


Abstract
The genome-wide identification of microRNA transcription start sites (miRNA TSSs) is essential for understanding how miRNAs are regulated in development and disease. In this study, we developed mirSTP (mirna transcription Start sites Tracking Program), a probabilistic model for identifying active miRNA TSSs from nascent transcriptomes generated by global run-on sequencing (GRO-seq) and precision run-on sequencing (PRO-seq). MirSTP takes advantage of characteristic bidirectional transcription signatures at active TSSs in GRO/PRO-seq data, and provides accurate TSS prediction for human intergenic miRNAs at a high resolution. MirSTP performed better than existing generalized and experiment specific methods, in terms of the enrichment of various promoter-associated marks. MirSTP analysis of 27 human cell lines in 183 GRO-seq and 28 PRO-seq experiments identified TSSs for 480 intergenic miRNAs, indicating a wide usage of alternative TSSs. By integrating predicted miRNA TSSs with matched ENCODE transcription factor (TF) ChIP-seq data, we connected miRNAs into the transcriptional circuitry, which provides a valuable source for understanding the complex interplay between TF and miRNA. With mirSTP, we not only predicted TSSs for 72 miRNAs, but also identified 12 primary miRNAs with significant RNA polymerase pausing alterations after JQ1 treatment; each miRNA was further validated through BRD4 binding to its predicted promoter. MirSTP is available at http://bioinfo.vanderbilt.edu/mirSTP/.
INTRODUCTION
MicroRNAs (miRNAs) are a growing class of small, non-coding RNAs that play significant roles in cell identity, development and disease (1). miRNAs mainly work together with transcription factors to tune gene expression in response to environmental changes (2). To fully understand miRNA function, it is essential to connect miRNAs to their transcriptional circuitry, including their upstream regulators as well as their downstream targets. Compared to the increasing knowledge of miRNAs modulating gene expression, our understanding of the regulation of miRNA transcription is lagging far behind, mainly due to the fact that miRNA transcription start sites are largely unknown. miRNAs are typically generated from long primary miRNAs (pri-miRNAs). Pri-miRNAs are rapidly cleaved by the enzyme Drosha (3), which creates a challenge for conventional transcription start site mapping approaches that rely on the full complete RNA. Approximately 2000 miRNAs are annotated in humans (4,5), but only a handful of microRNA transcription start sites (miRNA TSSs) have been identified (6–11). The sparse annotation of miRNA TSSs limits our ability to locate promoter regions and to associate transcription factor binding events with miRNA transcription.
Computational methods for genome-wide miRNA TSSs recognition have been developed to address this limitation (12–24). Early approaches utilized sequence features derived from known promoters to scan regions upstream of miRNAs for hallmarks of transcription start sites, such as the TATA-box, over-represented DNA motifs, TF binding profiles, evolutionary conservation and CpG content (12–15). For example, discriminative models were trained by core promoter sequences with all possible k-mers as input features (14). Comparative genomics methods were also used to search for highly conserved blocks within upstream regions of miRNA genes (12). S-Peaker, a probabilistic model to predict TSSs of single-peak transcription initiation, was based solely on known transcription factor binding affinity (13). The recent advances of high-throughput sequencing technologies led to significant improvements for miRNA TSSs prediction. For instance, ChIP-seq (Chromatin Immunoprecipitation coupled with sequencing) for RNA Pol II and H3K4me3 can demarcate active promoters, while CAGE-seq (Cap Analysis of Gene Expression followed by sequencing) detects the 5΄ end of capped molecules and maps the location of TSSs genome-wide. Several methods that successfully identify TSSs for numerous miRNAs have been developed by integrating multiple types of data and experiments from multiple cell types (16–19,22). For example, 847 human miRNA TSSs were predicted using miRStart, a support vector machine model by combining ChIP-seq of H3K4me3 from CD4+ T cells, TSS-seq from six human cell lines and CAGE-seq from 124 human samples (19). In addition, PROmiRNA is a semi-supervised statistical method based on the CAGE-seq data of 33 human RNA libraries generated from FANTOM project and sequence features, including TATA box, conservation and CpG density. PROmiRNA has successfully identified TSSs for 1228 miRNAs, among which 389 are intergenic miRNAs (22). Pooling data from multiple conditions/experiments provide researchers with the ability to discover all potential miRNAs; however, it is difficult to predict cell/experiment-specific TSSs, which is very important since miRNAs have been reported to undergo tight tissue-specific regulation (25,26).
In order to detect condition/experiment-specific miRNA TSSs, microTSS was developed to integrate deep RNA-seq with active transcription marks, including H3K4me3, RNA Pol II occupancy and DNase-based TF footprints. MicroTSS provided accurate TSS predictions for 86 and 82 intergenic miRNAs in hESC and IMR90 cells, respectively. However, the performance of microTSS is dependent on sequencing depth since low-abundant pri-miRNAs are transient and can only be detected in deep-coverage RNA-seq data (23). An alternative workflow to identify cell-specific miRNA TSSs combines active promoter marks, such as H3K4me3, chromatin accessibility from DNase-seq and sequence features (24). This strategy has successfully discovered TSSs for 663 intragenic miRNAs, and 620 intergenic miRNAs in 54 cell lines. The workflow's major limitation is its low-resolution and broad predictions due to the underlying features of histone and open chromatin marks, which reduces the sensitivity in detecting alternative TSSs. It is noteworthy that microTSS and the workflow presented by Hua et al. both require multiple types of data in the same cell/condition, which are expensive and not easily available for new cells/conditions, thereby hindering a wide application of these methods.
Global nuclear run-on sequencing (GRO-seq) (27) and precision nuclear run-on sequencing (PRO-seq) (28) are techniques used to measure active RNA polymerases by quantifying nascent transcription. These methods allow for the calculation of transcription rates, and the assessment of polymerase pausing and elongation; they also provide continuous signals throughout the entire transcription unit. Both GRO-seq and PRO-seq data show sharp peaks around TSSs in both the sense and antisense directions (27–29). Utilizing these features, we developed mirna transcription Start site Tracking Program (mirSTP), which provides a sensitive, high resolution and highly accurate approach to recognize condition-specific miRNA TSSs genome-wide. MirSTP requires just one GRO/PRO-seq experiment rather than multiple data types and focuses on the TSS identification for intergenic miRNAs. We first evaluated the performance of mirSTP using known gene TSSs, and then compared the performance with existing methods in terms of active promoter marks. Predicted miRNA TSSs were further used to associate TF binding events with miRNA regulation. Using mirSTP, we not only identified miRNA TSSs, but also quantified pri-miRNAs expression and compared pri-miRNA transcriptional pausing between JQ1 treatment and control PRO-seq data. These estimations suggested that Pri-miRNAs with significant transcriptional pausing alterations after JQ1 treatment were directly regulated by the BET family, all of which were further validated by BRD4 ChIP-seq data. The source code of mirSTP and the original PRO-seq data generated in this study are freely available at http://bioinfo.vanderbilt.edu/mirSTP/.
MATERIALS AND METHODS
Description of mirSTP algorithm
The development of mirSTP was motivated by two characteristic features of GRO/PRO-seq data, one of which is divergent transcription near transcription start sites. Specifically, there are sharp peaks in both the sense (∼50 bp downstream of TSS) and antisense directions (∼250 bp upstream of TSS). The other feature is a continuous signal over pri-miRNA region, since GRO/PRO-seq captures all elongation-competent RNA polymerase. MirSTP has two steps. In the first step, mirSTP discriminated candidate TSS sites from local background (non-TSS sites) based on Poisson distribution. The log likelihood of a candidate region i to be a TSS site was estimated by the observed read distribution within ±500 bp region in both sense (+) and antisense (−) directions:
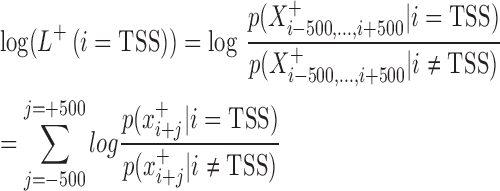 |
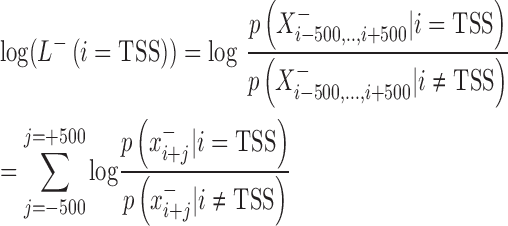 |
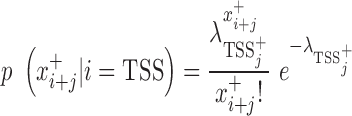 |
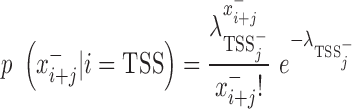 |
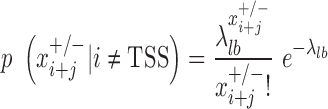 |
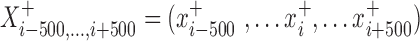 is a vector of the observed read counts from the upstream to the downstream 500 bp of the candidate site i in the sense direction relative to the direction of precursor miRNA, while
is a vector of the observed read counts from the upstream to the downstream 500 bp of the candidate site i in the sense direction relative to the direction of precursor miRNA, while 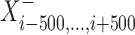 is the same, but in the antisense direction.
is the same, but in the antisense direction.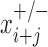 is the observed read count of the position j relative to the candidate site i in the sense/antisense direction, where j ranges from −500 to +500. The read count was calculated in 10-bp window. The expected read density at position j relative to TSSs,
is the observed read count of the position j relative to the candidate site i in the sense/antisense direction, where j ranges from −500 to +500. The read count was calculated in 10-bp window. The expected read density at position j relative to TSSs, 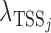 , was estimated from the known TSSs of genes. GRO-seq and PRO-seq have their own TSS models, i.e.
, was estimated from the known TSSs of genes. GRO-seq and PRO-seq have their own TSS models, i.e. 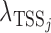 , which differs between GRO and PRO-seq data. The non-TSS model was based on the read density estimated from the local neighboring regions (+1 kb to +2kb downstream of TSSs) and denoted by
, which differs between GRO and PRO-seq data. The non-TSS model was based on the read density estimated from the local neighboring regions (+1 kb to +2kb downstream of TSSs) and denoted by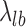 . Different cutoffs were set to determine whether a site is a TSS in both the sense and antisense directions (
. Different cutoffs were set to determine whether a site is a TSS in both the sense and antisense directions (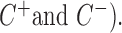 The region i was identified to be a candidate TSS site if
The region i was identified to be a candidate TSS site if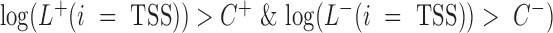 . To select appropriate cutoffs, we applied mirSTP to identify TSSs for active genes on K562 PRO-seq data. A higher cutoff will detect fewer, but more accurate TSSs, while a lower cutoff will identify more, but less accurate TSSs. To balance sensitivity and specificity, we defined three cutoff levels: stringent, medium and relaxed. The stringent cutoff was chosen where only ∼80% of active TSSs were identified, while the relaxed cutoff was selected where ∼95% of active TSSs were identified and the medium cutoff where ∼85% TSSs were identified. Although the stringent level had the lowest sensitivity, we expected that it would be the most accurate predictor of TSSs. In contrast, the relaxed level achieved the highest sensitivity, but obtained the least accuracy of TSSs.
. To select appropriate cutoffs, we applied mirSTP to identify TSSs for active genes on K562 PRO-seq data. A higher cutoff will detect fewer, but more accurate TSSs, while a lower cutoff will identify more, but less accurate TSSs. To balance sensitivity and specificity, we defined three cutoff levels: stringent, medium and relaxed. The stringent cutoff was chosen where only ∼80% of active TSSs were identified, while the relaxed cutoff was selected where ∼95% of active TSSs were identified and the medium cutoff where ∼85% TSSs were identified. Although the stringent level had the lowest sensitivity, we expected that it would be the most accurate predictor of TSSs. In contrast, the relaxed level achieved the highest sensitivity, but obtained the least accuracy of TSSs.
The second step of mirSTP was to filter inactive pri-miRNAs. A pri-miRNA is active if there is a continuous signal over the whole gene body (gb) at the sense direction. After counting the total number of reads from the downstream 1k of the TSS to the precursor miRNA (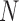 is the number of reads, l is the length of this gb region), mirSTP calculated the probability
is the number of reads, l is the length of this gb region), mirSTP calculated the probability 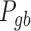 that we would observe at least
that we would observe at least 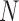 reads based on the Poisson distribution of the global background density
reads based on the Poisson distribution of the global background density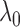 (27). The global background density
(27). The global background density 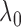 was estimated using the method (the default is 0.04 reads/kb) (27).
was estimated using the method (the default is 0.04 reads/kb) (27).
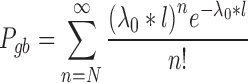 |
In addition, mirSTP calculated the minimum number of reads in non-overlapping sliding windows of 5kb, 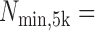 min (
min (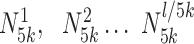 ). If
). If 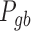 was <0.0001 and
was <0.0001 and 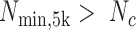 , the pri-miRNA was called active.
, the pri-miRNA was called active. 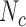 was set to 10 at the stringent cutoff level, five at the medium cutoff level, and two at the relaxed cutoff level.
was set to 10 at the stringent cutoff level, five at the medium cutoff level, and two at the relaxed cutoff level.
If multiple active TSS sites were identified for one precursor miRNA, the site with the maximum score 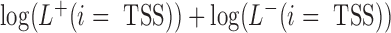 was the representative TSS for the miRNA.
was the representative TSS for the miRNA.
PRO-seq data library preparation
Most PRO-seq or GRO-seq datasets were downloaded from GEO (Supplementary Table S1). PRO-seq data from Kasumi-1, OCI-LY1, MV4-11, U936 and Daudi cell lines were generated in our lab according to previously published methods with minor modifications (28,30). Briefly, 20 million nuclei were isolated and in vitro nuclear run-on assays were performed using regular ATP, UTP, GTP and biotinylated CTP at 30°C for 3 min, so that newly synthesized RNA was labeled with biotin. Total nuclear RNA was isolated using TRIzol, then was fragmented by base hydrolysis in 0.2 N NaOH and run through P-30 columns for buffer exchange. Fragmented biotinylated nascent RNA was purified using streptavidin beads and 3΄ RNA adaptor was ligated. After the second bead purification, 5΄ end repair was performed and 5΄ RNA adaptor was ligated. After the third bead purification, reverse transcription was performed to generate cDNA, which was followed by polymerase chain reaction amplification with indexed primers for sequencing. Libraries were submitted to the Vanderbilt Technologies for Advanced Genomics (VANTAGE) for sequencing.
GRO/PRO-seq analysis
We trimmed the adapter sequences using cutadapt (version 1.9.1) (31) and any reads less than 15 bp were removed. PRO-seq reads were reverse complemented. GRO-seq reads and reverse complemented PRO-seq reads were aligned to the human genome hg19 using Bowtie2 (version 2.1.0) (32). Reads mapped to rRNA loci and reads with a mapping quality <10 were removed. Only the 5΄ end of GRO-seq and the 3΄ end of PRO-seq reads were kept.
GRO-cap and ChIP-seq data
Predicted TSSs were evaluated by the GRO-cap data, Pol II binding and various histone modification profiles. The GRO-cap data of K562 cells were obtained from GEO (GSM1480321) (29). Pol II, H3k4me2, H3k4me3, H3k9ac, H3k27ac, H3K9me3, H3K27me3, H3k79me2 and H3K63me3 ChIP-seq data of K562 cells were obtained from the ENCODE project, which are available at https://www.encodeproject.org/ (33). The GRO-cap enrichment (normalized to 10 Mb) within the region ±100 bp centered by the predicted TSSs with the bin size of 20 bp was generated. Pol II, H3k4me2, H3k4me3, H3k9ac and H3k27ac, H3K9me3, H3K27me3, H3k79me2 and H3K63me3 signals within the region ±2k bp around the predicted TSSs with the bin size of 200 bp were analyzed.
The uniform peaks of ChIP-seq data of transcription factors in K562, GM12878 and H1-hesc cells were downloaded from the ENCODE project (33), http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeAwgTfbsUniform/. Peaks were assigned to the closest known and predicted TSSs by HOMER (http://homer.salk.edu/) (34). Peaks more than 2 kb away from TSSs were not considered.
TCGA miRNA expression data
miRNA expression data from 10 TCGA cancer types, including head and neck squamous cell carcinoma, uterine corpus endometrial carcinoma (UCEC), thyroid carcinoma, stomach adenocarcinoma (STAD), lung squamous cell carcinoma (LUSC), prostate adenocarcinoma (PRAD), colon and rectum adenocarcinoma, breast invasive carcinoma (BRCA), ovarian serous cystadenocarcinoma (OV) and kidney renal clear cell carcinoma, were downloaded from Firehose, developed by the Broad GDAC (https://confluence.broadinstitute.org/display/GDAC/Dashboard-Stddata). Log2 transformed RPM values (reads per million miRNA matures reads) were used to calculate the Pearson correlation between two miRNAs.
RESULTS
Identification of TSSs at a high resolution
We applied mirSTP to predict TSSs for active genes in K562 PRO-seq (∼388M unique mapped reads) and K562 GRO-seq data (∼20M unique mapped reads). MirSTP scanned and scored upstream regions of active genes and identified the representative TSS for each gene. Three levels of thresholds were used to select representative TSSs: stringent, medium and relaxed. Stringent is the highest cutoff level for TSS identification, while relaxed is the lowest. TSSs from ∼80% of active genes were identified using the stringent cutoff, while ∼85% of active genes were detected at the medium cutoff, and ∼95% of active genes were discovered at the relaxed cutoff level. Although the stringent cutoff level obtained the lowest sensitivity, it was expected to deliver the highest accuracy. We evaluated the performance of mirSTP based on known TSS annotations from Refseq, UCSC and Ensembl. In general, the median distance of predicted TSSs from known TSSs was around 35–102 nt in both PRO-seq (Figure 1A) and GRO-seq data (Figure 1B) no matter which annotations (Refseq, UCSC or Ensembl) were compared and which cutoff levels (stringent, medium or relaxed) were chosen. This result is comparable to the microTSS method, which benefits from high-resolution and deep-sequencing RNA-seq data and outperformed the existing methods in prediction accuracy in the cell-specific comparison (23,24). The result also demonstrated that mirSTP worked well in both GRO and PRO-seq data, although GRO-seq (∼50 bp resolution) provides a lower resolution than PRO-seq (single bp resolution) (35). We observed shorter distances between predicted and known TSSs for Ensembl and UCSC, than with Refseq, mainly due to the fact that Ensembl and UCSC have more transcript isoforms than Refseq. The median distance of predicted TSSs from the Ensembl annotated TSSs was only 35 nt at the stringent cutoff level. Many alternative TSSs that are annotated in Ensembl or UCSC, but not in Refseq, were confirmed by mirSTP. For example, mirSTP predicted that the TSS of ARHGAP6 was located at chrX: 11 445 343 (hg19) in K562 GRO/PRO-seq data. Although the predicted site is ∼283k bp away from the Refseq TSS annotation (chrX: 11 683 821, Figure 1C), there are TSSs annotated in UCSC (uc004cun.1) and Ensembl (ENST00000380376) (chrX: 11 445 893) that match mirSTP's prediction. The TSS was also supported by K562 GRO-cap and RNA-seq data (Figure 1C), further demonstrating that it is a real TSS and probably a K562-specific TSS.
Figure 1.

Prediction distance from known gene TSSs annotated by RefSeq, UCSC or Ensembl databases at relax, medium or stringent cutoffs in K562 PRO-seq data (A) and K562 GRO-seq data (B). Genomic characteristics of the region around the predicted ARHGAP6 TSS by mirSTP in the K562 cell line, including PRO-seq, GRO-seq, GRO-cap and RNA-seq. The predicted TSS was supported by GRO-cap and RNA-seq data. Although the TSS was not annotated by Refseq, it was included in both UCSC and Ensembl (C). MirSTP performance on variable K562 PRO-seq depth (D). Y-axis is the prediction distance from known gene TSSs annotated by Ensembl at the medium cutoff, while x-axis is the random subsampled PRO-seq size.
To assess the effect of sequencing depth on mirSTP performance, we randomly sampled K562 PRO-seq data (∼388 M unique mapped reads), resulting in 4 subsets of 100M, 50M, 30M and 10M reads. We evaluated the performance of mirSTP on each subset based on known TSS annotations from Ensembl at the medium cutoff level. The median distance between the predicted and known TSS annotations are similar across different sequencing depth (Figure 1D). Supplementary Figure S1 displays the distances against all three databases (RefSeq, Ensembl or UCSC) at each cutoff level (stringent, medium or relaxed). Even at a very low sequencing depth (10M reads), mirSTP provides accurate TSS predictions, suggesting that sequencing depth has a minor effect on mirSTP performance.
Comparison with existing methods
A total of 1595 human annotated miRNAs were obtained from miRBase v19 (4,36), of which 592 were classified as intergenic, as they are not located inside of any Refseq annotated genes. MiRNAs that have a downstream Refseq annotated gene within 2000 bp on the opposite strand were removed to avoid false TSS signatures caused by GRO/PRO-seq signals of downstream genes extending to miRNA promoter regions. After the filtration, 572 miRNAs were included in the analysis. We applied mirSTP to scan upstream regions of these 572 intergenic miRNAs on K562 PRO-seq data, and identified 104 active miRNA TSSs at the relaxed cutoff level, corresponding to 135 miRNA precursors (Supplementary Data). Due to the lack of a large benchmark set of miRNA promoters, it is difficult to make a direct comparison between mirSTP and other methods. Therefore, we performed an indirect comparison based on promoter-associated signals, such as GRO-cap, Pol II binding and chromatin features. GRO-cap, a modified form of GRO-seq used to sequence the 5΄ end of cap-protected nascent RNAs, provides a comprehensive and precise map of TSS locations (29). RNA polymerase II occupancy is associated with gene transcription, which shows peaks centered near TSSs (37). The enrichment of H3K4me3 is also a hallmark of actively transcribed promoters (38,39). In addition, H3K4me2 and H3ac have been reported to be strongly enriched around TSSs of genes (40).
We first compared mirSTP with two generalized methods: miRStart (19) and PROmiRNA (22). Both of these methods used data pooled from multiple cell lines to infer all putative miRNA TSSs without being constrained to a specific condition. MiRStart combined CAGE tags from 124 human samples, TSS seq tags from six cell lines and H3K4me3 modifications from CD4+ T cells, while PROmiRNA trained a model based on deep CAGE data from 33 human RNA libraries and promoter features. Since PROmiRNA offered multiple TSS predictions for each miRNA, we compared the TSS with the highest score (-H) and the TSS closest to precursor miRNA (-C). TSSs of 44 miRNAs were commonly detected by mirSTP, PROmiRNA and miRStart. Analyzing a ±100 bp window around the 44 TSSs predicted by mirSTP, PROmiRNA-H, PROmiRNA-C and miRStart, we found that TSSs by mirSTP were more enriched for the GRO-Cap signal than for other methods (Figure 2). We analyzed a ±2 kb window around the TSSs, and discovered that TSSs by mirSTP were more enriched for Pol II ChIP-seq data with a C-terminal domain antibody specific for phosphorylated Ser5, which is associated with transcription initiation and polymerase pausing and mainly detects Pol II at the 5΄ ends of genes (Figure 2). Additionally, we observed stronger activating methylation marks (H3K4me2 and H3K4me3) and acetylation marks (H3K9ac and H3K27ac) around TSSs by mirSTP (Figure 2). In contrast, TSSs from different methods presented similar signals associated with repressive histone methylation marks (H3K9me3 and H3K27me3, Supplementary Figure S2) and marks of transcriptional elongation (H3K79me2 and H3K36me3, Supplementary Figure S2). The strong enrichment for various promoter-associated marks around our predicted TSSs demonstrated the power of mirSTP to identify miRNA TSSs.
Figure 2.

Comparison between mirSTP, PROmiRNA and miRStart in terms of GROcap, Pol II occupancy and promoter-associated histone modification profiles, including H3K4me2, H3K4me3, H3K9ac and H3K27ac. Promi-H and Promi-C represent the highest scored and the closest distance to pre-miRNAs.
We then compared mirSTP with the prior method developed by Hua et al. for predicting cell-specific miRNA TSSs (24). Integrating H3K4me3 ChIP-seq and DNase-seq, as well as sequence features, Hua et al. identified TSSs for 122 intergenic miRNAs in the K562 cell line. Although both methods successfully recognized TSSs for 53 miRNAs (Figure 3A), their predicted sites were different. We found that the 53 TSSs identified by mirSTP showed a stronger enrichment for GRO-cap signals than those identified by Hua et al.'s method, suggesting that mirSTP is more accurate. TSSs of 82 miRNAs were uniquely identified by mirSTP (Figure 3B). We observed strong GRO-cap signals around the 82 TSSs as well, which indicated that they were highly likely to be true TSSs (Figure 3C). For example, mirSTP detected the TSS of miR-146b, about 900 bp upstream from the precursor (Figure 3E). The TSS, marked by strong divergent GRO-Cap and Pol II binding signals, has been experimentally validated by a 5΄ RACE technique (10). Other examples of TSSs are illustrated in Supplementary Figure S3. TSSs of 69 miRNAs were uniquely detected by Hua et al. Although there were comparable GRO-cap signals around the predicted TSSs (Figure 3C), PRO-seq and H3K36me3 signals around the ±2 kb window of the corresponding precursor miRNAs were much weaker than those identified by mirSTP (Figure 3D), which suggested that the miRNAs were not active in the K562 cell line. Taking a detailed look at GRO-cap data, we found that high GRO-cap signals were actually generated by their neighboring active genes, proving that the unique predicted TSSs by Hua et al. included several false positives. For example, the predicted TSS of miR3142/miR146a is actually the TSS of PTTG1 (Supplementary Figure S4). Several other examples are given in Supplementary Figure S4.
Figure 3.

Comparison between mirSTP and method by Hua et al., which also provides experiment-specific miRNA TSSs. Venn diagram of the number of predicted TSSs for intergenic miRNAs in the K562 cell line (A). GRO-cap signal around the predicted TSSs for the common 53 intergenic miRNAs (B). GRO-cap signal around the predicted unique TSSs for 82 intergenic miRNAs by mirSTP and 69 intergenic miRNAs by Hua et al. (C). The PRO-seq and H3K36me3 profiles for +/− 2k regions around 82 intergenic miRNAs whose TSS were predicted by mirSTP and 69 by Hua et al. (D). Genomic characteristics of the region around the predicted miR146b TSS by mirSTP in K562 cell line, including PRO-seq, GRO-cap and Pol II ChIP-seq. The predicted TSS was supported by GRO-cap and Pol II occupancy (E).
Identification of intergenic miRNA TSSs in 27 cell types
We applied mirSTP to 183 GRO-seq and 28 PRO-seq experiments in 27 human cell lines (Supplementary Table S1). In total, mirSTP identified putative TSSs for 480 of 572 intergenic miRNAs. The number of predicted miRNA TSSs varied greatly across different datasets. Only 47 miRNA TSSs corresponding to 50 miRNA precursors were identified in HelaS3 GRO-seq data, while 269 miRNA TSSs corresponding to 353 miRNA precursors were recognized in CyT49 GRO-Seq data (Supplementary Data).
Predicted miRNA TSSs suggested the existence of alternative TSS usage in different cell lines. An example of cell-specific TSS usage for miR200b cluster (miR-200b, -200a and -429) is illustrated in Figure 4A. In the MCF7 cell line, the TSS for the miR200b cluster was predicted to be located 4 kb upstream of miR200b (chr1: 1 098 244, hg19), which had been validated by previous studies (41,42). A strong RNA Pol II and H3K4me3 signal overlapping the predicted TSS also indicated preferential transcription from this site in MCF7 cells. However, in the K562 cell line the predicted TSS was located 9kb upstream of miR200b (chr1:1 093 024, hg19). Although this TSS has not been previously reported, the site was supported by the enrichment of RNA Pol II and H3K4me3 signatures. The GRO-cap data of the K562 cells showed divergent transcription around this site. Moreover, these two sites were both supported by the strict TSS predictions based on CAGE peaks in the FANTOM5 project (43,44). Comparing the similarity of the predicted TSSs across experiments, we observed the expected high similarity of the TSS usage in the MCF7 cell line from multiple GRO-seq datasets. Most interestingly, we found the tissue-specific usage of alternative TSSs (Supplementary Figure S5). For example, most of the blood cells clustered together (including B-cell, CD4, Kasumi-1, MV4-11, Daudi, U936 and Jurkat), suggesting that TSS usage was more similar within cells in blood than cells from other tissues. As another example, CyT49 and H1-Esc, two stem cell lines, were more similar in the TSS usage than other cell lines. The tissue-specific TSS usage may contribute to tissue-specific expression of miRNAs.
Figure 4.

Genomic characteristics of the region around the predicted miR200a/200b/429 TSS by mirSTP in MCF7 and K562 cell lines, including GRO-seq, Pol II occupancy and H3K4me3. The predicted TSSs in MCF7 and K562 cell lines were both supported by Pol II occupancy and H3K4me3 enrichment. The TSS in K562 cell line was also supported by the corresponding GRO-cap data (A). The maximum expression correlation value between mature miRNAs within each polycistronic miRNA across 10 TCGA cancer types (B). The number of TF ChIP-seq peaks within 500 bp, 500∼1k, 1k∼2k or 2k∼5p away from predicted miRNA TSSs (miR_TSS) or precursor miRNAs (miR) in K562, GM12878 and H1-hesc cell lines (C). The number of intergenic miRNA targets for each TF (D).
Some intergenic miRNAs shared the same TSSs across multiple cell lines, which suggested they were transcribed from single polycistronic transcripts. We identified the 22 most frequently occurring polycistronic miRNAs (Supplementary Table S2), including miR106a∼363, miR130b/301b and mir200a/b/429. To investigate whether they derive from a common primary transcript, we explored the potential of miRNA member co-expression in the same cluster across 10 TCGA cancer types using miRNA-seq data. A highly correlated expression is expected if they come from a common primary transcript. Of 22 miRNA clusters, three (miR3179∼miR3670, miR3674/miR596 and miR4421/6650) were not detected in any cancer types. Additionally, no correlation coefficients were obtained for four miRNA clusters since one miRNA member was either lowly or not expressed in all cancer types. For the remaining 15 miRNA clusters, members were highly co-expressed in most cancer types (Figure 4B). In some cases, all members of the same cluster shared similar expression patterns across all cancer types. For example, miRNA clusters, like miR192/194-2, miR221/222, miR23a/27a, miR29b-2/29c and miR30b/30d, were co-expressed in all 10 TCGA cancer types. In other cases, co-clustered miRNAs presented different expression patterns across cancer types, where miRNA members were highly co-expressed in some cancer types but the correlation was lost in other cancer types. For instance, the expressions of miR130b and miR301b were highly correlated in UCEC, STAD, LUSC and BRCA (R > 0.7), yet they were poorly correlated in OV (R = 0.3, Figure 4B and Supplementary Figure S6). As another example, miR940 and miR3677 were highly co-expressed in BRCA, STAD and UCEC (R > 0.7), but they correlated poorly in PRAD (R = 0.34, Figure 4B and Supplementary Figure S7). These results indicated that although they are indeed polycistronic miRNA transcripts, there are cancer/tissue-specific post-transcriptional regulatory mechanisms that disturbed the co-expression of miRNA members.
Prediction of TF–miRNA interactions
Knowing miRNA TSSs makes it possible to accurately connect miRNA to the transcription regulatory network. To identify transcription factors (TF) regulating these 480 miRNAs, we downloaded the TF ChIP-seq Uniform Peaks from ENCODE in the Tier 1 cell lines (K562, GM12878 and H1-hesc), and linked the peaks to cell-specific miRNA TSSs. We discovered that the peaks were much more enriched in the predicted TSSs than in the miRNA precursor loci (Figure 4C). In the K562 cell line, for example, most peaks (1815 peaks, 54%) were within 500 bp of the predicted TSSs, while only 52 peaks were within 500 bp of the miRNA loci (Figure 4C). We observed a similar pattern in the GM12878 and H1-hesc cell lines, which supported the accuracy of the predicted TSSs (Figure 4C).
If the TF ChIP-seq peak was within 2 kb from a miRNA TSS, we considered it a TF–miRNA interaction. In the K562 cell line, 2678 ChIP-seq peaks were assigned to 135 intergenic miRNA/miRNA clusters. After removing the redundancy, 1893 TF–miRNA interactions were discovered, including 97 TFs and 101 miRNA/miRNA clusters (Supplementary Table S3). Max and Myc were the most commonly discovered DNA binding TFs, and POLR2A transcribed the majority of these miRNAs/miRNA clusters (65 miRNA TSSs), (Figure 4D). These predicted TF–miRNA regulations include both previously described and novel interactions. Many of the 54 miRNAs/miRNA clusters targeted by Myc have been identified by previous studies, which verified the reliability of our prediction method. For example, Myc regulates miR-101 (45), miR-129 (46), miR-148a (47), miR-23a/b (48), miR-200 members (49) and miR-29 (50). The reliable TF–miRNA interactions provided us with a valuable source for understanding the miRNA function in complex regulatory networks. In addition, we found statistically co-associated TFs regulating the 101 miRNAs in the K562 cell line (Supplementary Table S4). TF co-binding across the genome was compiled by the Encode project (51,52), which was also confirmed by our study; TF factors included Stat1 and Stat2 (53), Maff and Mafk (54), Max-Myc-Maz (55), Jun and Fos (56) and the RNA Polymerase III preinitiation complex (Pol III, Bdp1 and Brf1) (57) (Supplementary Figure S8). Our results indicated that known TF complexes also co-bind to the promoter regions of intergenic miRNAs.
A major advantage of using GRO/PRO-seq data is that it can track miRNA TSSs, and quantify primary miRNA transcription rates simultaneously in a single experiment. By applying mirSTP to PRO-seq datasets in the Kasumi-1 cell line (30), we discovered TSSs for 72 intergenic miRNAs at the stringent cutoff, corresponding to 59 pri-miRNAs. We quantified the pri-miRNA expression by counting the PRO-seq reads in the sense strand from the predicted TSSs to the precursor miRNAs, and calculated the pausing index for each pri-miRNA as the ratio of promoter-proximal density (pp) divided by gb density (27,58–60). We evaluated the significance of pausing index alteration in BET-inhibitor-treated versus DMSO samples using Fisher's exact test followed by multiple testing adjustments (27). After JQ1 treatment, 10 pri-miRNAs showed a significant increase in pausing index (FDR < 0.01), while only two pri-miRNAs exhibited a decrease in pausing index (FDR < 0.01, Supplementary Table S5), which suggested that these 12 pri-miRNAs are direct targets of the BET family (Figure 5A).
Figure 5.

The table lists 12 pri-miRNAs with significant pausing alteration (FDR < 0.01) after JQ1 treatment, all of which have BRD4 binding peaks close to their predicted TSSs (A). Predicted TSS for pri-miR221/222 and PRO-seq profiles in promoter-proximal (pp) and gene body (gb) regions of pri-miR221/222 in DMSO and JQ1 treatment samples. Pri-miR221/222 showed an increased signal in pp regions but decreased signal in gb regions, resulting in a significant elevation of pausing after JQ1 treatment. BRD4 ChIP-seq data revealed its binding to the TSS region, further demonstrating pri-miR221/222 is a direct target of BRD4 (B).
As a further support of the PRO-seq results, we explored BRD4 ChIP-seq derived from MUTZ3 (an AML cell line, Array Express accessions: ERR412006) and found that all 12 pri-miRNAs were bound by BRD4 except miR29b2/miR29c (distance < 500 bp). Although no binding signal was found for miR29b2/miR29c in the MUTZ3 cell line, BRD4 binding to miR29b2/miR29c was verified by ChIP assays in the Kasumi-1 cell line (30). MiR221/222 was the second most affected by BET inhibitors with increased proximal-promoter (pp) and decreased gb density, which had not been previously recognized (30). The predicted TSS for miR221/222 by mirSTP was located ∼ 23 kb upstream of the precursor and showed a strong increase in pausing index after the addition of BET inhibitors (log2FC = 1.12, FDR < 2e-24) (Figure 5B). Meanwhile, ChIP-seq revealed BRD4 binding to the promoter regions of miR221/222 (Figure 5B), which suggested that miR221/222 was a direct target of BRD4. The tight association between the PRO-seq and ChIP-seq results demonstrated that the accurate identification of miRNA TSSs and the quantification of pri-miRNA transcription from GRO/PRO-seq data are very useful for defining the transcriptional regulation of miRNAs.
DISCUSSION
The accurate identification of miRNA TSSs is crucial for locating the core promoters of miRNAs, and integrating the control of miRNA transcription into complex regulatory networks. MiRNAs are initially transcribed into large pri-miRNAs, which undergo sequential processing steps to generate mature miRNAs. It is difficult for traditional transcriptome profiling, such as RNA-seq, to capture pri-miRNAs, because of their transient nature. GRO/PRO-seq techniques, which directly map elongation-competent RNA polymerases (including RNAPI, II and III) across the entire genome to detect RNA transient transcription on a genome-wide scale (including the transient pri-miRNAs), provide an ideal method for mapping pri-miRNA TSSs and link them to transcription rates. In this study, we developed a novel method, mirSTP, for predicting intergenic miRNA TSSs by taking advantage of two major features characterized by GRO/PRO-seq data: divergent transcription around TSSs and continuous transcription across gb regions. MirSTP provides accurate, experiment-specific and high-resolution miRNA TSS predictions, which are strongly supported by GRO-cap signals and other TSS-associated histone markers. MirSTP compares well with existing CAGE- or chromatin-based, generalized or experiment-specific miRNA TSS prediction methods.
MirSTP is readily applicable to any cell line or condition with available GRO/PRO-seq data. Compared to microTSS, which also provides experiment-specific and highly accurate predictions for intergenic miRNA TSSs, mirSTP has much lower requirements regarding data types and sequencing depth. MicroTSS requires three datasets to perform a prediction, including deep sequenced RNA-seq, ChIP-Seq and DNase-Seq, which can be very costly to generate. In contrast, mirSTP only uses one dataset (GRO/PRO-Seq) and takes advantage of the sharp divergent peaks near the TSS to provide highly accurate predictions without the requirement of deep sequencing. Algorithms based on GRO-cap and CAGE data, like PROmiRNA and miRStart, also provide high-resolution predictions. However, GRO-cap and CAGE only measure the 5΄ end of the transcript, making it difficult to decide whether the observed TSS belongs to the miRNA without the continuous signal to the precursor. In contrast, GRO/PRO-seq, with the ability to capture all active transcription, is able to track the whole primary transcript. Another advantage of GRO/PRO-seq data is its ability to quantify pri-miRNA expression. After TSS prediction, researchers can complete the same analysis for pri-miRNAs, just as they would for genes, including quantifying the transcriptional changes in promoter-proximal and gb regions, and estimating pausing index alteration. After BET inhibitor treatment, pri-miRNAs with significant pausing index changes were highly likely to be BRD2/3/4 targets, and, in fact, each contains BRD4 binding in the predicted promoter regions. Therefore, in a single experiment, researchers can both identify miRNA TSSs, and provide a direct readout of pri-miRNA activity, which is very helpful for unveiling targets of the regulator of interest. One limitation of mirSTP is that it predicts the representative TSS for each miRNA, rather than all possible TSSs. This limitation could be overcome by pinpointing all of the divergent transcription sites instead of the maximal one by mirSTP. However, this strategy could greatly increase the number of false TSSs. Additional efforts would be needed to choose an appropriate cutoff or incorporate TSS-relevant sequence features to balance the sensitivity and specificity.
By applying mirSTP to 183 GRO-seq and 28 PRO-seq experiments in 27 human cell lines, we identified TSSs for 84% of intergenic miRNAs. No putative TSSs were discovered for some miRNAs, which is mainly due to two reasons. One reason is that the miRNAs were not expressed or expressed at low levels in the 27 cell lines. Applying mirSTP to a variety of cell lines would probably uncover TSSs for those miRNAs. The second reason is due to the lack of divergent transcription around TSSs for some miRNAs. Previous studies have found that not all active genes display significant divergent transcription around promoter-proximal regions (27). MirSTP cannot identify those TSSs without divergent transcription since it highly depends on this feature to recognize TSSs. In this case, other types of data such as CAGE-seq, histone modification profiles or sequence features would be helpful for identifying TSSs.
By comparing TSSs detected in different cell lines, we found a wide usage of tissue-specific alternative start sites, which further emphasizes the advantage of experiment-specific TSSs methods versus generalized TSS prediction methods. The identification of TSSs in the cell type or condition of interest allows researchers to build condition-specific regulatory networks to interpret the regulation of gene expression more accurately. MirSTP results also discover known and novel polycistronic miRNA transcripts. Most miRNAs derived from the same transcripts are either highly co-expressed or co-silenced across all 10 TCGA cancer types, demonstrating that they are truly co-regulated. For example, all members are highly co-expressed in some cancer types, while co-expression is lost in other types of cancer. Such an observation suggests the existence of a cancer-specific post-transcriptional regulation mechanism that blocks individual members of polycistronic transcripts from the maturation process. This agrees with a previous study, which reported that ADARs (Adenosine deaminases acting on RNAs) are responsible for the differential expression of polycistronic miRNA clusters by altering the structural conformation of pri-miRNA (61).
Beyond the development of mirSTP, we also integrated Encode TF ChIP-seq data (51) with the predicted TSSs in the same cell type to predict TF–miRNA regulations. We found that the peaks were highly enriched in the predicted TSS regions. Many TF–miRNA interactions have been reported by previous studies, which demonstrated that the accurate identification of TSSs is essential for reliably defining the regulation of miRNAs. Increasing evidence has indicated the reciprocal regulation between TF and miRNAs, which play very important roles in biological processes such as development, homeostasis and pathology (47,62–64). Previous studies have reported instances of a MYC-miRNA circuitry where MYC targets a number of miRNAs, and, simultaneously, miRNAs regulate MYC, thereby creating double negative feedback loops (62,63,65). For instance, the MYC-miRNA circuit acts as a mechanism to sustain a MYC oncogenic signal and to drive lymphoma progression (62). Twenty-three miRNA ↔ TF composite feedback loops were found in Caenorhabditis elegans that provide for a highly coordinated and adaptable control of gene expression (64). Our highly reliable TF–miRNA interactions would dramatically extend the interplay between TF and miRNAs. For example, some known associations between TF and miRNAs were identified, such as Myc ↔ miR-101 and Myc ↔ miR-200. Novel reciprocal interactions could be revealed if we combined TF–miRNA interactions with miRNA target prediction. The TF–miRNA interactions add an additional layer to these regulatory networks and provide an invaluable source for understanding how TF and miRNAs interact to achieve the precise regulation of gene expression.
AVAILABILITY
MirSTP is available at http://bioinfo.vanderbilt.edu/mirSTP/.
ACCESSION NUMBER
PRO-seq data generated in this study have been deposited in the GEO database under the accession code: GSE97143.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Michael Smith for his editorial work on this manuscript, and thank reviewers for their great suggestions and comments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Institute [5U01 CA163056-05 to YS, 5R01 CA178030-02 to SH, 2R01 CA064140-21A1 to S.H.]; Cancer Center Support Grant [2P30 CA068485-19 to Y.S.]; NCI SPORE in GI Cancer Career Development Award [P50 CA095103 to Q.L.]; Institutional Funds. Funding for open access charge: Institutional Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. He L., Hannon G.J.. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004; 5:522–531. [DOI] [PubMed] [Google Scholar]
- 2. Nazarov P.V., Reinsbach S.E., Muller A., Nicot N., Philippidou D., Vallar L., Kreis S.. Interplay of microRNAs, transcription factors and target genes: linking dynamic expression changes to function. Nucleic Acids Res. 2013; 41:2817–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ha M., Kim V.N.. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014; 15:509–524. [DOI] [PubMed] [Google Scholar]
- 4. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kozomara A., Griffiths-Jones S.. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39:D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee Y., Jeon K., Lee J.T., Kim S., Kim V.N.. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002; 21:4663–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bracht J., Hunter S., Eachus R., Weeks P., Pasquinelli A.E.. Trans-splicing and polyadenylation of let-7 microRNA primary transcripts. RNA. 2004; 10:1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai X., Hagedorn C.H., Cullen B.R.. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004; 10:1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Houbaviy H.B., Dennis L., Jaenisch R., Sharp P.A.. Characterization of a highly variable eutherian microRNA gene. RNA. 2005; 11:1245–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taganov K.D., Boldin M.P., Chang K.J., Baltimore D.. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang T.C., Wentzel E.A., Kent O.A., Ramachandran K., Mullendore M., Lee K.H., Feldmann G., Yamakuchi M., Ferlito M., Lowenstein C.J. et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell. 2007; 26:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fujita S., Iba H.. Putative promoter regions of miRNA genes involved in evolutionarily conserved regulatory systems among vertebrates. Bioinformatics. 2008; 24:303–308. [DOI] [PubMed] [Google Scholar]
- 13. Megraw M., Pereira F., Jensen S.T., Ohler U., Hatzigeorgiou A.G.. A transcription factor affinity-based code for mammalian transcription initiation. Genome Res. 2009; 19:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou X., Ruan J., Wang G., Zhang W.. Characterization and identification of microRNA core promoters in four model species. PLoS Comput. Biol. 2007; 3:e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saini H.K., Enright A.J., Griffiths-Jones S.. Annotation of mammalian primary microRNAs. BMC Genomics. 2008; 9:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marson A., Levine S.S., Cole M.F., Frampton G.M., Brambrink T., Johnstone S., Guenther M.G., Johnston W.K., Wernig M., Newman J. et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008; 134:521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ozsolak F., Poling L.L., Wang Z., Liu H., Liu X.S., Roeder R.G., Zhang X., Song J.S., Fisher D.E.. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008; 22:3172–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barski A., Jothi R., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Zhao K.. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009; 19:1742–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chien C.H., Sun Y.M., Chang W.C., Chiang-Hsieh P.Y., Lee T.Y., Tsai W.C., Horng J.T., Tsou A.P., Huang H.D.. Identifying transcriptional start sites of human microRNAs based on high-throughput sequencing data. Nucleic Acids Res. 2011; 39:9345–9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X., Xuan Z., Zhao X., Li Y., Zhang M.Q.. High-resolution human core-promoter prediction with CoreBoost_HM. Genome Res. 2009; 19:266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corcoran D.L., Pandit K.V., Gordon B., Bhattacharjee A., Kaminski N., Benos P.V.. Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One. 2009; 4:e5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsico A., Huska M.R., Lasserre J., Hu H., Vucicevic D., Musahl A., Orom U., Vingron M.. PROmiRNA: a new miRNA promoter recognition method uncovers the complex regulation of intronic miRNAs. Genome Biol. 2013; 14:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Georgakilas G., Vlachos I.S., Paraskevopoulou M.D., Yang P., Zhang Y., Economides A.N., Hatzigeorgiou A.G.. microTSS: accurate microRNA transcription start site identification reveals a significant number of divergent pri-miRNAs. Nat. Commun. 2014; 5:5700. [DOI] [PubMed] [Google Scholar]
- 24. Hua X., Chen L., Wang J., Li J., Wingender E.. Identifying cell-specific microRNA transcriptional start sites. Bioinformatics. 2016; 32:2403–2410. [DOI] [PubMed] [Google Scholar]
- 25. Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stahler C., Meese E. et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016; 44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo Z., Maki M., Ding R., Yang Y., Zhang B., Xiong L.. Genome-wide survey of tissue-specific microRNA and transcription factor regulatory networks in 12 tissues. Sci. Rep. 2014; 4:5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Sci. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kwak H., Fuda N.J., Core L.J., Lis J.T.. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013; 339:950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Core L.J., Martins A.L., Danko C.G., Waters C.T., Siepel A., Lis J.T.. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 2014; 46:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao Y., Liu Q., Acharya P., Stengel K.R., Sheng Q., Zhou X., Kwak H., Fischer M.A., Bradner J.E., Strickland S.A. et al. High-resolution mapping of RNA polymerases identifies mechanisms of sensitivity and resistance to BET inhibitors in t(8;21) AML. Cell Rep. 2016; 16:2003–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011; 17:10–12. [Google Scholar]
- 32. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jonkers I., Lis J.T.. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015; 16:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol. Biol. 2006; 342:129–138. [DOI] [PubMed] [Google Scholar]
- 37. Quinodoz M., Gobet C., Naef F., Gustafson K.B.. Characteristic bimodal profiles of RNA polymerase II at thousands of active mammalian promoters. Genome Biol. 2014; 15:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim T.H., Barrera L.O., Zheng M., Qu C., Singer M.A., Richmond T.A., Wu Y., Green R.D., Ren B.. A high-resolution map of active promoters in the human genome. Nature. 2005; 436:876–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007; 39:311–318. [DOI] [PubMed] [Google Scholar]
- 40. Koch C.M., Andrews R.M., Flicek P., Dillon S.C., Karaoz U., Clelland G.K., Wilcox S., Beare D.M., Fowler J.C., Couttet P. et al. The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res. 2007; 17:691–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wee E.J., Peters K., Nair S.S., Hulf T., Stein S., Wagner S., Bailey P., Lee S.Y., Qu W.J., Brewster B. et al. Mapping the regulatory sequences controlling 93 breast cancer-associated miRNA genes leads to the identification of two functional promoters of the Hsa-mir-200b cluster, methylation of which is associated with metastasis or hormone receptor status in advanced breast cancer. Oncogene. 2012; 31:4182–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bracken C.P., Gregory P.A., Kolesnikoff N., Bert A.G., Wang J., Shannon M.F., Goodall G.J.. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008; 68:7846–7854. [DOI] [PubMed] [Google Scholar]
- 43. Lizio M., Harshbarger J., Shimoji H., Severin J., Kasukawa T., Sahin S., Abugessaisa I., Fukuda S., Hori F., Ishikawa-Kato S. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015; 16:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. FANTOM Consortium and the RIKEN PMI and CLST (DGT) Forrest A.R., Kawaji H., Rehli M., Baillie J.K., de Hoon M.J., Haberle V., Lassmann T. et al. A promoter-level mammalian expression atlas. Nature. 2014; 507:462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang L., Zhang X., Jia L.T., Hu S.J., Zhao J., Yang J.D., Wen W.H., Wang Z., Wang T., Zhao J. et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology. 2014; 59:1850–1863. [DOI] [PubMed] [Google Scholar]
- 46. Han H., Li W., Shen H., Zhang J., Zhu Y., Li Y.. microRNA-129-5p, a c-Myc negative target, affects hepatocellular carcinoma progression by blocking the Warburg effect. J. Mol. Cell Biol. 2016; 8:400–410. [DOI] [PubMed] [Google Scholar]
- 47. Ma Y., Wang B., Jiang F., Wang D., Liu H., Yan Y., Dong H., Wang F., Gong B., Zhu Y. et al. A feedback loop consisting of microRNA 23a/27a and the beta-like globin suppressors KLF3 and SP1 regulates globin gene expression. Mol. Cell. Biol. 2013; 33:3994–4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gao P., Tchernyshyov I., Chang T.C., Lee Y.S., Kita K., Ochi T., Zeller K.I., De Marzo A.M., Van Eyk J.E., Mendell J.T. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009; 458:762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin C.H., Jackson A.L., Guo J., Linsley P.S., Eisenman R.N.. Myc-regulated microRNAs attenuate embryonic stem cell differentiation. EMBO J. 2009; 28:3157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang X., Zhao X., Fiskus W., Lin J., Lwin T., Rao R., Zhang Y., Chan J.C., Fu K., Marquez V.E. et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012; 22:506–523. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51. Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R. et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bluyssen H.A., Muzaffar R., Vlieststra R.J., van der Made A.C., Leung S., Stark G.R., Kerr I.M., Trapman J., Levy D.E.. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:5645–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Motohashi H., Katsuoka F., Engel J.D., Yamamoto M.. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:6379–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bossone S.A., Asselin C., Patel A.J., Marcu K.B.. MAZ, a zinc finger protein, binds to c-MYC and C2 gene sequences regulating transcriptional initiation and termination. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:7452–7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Angel P., Karin M.. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta. 1991; 1072:129–157. [DOI] [PubMed] [Google Scholar]
- 57. Khoo S.K., Wu C.C., Lin Y.C., Lee J.C., Chen H.T.. Mapping the protein interaction network for TFIIB-related factor Brf1 in the RNA polymerase III preinitiation complex. Mol. Cell. Biol. 2014; 34:551–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zeitlinger J., Stark A., Kellis M., Hong J.W., Nechaev S., Adelman K., Levine M., Young R.A.. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat. Genet. 2007; 39:1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Reppas N.B., Wade J.T., Church G.M., Struhl K.. The transition between transcriptional initiation and elongation in E. coli is highly variable and often rate limiting. Mol. Cell. 2006; 24:747–757. [DOI] [PubMed] [Google Scholar]
- 60. Muse G.W., Gilchrist D.A., Nechaev S., Shah R., Parker J.S., Grissom S.F., Zeitlinger J., Adelman K.. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007; 39:1507–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chawla G., Sokol N.S.. ADAR mediates differential expression of polycistronic microRNAs. Nucleic Acids Res. 2014; 42:5245–5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tao J., Zhao X.. c-MYC-miRNA circuitry: a central regulator of aggressive B-cell malignancies. Cell Cycle. 2014; 13:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Daneshvar K., Nath S., Khan A., Shover W., Richardson C., Goodliffe J.M.. MicroRNA miR-308 regulates dMyc through a negative feedback loop in Drosophila. Biol. Open. 2013; 2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martinez N.J., Ow M.C., Barrasa M.I., Hammell M., Sequerra R., Doucette-Stamm L., Roth F.P., Ambros V.R., Walhout A.J.. A C. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity. Genes Dev. 2008; 22:2535–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Han H., Sun D., Li W., Shen H., Zhu Y., Li C., Chen Y., Lu L., Li W., Zhang J. et al. A c-Myc-MicroRNA functional feedback loop affects hepatocarcinogenesis. Hepatology. 2013; 57:2378–2389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.