat. Struct. Biol. 1998; 5:707–713. [DOI] [PubMed] [Google Scholar]
- 102. Garcia-Ortega L., Masip M., Mancheno J.M., Onaderra M., Lizarbe M.A., Garcia-Mayoral M.F., Bruix M., Martinez del Pozo A., Gavilanes J.G.. Deletion of the NH2-terminal beta-hairpin of the ribotoxin alpha-sarcin produces a nontoxic but active ribonuclease. J. Biol. Chem. 2002; 277:18632–18639. [DOI] [PubMed] [Google Scholar]
- 103. Lopes A.P., Lopes L.M., Fraga T.R., Chura-Chambi R.M., Sanson A.L., Cheng E., Nakajima E., Morganti L., Martins E.A.. VapC from the leptospiral VapBC toxin-antitoxin module displays ribonuclease activity on the initiator tRNA. PLoS One. 2014; 9:e101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zaychikova M.V., Zakharevich N.V., Sagaidak M.O., Bogolubova N.A., Smirnova T.G., Andreevskaya S.N., Larionova E.E., Alekseeva M.G., Chernousova L.N., Danilenko V.N.. Mycobacterium tuberculosis Type II toxin-antitoxin systems: genetic polymorphisms and functional properties and the possibility of their use for genotyping. PLoS One. 2015; 10:e0143682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Demidenok O.I., Kaprelyants A.S., Goncharenko A.V.. Toxin-antitoxin vapBC locus participates in formation of the dormant state in Mycobacterium smegmatis. FEMS Microbiol. Lett. 2014; 352:69–77. [DOI] [PubMed] [Google Scholar]
- 106. Agarwal S., Mishra N.K., Bhatnagar S., Bhatnagar R.. PemK toxin of Bacillus anthracis is a ribonuclease an insight into its active site, structure, and functION. J. Biol. Chem. 2010; 285:7254–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chopra N., Agarwal S., Verma S., Bhatnagar S., Bhatnagar R.. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided Mol. Des. 2011; 25:275–291. [DOI] [PubMed] [Google Scholar]
- 108. Williams J.J., Hergenrother P.J.. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012; 20:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Labbe C.M., Laconde G., Kuenemann M.A., Villoutreix B.O., Sperandio O.. iPPI-DB: a manually curated and interactive database of small non-peptide inhibitors of protein-protein interactions. Drug Discov. Today. 2013; 18:958–968. [DOI] [PubMed] [Google Scholar]
- 110. Kim Y.W., Grossmann T.N., Verdine G.L.. Synthesis of all-hydrocarbon stapled alpha-helical peptides by ring-closing olefin metathesis. Nat. Protoc. 2011; 6:761–771. [DOI] [PubMed] [Google Scholar]
- 111. Wagstaff K.M., Jans D.A.. Protein transduction: cell penetrating peptides and their therapeutic applications. Curr. Med. Chem. 2006; 13:1371–1387. [DOI] [PubMed] [Google Scholar]
- 112. Zorko M., Langel U.. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005; 57:529–545. [DOI] [PubMed] [Google Scholar]
- 113. Reinhardt A., Neundorf I.. Design and application of antimicrobial peptide conjugates. Int. J. Mol. Sci. 2016; 17:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Zaman M., Ahmad E., Qadeer A., Rabbani G., Khan R.H.. Nanoparticles in relation to peptide and protein aggregation. Int. J. Nanomed. 2014; 9:899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.