


Abstract
Gene expression in Plasmodium falciparum is tightly regulated to ensure successful propagation of the parasite throughout its complex life cycle. The earliest transcriptomics studies in P. falciparum suggested a cascade of transcriptional activity over the course of the 48-hour intraerythrocytic developmental cycle (IDC); however, the just-in-time transcriptional model has recently been challenged by findings that show the importance of post-transcriptional regulation. To further explore the role of transcriptional regulation, we performed the first genome-wide nascent RNA profiling in P. falciparum. Our findings indicate that the majority of genes are transcribed simultaneously during the trophozoite stage of the IDC and that only a small subset of genes is subject to differential transcriptional timing. RNA polymerase II is engaged with promoter regions prior to this transcriptional burst, suggesting that Pol II pausing plays a dominant role in gene regulation. In addition, we found that the overall transcriptional program during gametocyte differentiation is surprisingly similar to the IDC, with the exception of relatively small subsets of genes. Results from this study suggest that further characterization of the molecular players that regulate stage-specific gene expression and Pol II pausing will contribute to our continuous search for novel antimalarial drug targets.
INTRODUCTION
As one of the world's deadliest infectious diseases, malaria is responsible for ∼438 000 deaths annually, the vast majority of which occur among children under the age of five (1). Of the five Plasmodium species that can cause malaria in humans, P. falciparum is responsible for the most severe form of malaria and causes about 90% of all malarial deaths (1). Currently, no approved efficient protective vaccine is available for disease prevention, and the rapid development of parasite resistance to current antimalarial drugs is a major challenge for the control of malaria. Therefore, a better understanding of the parasite's biological system is required to identify novel drug targets and to further combat the disease.
Plasmodium falciparum has a complex life cycle involving multiple phases in its human and mosquito hosts. The stage responsible for clinical malaria is the intraerythrocytic developmental cycle (IDC). In this cycle, the parasite replicates asexually inside red blood cells and develops through ring, trophozoite and schizont stages to multiply into 16–32 daughter parasites (2). During the IDC, environmental stress can induce sexual differentiation of parasites into male and female gametocytes. Gametocytes are morphological and functionally different from asexual parasites. Mature gametocytes, ingested by a mosquito, undergo sexual replication in the mosquito midgut and further develop into the salivary gland sporozoites that can be transmitted to a new human host. Formation and carriage of gametocytes is key to disease transmission.
This multi-stage life cycle of the parasite is highly fine-tuned, presumably by strict control of stage-specific gene expression. In eukaryotes, stage-specific regulation of gene expression can be a combined effect of transcriptional, post-transcriptional and translational control. In P. falciparum, the nature and the contribution of mechanisms regulating gene expression are still poorly understood. Compared to organisms with similar genome size, only one-third of the expected number of specific transcription factors (TFs) and few mediator subunits have been uncovered in the P. falciparum genome (3,4). Recently, an apicomplexan-specific family of proteins containing AP2 DNA binding domains (ApiAP2) has been identified in apicomplexan parasites as the major group of putative sequence-specific transcription factors (5–8). While their number is relatively small (27 in the P. falciparum genome), they are likely to act as master regulators of transcription during parasite development. However, it remains unclear how such a limited number of TFs can generate complex patterns of gene expression in multiple life cycle stages.
Accumulating evidence suggests that P. falciparum uses chromatin structure as a basal control for transcriptional initiation. Genome architecture studies (9,10) showed that chromatin is relatively closed during the ring and schizont stages, but opens substantially during the trophozoite stage, providing a transcriptional permissive state. This open-and-closed binary chromatin activity is also reflected in nucleosome occupancy studies (11–13) and histone abundance levels (14–16). Nucleosome density is relatively low at the trophozoite stage, but maintained high at early ring and late schizont stages. In addition, studies using chromatin immunoprecipitation directed against RNA polymerase II (Pol II) and coupled to genomic DNA microarrays (ChIP-on-chip) indicate that Pol II is divided into a bi-phasic occupancy throughout the parasite IDC (17).
Controversially, such chromatin structure re-arrangement activity and Pol II profiling do not correlate well with previously observed complex patterns of gene expression profiles constructed from steady-state mRNA (18–22). The cascade of gene expression observed at the steady-state mRNA level led to a ‘just-in-time’ model suggesting mRNA is produced when the encoded protein is required during the cell cycle. However, comparative genomics analysis of steady state mRNA and protein profiles of different P. falciparum stages showed a significant delay between peak of mRNA and protein levels for 30–40% of the analyzed genes (14,23), supporting a model of ‘just-in-time’ translation for some mRNAs. Recently published novel genome-wide approaches that compared steady-state mRNA with polysome-associated mRNA or ribosomal occupancy of mRNAs have also provided a good indicator of active protein production and further validated the model of ‘just-in-time’ translation for specific subsets of genes (19,24). In particular, this was true for proteins involved in remodeling of the erythrocyte just after parasite invasion.
The paucity of transcription factors (25), the lack of identified DNA regulatory elements (26) together with the weak correlation between chromatin-remodeling events, Pol II occupancy, and steady-state mRNA levels, suggested significant post-transcriptional mechanisms regulating the parasite development. However, when Plasmodium genes are exactly transcribed and how many of them are regulated at the post-transcriptional level to generate the cascade of steady-state mRNA observed throughout the parasite life cycle remains to be determined.
MATERIALS AND METHODS
Parasite culture
Parasite strain, P. falciparum 3D7, was cultured at ∼8% parasitemia in human erythrocytes at 5% hematocrit in a total culture volume of 25 ml as described in (27). To obtain highly synchronized cultures for the asexual stages, two 5% d-sorbitol treatments were performed eight hours apart at the ring stage. Parasites were collected every 6 h covering early ring, late ring, early trophozoite, late trophozoite, early schizont and late schizont stages. Giemsa-stained blood smears were used to assess parasite developmental stages. To obtain gametocyte-stage parasites, P. falciparum strain NF54 was cultured as described previously (28). In brief, parasites were first synchronized using 5% sorbitol lysis buffer and diluted to reach 0.5% parasitemia at 8.3% hematocrit the following day. A reduction of culture media to 10 ml for three subsequent days was used as a way to stress the parasites and induce gametocyte production. Culture volume was then returned to 25 ml per flask. During the next 5 days, cultures were maintained by daily media exchange using media containing 10 ml of 50 mM N-acetyl glucosamine (NAG) to remove asexual stage parasites. Gametocyte cultures at 2% parasitemia were harvested 10 days (stage III) or 14 days (stage V) after the start of this procedure.
Nuclear isolation
Nuclear isolation was performed as described in (13,29). Parasite pellets were resuspended in 1 ml of nuclear extraction buffer (10 mM Tris–HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 250 units of SUPERaseIn (Ambion), 10% glycerol and 0.5% Igepal CA-360 (Sigma-Aldrich, St. Louis, MO, USA)) and incubated on ice for 10 min. Parasites were then mechanically lysed by passing the suspension fifteen times through a 26G  inch needle. Nuclei were pelleted by centrifugation for 20 min at 2500 × g at 4°C and resuspended in 1 ml of nuclear extraction buffer, followed by gently pipetting up and down 10 times. Nuclei were centrifuged for 20 min at 2500 × g at 4°C and resuspended in 100 μl of storage solution (50 mM Tris–Cl pH 8, 5 mM MgCl2, 0.1 mM EDTA, 40% glycerol and 50 units of SUPERaseIn).
inch needle. Nuclei were pelleted by centrifugation for 20 min at 2500 × g at 4°C and resuspended in 1 ml of nuclear extraction buffer, followed by gently pipetting up and down 10 times. Nuclei were centrifuged for 20 min at 2500 × g at 4°C and resuspended in 100 μl of storage solution (50 mM Tris–Cl pH 8, 5 mM MgCl2, 0.1 mM EDTA, 40% glycerol and 50 units of SUPERaseIn).
Nuclear run-on reaction
Nuclei (100 μl) were incubated with 600 μl of nuclear run-on reaction buffer (10 mM Tris–Cl pH 8.0, 5 mM MgCl2, 1 mM DTT, 300 mM KCl and 200 units of SUPERaseIn, 1% sarkosyl, 4 mM ATP, 1 mM CTP, 1 mM GTP, 200 mM ethylene uridine (EU) (Click-it Nascent RNA Capture Kit, Thermo Fisher), 400 mM creatine phosphate and 0.2 mg/ml creatine kinases) adopted and modified from (30,31). Reaction mixtures were incubated for 30 min at 37°C followed by nuclear RNA isolation.
Base hydrolysis of nuclear RNA
Base hydrolysis was performed as described in (30). For each 20 μl of RNA, 5 μl of 1M NaOH is added and incubated for 15 min on ice. The reaction was neutralized with 25 μl of 1 M Tris–Cl pH 6.8. Fragmented RNA was precipitated by adding 4 μl glycogen, 75 μl 5 M ammonium acetate and 700 μl 100% ethanol).
Nascent RNA purification and cDNA preparation
Nascent RNA was purified from total nuclear RNA samples using the Click-iT Nascent RNA Capture Kit (Thermo Fisher) according to the manufacturer's instructions. In brief, biotin-azide was attached to ethylene-groups of the EU-labeled RNA using click-it chemistry. The EU-labeled nascent RNA was purified using MyOne Streptavidin T1 magnetic Dynabeads (Life Technologies). The preparation of cDNA was performed using nascent RNA captured on the beads. cDNA synthesis reaction mix (6 μg of random hexamer (integrated DNA technologies, Coralville, IA, USA), 2 μg of anchored oligo (dT)20 (Integrated DNA Technologies), 2 μl 10 mM dNTP mix (Life Technologies) and 14 μl buffer J from Click-iT Nascent RNA Capture Kit (Thermo Fisher) in a total of 20 μl volume) was added to the beads and incubated for 10 min at 70°C, and then chilled on ice for 5 min. Next, a mix of 4 μl 10X RT buffer, 8 μl 20 mM MgCl2, 4 μl 0.1 M DTT, 2 μl 20 U/μl SuperaseIn and 2 μl 200 U/μl SuperScript III Reverse Transcriptase (all from Life Technologies) was added to the mixture and incubated for 10 min at 25°C, 50 min at 50°C, and finally 5 min at 85°C for first strand cDNA synthesis. To digest RNA and release the first-strand cDNA, 2 μl 2 U/μl Escherichia coli RNase H (Life Technologies) was added, followed by a 20 min incubation at 37°C. The beads then were removed using a magnet and first-strand cDNA was used for second-strand cDNA synthesis by adding 70 μl 5× nuclease-free water (Life Technologies), 30 μl second-strand buffer (Life Technologies), 3 μl 10 mM dNTP mix (Life Technologies), 4 μl 10 U/μl E. coli DNA Polymerase (NEB) and 1 μl 10 U/μl E. coliDNA ligase (NEB). The mixture was then incubated for 2 h at 16°C. Finally, double-stranded cDNA was purified using 1.8× Agencourt AMPure XP beads (Beckman Coulter). Validation PCRs were performed using the primers listed in Supplemental Table S1.
Library preparation and sequencing
Libraries were prepared using the KAPA Biosystems Library Preparation Kit (KAPA Biosystems, Woburn, MA, USA) according to the manufacturer's instructions with the following modifications for the high AT-content of the P. falciparum genome: the libraries were amplified for 15 PCR cycles (45 s at 98°C followed by 15 cycles of [15 s at 98°C, 30 s at 55°C, 30 s at 62°C], 5 min 62°C). Libraries were sequenced on the Illumina HiSeq2500 (Illumina, San Diego, CA, USA) generating 50 bp paired-end sequence reads or the NextSeq500 generating 75 bp paired-end sequence reads.
Sequence mapping
The first 10 bases and the last base or last 20 bases were systematically trimmed from 50 and 75 bp reads, respectively, using FastQ Trimmer from FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Poly-A/T repeats and contaminating adaptor reads were removed using Scythe (https://github.com/ucdavis-bioinformatics/scythe) (32). Reads containing bases with a quality score below 25 and Ns, reads that were unpaired, and reads shorter than 18 bases were also filtered using Sickle (https://github.com/najoshi/sickle) (33). In addition, high quality single reads that lost their mate pair during read processing were kept and mapped as single-end reads in parallel with paired-end reads. All trimmed reads were first mapped to the human genome version hg19 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/) using Bowtie 2 (34), and all non-human reads were further mapped to P. falciparum 3D7 genome v13.0 (www.plasmoDB.org) using TopHat2 (35) allowing a maximum of one mismatch per read segment and a segment length of 18. Finally, reads that mapped to multiple locations in the genome (samtools v0.1.19), paired-end reads that were not properly paired (samtools v0.1.19), reads that were PCR duplicates (MarkDuplicates, Picard Tools v1.114), and reads that mapped to ribosomal RNA or transfer RNA were discarded from the final working reads.
Calculation of normalized gene expression values
Raw genome-wide coverage profiles were generated using BEDtools (36). For each stage, numbers of mapped reads from both single-ended mapping and paired-end mapping were combined. For each gene, we then calculated the number of reads that mapped to its exons, and normalized these read counts by GC content and the sum of exon lengths using R package EDASeq (37). Spearman correlations between biological replicates were calculated using the EDASeq normalized exon counts. For biological replicates that were highly correlated (Spearman R > 0.85), bam files were merged using Samtools v0.1.19 (38) and the normalized exon count was recalculated. Genes with an average exon read count <2 at all stages were considered not expressed and were removed from the data set. To accurately measure transcriptional activity at each stage, we normalized the exon read count to the amount of RNA yield per parasite. A stage-specific scaling factor was calculated for each library by dividing the total number of filtered reads by the amount of RNA extracted per 1 flask of parasite-infected culture. In addition, we corrected for differences in parasitemia by multiplying by a parasitemia factor that was calculated as the highest parasitemia in any stage divided by the parasitemia in the stage of interest. In order to compare libraries between stages, we re-standardized the normalized read counts of each library X relative to the normalized read counts of the library with the smallest number of filtered reads. All of these calculations were performed using the following equation:
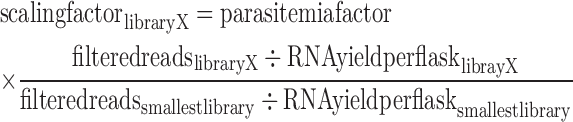 |
The final abundance value of each gene was presented as the normalized exon read count per kilobase gene model divided by the scaling factor of that stage (Supplemental Table S1). A gene with an abundance value <15% of the median at all stages was considered not expressed and was discarded for further analysis.
Cluster and GO enrichment analysis
All genes that showed >2-fold change in the normalized exon read count across the IDC stages were used for cluster analysis. For each gene, abundance values at the six different stages were z-scored, followed by k-means clustering in R (version 3.2.4) with a maximum of 1000 iterations. The number of clusters used in this analysis was guided by the percent of variance captured (within group sum of squares). The optimal number of cluster was determined as the smallest number of clusters that captured at least 75% of the variance. For each cluster, gene ontology enrichment was performed using R package goseq (39). All GO terms with a P-value <0.001 were reported.
PCR validataion for GRO-seq cluster analysis
Four sets of primers were designed to amplify genes that are highly transcribed at the ring stage cluster (membrane associated histidine-rich protein, MAHRP1, PF3D7_1370300), trophozoite stage (trophozoite exported protein 1, TEX1, PF3D7_0603400), schizont stage (rhoptry-associated membrane antigen, RAMA, PF3D7_0707300), and a gene that did pass through filtration threshold (putative pyridine nucleotide transhydrogenase, PNT, PF3D7_1453500). PCR amplification was performed using cDNA library samples from early ring (ER), late ring (LR), late trophozoite (LT) and late schizont (LS) stage alone with a control sample with no DNA template (No Temp). As different amount of parasite was used for nascent RNA isolation at each stage, we first diluted each library sample parasite extracted from two culture flasks (∼209 parasites). All four PCRs were performed using 1 μl of the diluted cDNA library sample with ∼10 pmol of both forward and reverse primers. DNA was incubated for 5 min at 95°C, then 30 s at 98°C, 30 s at 55°C, 30 s at 62°C for 35 cycles. 5 μl of each PCR sample was used for agarose gel electrophoresis. For each primer set, PCR efficiency was tested using genomic DNA under the same amplification conditions as described above. All primer used for PCR validation are listed in the Supplemental Table S1.
Immunofluorescence microscopy
Plasmodium falciparum asexual stage parasites were fixed onto slides using 4% paraformaldehyde for 30 min at RT. Slides were washed three times using 1× PBS. The parasites were permeabilized with 0.1% Triton-X for 30 min at RT, followed by three washes with 1× PBS. Samples were blocked overnight at 4°C in IFA buffer (2% BSA, 0.05% Tween-20, 100 mM glycine, 3 mM EDTA, 150 mM NaCl and 1× PBS). Slides were incubated for 1 h at RT with anti-RNA polymerase II CTD phospho serine 2 antibody (Abcam ab5095; 1:250) followed by an incubation with donkey anti-rabbit dylight 550 antibody (Abcam ab98489; 1:500) for 1 h at RT. Slides were washed with 1× PBS and mounted using Vectashield mounting medium with DAPI. Images were acquired using the Olympus BX40 epifluorescence microscope.
Chromatin immunoprecipitation
Synchronized parasite cultures were collected and subsequently lysed by incubating in 0.15% saponin for 10 min on ice. Parasites were centrifuged at 3234 × g for 10 min at 4°C, and washed three times with PBS. For each wash, parasites were resuspended in cold PBS and centrifuged for 10 min at 3234 × g at 4°C. Subsequently, parasites were crosslinked for 10 min with 1% formaldehyde in PBS at 37°C. Glycine was added to a final concentration of 0.125 M to quench the crosslinking reaction, and incubated for 5 min at 37°C. Parasites were centrifuged for 5 min at 2500 × g at 4°C, washed twice with cold PBS and stored at –80°C.
For chromatin immunoprecipitation, parasites were first incubated on ice in nuclear extraction buffer (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), EDTA-free protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche)). After 30 min, Igepal CA-360 (Sigma-Aldrich) was added to a final concentration of 0.25% and the parasites were lysed by passing the suspension through a 26 G  inch needle seven times. Parasite nuclei were centrifuged at 4°C for 20 min at 2500 × g. Parasite nuclei were resuspended in shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris–HCl pH 7.5, EDTA-free protease inhibitor cocktail and phosphatase inhibitor cocktail). Chromatin was fragmented using the Covaris Ultra Sonicator (S220) for 10 min with the following settings; 5% duty cycle, 140 intensity peak incident power, 200 cycles per burst). To remove insoluble material, samples were centrifuged for 10 min at 17 000 × g at 4°C.
inch needle seven times. Parasite nuclei were centrifuged at 4°C for 20 min at 2500 × g. Parasite nuclei were resuspended in shearing buffer (0.1% SDS, 1 mM EDTA, 10 mM Tris–HCl pH 7.5, EDTA-free protease inhibitor cocktail and phosphatase inhibitor cocktail). Chromatin was fragmented using the Covaris Ultra Sonicator (S220) for 10 min with the following settings; 5% duty cycle, 140 intensity peak incident power, 200 cycles per burst). To remove insoluble material, samples were centrifuged for 10 min at 17 000 × g at 4°C.
Fragmented chromatin was diluted 1:1 in ChIP dilution buffer (30 mM Tris–HCl pH 8, 3 mM EDTA, 0.1% SDS, 300 mM NaCl, 1.8% Triton X-100, EDTA-free protease inhibitor cocktail and phosphatase inhibitor cocktail). Samples were precleared with Protein A Agarose beads to reduce non-specific background and incubated overnight at 4°C with 2 μg of anti-RNA pol II antibody (ab5095, Abcam). A sample with no antibody was also incubated overnight at 4°C to be used as the negative control. Antibody–protein complexes were recovered using Protein A Agarose beads, followed by extensive washes with low salt immune complex wash buffer, high salt immune complex was buffer, LiCl immune complex wash buffer and TE buffer. Chromatin was eluted from the beads by incubating twice with freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3) for 15 min at RT. Samples were reverse crosslinked overnight at 45°C by adding NaCl to a final concentration of 0.5 M. RNase A (Life Technologies) was added to the samples and incubated for 30 min at 37°C followed by a 2 h incubation at 45°C with the addition of EDTA (final concentration 8 mM), Tris–HCl pH 7 (final concentration 33 mM) and proteinase K (final concentration 66 μg/ml; New England Biolabs). DNA was extracted by phenol:chloroform:isoamylalcohol and ethanol precipitation. Extracted DNA was purified using 1.8× Agencourt AMPure XP Beads (Beckman Coulter). Validation PCRs were performed using the primers listed in Supplemental Table S4.
Libraries from the ChIP samples were prepared using the KAPA Library Preparation Kit (KAPA Biosystems). Libraries were amplified for a total of 12 PCR cycles (12 cycles of [15 s at 98°C, 30 s at 55°C, 30 s at 62°C]) using the KAPA HiFi HotStart Ready Mix (KAPA Biosystems). Libraries were sequenced on the Illumina NextSeq500. Read coverage mapping to exonic regions were calculated for both positive and negative libraries, then normalized by dividing these numbers with million number of reads for each library. Finally, the signals obtained from the negative controls were subtracted from the ChIP-Seq library of the same stage.
Histone variants and nucleosome occupancy analysis
Sequence read files of MNase-digested chromatin (input), H2A.Z, H3K4me3 and H3K9ac ChIP-seq data sets (GSE23787) (40) and a nucleosome occupancy data set (SRP026365) (13) were downloaded from NCBI SequenceRead Archive. For H2A.Z, H3K4me3, K3K9ac and input data sets, reads were mapped directly to P. falciparum 3D7 genome v13.0 (www.plasmoDB.org) using Bowtie 2 (34). Non-uniquely mapped reads and PCR duplicates were discarded from final working reads. Coverage depth was first normalized to million mapped reads and was expressed as the ratio between sample and input. The nucleosome occupancy data set was mapped and normalized as described in the original publication (13). The normalized read coverage in the 500 bp upstream of the annotated ATG was calculated and used to generate heatmaps using the command pheatmap in R.
Motif identification
Two motif-discovery programs were used in this study to identify over-represented DNA motifs upstream of gene start site (ATG) of genes within each nascent cluster. A region of 1,000 bp upstream of the coding region was used for this search, based on the reported distribution of transcription start sites (41) (81% of TSS are located within 1000 bp of the coding region). Genes that were located <1000 bp from their 5΄ neighboring gene were removed from the analysis. MEME-ChIP runs two de novo motif identification algorithms, MEME and DREME. The parameters used for MEME algorithm were minw = 7, maxw = 12 in zoops mode with E-value ≤ 1e–01. The parameters used for DREME and CentriMo, a motif enrichment analysis algorithm included in the MEME-ChIP package, were the default parameters:-dreme-e 0.05 -centrimo-score 5.0 -centrimo-ethresh 10.
RESULTS
Generation of nascent RNA profiles for the P. falciparum blood stages
In this study, we explored gene expression in P. falciparum at the initiation level using a modified global run-on sequencing (GRO-seq) methodology (30,31) that specifically captures newly transcribed RNA (nascent RNA) in a genome-wide manner. An overview of the GRO-seq methodology is presented in Figure 1A. We have generated eight genome-wide nascent RNA profiles covering six asexual stages across the IDC, and early (stage II/III) and late (stage IV/V) gametocyte stages. To optimize the protocol, we determined that a minimum of 30 min incubation period was required to obtain sufficient nascent RNA from the in vitro transcription reaction (Supplemental Figure S1A). Several additional quality controls were implemented to further validate the GRO-seq methodology. First, experimental noise was measured by performing the nuclear run-on reaction in the presence of unmodified uridine (Supplemental Figure S1B). This negative control yielded extremely low amounts of RNA (Supplemental Figure S1B) and low genome coverage (<1-fold) after sequencing (Supplemental Figure S1C), confirming minimal DNA and non-nascent RNA contamination using GRO-seq methodology. Second, to confirm that the nuclear run-on reaction generates nascent RNA in a stage-specific manner, we performed our assay on tightly synchronized trophozoite-stage parasites and observed that TEX1 gene (trophozoite exported protein 1; PF3D7_0603400) was highly transcribed, while no signal was obtained for a sporozoite-specific gene, STP (putative serine/threonine protein kinase; PF3D7_0107600) (Supplemental Figure S1B). Finally, we generated two biological replicates for five of the IDC stages, which showed high Spearman correlation coefficients ranging from 0.88 to 0.95 (Supplemental Figure S1D), confirming the reproducibility of the GRO-seq methodology. Together, these results indicate that our experimental approach is efficient, reproducible, and has minimal background noise.
Figure 1.

Global nuclear Run-On coupled to next-generation sequencing (GRO-seq) in the malaria parasite P. falciparum. (A) Schematic overview of the GRO-seq methodology. In brief, parasites were extracted from highly synchronized cultures followed by extraction of the nuclei. Transcription was allowed to take place for 30 min in the presence of 5-ethynyl uridine (EU). EU-labeled RNA was then purified and prepared for Illumina sequencing. (B) Genome browser view of normalized nascent RNA profiles that show the varying levels of transcriptional activity during the blood stages. The majority of genes were most highly transcribed at the trophozoite stage, and a representative gene (PF3D7_0716300) is shown in the top panel. A subset of genes was most highly transcribed at the schizont stage or gametocyte stages. An example of a gene that is highly transcribed at the schizont stage (PF3D7_0905400) is shown in the middle panel, whereas a gene with a profile of high transcription at the gametocyte stage (PF3D7_1327300) is shown in the bottom panel. (C) Immunofluorescence analysis showing RNA polymerase II activity at the asexual IDC stages. A strong signal was detected at the early and late trophozoite stages. At the early schizont stage, higher activity of RNA polymerase II was detected in some nuclei compared to others within a single parasite. No RNA polymerase II signal was observed at the early ring and late schizont stages. (D) A total of 5,221 genes were identified to be expressed during the IDC and were grouped into 9 clusters based on their expression patterns. A selection of enriched GO-terms is listed on the right of each cluster. ER, early ring; LR, late ring; ET, early trophozoite; LT, late trophozoite; ES, early schizont; LS, late schizont; EG, early gametocyte stage; LG, late gametocyte stage.
Upon sequencing of the GRO-seq libraries, we obtained between 670 456 and 11 903 568 mapped and filtered reads per stage after combining biological replicates, corresponding to 1.46–25.88-fold exome-coverage. To be able to directly compare gene expression levels between the various stages, we corrected for differences in the number of parasites used as input for the nuclear run-on reaction. This normalization was achieved by dividing the coverage read depth at each base pair by a stage-specific scaling factor that was based on, among others, the culture volume and parasitemia at the time point of harvest (see Material and Methods and Supplemental Table S1).
A global picture of transcriptional activity in the blood stages
Overall, GRO-seq data revealed that transcriptional activity exhibited a bell curve shape during asexual cycle, from extremely low global transcriptional activity at the early ring stage, via slightly increased transcription at the late ring stage to a strong peak of transcription at the early and late trophozoite stages. Finally, transcriptional activity decreases again as the parasite progressed into the late schizont stage (Figure 1B, Supplemental Figure S2 and Supplemental Figure S3, and Supplemental Table S2). This bell-curved pattern was validated using Immunofluorescence microscopy capturing RNA polymerase II abundance levels at single-cell resolution (Figure 1C). In addition, our GRO-seq data indicates that transcriptional activity was high in early gametocytes and subsequently decreased in late gametocytes (Figure 1B, Supplemental Figure S2 and Supplemental Figure S3, and Supplemental Table S2). A total of 5207 genes (99% of all protein-coding genes) were detected in at least one of the eight stages sampled in this study (Supplemental Table S1), while 77 genes did not reach our threshold. These genes included genes expressed on the RBC surface (var, surfin) and genes expressed in other stages of the parasite life cycle, such as sporozoite invasion-associated protein 1 and liver specific protein 1 putative (LISP1).
Cluster analysis of transcriptional profiles across the P. falciparum asexual cell cycle
We identified a total of 5,187 genes expressed at any time point during the IDC, which were grouped into nine distinct clusters based on their nascent transcriptional profile across the IDC (Figure 1D and Supplemental Table S1). The large majority of genes (n = 4,607; 89%) were most abundantly transcribed at the trophozoite stage, while 532 genes (10%) showed a high level of transcription at the schizont stage and 48 genes (1%) were most highly transcribed at the ring stage. We observed enrichment in Gene Ontology (GO) terms associated with host cell remodeling among the earliest transcribed genes (clusters A1 and A2) including several PHISTb and early transcribed membrane proteins (ETRAMPs) (Figure 1D, Supplemental Table S1, and Supplemental Table S3). Six out of 25 AP2 TFs detected during the IDC were most highly transcribed at these early stages, suggesting that these TF could be involved in driving this first wave of transcriptional activity. Genes that were most highly transcribed at the late trophozoite stage (clusters A3–A7) were associated with biological processes that are known to occur at this stage, such as translation and DNA replication. The earliest of these subsets of genes (cluster A3) included PfAlba1, as well as 16 putative RNA-binding proteins, and showed GO enrichment for ‘nucleic acid binding’ and ‘RNA binding’. Genes involved in pathogenesis associated with GO terms such as ‘cell adhesion molecule binding’ and ‘infected host cell surface knob’ were enriched at the early schizont stage (cluster A8). Finally, a relatively small number of genes (cluster A9) with strong enrichment for involvement in host cell invasion, such as merozoite surface proteins and rhoptry-associated proteins, were most abundantly transcribed at the late schizont stage. To validate these cluster assignments, we determined the relative transcript abundance for several genes detected at different stages of the IDC using semi-quantitative PCR. Ring, trophozoite and schizont-stage genes all showed a good concordance between PCR results and the GRO-seq cluster analysis. In addition, a gene that did not pass our threshold for expression was also not detected by PCR (Supplemental Figure S4). Together, these results indicate that most genes are highly transcribed simultaneously at the trophozoite stage, while only a subset of genes is differentially regulated and transcribed either early in the cell cycle to enable the parasite to establish a hospitable environment inside the erythrocyte, or late in the cell cycle in preparation for merozoite egress and re-invasion.
RNA polymerase II occupancy confirms nascent transcriptional profiles
To further validate our observation of widespread transcriptional activity in trophozoites and targeted transcription of mostly invasion-related genes in schizonts, we determined Pol II occupancy at the early ring, early trophozoite, and late schizont stages using chromatin immunoprecipitation followed by next-generation sequencing (ChIP-seq; Supplemental Figure S5, and Supplemental Table S4). Pol II complexes were purified using an antibody that specifically binds to the C-terminal repeat domain with phosphorylated serines in position 2 (Ser2), a sign that Pol II is in a state of active transcription (42,43). Using semi-quantitative PCR, we verified enrichment of coding regions but not intergenic regions and low experimental noise in our ChIP procedure (Figure 2A).
Figure 2.

Confirmation of GRO-seq results by Pol II ChIP-seq. (A) Validation of Pol II ChIP using semi-quantitative PCR, showing enrichment of the coding region of elongation factor 2 (EF2) at the trophozoite and schizont stages as compared to an intergenic region (INT) and the negative (no antibody) control. (B) High level genome browser view (Pf3D7_14_v3:398 913–442 486) of normalized ChIP-seq data (top panel). The bottom panel shows a comparison between ChIP-seq (turquoise) and GRO-seq (dark red) data sets for a smaller region of chr14 as indicated by a black rectangle in the top panel. A rhoptry-associated membrane antigen gene (PF3D7_1410400, indicated by the blue rectangle) showed high expression at the schizont stage in both ChIP-seq and GRO-seq data sets. (C) Average Pol II ChIP-seq coverage plots of genes from GRO-seq ring-stage (A1), trophozoite-stage (A2), schizont-stage (A9) clusters, and non-expressed genes around gene start (ATG) and gene end. At the schizont stage, late schizont-stage genes (GRO-seq cluster A9) show higher Pol II occupancy in our ChIP-seq data set as compared to genes with other GRO-seq expression profiles, consistent with the GRO-seq results. (D) Comparison of gene expression profiles as observed in GRO-seq, Pol II Chip-seq, and RNA-seq (19) data sets, highlighting the discrepancies between transcriptional activity (measured by GRO-seq and ChIP-seq) and steady-state mRNA abundance. Genes (n = 4888) are ranked in the same order in each heatmap. ER, early ring; LR, late ring; ET, early trophozoite; LT, late trophozoite; ES, early schizont; LS, late schizont.
Pol II occupancy at the early ring stage was extremely low as compared to other stages, in agreement with the lack of nascent RNA signal at this stage of the cell cycle (Figure 2B). Similar to the nascent gene expression profiles, the majority of genes showed the highest Pol II occupancy at the trophozoite stage, while a subset of genes was most highly occupied by Pol II at the schizont stage (Figure 2B). A metagenomic analysis showed that the average Pol II occupancy of genes with late schizont expression profiles in GRO-seq (cluster A9) is higher in schizonts as compared to genes in other clusters (Figure 2C). Finally, the expression patterns of the Pol II ChIP-seq and GRO-seq data sets are more similar than for either GRO-seq or Pol II ChIP-seq and a publicly available steady-state mRNA-seq data set (19) (Figure 2D). Together, these results validate our GRO-seq results and confirm that steady-state mRNA levels do not strictly reflect transcriptional activity, but may be subject to post-transcriptional processes, such as degradation and storage.
Transcriptional activity in gametocytes
In early and late gametocytes, the most highly expressed genes encoded ribosomal proteins and proteins involved in movement and motor activity, while the bottom 20% of the genes were enriched for involvement in pathogenesis, erythrocyte remodeling, and antigenic variation (Supplemental Figure S3 and Supplemental Table S2). To validate our results for the gametocyte stage, we examined 27 genes that have been shown to be essential for gametocytogenesis (6). The majority of these genes (n = 18, 67%) showed the highest transcriptional activity in the gametocyte stages, including the well-established gametocyte-specific makers P. falciparum gamete antigen 27/25 (PF3D7_1302100) and sexual stage-specific protein precursor Pfs16 (PF3D7_0406200) (Supplemental Figure S6). The remaining nine genes were most highly transcribed at one of the asexual stages, suggesting that the encoded proteins may play an essential role at the earlier stages of gametocytogenesis, but are not highly transcribed after early gametocyte differentiation. We also studied the transcriptional profiles for 686 homologs of P. berghei genes that were known to be transcribed at the gametocyte stage and are subject to translational repression by RNA-binding proteins DOZI and CITH (44,45). The majority of these genes were in the top 50% of transcriptional activity at either the early or the late gametocyte stage (n = 537, 78%, P = 0.0001, Chi-square test), while 342 genes (50%, P = 0.0001, Chi-square test) were in the top 25% of transcriptional activity (Supplemental Table S5), confirming that these genes are indeed active in gametocytes.
To further compare transcriptional activity between gametocyte stages and asexual stages, we calculated for each gene the fold change between the average nascent RNA abundance values of gametocyte stages and asexual stages, and subsequently divided genes into five groups based on this ratio (Supplemental Table S1). Clusters B1 and B2 contain genes that show >4-fold (n = 403) or two-fold (n = 1536) higher transcriptional activity in gametocytes than in the asexual stages, respectively. These groups showed enrichment for genes associated with motility (Figure 3 and Supplemental Table S6), such as several genes encoding dynein subunits and actin-related proteins. Interestingly, many of these genes also showed transcriptional activity during the IDC, albeit at a lower level, but are not detected or present at much lower levels in steady-state mRNA (46) (Figure 3), suggesting that these transcripts may be degraded during the IDC when they are not needed. Cluster B3 contains a large group of genes (n = 3107) for which transcriptional activity does not change by more than two-fold in the transition from IDC to gametocytes, indicating that overall, transcriptional programs of asexual parasites and gametocytes are not very different. This is also demonstrated by the relatively high correlation in GRO-seq data between the trophozoite and gametocyte stages (Spearman R 0.74–0.84, Supplemental Figure S1D). Finally, genes that were turned off in gametocytes as compared to the asexual stages were enriched for GO terms associated with pathogenesis and cell invasion, in line with our understanding of parasite biology (clusters B4 and B5; Figure 3).
Figure 3.

Differences in transcriptional profiles between asexual parasites and gametocytes. Genes were grouped based on the ratio between their average GRO-seq expression level in the asexual and in gametocytes. The average expression profiles for GRO-seq and RNA-seq (46) within each group are shown as blue and red lines, respectively. GO enrichment in each of the clusters is shown on the right. E, early; L, late; R, ring; T, trophozoite; S, schizont; G, gametocyte; O, ookinete.
Among the genes that were highly upregulated in gametocytes as compared to the IDC (clusters B1) were two AP2 transcription factors: the ookinete-specific transcription factor AP2-O and an AP2 TF with unknown function, PF3D7_1429200 (Supplemental Table S1). Six out of eight CPW-WPC proteins that are involved in chromatin remodeling showed >4-fold higher levels of transcription in gametocytes than in asexual stages. In addition, several mRNA-binding proteins were upregulated, including PUF1, PUF2, five RAP proteins with RNA-binding domains that are almost exclusively found in Apicomplexans, and putative RNA-binding protein PF3D7_0716000, which may be involved in posttranslational regulation and stabilization of >10% of the transcriptome that is known to occur in the transition from gametocytes to ookinetes (44,45). Finally, a relatively large fraction of genes in cluster B1 were conserved proteins with unknown function (n = 199, 49.4%, P = 0.0001, Fisher's exact test), indicating that we still lack a significant understanding of many of the parasite-specific processes that take place during sexual differentiation. To determine if a common transcription factor motif could be identified in promoters of the genes that were upregulated during gametocytogenesis, we performed a motif search on the 1000 bp upstream of the annotated ATG of genes in clusters C1 and C2 using MEME. When performing the search on the total set of 1939 genes, we identified motifs TGTDC and CATDCA, which both have overlap with previously identified AP2 TF binding motifs (47) (Supplemental Table S7).
Nascent transcriptional activity and epigenetic landscape
In other eukaryotes, histone variants and activating histone post-translational modifications (PTMs) are associated with actively transcribed genes. In addition, nucleosome depletion around the transcription start site (TSS) has been shown to be associated with genes that are highly transcribed (11,13,48–51). To further investigate mechanisms that contribute to regulation of transcriptional initiation, we therefore analyzed previously published H2A.Z, H3K9ac, H3K4me3 ChIP-seq datasets (40) and a nucleosome landscape MNase-seq dataset (13). These data sets have previously been analyzed for correlation with steady-state mRNA abundance, but not with GRO-seq data. Similar to previous findings (40), no significant correlation was observed between transcriptional activity and H2A.z or H3K4me3 data sets (Supplemental Figure S7); however, a side-by-side comparison of GRO-seq and H3K9ac abundance heatmaps (Figure 4A) showed that genes with schizont-stage transcriptional activity tend to have higher H3K9ac marks at the later stage. Nucleosome occupancy in the 500bp upstream of the coding region is relatively high in ring and schizont-stage parasites, while global nucleosome depletion occurs in trophozoites and gametocytes (Figure 4A, Supplemental Figure S7, and Supplemental Table S8), as described previously (11,13). Taken together, high levels of transcriptional activity at the trophozoite and gametocyte stages correlate well with an open chromatin structure. However, the subset of genes transcribed at the schizont stage does not seem to be regulated by the same changes in chromatin organization that occur at the trophozoite stage.
Figure 4.

Association of transcriptional activity with chromatin structure, Pol II pausing, and steady-state mRNA expression. (A) A comparison of transcriptional activity with H3K9ac abundance, and global nucleosome occupancy during the IDC. Genes were ranked according to transcriptional profile during the IDC in the same order as in Figure 1C. (B) Averaged Pol II pausing index for all genes in GRO-seq data at each stage. (C) Comparison of gene expression profiles in GRO-seq and RNA-seq. Nascent RNA and steady-state mRNA data sets were z-scored by gene individually and then clustered based on the combined data. Clusters C4, and C5 show large differences in the moment of peak transcript abundance between the two data sets, suggestive of strong post-transcriptional regulation. Enriched GO terms are indicated on the right.
Nascent transcriptional activity and RNA polymerase II pausing
Recruitment of Pol II and the formation of the pre-initiation complex (PIC) are critical steps in gene activation and subject to strict regulation. However, evidence is emerging that Pol II can also be regulated at the level of early transcription elongation (30,42,52–57). This elongation control is achieved by pausing of Pol II 30–50 nucleotides downstream of the promoter, and subsequently requires additional positive signals before elongation can be continued. Pol II pausing has previously been studied using GRO-seq data (30,55,58) and Pol II ChIP-seq data (mainly on Pol II with Serine 5 phosphorylated CTDs) (58,59). To find evidence for Pol II pausing in P. falciparum, we focused on the GRO-seq read coverage profiles in 5΄ UTR regions. In total, 60–70% of all GRO-seq reads mapped to intergenic regions, including the 5΄ UTRs (Supplemental Figure S8A). Similar to previously published GRO-seq datasets from other eukaryotes, we observed a peak in read coverage around the gene start (Supplemental Figure S8B) that was positively correlated with transcriptional activity (Supplemental Figure S8C). This pattern has been described to be the result of paused Pol II complexes that were activated during the nuclear run-on procedure. Next, we calculated a ratio, hereafter called pausing index, of sequence read density in the 5΄ UTR (defined as 500 bp upstream of ATG) to that in the gene body (500 bp downstream of ATG) for each gene at each developmental stage as described by Core et al. (30). We observed that the average of pausing indexes for all genes is highest at the ring stage, followed by a sharp decrease at the trophozoite stage and a small increase as the parasite enters the schizont stage (Figure 4B). These data suggest that Pol II is starting to be engaged with the genome in the ring stage, but is prevented from continuing transcriptional elongation until activation in the trophozoite stage, resulting in a global transcriptional burst. For genes with stage-specific transcription patterns, the Pol II pausing index was lowest in the stage with the highest transcriptional activity (Supplemental Figure S9A). The negative association between the Pol II pausing index and transcriptional activity are similar to observations in human cells and are believed to be indicative of transcriptional control, where the rate of Pol II engagement to the promoter is higher than the rate of Pol II entering its elongation phase.
In addition to the extended coverage at 5΄ UTR, we also observed a high read coverage at the 3΄ UTR regions, again similar to GRO-seq profiles from other eukaryotes (Supplemental Figure S9B). In yeast (60), Pol II pausing at the 3΄ UTR was found to be associated with splicing events. In P. falciparum, we observed that 3΄ UTR GRO-seq coverage was approximately 1.5-fold higher in multi-exon genes as compared to single exon genes at the late trophozoite stage (P = 1.283e–15, Mann–Whitney U test) (Supplemental Figure S9B). These results suggest that splicing may indeed contribute to Pol II pausing at the 3΄ UTR, but is unlikely to be the only event that triggers pausing of the transcriptional complex at this location.
Comparison between nascent RNA and steady-state mRNA abundance
To measure the degree in which transcriptional and post-transcriptional regulatory mechanisms contribute to global gene expression, we further clustered genes based on both nascent RNA and steady-state mRNA expression profiles, resulting in six distinct clusters (Figure 4C and Supplemental Table S9). For this analysis, we only used the GRO-seq data that exactly matched the stages available in the steady-state mRNA data set (19). Out of the 4881 genes that changed in abundance during the cell cycle in both data sets, 2503 genes (58%) showed nearly identical profiles for both data sets (clusters C2, C3 and C6, respectively). In contrast, cluster C4 contains 1126 genes for which transcription peaked in late trophozoite stage, but that were continuously detected until the late schizont stage in steady-state mRNA, indicating that these transcripts undergo partial stabilization. This cluster showed enrichment for genes involved in protein metabolism (Supplemental Table S9). In addition, cluster C5 (n = 719) and C1 (n = 197) showed an even larger discrepancy between the moment of transcription and the time point of highest abundance in steady-state mRNA, suggesting that these transcripts are the subjects of strong post-transcriptional regulation. The small group of genes that was previously identified as ring-stage specific (cluster C1) enriched for involvement in erythrocyte remodeling also showed transcriptional activity at the trophozoite stage, suggesting that these transcripts might be transcribed later in the cell cycle and stored long-term until the next round of erythrocyte invasion.
For the clusters with genes that do not seem to be subject to post-transcriptional regulation and are most likely controlled at the level of transcription (clusters C2, C3 and C6), we mined the region upstream of the gene start for transcription factor binding motifs (Supplemental Table S7). Motifs that almost exclusively contained A's or T's were identified for all clusters. Such stretches of AT could be reminiscent of the TATA box in higher eukaryotes, but were not further considered due to the high AT content of the P. falciparum genome. In clusters C2 and C3, we identified the short motifs STTC and SYTC, respectively. In addition, we observed enrichment for motifs GTG, GWG, and RTGT in clusters C3, C4 and C5, respectively. The reverse complements of these short motifs (CACACA and ACACAC) have previously been shown to be associated with DNA replication (61) and possibly interact with AP2 TFs PF3D7_0802100 and PF3D7_1456000 (47). In addition, genes in cluster C6 showed enrichment for motif GTGHA, which has previously been described as the binding sequence of AP2 TF PF3D7_1007700 and is associated with invasion genes (47,61–63).
For genes in clusters showing evidence of post-transcriptional regulation (clusters C1, C4 and C5), we searched the regions upstream of the gene start and downstream of the gene stop for transcription factor or RNA-binding protein motifs. GTG and ACAC motifs were identified in the 500 bp downstream of the gene stop in cluster C5, similar to motifs identified in the 5΄UTRs of clusters C3, C4 and C5.These results suggests that the GTG/CAC motif is very common and may be similar to frequently observed TATA stretches. Overall, the lack of specific motifs identified in our analyses emphasizes the need for developing novel experimental designs to discover mechanisms regulating transcripts at the transcriptional and post-transcriptional level.
DISCUSSION
Over the past few years, various studies have analyzed steady-state mRNA abundance throughout the blood stages of malaria parasites (18–21,46). However, steady-state mRNA is the product of transcription, stability, and degradation, and may therefore not accurately reflect transcriptional activity. Here, we present the first genome-wide study that specifically measures the timing and level of transcription during the P. falciparum asexual and sexual blood stages by performing global run-on sequencing. This data set is an invaluable asset towards a better understanding of gene regulation at the transcriptional and post-transcriptional levels during the life cycle of P. falciparum.
The results of our study suggest that transcriptional activity at the ring stage is limited to a small subset of genes encoding erythrocyte-remodeling proteins. However, at this stage of the life cycle, Pol II is already engaged with nearly every promoter in the genome, waiting for an activation signal that initiates a massive burst of transcription at the trophozoite stage. In line with this model, two general transcription factors, PfTBP and PfTFIIE, were previously shown to interact with both active and inactive promoters at the ring stage (64). Once transcriptional elongation commences at the trophozoite stage, a large proportion of the genome is transcribed in agreement with a study that used the nuclear run-on methodology on individual P. falciparum genes (31). This massive transcription event seems to be somewhat ‘leaky’, resulting in low-level transcription of genes that are specific for other life cycle stages of the parasite. Since these non-IDC genes are typically not detected in steady-state mRNA, we conclude that their transcripts may be quickly degraded. At the schizont stage, transcription is turned down, except for a subset of invasion-related genes that show upregulation of transcriptional activity, in agreement with a previous ChIP-on-ChIP analysis of Pol II (17). Finally, as the parasite differentiates into a gametocyte, the transcriptional program remains largely unchanged as compared to the trophozoite stage with some exceptions, including invasion genes that are turned off and motility-related genes that are turned on.
The only genes for which we have been able to confirm an AP2 TF motif are the invasion genes, and together with ring-stage specific genes and virulence genes, these are the only genes that seem to be differentially regulated during the IDC. For the invasion genes, mechanisms that control gene expression include the binding of a specific transcript factor to the promoter region and the attachment of a bromodomain protein, PfBDP1, to acetylated histone H3 (65). Virulence genes, in particular var genes are controlled by a combination of repressive histone modifications, long non-coding RNAs and localization away from the rest of the genome in perinuclear heterochromatin. In contrast, the massive transcriptional events at both the trophozoite and gametocyte stages are associated with a genome-wide depletion of nucleosomes (12,13). In addition, at the trophozoite stage, the promoters of the majority of genes are marked by activating histone PTM H3K9ac. Taken together, these data suggest that a large part of the genome is not regulated by classical eukaryotic mechanisms of transcription initiation that involve local chromatin changes and the presence of specific transcription factors that drive expression of a subset of genes. Instead, the majority of promoters are occupied by paused Pol II, activation of which coincides with genome-wide changes in chromatin structure, including nucleosome depletion (12,13), increased chromosomal intermingling (10), nuclear expansion and an increase in the number of nuclear pores (66). In this model of all-at-once transcription, there is no need for a large array of specific transcription factors and corresponding motifs, which may explain why a larger set of specific transcription factors has remained elusive in Plasmodium spp. to date. This lack of a need for finely tuned transcriptional activity may also explain how the parasite can quickly divide and form 16 – 32 daughter cells in a relatively short time frame (<12 h).
The global Pol II pausing that takes place at the ring stage prior to massive transcriptional activity at the trophozoite stage may function as checkpoint before transcription elongation. In metazoans, the release of paused Pol II is mediated by phosphorylation of various proteins, including DRB sensitivity-inducing factor (DSIF, consisting of subunits SPT4 and SPT5), negative elongation factor (NELF), and the carboxyl terminal domain of the large subunit of Pol II at Ser2, by positive transcription elongation factor-b (P-TEFb) complex (67–69). Inhibition of mammalian P-TEFb results a nearly complete block of transcription, suggesting that most active genes experience pausing events that require P-TEFb for elongation activation (55,56,59). These results indicate that P-TEFb is a key regulator for transcription. In Plasmodium, many of the critical regulators, such as subunits of P-TEFb, DSIF subunits, and NELF, involved in Pol II pausing have been identified. The major P-TEFb subunits are cyclin-dependent kinase 9 (CDK9) and cyclin proteins (T1, T2 and K). Four cyclin genes have been described in P. falciparum (PfCYC1-4) (70,71), of which only PfCYC4 shows homology to human cyclin T1, T2 and K. In addition, CDK9 is homologous to several parasite kinases, of which CDC2-related protein kinase 1 (PfCRK1) and protein kinase 5 (PfPK5) show the strongest similarity (Blastp E-value <10−66). Studies in higher eukaryotes suggest that the nucleosome landscape, such as the positioning of the +1 nucleosome, could play a regulatory role in pausing by providing an energy barrier for elongating Pol II (72,73). The most strongly positioned nucleosomes in P. falciparum are at the start of the coding regions and could act as a barrier for RNA Pol II pausing. Furthermore, Pol II pausing and releasing have also been linked to nascent RNA hairpin structure, RNAs transcribed from enhancers (74,75), promoter elements, and template DNA motifs, such as the downstream promoter element (DPE), TATA box and GAGA motif (74,76–82). Unfortunately, due to low sequence homology and AT-richness of the P. falciparum genome, many regulatory mechanisms involved in RNA pol II regulation and pausing, such as enhancers, mediators, chromatin modifiers, and promoter elements, remain undefined. In addition, compared to mammalian polymerases that contain a C-terminal domain (CTD) with 52 identical heptad repeats, the Plasmodium CTD tail of Pol II displays wide variation in terms of length and composition (83,84). For example, primate malaria parasites, such as P. knowlesi, P. vivax, P. falciparum, and P. cynomolgi have an increased number of heptads with a high level of variability as compared to malaria parasites infect other species (85). Additional work will be needed to truly understand how Pol II pausing is established and controlled in Plasmodium parasites. However, the present work established that while more classical regulatory mechanisms of transcription only control subsets of genes, such as invasion genes or var genes, the activation of paused Pol II complexes appears to be an essential genome-wide event during the IDC in P. falciparum. The identification of compounds that can specifically inhibit the activity of P-TEFb in the parasite will be a potentially powerful approach toward novel highly effective antimalarial drugs.
The large transcriptional activity that we observed here at the trophozoite and early schizont stages is in partial disagreement with the cascade of transcript abundance that has been observed in steady-state mRNA data sets (18–21,46), but can be explained by a role for post-transcriptional gene regulation. In mature gametocytes, translational latency has been well documented and entails the temporary storage of hundreds of transcripts in ribonucleoprotein complexes of female gametocytes until translation once the parasite has developed into an ookinete inside the mosquito. Evidence is emerging that similar mechanisms control subsets of genes during the IDC. For example, Vembar et al. showed the targeted capture and stabilization of transcripts encoding invasion-related proteins by PfAlba1, followed by release and translation of these proteins at a later time point during the cell cycle (86). Interestingly, our study shows that invasion-related genes are also regulated at the level of transcription initiation, suggesting that a single group of genes can be subject to regulation at multiple levels. We recently reported that P. falciparum encodes a relatively large number of RNA-binding proteins, and that many of the known translational regulators that act during other stages of the life cycle, such as DOZI and CITH, are also associated with mRNA during the IDC (87). These results are indicative of widespread control of gene expression at the post-transcriptional level. In addition, post-transcriptional gene regulation could also explain how the similar transcriptional profiles in trophozoites and gametocytes can give rise to widely different cell types.
This study reveals for the first time the transcriptional activity of genes during the intraerythrocytic developmental cycle and gametocyte differentiation. Our main findings are that (1) most genes are actively transcribed at the trophozoite stage, (2) the transcriptional profile of gametocytes is surprisingly similar to trophozoites with the exception of downregulation of invasion genes and upregulation of genes related to motor activity, and (3) Pol II pausing acts as major control mechanism during the IDC, halting transcriptional elongation in the ring stage and once lifted, giving rise to the transcriptional burst at the trophozoite stage. Together, these results provide a much-needed increase in our understanding of P. falciparum biology and suggest that proteins involved in transcriptional elongation may be highly effective targets for anti-malarial therapy.
ACCESSION NUMBERS
The Gene Expression Omnibus accession number for GRO-seq and ChIP-seq datasets reported in this study is GSE85478.
Supplementary Material
ACKNOWLEDGEMENTS
The following reagents were obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH: Plasmodium falciparum strains 3D7 (MRA-102) deposited by D.J. Carucci and NF54 (MRA-1000) deposited by Megan Dowler, Walter Reed Army Institute of Research.
Footnotes
Present address: Michael Lee, Department of Diabetes, Complications, and Metabolism, Irell and Manella Graduate School of the City of Hope, Duarte, CA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01 AI85077-01A1, R01 AI06775-01 to K.G.L.R.]; National Science Foundation [IIS-1302134 to K.G.L.R.]; University of California, Riverside [NIFA-Hatch-225935 to K.G.L.R.]; University of Texas Health Science Center at San Antonio (to E.M.B.). Funding for open access charge: NIH [R01 AI06775-01].
Conflict of interest statement. None declared.
REFERENCES
- 1. WHO World Malaria Report. 2015; 2015, http://www.who.int/malaria/publications/world-malaria-report-2015/report/en/. [Google Scholar]
- 2. Wright G.J., Rayner J.C.. Plasmodium falciparum erythrocyte invasion: combining function with immune evasion. PLoS Pathog. 2014; 10:e1003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baum J., Papenfuss A.T., Mair G.R., Janse C.J., Vlachou D., Waters A.P., Cowman A.F., Crabb B.S., de Koning-Ward T.F.. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. 2009; 37:3788–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coulson R.M., Hall N., Ouzounis C.A.. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res. 2004; 14:1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Painter H.J., Campbell T.L., Llinas M.. The Apicomplexan AP2 family: integral factors regulating Plasmodium development. Mol. Biochem. Parasitol. 2011; 176:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Josling G.A., Llinas M.. Sexual development in Plasmodium parasites: knowing when it's time to commit. Nat. Rev. Microbiol. 2015; 13:573–587. [DOI] [PubMed] [Google Scholar]
- 7. De Silva E.K., Gehrke A.R., Olszewski K., Leon I., Chahal J.S., Bulyk M.L., Llinas M.. Specific DNA-binding by apicomplexan AP2 transcription factors. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8393–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sinha A., Hughes K.R., Modrzynska K.K., Otto T.D., Pfander C., Dickens N.J., Religa A.A., Bushell E., Graham A.L., Cameron R. et al. . A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 2014; 507:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ay F., Bunnik E.M., Varoquaux N., Vert J.P., Noble W.S., Le Roch K.G.. Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum: Gene regulation via histone modifications, nucleosome positioning and nuclear architecture in P. falciparum. Bioessays. 2015; 37:182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ay F., Bunnik E.M., Varoquaux N., Bol S.M., Prudhomme J., Vert J.P., Noble W.S., Le Roch K.G.. Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 2014; 24:974–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ponts N., Harris E.Y., Lonardi S., Le Roch K.G.. Nucleosome occupancy at transcription start sites in the human malaria parasite: a hard-wired evolution of virulence. Infect. Genet. Evol. 2011; 11:716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ponts N., Harris E.Y., Prudhomme J., Wick I., Eckhardt-Ludka C., Hicks G.R., Hardiman G., Lonardi S., Le Roch K.G.. Nucleosome landscape and control of transcription in the human malaria parasite. Genome Res. 2010; 20:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bunnik E.M., Polishko A., Prudhomme J., Ponts N., Gill S.S., Lonardi S., Le Roch K.G.. DNA-encoded nucleosome occupancy is associated with transcription levels in the human malaria parasite Plasmodium falciparum. BMC Genomics. 2014; 15:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le Roch K.G., Johnson J.R., Florens L., Zhou Y., Santrosyan A., Grainger M., Yan S.F., Williamson K.C., Holder A.A., Carucci D.J. et al. . Global analysis of transcript and protein levels across the Plasmodium falciparum life cycle. Genome Res. 2004; 14:2308–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oehring S.C., Woodcroft B.J., Moes S., Wetzel J., Dietz O., Pulfer A., Dekiwadia C., Maeser P., Flueck C., Witmer K. et al. . Organellar proteomics reveals hundreds of novel nuclear proteins in the malaria parasite Plasmodium falciparum. Genome Biol. 2012; 13:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saraf A., Cervantes S., Bunnik E.M., Ponts N., Sardiu M.E., Chung D.D., Prudhomme J., Varberg J.M., Wen Z., Washburn M.P. et al. . Dynamic and combinatorial landscape of histone modifications during the intraerythrocytic developmental cycle of the malaria parasite. J. Proteome Res. 2016; 15:2787–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rai R., Zhu L., Chen H., Gupta A.P., Sze S.K., Zheng J., Ruedl C., Bozdech Z., Featherstone M.. Genome-wide analysis in Plasmodium falciparum reveals early and late phases of RNA polymerase II occupancy during the infectious cycle. BMC Genomics. 2014; 15:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bozdech Z., Llinas M., Pulliam B.L., Wong E.D., Zhu J., DeRisi J.L.. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003; 1:E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bunnik E.M., Chung D.W., Hamilton M., Ponts N., Saraf A., Prudhomme J., Florens L., Le Roch K.G.. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 2013; 14:R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Le Roch K.G., Zhou Y., Blair P.L., Grainger M., Moch J.K., Haynes J.D., De La Vega P., Holder A.A., Batalov S., Carucci D.J. et al. . Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003; 301:1503–1508. [DOI] [PubMed] [Google Scholar]
- 21. Otto T.D., Wilinski D., Assefa S., Keane T.M., Sarry L.R., Bohme U., Lemieux J., Barrell B., Pain A., Berriman M. et al. . New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Mol. Microbiol. 2010; 76:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sorber K., Dimon M.T., DeRisi J.L.. RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 2011; 39:3820–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foth B.J., Zhang N., Chaal B.K., Sze S.K., Preiser P.R., Bozdech Z.. Quantitative time-course profiling of parasite and host cell proteins in the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteomics. 2011; 10, doi:10.1074/mcp.M110.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Caro F., Ahyong V., Betegon M., DeRisi J.L.. Genome-wide regulatory dynamics of translation in the Plasmodium falciparum asexual blood stages. Elife. 2014; 3, doi:10.7554/eLife.04106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balaji S., Babu M.M., Iyer L.M., Aravind L.. Discovery of the principal specific transcription factors of Apicomplexa and their implication for the evolution of the AP2-integrase DNA binding domains. Nucleic Acids Res. 2005; 33:3994–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bischoff E., Vaquero C.. In silico and biological survey of transcription-associated proteins implicated in the transcriptional machinery during the erythrocytic development of Plasmodium falciparum. BMC Genomics. 2010; 11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trager W., Jensen J.B.. Human malaria parasites in continuous culture. Science. 1976; 193:673–675. [DOI] [PubMed] [Google Scholar]
- 28. Ifediba T., Vanderberg J.P.. Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature. 1981; 294:364–366. [DOI] [PubMed] [Google Scholar]
- 29. Core L.J., Lis J.T.. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008; 319:1791–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sims J.S., Militello K.T., Sims P.A., Patel V.P., Kasper J.M., Wirth D.F.. Patterns of gene-specific and total transcriptional activity during the Plasmodium falciparum intraerythrocytic developmental cycle. Eukaryot. Cell. 2009; 8:327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buffalo V. 2011; pp. Scythe - A very simple adapter trimmer. [Google Scholar]
- 33. Joshi NA F.J. 2011; pp. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files. [Google Scholar]
- 34. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Risso D., Schwartz K., Sherlock G., Dudoit S.. GC-content normalization for RNA-Seq data. BMC Bioinformatics. 2011; 12:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Young M.D., Wakefield M.J., Smyth G.K., Oshlack A.. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010; 11:R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bartfai R., Hoeijmakers W.A., Salcedo-Amaya A.M., Smits A.H., Janssen-Megens E., Kaan A., Treeck M., Gilberger T.W., Francoijs K.J., Stunnenberg H.G.. H2A.Z demarcates intergenic regions of the plasmodium falciparum epigenome that are dynamically marked by H3K9ac and H3K4me3. PLoS Pathog. 2010; 6:e1001223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gissot M., Refour P., Briquet S., Boschet C., Coupe S., Mazier D., Vaquero C.. Transcriptome of 3D7 and its gametocyte-less derivative F12 Plasmodium falciparum clones during erythrocytic development using a gene-specific microarray assigned to gene regulation, cell cycle and transcription factors. Gene. 2004; 341:267–277. [DOI] [PubMed] [Google Scholar]
- 42. Jonkers I., Lis J.T.. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell. Biol. 2015; 16:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bowman E.A., Kelly W.G.. RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: a tail of two kinases. Nucleus. 2014; 5:224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guerreiro A., Deligianni E., Santos J.M., Silva P.A., Louis C., Pain A., Janse C.J., Franke-Fayard B., Carret C.K., Siden-Kiamos I. et al. . Genome-wide RIP-Chip analysis of translational repressor-bound mRNAs in the Plasmodium gametocyte. Genome Biol. 2014; 15:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mair G.R., Braks J.A., Garver L.S., Wiegant J.C., Hall N., Dirks R.W., Khan S.M., Dimopoulos G., Janse C.J., Waters A.P.. Regulation of sexual development of Plasmodium by translational repression. Science. 2006; 313:667–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lopez-Barragan M.J., Lemieux J., Quinones M., Williamson K.C., Molina-Cruz A., Cui K., Barillas-Mury C., Zhao K., Su X.Z.. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011; 12:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Campbell T.L., De Silva E.K., Olszewski K.L., Elemento O., Llinas M.. Identification and genome-wide prediction of DNA binding specificities for the ApiAP2 family of regulators from the malaria parasite. PLoS Pathog. 2010; 6:e1001165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mellor J. Dynamic nucleosomes and gene transcription. Trends Genet. 2006; 22:320–329. [DOI] [PubMed] [Google Scholar]
- 49. Nocetti N., Whitehouse I.. Nucleosome repositioning underlies dynamic gene expression. Genes Dev. 2016; 30:660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Voss T.C., Hager G.L.. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014; 15:69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kensche P.R., Hoeijmakers W.A., Toenhake C.G., Bras M., Chappell L., Berriman M., Bartfai R.. The nucleosome landscape of Plasmodium falciparum reveals chromatin architecture and dynamics of regulatory sequences. Nucleic Acids Res. 2016; 44:2110–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Adelman K., Lis J.T.. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat. Rev. Genet. 2012; 13:720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gaertner B., Zeitlinger J.. RNA polymerase II pausing during development. Development. 2014; 141:1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kwak H., Fuda N.J., Core L.J., Lis J.T.. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013; 339:950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jonkers I., Kwak H., Lis J.T.. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. eLife. 2014; 3:e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Henriques T., Gilchrist D.A., Nechaev S., Bern M., Muse G.W., Burkholder A., Fargo D.C., Adelman K.. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol. Cell. 2013; 52:517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Min I.M., Waterfall J.J., Core L.J., Munroe R.J., Schimenti J., Lis J.T.. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev. 2011; 25:742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Williams L.H., Fromm G., Gokey N.G., Henriques T., Muse G.W., Burkholder A., Fargo D.C., Hu G., Adelman K.. Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol. Cell. 2015; 58:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rahl P.B., Lin C.Y., Seila A.C., Flynn R.A., McCuine S., Burge C.B., Sharp P.A., Young R.A.. c-Myc regulates transcriptional pause release. Cell. 2010; 141:432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carrillo Oesterreich F., Preibisch S., Neugebauer K.M.. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol. Cell. 2010; 40:571–581. [DOI] [PubMed] [Google Scholar]
- 61. Young J.A., Johnson J.R., Benner C., Yan S.F., Chen K., Le Roch K.G., Zhou Y., Winzeler E.A.. In silico discovery of transcription regulatory elements in Plasmodium falciparum. BMC Genomics. 2008; 9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Iengar P., Joshi N.V.. Identification of putative regulatory motifs in the upstream regions of co-expressed functional groups of genes in Plasmodium falciparum. BMC Genomics. 2009; 10:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Essien K., Stoeckert C.J. Jr. Conservation and divergence of known apicomplexan transcriptional regulons. BMC Genomics. 2010; 11:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gopalakrishnan A.M., Nyindodo L.A., Ross Fergus M., Lopez-Estrano C.. Plasmodium falciparum: preinitiation complex occupancy of active and inactive promoters during erythrocytic stage. Exp. Parasitol. 2009; 121:46–54. [DOI] [PubMed] [Google Scholar]
- 65. Josling G.A., Petter M., Oehring S.C., Gupta A.P., Dietz O., Wilson D.W., Schubert T., Langst G., Gilson P.R., Crabb B.S. et al. . A Plasmodium falciparum bromodomain protein regulates invasion gene expression. Cell Host Microbe. 2015; 17:741–751. [DOI] [PubMed] [Google Scholar]
- 66. Weiner A., Dahan-Pasternak N., Shimoni E., Shinder V., von Huth P., Elbaum M., Dzikowski R.. 3D nuclear architecture reveals coupled cell cycle dynamics of chromatin and nuclear pores in the malaria parasite Plasmodium falciparum. Cell Microbiol. 2011; 13:967–977.21501361 [Google Scholar]
- 67. Peterlin B.M., Price D.H.. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell. 2006; 23:297–305. [DOI] [PubMed] [Google Scholar]
- 68. Zhou Q., Li T., Price D.H.. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012; 81:119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lis J.T., Mason P., Peng J., Price D.H., Werner J.. P-TEFb kinase recruitment and function at heat shock loci. Genes Dev. 2000; 14:792–803. [PMC free article] [PubMed] [Google Scholar]
- 70. Le Roch K., Sestier C., Dorin D., Waters N., Kappes B., Chakrabarti D., Meijer L., Doerig C.. Activation of a Plasmodium falciparum cdc2-related kinase by heterologous p25 and cyclin H. Functional characterization of a P. falciparum cyclin homologue. J. Biol. Chem. 2000; 275:8952–8958. [DOI] [PubMed] [Google Scholar]
- 71. Merckx A., Le Roch K., Nivez M.P., Dorin D., Alano P., Gutierrez G.J., Nebreda A.R., Goldring D., Whittle C., Patterson S. et al. . Identification and initial characterization of three novel cyclin-related proteins of the human malaria parasite Plasmodium falciparum. J. Biol. Chem. 2003; 278:39839–39850. [DOI] [PubMed] [Google Scholar]
- 72. Li J., Gilmour D.S.. Distinct mechanisms of transcriptional pausing orchestrated by GAGA factor and M1BP, a novel transcription factor. EMBO J. 2013; 32:1829–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Weber C.M., Ramachandran S., Henikoff S.. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell. 2014; 53:819–830. [DOI] [PubMed] [Google Scholar]
- 74. Liu X., Kraus W.L., Bai X.. Ready, pause, go: regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem. Sci. 2015; 40:516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ghavi-Helm Y., Klein F.A., Pakozdi T., Ciglar L., Noordermeer D., Huber W., Furlong E.E.. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014; 512:96–100. [DOI] [PubMed] [Google Scholar]
- 76. Artsimovitch I., Landick R.. Pausing by bacterial RNA polymerase is mediated by mechanistically distinct classes of signals. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Toulme F., Mosrin-Huaman C., Artsimovitch I., Rahmouni A.R.. Transcriptional pausing in vivo: a nascent RNA hairpin restricts lateral movements of RNA polymerase in both forward and reverse directions. J. Mol. Biol. 2005; 351:39–51. [DOI] [PubMed] [Google Scholar]
- 78. Gaertner B., Johnston J., Chen K., Wallaschek N., Paulson A., Garruss A.S., Gaudenz K., De Kumar B., Krumlauf R., Zeitlinger J.. Poised RNA polymerase II changes over developmental time and prepares genes for future expression. Cell Rep. 2012; 2:1670–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Amir-Zilberstein L., Ainbinder E., Toube L., Yamaguchi Y., Handa H., Dikstein R.. Differential regulation of NF-kappaB by elongation factors is determined by core promoter type. Mol. Cell. Biol. 2007; 27:5246–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hendrix D.A., Hong J.W., Zeitlinger J., Rokhsar D.S., Levine M.S.. Promoter elements associated with RNA Pol II stalling in the Drosophila embryo. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:7762–7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nechaev S., Fargo D.C., dos Santos G., Liu L., Gao Y., Adelman K.. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science. 2010; 327:335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee C., Li X., Hechmer A., Eisen M., Biggin M.D., Venters B.J., Jiang C., Li J., Pugh B.F., Gilmour D.S.. NELF and GAGA factor are linked to promoter-proximal pausing at many genes in Drosophila. Mol. Cell. Biol. 2008; 28:3290–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kishore S.P., Perkins S.L., Templeton T.J., Deitsch K.W.. An unusual recent expansion of the C-terminal domain of RNA polymerase II in primate malaria parasites features a motif otherwise found only in mammalian polymerases. J. Mol. Evol. 2009; 68:706–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ukaegbu U.E., Deitsch K.W.. The emerging role for RNA polymerase II in regulating virulence gene expression in malaria parasites. PLoS Pathog. 2015; 11:e1004926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yamaguchi Y., Inukai N., Narita T., Wada T., Handa H.. Evidence that negative elongation factor represses transcription elongation through binding to a DRB sensitivity-inducing factor/RNA polymerase II complex and RNA. Mol. Cell. Biol. 2002; 22:2918–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vembar S.S., Macpherson C.R., Sismeiro O., Coppee J.Y., Scherf A.. The PfAlba1 RNA-binding protein is an important regulator of translational timing in Plasmodium falciparum blood stages. Genome Biol. 2015; 16:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bunnik E.M., Batugedara G., Saraf A., Prudhomme J., Florens L., Le Roch K.G.. The mRNA-bound proteome of the human malaria parasite Plasmodium falciparum. Genome Biol. 2016; 17:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.