


Abstract
One of the fastest cellular responses to genotoxic stress is the formation of poly(ADP-ribose) polymers (PAR) by poly(ADP-ribose)polymerase 1 (PARP1, or ARTD1). PARP1 and its enzymatic product PAR regulate diverse biological processes, such as DNA repair, chromatin remodeling, transcription and cell death. However, the inter-dependent function of the PARP1 protein and its enzymatic activity clouds the mechanism underlying the biological response. We generated a PARP1 knock-in mouse model carrying a point mutation in the catalytic domain of PARP1 (D993A), which impairs the kinetics of the PARP1 activity and the PAR chain complexity in vitro and in vivo, designated as hypo-PARylation. PARP1D993A/D993A mice and cells are viable and show no obvious abnormalities. Despite a mild defect in base excision repair (BER), this hypo-PARylation compromises the DNA damage response during DNA replication, leading to cell death or senescence. Strikingly, PARP1D993A/D993A mice are hypersensitive to alkylation in vivo, phenocopying the phenotype of PARP1 knockout mice. Our study thus unravels a novel regulatory mechanism, which could not be revealed by classical loss-of-function studies, on how PAR homeostasis, but not the PARP1 protein, protects cells and organisms from acute DNA damage.
INTRODUCTION
Poly(ADP-ribosyl)ation (PARylation) is a post-translational modification that is mainly carried out by poly(ADP-ribose) polymerase 1 (PARP1, also known as ARTD1). Upon genotoxic stress, PARP1 is activated to catalyse, using NAD+, the formation of poly(ADP-ribose) polymers (PAR) covalently or non-covalently onto many proteins, but mainly onto itself (1–3). PARP1-activating DNA lesions include primarily single strand breaks (SSBs) and stalled replication forks, as well as DNA double strand breaks (DSBs) (1,4,5). As binding to DNA breaks rapidly activates PARP1, PARylation is regarded as an early DNA damage response (DDR). PARylation or PAR can target a variety of proteins that bind PAR via different PAR-binding motifs, which are thought to provide a spatiotemporal interaction scaffold (4,6,7). PAR is short-lived, because it is rapidly degraded by poly(ADP-ribose) glycohydrolase (PARG) (2,8).
PARP1 activation leads to auto-PARylation of PARP1, which can aid in the recruitment of the scaffold protein XRCC1 (and others) to conduct the base excision repair (BER). Thus, PARP1 and PARylation are involved in BER (see (9) for review). In addition, replication stress or replication fork stalling can activate PARP1 (6,10–12). In response to S-phase poisons, PARP1 or PARylation interact with ATR to induce the S-phase checkpoint (12,13). We recently showed that PAR binding to Chk1 is required for a full activation of the intra-S checkpoint, independent of ATR (6). Moreover, PARP1 and PAR can slow down the progression of replication forks, thus reducing the generation of DSBs during S-phase (11,14). As a consequence of the manifold defects in handling DNA damage, PARP1-deficient cells and mice are hypersensitive to alkylating agents and exhibit increased genomic instability and deleterious defects (15–19). We previously showed similar defects in the genotoxic stress response in mice and cells lacking the nuclear isoform of PARG, which exhibit a defective PAR turnover and, hence, hypo-PARylation (20–22).
The biological function of the PARylation system has been investigated extensively using chemical inhibitors and, importantly, animal models in which PAR forming and degrading enzymes are deleted. These studies have established that PARP1 and PARylation regulate chromatin remodeling, transcription, replication, genome maintenance and cell death (as reviewed in (1–3,9,23)). Nonetheless, the biology of PARylation is still enigmatic, because the findings and interpretations are often inconsistent. For example, studies using Drosophila demonstrated a PARP-mediated ‘loosening’ of chromatin at the ‘puff’ loci (24), while nucleosome-PARylation-independent condensation of chromatin was reported (25). Secondly, it is still under debate whether PARP1 as a co-activator of NF-κB requires its enzymatic activity (26–29). Thirdly, PARP1 can promote (10,30–32) or inhibit homologous recombination (HR) (15,17,33–35,36–38).
The causes for these discrepancies might well be influenced by the spatiotemporal level of PAR formation during various experimental and physiological conditions. Also, the impact of PAR and/or PARP1 seems to be dependent on the type and the intensity of the inflicted damage type or signaling. More importantly, the PAR readers consist of numerous proteins, which fulfill a plethora of functions (7). Unfortunately, reports on the regulation of the PARP1 enzymatic activity in vivo to date have been very sparse (reviewed in (2,39,40)). One technical limitation hinders the employed genetic and pharmaceutical approaches: While the knockout (KO) or knock-down of PARP1 eliminates the PARP1 protein together with more than 90% of the generated PAR (41), PARP inhibitors target unspecifically other PARP family members, which harbor diversified biochemical and biological functions (42). Another problem is that other PARP family members (e.g. PARP2) may compensate for some of the functions of PARP1 if the entire protein is eliminated. Thus, the previous approaches could not distinguish between the impact of PARP1 and that of its enzymatic product, i.e. PAR. As a consequence, the inter-dependent nature of the PARP1 protein and the generated PAR (43), as well as the biological significance of the dynamics and the homeostasis of PARylation thus remains elusive, partially due to the lack of appropriate experimental models.
In the present study, we sought to clarify the specific functions of PARP1’s enzymatic activity in vivo by generating a separation-of-function mutant PARP1 knock-in (Ki) mouse model mutating Asp (D) 993 to Ala (A) of the PARP1 protein. The D933A mutation compromises the kinetics of the PARylation activity and the complexity of the PAR chains. This mutation is compatible with the development and tissue homeostasis of mice and the viability of cells under unperturbed conditions. However, homozygous PARP1D993A/D993A cells and mice are hypersensitive to alkylation or oxidative stress - most likely due to defects in BER and DDR defects in S-phase, which enhance cell death and cellular senescence. This PARP1 Ki model classifies PARP1 functions by its requirement for an acute synthesis of PAR polymers and differentiates the functions of the PARP1 activity in acute DDR and physiological development.
MATERIALS AND METHODS
Generation of PARP1D993A/D993A mice
The gene-targeting vector containing the point mutation in exon 23 (Supplementary Figure S1A) was electroporated into E14.1 embryonic stem (ES) cells. Southern blot analysis of selected ES clones confirmed targeted (Tg) and knock-in (Ki) allele mutation in the Adprt locus before and after transfection with Cre-recombinase, respectively. For identification of the Tg allele, SB was performed with genomic DNA from ES cells digested with XbaI and BspH1 using the probe 6.4 (Supplementary Figure S1A) for hybridization, which yields a fragment of 8.5 kb for the wild type (WT) allele, and 6.6 kB for the Tg allele (Supplementary Figure S1B). To verify the Ki allele, genomic DNA was digested with XbaI and BspH1 and subjected to SB analysis using the probe 7.6 (Supplementary Figure S1A) which generates a fragment of 8.5 kb for the WT allele, and 2.9 kB for the Tg allele and 1.9 kB for the Ki allele (Supplementary Figure S1C). The heterozygous PARP1 Ki (PARP1+/D993A) ES clones were injected into blastocysts to generate chimeras, which were subsequently crossed with C57BL/6 mice to obtain PARP1+/D993A founder lines. Genotyping of the animals was performed by polymerase chain reaction (PCR) using the following primers. PARP1 KO: OVLI (GTTGTGAACGACCTTCTGGG) OVLIR (CCTTCCAGAAGCAGGAGAAG) and NeoIIR (GCTTCAGTGACAACGTCGAG). PARP1 Ki: D993A F2 (ATGAGTATCCTTTCTTGGCTATG) and D993A: R2 (CTGAGCAATGGCGTAGACA). All sequences are given from 5′ to 3′ orientation.
Genotoxic treatment of mice
The desired amount of methyl-nitroso-guanidine MNU (Sigma-Aldrich, Taufkirchen, Germany) was solved freshly in 0.9% (w/v) NaCl (pH < 5) and sterile filtered prior to use. The body weight of the animals was measured and the injection volume was calculated accordingly. The MNU solution or solvent was administered intraperitoneally into adult, young mice (2–3 months of age).
Collection of tissues from mice and histology
Adult mice were sacrificed and the organs were subjected to RNA isolation or histological analysis. For histology, small intestines were flushed once with PBS and once with 4% paraformaldehyde (PFA; Sigma-Aldrich) before fixation at 4°C overnight. For paraffin sectioning, fixed tissues were washed twice with PBS for 10 min each and then sequentially embedded in paraffin. Tissue sections (7 μm) were prepared for H&E staining, which were examined by microscopy (AxioImager M1 with AxioCam Mrm camera operated by Axiovision software, Zeiss, Jena, Germany).
RNA isolation, cDNA synthesis and semi-quantitative PCR
RNA isolation, reverse transcription (RT) and semi-quantitative RT-PCR was carried out as described previously (44). RNA from tissue was isolated using Trizol reagent (Thermo Scientific, Darmstadt, Germany) according to the manufacturers' suggestions. First strand cDNA was then synthesized from 0.5 μg of RNA using Affinity Script Multiple Temperature cDNA synthesis Kit according to the manual (Agilent, Waldbronn, Germany). Primer pairs for GAPDH were GCACAGTCAAGGCCGAGAAT and GCCTTCTCCATGGTGGTGAA and those for PARP1 were GGATCCCATCTGGTGTCAAC (forward primer annealing in exon 22) and GTGCAGAGGCACTAGGGAGA (reverse primer annealing in exon 23). The sequencing primer was TTCACTGGGTTAGAGCATTG. All sequences are given from 5′ to 3′ orientation.
Purification of PARP1 proteins
The protein baculovirus expression vectors pQE-TriSystem (Qiagen, Hombrechtikon, Switzerland) and BacPak8 (Clontech, Saint-Germain-en-Laye, France) were used for the expression of recombinant proteins in Sf21 insect cells as described previously (45,46). Wild-type (WT) human PARP1 or the D993A mutant were cloned and expressed as carboxyl-terminal His-tagged proteins. All recombinant proteins were purified by one step affinity chromatography using ProBond resin according to the manufacturer's recommendations (Invitrogen). Expression and purification of all recombinant proteins were analysed by SDS-PAGE followed by Coomassie staining.
ADP-ribosylation assay in vitro
10 pM recombinant His-tagged human WT and D993A mutant PARP1 were incubated with 150 nM [32P]NAD+ in the presence of the indicated concentration of unlabelled NAD+ (PerkinElmer, Schwerzenbach, Switzerland) for 2 or 15 min at 37°C in reaction buffer (50 mM Tris–HCl, pH 8.0, 4 mM MgCl2, 0.25 mM DTT,) and 5 pM annealed double-stranded oligomer (5′-GGAATTCC-3′), in a total reaction volume of 25 μl. The reaction was stopped by adding the SDS sample buffer and boiling (5 min, 95°C). Samples were separated on a 8% SDS-PAGE, the protein content was visualized by coomassie staining and the autoradiographic signal quantified by using GelEval (http://www.frogdance.dundee.ac.uk).
PAR synthesis and purification
PAR was synthesized according to Fahrer et al. (47). Briefly, a reaction mixture including 100 mM Tris–HCl pH 7.8, 10 mM MgCl2, 1 mM DTT, 300 μg/ml histone HIIa, 50 μg/ml oligonucleotide (GGAATTCC), 1 mM NAD+ and 150 nM PARP1 was incubated at 37°C for 45 min. The reaction was stopped by 20% ice-cold trichloro-acetic acid (TCA) and precipitated PAR was pelleted by centrifugation. To detach PAR from proteins, it was incubated in 0.5 M KOH/50 mM EDTA for 10 min. To stop the reaction the pH was adjusted to 7.5–8.0. After DNA and protein digestion at 37°C overnight, PAR was purified by phenol–chloroform–isoamyl alcohol extraction, which was followed by ethanol precipitation. Finally, PAR was dissolved in water and the concentration was determined by UV absorbance at 259 nm.
Characterization of PAR chain length and branching
The PAR chain length distribution was analyzed using an Agilent 1100 HPLC system equipped with a DNA Pac PA-100 analytical column (4 × 250 mm). 50 nM of the purified PAR was loaded onto the column equilibrated with buffer A (25 mM Tris–HCl pH 9.0) and the polymers were eluted using a multistep gradient of buffer B (25 mM Tris–HCl pH 9.0, 1 M NaCl). The method was adapted from Fahrer et al. (47) and set as follows: 0 min (0% B), 3 min (20% B), 20 min (35% B), 40 min (42% B), 70 min (47% B), 110 min (53% B), 120 min (61% B), 131 min (70% B), 132 min (100% B), 152 min (100% B). PAR molecules were detected by UV absorbance at 259 nm.
Mass spectrometric analysis of PAR branching
Mass spectrometric analysis of PAR branching was performed as reported previously (48,49). Briefly, MEFs were challenged with 250 μM H2O2 for 10 min and PAR was sampled for the UPLC–MS/MS analysis. Cells were washed with ice-cold PBS and lyzed with 1 ml 20% TCA. The lyzed cells were harvested using a cell scraper and centrifuged for 5 min at 3000 × g and 4°C. The pellet washed twice with 500 μl ice-cold 70% ethanol, and centrifuged for 5 min at 3000 × g at 4°C. The pellet was air-dried at 37°C, resuspended in 255 μl 0.5 M KOH and neutralized with 50 μl 4.8 M MOPS buffer. DNA concentration was measured with an extinction wavelength of 360 nm and an emission wavelength of 460 nm utilizing a VarioskanFlash Fluorescence Reader (Thermo Scientific). The DNA concentration of a sample was calculated using a standard curve from defined amounts of calf thymus DNA (Sigma-Aldrich). Heavy-isotope labelled, undigested PAR (12 pmol) was added as an internal standard. DNA and RNA were digested for 3 h at 37°C by incubating samples with 0.1 mg/ml DNase 1 (Roche, Mannheim, Germany), 0.1 mg/ml RNase A (Sigma-Aldrich), 50 mM MgCl2 and 100 mM CaCl2. Then, 1.25 μl of 40 mg/ml proteinase K (Roche) were added and samples were incubated at 37°C over night. Thereafter, PAR was purified using the High Pure miRNA Isolation kit (Roche) according to the manufacturer's instructions and was eluted in 100 μl RNase-free water and then digested into its subunits with 10 U PDE1 (Affymetrix, Santa Clara, USA) and 0.5 U alkaline phosphatase (Sigma-Aldrich) for 3 h at 37°C. Next, the samples were filtered through a 10-kD Nanosep filter and subsequently dried in a speedvac. The samples were then resolved in 25 μl MilliQ water and subjected to UPLC–MS/MS analysis as described in (49).
Cell culture
Primary mouse embryonic fibroblasts (pMEFs) were isolated from E13.5 embryos derived from intercrosses of PARP1+/− and PARP1Ki/+ mice in the 129/Sv/B6 mixed background, or immortalized following a 3T3 protocol as described previously (50). MEFs were maintained in Dulbecco's modified eagles medium (DMEM), containing 10% fetal calf serum, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin and 0.1 mM β-mercaptoethanol (Thermo Scientific) at 37°C and 5% CO2. Cellular experiments were performed with pMEFs (< passage 4), unless otherwise stated. For immunohistochemistry and HiMAC assays (see below), cells were seeded on black flat-bottom 96-well plates (Corning, Wiesbaden, Germany) coated with 0.1% gelatin (Sigma-Aldrich) in PBS.
Inhibitor treatments
pMEFs were pre-treated for 1 h with the respective inhibitors prior to the genotoxic treatments at 37°C, 3% O2, 5% CO2. The inhibitors were present during the genotoxic treatments, the damage recovery period as well as the EdU pulse labeling. The final concentrations of the inhibitors are: 500 nM Aphidicolin (Sigma-Aldrich); 5 μM PARPi (Rucaparib, SelleckChem, Houston, TX, USA); 5 mM Caffeine (Sigma-Aldrich); 10 μM ATMi (Ku-5593; Abcam, Cambridge, UK).
Cellular survival assay
Defined number of pMEFs of the indicated genotypes (300 for WT and D993A/D993A, and 400 of –/–) were plated on black flat-bottom 96-well plates pre-coated with 0.1% gelatin in PBS and cultured at 37°C, 5% CO2, 3% O2. Forty eight hours after seeding, pMEFs were treated with the indicated drugs for the indicated durations, and released into fresh MEF medium. At 6 and 9 days of recovery in the MNNG and MNU or CPT experiments, respectively, the cells were subject to the HiMAC assay. Nuclei after DAPI staining were counted by high content microscopy (Cellomics Arrayscan VTI, Thermo Scientific) and the survival was calculated as the number of nuclei in each condition relative to the respective DMSO-treated condition.
Clonogenic survival assay
pMEFs were seeded on 10 cm dishes (15 000 cells per dish) 24 h before a 30 min pulse treatment with MNNG or DMSO and were released into drug-free culture. After 10 days, cells were fixed with 70% EtOH for 30 min and then stained with 0.5% crystal violet in 1% EtOH for 30 min. The plates were scanned and the crystal violet signals were quantified with MultiGauge (Fuijifilm, Düsseldorf, Germany). Survival was calculated as the average signal intensity of a particular condition divided by the average signal intensity of the respective DMSO-treated condition.
Senescence-associated β-galactosidase assay (SA-β-Gal)
Twenty thousand pMEFs (< passage 2) were seeded per well of a six-well dish (Nunc, Sigma-Aldrich) before treatment with MNNG. A commercially available Sa-β-Gal kit (Cell Signaling, Danvers, USA) was used according to the manufacturers′ instructions. SA-β-Gal positive cells were scored by microscopy (Axiovert 40CFL with an AxioCam MRc5 camera, Zeiss, Jena, Germany). Data were expressed as ratios of positive cells at a certain condition (i.e. MNNG treatment and genotypes) relative to the untreated WT cells.
High content microscopy-assisted cell cycle phenotyping (HiMAC)
HiMAC was essentially performed as described previously (51). Briefly, pMEFs were seeded to gelatin-coated (see above), black flat-bottom 96-well plates and allowed to adhere for at least 24 h. For H2O2 treatments, culture medium was removed and cells were treated with 100 μM H2O2 (Sigma-Aldrich) in PBS supplemented with 1 mM MgCl2 (Mg-PBS) for 8 min at 37°C, 5% CO2, 3% O2. After treatments, H2O2 solution was removed, the cells washed once with fresh culture medium, and then released into drug-free culture for the indicated durations. Prior to fixation at the indicated time points, the cells were pulse-labeled with 4 μM EdU (Thermo Scientific) for 45 min. At time points, pMEFs were fixed in PBS containing 3% PFA (Sigma-Aldrich), 0.025% glutaraldehyde (Applichem, Darmstadt, Germany) and 0.1% Triton X-100 (Sigma-Aldrich). Blocking and antibody incubation was carried out in 1.5% BSA (Sigma-Aldrich), 0.4% Triton X-100 in TBS. and secondary antibodies. Primary antibodies applied over night at 4°C: γH2AX (JBW301, Millipore, Darmstadt, Germany; 1:300), 53BP1 (NB100-304, Novus Biologicals, Littleton, USA; 1:300 for Lot A3 and 1:4000 for Lot A4 and A5). Secondary antibodies incubated for 1 h at RT: anti-rabbit IgG-Cy3, anti-mouse IgG-FITC, anti-mouse IgG-Cy3 (All from Sigma-Aldrich; used 1:300, 1:200; 1:400, respectively). EdU click reaction was then allowed for 1 h at RT (51) and the cells were stained with 1 μg/ml DAPI (Sigma-Aldrich) in PBS for 30 min at RT. Images were acquired in an automated manner using a BD pathway 435 system (Beckton Dickinson, Heidelberg, Germany), data processing by CellProfiler software (52), and analysis using the HiMAC analysis template (51).
High content analysis of cell cycle exit
One thousand pMEFs were seeded per well of gelatin-coated black flat-bottom 96-well plates 24 h before treatment with the indicated doses of MNNG and released into drug-free culture for the indicated durations. Twenty four hours before fixation, the cells were labelled with 4 μM EdU for 24 h. After fixation, the samples were processed and analysed as described for HiMAC (see above). The antibodies used were: α-Ki67 (SP6, Thermo Scientific; 1:300) and α-rabbit IgG-Cy3 (Sigma-Aldrich; 1:300). Ki67 intensity thresholds were empirically determined.
High content analysis of poly(ADP-ribose) by immunofluorescence (PAR-IF)
pMEFs were seeded to 96-well plates as described for HiMAC. Genotoxic treatment with 1.5 mM H2O2 or the indicated concentrations of MNNG, MNU or CPT for the indicated durations were performed as described above. At time points, the cells were fixed with 100 μl ice-cold 100% MetOH for 8 min at 4°C. After blocking the cells with 5% non-fat dry milk in TBS containing 0.01% Tween-20 at 30°C for 30 min, anti-PAR antibody (10H, Millipore; 1:300) and anti-mouse IgG-FITC in the H2O2 experiments, or anti-mouse IgG-Cy3 in the MNNG, MNU and CPT experiments (both from Sigma-Aldrich; 1:200 and 1:400, respectively), were applied in blocking solution for 45 min at 30°C. Subsequent DAPI staining, microscopy and image analysis was carried out as described above (HiMAC). The nuclear intensity of PAR signals was normalized against the DAPI sum of each individual cell to account for changes in the DNA content during cell cycle progression. Highly condensed nuclei were excluded from the analysis.
Alkaline single cell electrophoresis (Comet assay)
The alkaline comet assay was carried out essentially as described earlier (53). Briefly, pMEFs were treated for 8 min at 37°C with 100 μm H2O2 followed by the indicated durations in drug-free culture to allow for DNA repair. At time points, pMEFs were harvested and suspended at a density of 1 × 106 cells per ml in PBS and mixed 1:4 with molten low-melting point agarose (Sigma-Aldrich) at 41°C. This mixture was overlaid on the pre-warmed frosted microscope slides with window (Erie scientific, Portsmouth, USA), which had been coated with 1% Agarose Type II (Sigma-Aldrich). Next, the slides were incubated at 4°C for 60 min in alkaline lysis buffer containing 10% DMSO (Thermo Scientific) 1% Triton-X-100, 10 mM Tris, 100 mM EDTA, 2.4 M NaCl, 1% (w/v) N-lauryl-sarcosine (all from Sigma-Aldrich) at pH 10. The slides were then equilibrated in electrophoresis buffer (333 mM NaOH, 1 mM EDTA, pH 13) at 4°C for 25 min and subsequently electrophoresed at 4°C at 1 V/cm (480 mA) for 25 min. After neutralization for 5 min in 0.4 M Tris (pH 7.5), washing with 100% EtOH and staining with 2.5 μg/ml propidium iodide (Sigma-Aldrich) in PBS, the samples were analysed using an Axioplan 2 microscope (Zeiss) equipped with a stingray camera (Allied Vision Technologies, Stadtroda, Germany) operated by Comet Assay IV software (Perceptive instruments, Edmonds, UK).
Recruitment of GFP-XRCC1 following laser damage
For measurement of the recruitment of GFP-XRCC1, cells were cultured on Nunc™ Lab-Tek® Chamber Slides (Nunc, Thermo Scientific). One day before laser damage, adherent cells were transfected with GFP-XRCC1 vector using Lipofectamine 2000 (Thermo Scientific) according to the manufacturers' instructions. For laser damage induction, the pulsed UV-A laser was coupled into a confocal laser-scanning microscope (LSM 510, Zeiss) via epifluorescence illumination path (53). Cells were maintained in an incubation chamber supplied with a 5% CO2, 37°C by a Tempcontrol 37-2 digital and CTI-Controller 3700 digital (both Carl Zeiss, Jena, Germany). Automated movement of the motorized x, y table during procedure caused laser tracks, irradiating the cells with a 2 μJ single laser pulses every 1.5 μm. No sensitisation was used. Confocal imaging for living cells was performed using the LSM510 laser scanning microscope (Zeiss) equipped with a HeNe and an Argon ion lasers and emission filter set for the detection of FITC signals (BP530/20). Scanning and the time course of intensity changes of fusion proteins were recorded via time series of the Zeiss LSM software version 3.2. Mean fluorescence intensity was measured in three regions of interest (ROIs): irradiated area (I), in the nucleus outside irradiated area for photobleaching correction (C) and outside the nucleus for background value (B). Fold change of intensity was calculated as follows: fold change of intensity = (I – B)/(C – B).
Western blotting
Western blotting was carried out as described previously (44). Briefly, pMEFs treated as indicated were harvested by trypsinization, washed once in ice-cold PBS containing 1× protease and 1× phosphatase inhibitor cocktails (Sigma-Aldrich) and then lysed in 20 mM HEPES pH 7.6, 20% glycerol, 0.5 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA pH 8.0, 0.5% NP-40, 1 mM DTT, 1 mM PMSF, 5 mg/ml leupeptin, 2 mg/ml aprotinin, 1 mM β-glycerophosphate, 1 mM Na3VO4 and 10 mM NaF (Sigma-Aldrich). Following SDS-PAGE, the membranes were blotted with the following antibodies: rabbit-anti-Cyclin A (Santa Cruz, Heidelberg, Germany; 1:500), mouse-anti-β-actin (Sigma-Aldrich, 1:5000) and horseradish peroxidase (HRP)-coupled secondary antibodies anti-mouse-IgG-HRP and anti-rabbit-IgG-HRP (both from DAKO/Agilent, Waldbronn, Germany, 1:5000).
RESULTS
PARP1D993A/D993Amice develop normally
Using the Cre-loxP gene targeting technology, we generated a PARP1 knock-in (Ki) mouse line harboring an amino acid exchange from aspartate (D) to alanine (A) in the codon 993 of PARP1 (PARP1D993A). We constructed a gene-targeting vector containing the point mutation in exon 23 (Supplementary Figure S1A) and electroporated it into E14.1 ES cells. Southern blot analysis of selected ES clones verified a targeted mutation in the Adprt locus (Supplementary Figure S1A and B) and the subsequent excision of the neo cassette by Cre-recombinase, generating the PARP1+/D993A ES clones (Supplementary Figure S1C). The PARP1+/Ki ES cells were used to generate PARP1+/Ki mice. Intercrosses of PARP1+/D993A mice resulted in homozygous Ki mice (designated PARP1D993A/D993A) at the expected Mendelian ratio (Supplementary Figure S1D). PARP1D993A/D993A mice were phenotypically normal throughout the observation period of two years (data not shown). The presence of the PARP1D993A mutation was confirmed by sequencing cDNA isolated from mouse livers (Supplementary Figure S1E). Semi-quantitative reverse transcription (RT)-PCR revealed no difference in the Parp1 transcripts of PARP1D993A/D993A mice compared to wild-type (WT) or PARP1+/D993A littermates (Supplementary Figure S1F).
PARP1D993A mutation alters the enzymatic kinetics of PARP1 and PAR chains
To characterize the impact of the PARP1D993A mutation on DNA damage-induced PARylation, we isolated and used primary embryonic fibroblasts (pMEFs) from embryos of the respective genotypes, to avoid the consideration that the immortalization procedure modifies the genotoxic stress response, to measure the kinetics and capacity of PARylation. We challenged the cells with the alkylating agent MNNG or H2O2, which are well-characterized PAR inducers, and analysed them by a high-content (HC) immunofluorescence (IF) microscopy assay (PAR-IF; Figure 1A and B). We detected a dose-response of the PAR formation capacity of the pMEFs after 30 min of treatment with MNNG. PARP1D993A/D993A pMEFs displayed less PAR compared to WT in all doses tested, particularly at 50 and 100 μM (Figure 1C). PARP1−/− cells expectedly induced negligible PAR (Figure 1B and C). In addition, the time-course showed that 200 μM MNNG robustly induced PAR already at 10 min in the WT pMEFs, while the PARP1D993A/D993A pMEFs were almost devoid of PAR in all tested doses at this time point. Even upon exposure to 200 μM MNNG, the PARP1D993A/D993A pMEFs induced only ∼10% of the WT level at 10 min, although it reached ∼80% of WT at 30 min (Figure 1C). Of note, obvious cell death (e.g. floating cells) was not detected during the given 30 min of treatment (data not shown). Similarly, PARP1D993A/D993A pMEFs also showed a lower and decelerated PARylation after treatment with H2O2, as well as with another alkylating agent, MNU (Supplementary Figure S2A–C). Because PARP1 can be activated by damaged replication forks (6,10,54), we next treated pMEFs with the topoisomerase inhibitor camptothecin (CPT) that induces replication fork damage and found similar PARylation defects (Supplementary Figure S2D).
Figure 1.

PARP1D993A mutation slows enzyme kinetics of PARP1 and impairs PAR complexity. (A) Scheme of a PAR formation assay by immunofluorescence (IF) staining of PAR (PAR-IF) and data analysis. (B) Representative pictures of the PAR-IF staining of pMEFs with the indicated PARP1 genotypes (wild type PARP1 (+/+); PARP1D993A/D993A (D993A) and PARP1 knockout (–/–)) after treatment with 200 μM MNNG or DMSO for 30 min. (C) Quantification of the PAR intensity in individual cells treated for 10 min or 30 min with the indicated doses of MNNG. The DMSO controls were analysed at both time points and only 30 min is shown. The data are the means ± SEM from at least 500 cells per condition normalized against the maximum wild type (WT) level. Similar results were obtained in three independent experiments using four pMEFs littermate pairs. Asterisks (*) indicate the difference of PARP1D993A/D993A versus WT (+/+); *P <0.05; **P <0.01; ***P <0.001, as determined by a two-way ANOVA with a Tukey′s post-test. (D) In vitro activity assay of recombinant WT and D933A mutant PARP1 proteins incubated for either 2 min or 5 min with oligonucleotides mimicking DNA strand breaks and the indicated concentrations of NAD+ containing 150 nM 32P-labeled NAD+. At each reaction time, an autoradiography (32P) and Coomassie staining of the gel (Com.) are shown. (E) Quantification of the 32P signals of the panel (d) after 2 min or 15 min of reaction time at the indicated concentrations of NAD+ expressed as ratio of D993A to WT. Data are the means ± SEM from three independent experiments. P-values were calculated by the Student's t-test. (F) Analysis of the PAR chain complexity by HPLC. HPLC-DAD chromatograms of in vitro synthesized PAR from recombinant PARP1 proteins (WT and D993A mutant). The individual peaks indicate PAR molecules of different chain lengths. The retention time at 5 min was set as the starting point (‘0 min’). The complexity of PAR increases with the relative retention time. (G) The retention times from the HPLC chromatograms in (F) are grouped based on their percentiles. The resulting thresholds in min (X-axis) are plotted against the respective averaged absorbance (‘mean absorbance’ on the Y-axis). Note that the dotted trend curve of PARP1D993A is shifted to the left, indicative of less PAR complexity. (H) and (I). UPLC–MS/MS analysis of branching levels of in vitro synthesized PAR from recombinant human PARP1 (WT and D993A mutant) (H) and PAR isolated from immortalized MEFs of the indicated genotype (I). The ratio of signal intensities from the digestion products di-ribosyladenosine (R2-Ado), which is specific for PAR branching points, and ribosyl-adenosine (R-ado), which is specific for the linear part of PAR, were used to analyse the degree of PAR branching. Data are the means ± SEM of three biological (H) and five technical replicates (I). The significance is determined by Student's t-test. **P < 0.01; ***P < 0.001.
The D993A mutation was reported to inactivate the human PARP1 catalytic activity (55). We performed an in vitro auto-PARylation assay on the recombinant human WT and D993A PARP1 proteins at different NAD+ concentrations. We found that D993A PARP1 formed less PARs than WT PARP1 in a 2-min reaction time (Figure 1D upper panel, E). However, a similar amount of PARs was formed by WT and D993A PARP1 at the 15 min reaction time (Figure 1D lower panel, E). At the same NAD+ supply, D993A PARP1 failed to form the same amount of PARs within a short reaction time, but eventually reached a similar level later on. We next characterized the quality of PARs formed by the D993A PARP1 by HPLC and mass spectrometry (MS). First, we analysed PARs synthesized by WT or D993A PARP1 by HPLC and found that the mutant PARP1 formed relatively shorter (HPLC) and less branched PAR chains (MS) compared to WT PARP1 (Figure 1F–H). Similarly, PARP1D993A/D993A pMEFs challenged with 250 μM H2O2 produced less-branched PARs than WT pMEFs (Figure 1I). Taken together, the PARP1D993A mutation delays DNA damage-induced PARylation and compromises the complexity of the PAR chains (hereof hypo-PARylation).
PARP1D993A mutation impairs base excision repair
To investigate the impact of hypo-PARylation in DNA repair, we conducted an alkaline comet assay and monitored the repair at a 60-min recovery time after 8 min exposure to H2O2. We observed a slightly higher level of the olive tail moments in PARP1D993A/D993A pMEFs compared to WT controls immediately after the treatment (0 min, Figure 2A and B). However, the difference was diminished at 30 min of the repair time. As expected, PARP1−/− pMEFs maintained higher comet tail moments compared to the other two genotypes even at 60 min of the repair. Apparently, shortly after damage induction, there was elevated damage in PARP1D993A/D993A and PARP1−/− cells, which likely represents a defect in early damage repair. However, this was compensated for in PARP1D993A/D993A cells at later time points. To further examine whether the altered PARylation of PARP1D993A/D993A cells has an impact on BER, we analyzed the behavior of the scaffold protein XRCC1, which is normally recruited to the damaged site by PARylated PARP1 (56). To this end, we engineered 3T3-immortalized MEFs and transfected them with GFP-tagged XRCC1 and monitored the chromatin recruitment of GFP-XRCC1 to the sites of laser-induced DNA breaks. As expected, the accumulation of GFP-XRCC1 at laser-induced DNA damage was completely abolished in PARP1−/− MEFs (Figure 2C and D). The retention of XRCC1 was significantly compromised as lower XRCC1 signals were detected in PARP1D993A/D993A MEFs at later time points, compared to WT (Figure 2D, note the slope of the curves from 100 s to 180 s). These results argue for an impaired BER after PARP1D993A mutation. We conclude that the level and kinetics of PAR formation, and probably also their structure, are important for the immediate response to base damage, though being dispensable under unperturbed physiological conditions.
Figure 2.

PARP1D993A mutation slows base excision repair. (A) Representative images of an alkaline comet assay of pMEFs with the indicated PARP1 genotypes (wild type PARP1 (+/+); PARP1D993A/D993A (D993A) and PARP1 knockout (–/–)). The cells were treated or not with 100 μM H2O2 for 8 min and analysed after the indicated recovery time. (B) Comet tail moments as quantified using Comet Assay IV software. The data are the means ± SEM from the indicated number of cells of each PARP1 genotype. **P <0.01; ***P <0.001; n.s.: not significant, as determined by a two-way ANOVA with a Sidak's post-test at the indicated time points. (C) Representative live-cell micrographs of MEFs of the indicated PARP1 genotype expressing GFP-XRCC1. The recruitment of GFP-XRCC1 to laser-induced DNA damage tracks at the indicated time. A white dashed frame marks the laser track. (D) Time-course of the fold change of GFP signals within the laser track normalized to the GFP background within an undamaged region. Color-coded asterisks (*) indicate the significance versus +/+ (WT) littermates, as determined by a two-way ANOVA with a Tukey's post-tests. N = 10. The area under the curve (AUC) is used to compare the full curves. The statistical analysis of AUC was performed using the Student's t-test. ##P < 0.01; ###P <0.001.
The hypo-PARylation inflicts replication-coupled DNA breaks
We next investigated the consequences of a reduced BER activity in PARylation-deficient cells, using the HiMAC assay, which enables tracing of the DDR during cell cycle progression in WT pMEFs (51,57). To this end, we first treated WT cells with a pulse of H2O2 before releasing them in fresh culture medium for various durations and labelled cells with EdU before sampling (see Figure 3A). IF revealed that in WT cells, oxidative stress induced the DNA damage marker γH2AX predominantly in the S-phase (EdU+ cells) after 1–2 h post-H2O2 (Figure 3B and C), which was accompanied by a progressive decline of DNA synthesis (EdU incorporation) (Figure 3D), indicating an activated intra-S checkpoint. A further HiMAC analysis revealed that γH2AX foci were increased in all three sub-phases of S-phase at 1–2 h post-H2O2 treatment (Supplementary Figure S3A and B). Of note, a high level of γH2AX signals in mid- and late S-phase coincided with a high DNA synthesis rate (EdU intensity) (Supplementary Figure S3C), suggesting that DNA damage accumulated in mid- and late S-phase and associated with DNA replication. To test this hypothesis, we treated cells with Aphidicolin that slows DNA replication, and found complete repression of γH2AX signals (Figure 3E). Moreover, a chemical inhibition by the ATM inhibitor and the pan-PIKK inhibitor Caffeine both abolished γH2AX signals (Figure 3E). These results suggest that DNA replication fork stalling triggered an ATM-dependent DDR in S-phase. Interestingly, the PARP inhibitor resulted in a much higher level of γH2AX foci in S-phase cells after H2O2 treatment compared to non-inhibitor treatment (Figure 3E). Thus, PARylation or PAR either cleans SSB/base lesions to prevent replication stress or modulates the repair of damaged replication forks, consistent with the notion of previous studies (6,10,54).
Figure 3.

H2O2-induced base lesions trigger PARylation-dependent replication stress. (A) Scheme of the experimental design for a HiMAC analysis in pMEFs. Briefly, cells were treated or not with 100 μM H2O2 for 8 min before recovery in a drug-free culture medium for various time points (marked as ‘X). 45 min prior to fixation and HiMAC at the given time points, the cells were pulse-labeled with EdU. (B) HiMAC analysis of WT pMEFs at 1 or 2 h, treated as described in A. The upper panel shows exemplary pictures. Mid panel: Cell cycle profiles obtained from individual cells from one representative experiment. Lower panel: yH2AX scatter plots showing a selective induction of yH2AX in S-phase cells. (C and D) Quantifications of γH2AX signal intensities in the indicated cell cycle phases (C) and of EdU signals of S-phase cells (D). The data are the means ± SEM from one representative experiment with >1000 individual cells analyzed per condition. Similar results were obtained in five independent experiments. (E) Wild type (WT) pMEF were treated or not with indicated inhibitors and analysed for their yH2AX foci. The data are the means ± SEM. N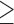 1000 cells. * marks the significance, as determined by a two-way ANOVA with a Tukey's post-test (C, E) and one-way ANOVA with a Holm-Sidak's post-test (D). * within the bars in (E) show a comparison to the respective PBS-treated samples. *P <0.05; **P <0.01; ***P <0.001. n.s.: not significant. The significance indicators inside the bars (E) show comparison to the respective PBS-treated controls. Similar results were obtained in five (A–D) and two (E) independent experiments in at least duplicates each.
1000 cells. * marks the significance, as determined by a two-way ANOVA with a Tukey's post-test (C, E) and one-way ANOVA with a Holm-Sidak's post-test (D). * within the bars in (E) show a comparison to the respective PBS-treated samples. *P <0.05; **P <0.01; ***P <0.001. n.s.: not significant. The significance indicators inside the bars (E) show comparison to the respective PBS-treated controls. Similar results were obtained in five (A–D) and two (E) independent experiments in at least duplicates each.
We then examined how the PARylation status would affect DNA damage accumulation in S-phase and subjected PARP1D993A/D993A, PARP1−/− and PARP1 WT pMEFs to oxidative damage by the H2O2 treatment. Among all genotypes, PARP1−/− pMEFs showed the highest numbers of γH2AX foci in the S-phase throughout the time course (Figure 4A and B). PARP1D993A/D993A pMEFs contained a mild, but a significantly higher level of γH2AX foci during the early recovery phase after damage (1 and 4 h post-H2O2) compared to WT controls (Figure 4B). Eventually, WT and PARP1D993A/D993A, as well as PARP1−/− pMEFs, could fully resolve the γH2AX foci after 26 h post-H2O2 (Figure 4B). Consistent with previous reports on the involvement of PARP1 and PAR formation in the repair of damaged replication forks (6,10,54), these data, together with Figure 3E, suggest that the S-phase-specific DDR under the present treatment regimen is likely ATM-dependent and triggered by DNA replication fork stalls, which are potently counteracted by proper PAR homeostasis.
Figure 4.

Hypo-PARylation primes replication stress and enhanced DSBs at replication restart upon H2O2 and MNNG. (A) Representative HiMAC images of pMEFs with the indicated PARP1 genotype (wild type PARP1 (+/+); PARP1D993A/D993A (D993A) and PARP1 knockout (–/–)) treated or not with 100 μM H2O2 for 8 min, released into fresh medium for the indicated durations and pulse labeled with EdU for 45 min before sampling (see Figure 3A for a schematic treatment regimen). (B) Quantification of γH2AX foci (upper panel) at the indicated time points and of EdU intensities (lower panel) in S-phase pMEFs (EdU+). (C) pMEFs of the indicated PARP1 genotype were treated or not with 20 μM MNNG for 30 min, released into fresh medium for the indicated durations and pulse labeled with EdU for 45 min before sampling. Quantification of the γH2AX signal intensities and the EdU intensities of pMEFs at the indicated time points are normalized against the respective DMSO controls. At least 400 EdU+ cells from +/+ and D993A, and 200 EdU+ cells from –/– per condition were scored and their mean γH2AX and EdU intensities plotted ± SEM. (D) Representative images of pMEFs with the indicated PARP1 genotypes treated as described in (A). (E) Quantification of the 53BP1 foci per cell at the indicated time points in S-phase cells from D. Color-coded asterisks (*) indicate the significance versus +/+ littermates, as determined by a two-way ANOVA with Tukey's post-tests in (B, E) and with Bonferroni′s post-tests in (C). *P < 0.05; **P < 0.01; ***P < 0.001. Data from (B) and (E) are the means ± SEM of at least three independent (PBS, H2O2, until 9 h post-H2O2) and two independent (16 and 26 h) experiments, using at least two littermate pairs of pMEFs in at least duplicates. (F) Correlation analysis of DNA synthesis (EdU) with γH2AX signals (top) and 53BP1 foci (bottom) of pMEFs with the indicated PARP1 genotypes at 4 h (left) and 9 h (right) post-H2O2 are shown. Single graphs are X/Y scatterplots (X: Re-scaled EdU; Y: DNA damage marker). The indicated percentiles of the EdU signals within the population are shown, based on the total EdU intensities of the individual cells (see also Supplementary Figure S5). Dark colored ‘smoothened curves’ show the corresponding single-cell data on the damage markers after smoothening by moving averages with a period of 40 to facilitate trend identification. Light-colored curves flanking the dark-colored smoothened curves represent the SEMs from at least 500 cells per condition. As PBS-treated cells showed no correlation, they were not presented here. For the full ranges of the EdU signals and of the corresponding DNA damage markers, please refer to Supplementary Figure S5.
To investigate the consequence of accumulated DNA damage in the S-phase, we also determined the DNA replication rate by quantifying the EdU incorporation of these cells. DNA synthesis in WT pMEFs decreased transiently from 1–4 h post-H2O2, indicating an activation of the intra-S checkpoint. Of note, the EdU incorporation rate closely mirrored the DNA damage profile and its recovery correlated with the decline of the γH2AX foci (Figure 4B), suggesting that DNA repair allows resumption of DNA replication. Furthermore, when exposed to MNNG, PARP1D993A/D993A and PARP1−/− pMEFs displayed a stronger induction of γH2AX signals compared to WT cells, correlating with a reduction of DNA synthesis, as judged by the reduction of EdU+ signals (Figure 4C). We next tested the DDR to S-phase poisoning by treating pMEFs with 125 nM CPT. This revealed a moderate increase of γH2AX signals in PARP1D993A/D993A pMEFs compared to WT and PARP1−/− cells and a reciprocal pattern of the EdU incorporation (Supplementary Figure S4A), suggesting that hypo-PARylation is more sensitive to S-phase damage. Altogether, a moderate, but consistent, defective response of the PARP1D993A mutation to different DNA damaging agents substantiate the importance of the full spectrum of PARylation to mitigate replication stress.
Full PARylation activity prevents replication restart-associated DSBs
To study whether these γH2AX foci were indeed converted to and thus represented DSBs, we analyzed by HiMAC the cells for 53BP1, an authentic marker for DSBs. PARP1−/− and PARP1D993A/D993A pMEFs displayed elevated 53BP1 foci from 9 h post-H2O2, following the γH2AX foci (Figure 4D and E). Interestingly, these 53BP1 foci started to appear when the replication had been stalled for a long time (4 h) and was just about to be resumed (compare at 4 and 9 h, Figure 4B and E). These observations suggest that the DSBs are either derived from a collapse of prolonged replication fork stalls or inflicted by replication restart. Therefore, we sought to distinguish which one of both processes caused DSBs. To this end, we performed correlation analyses to dissect the relationship between replication rate and the induction of the γH2AX and 53BP1 foci by monitoring the trend as to whether DNA damage evolution is coupled to DNA synthesis at 4 and 9 h post-H2O2 (Supplementary Figure S5).
As described in Supplementary Figure S5A, we first gated S-phase cells using EdU and DAPI signals (Supplementary Figure S5A(i)) and then classified these by their replication rates based on the EdU signal intensity and by re-scaling them from the lowest to the highest (Supplementary Figure S5A(ii), B–D). Of note, because of a global replication fork stall, much lower EdU signal intensities were measured at 4 h than at 9 h post-H2O2. Thus, we set up different gating strategies for both time points to gate for similar subsets of cells: At 4 h, we defined the population of cells with replication rates in the lower 30% range (0.3) as the ‘stalled’ replication, because their EdU signals were lower than most of the cells at 9 h post-H2O2. In addition, these cells contained less than one third of the EdU signals of untreated early S-phase cells (Supplementary Figure S5D). Accordingly, at 4 h, cells with EdU signal intensities at and slightly above that threshold underwent ‘replication restart’, while those far above exhibited ‘replication progression’ (Supplementary Figure S5A(ii), B and D). In contrast, at 9 h, we defined cells as replication ‘progression’ if their replication rate exceeded the lower 40% (0.4, Supplementary Figure S5A(ii)), because the EdU signals from this population were higher than those of untreated early S-phase cells and higher than in the subset of cells with ‘stalled’ replication at 4 h post-H2O2. Thus, the potential replication restart at 9 h post-H2O2 had to occur in cells with EdU signals below that threshold of the lower 40% (0.4, Supplementary Figure S5A(ii)). Then, we plotted the EdU signals of the individual cells against the DNA damage markers (i.e. γH2AX signal or the number of 53BP1 foci) and then smoothened the curves (Supplementary Figure S5A(iii), dark line). Figure 4F(i) shows the entire cell populations at 4 h post-H2O2 whereas Figure 4F(ii), (iii) and (iv) only depict the indicated subset of the cells in Supplementary Figure S5D. With this analysis, we found that the γH2AX induction first appeared in cells with marginal EdU signals in all genotypes (‘Stalling’ in Figure 4F(i)), indicating DNA damage induction occurs preferentially at stalled replication forks. While WT cells reached the plateau of the γH2AX induction already at low-to-intermediate EdU levels, PARP1−/− and, to some extent, PARP1D993A/D993A pMEFs displayed a further increase of γH2AX signals (Figure 4f(i)). To test whether the failure to successfully (re-) initiate DNA replication triggered DSBs in PARP1−/− and PARP1D993A/D993A pMEFs, we focused the subsequent analyses to cells potentially undergoing replication restart (indicated as ‘Replication restart’ in Figure 4F, Supplementary Figure S5A–C). Consistent with the γH2AX induction (Figure 4F(i)), DSBs, judged by the 53BP1 foci, were at higher levels in the subset of PARP1D993A/D993A and PARP1−/− cells undergoing a replication restart at 4 and 9 h (Figure 4F(ii), (iv)). Notably, this DSB induction in turn saturated (Figure 4F(ii)), or even decreased in cells that reached the intermediate-to-high replication rate (Figure 4F(iv); note the slopes of the curves), suggesting the damage-free progression of replication forks, once the replication restart was successful. Notably, the induction of 53BP1 foci in PARP1−/− pMEFs correlated with DNA replication only at 9 h (Figure 4F(iv)). Collectively, DSBs apparently occurred mainly in cells that intend to resume replication, but not in those cells that remain in a replication-arrested state. Thus, these correlation analyses indicate that the DSBs likely arose from a failed restart of replication forks, rather than being a consequence of prolonged replication fork stalling. In summary, these data suggest that the full activity of WT PARP1 is required to prevent DNA damage accumulation in the S-phase, which otherwise would progress to DSBs when the cells resume DNA synthesis.
Hypo-PARylation compromises the recovery from DNA damage
To investigate the cell fate response of PARP1D993A/D993A pMEFs to DNA damage, we exposed pMEFs to MNNG for 30 min and analyzed their survival at 6 days of recovery. We found an intermediate cell survival of PARP1D993A/D993A compared to WT and PARP1−/− pMEFs at doses of 5–10 μM MNNG (Figure 5A). Similarly, the MNU treatment also induced an intermediate cell death of PARP1D993A/D993A MEFs compared to WT controls and PARP1−/− pMEFs (Figure 5B). Moreover, we analyzed the cellular response after the CPT treatment and found that similar to the MNNG and MNU treatment, the CPT treatment resulted in an intermediate level of cell death of PARP1D993A/D993A MEFs, between WT and PARP1−/− pMEFs (Figure 5C). To substantiate these findings, we employed a sensitive colony formation assay using 10 and 20 μM MNNG to monitor the survival capacity of the cells. The clonogenic survival assay revealed a dramatically reduced colony formation of PARP1D993A/D993A pMEFs after MNNG when compared to WT, albeit not as strong as the PARP1−/− pMEFs (Figure 5D and E). These results demonstrate that a fully coordinated PARylation is required to protect cells from acute DNA alkylation damage.
Figure 5.

PARP1D993A mutation sensitizes cells to MNNG induced cell death and senescence. (A–C) Survival assay. pMEFs of the indicated PARP1 genotypes (wild type PARP1 (+/+); PARP1D993A/D993A (D993A) and PARP1 knockout (−/−)) were treated or not with the indicated concentrations of MNNG for 30 min (A), MNU for 60 min (B), or CPT for 60 min (C), released into fresh medium and then subjected to high-content microscopy-assisted cell counting at day 6 (A) and day 9 (B, C). ‘Survival’ fraction is calculated as % of DMSO-treated cells. The data are shown as a number of cells ± SEM of four (A) and three replicates (B, C). (D) Representative scans of clonogenic survival assays using pMEFs with the indicated PARP1 genotypes 10 days after 30 min-exposure to the indicated doses of MNNG. (E) Quantification of clonogenic assays. ‘Survival’ fraction is calculated as % of the signal intensities of DMSO-treated cells. The data are the means ± SEM derived from two independent experiments using two independent littermate pairs of pMEFs in duplicates. (F) Representative micrographs of senescence-associated-β-Galactosidase (SA-β-Gal) activity (blue) in the pMEFs of the indicated PARP1 genotypes at three days post-30 min exposure (3 dp) to 10 μM MNNG. Red arrowheads mark SA-β-Gal positive (+) cells. (G) Quantification of SA-β-Gal+ cells as induction (ratio) versus DMSO-treated +/+ littermates. (H) Scheme presents the experimental design of the cell cycle exit index (CCE) assay. (I) Representative images from the CCE assay. (J) Quantification of CCE relative to DMSO-treated controls according to the formula provided in (H). The data in (G) and (J) are the means ± SEM derived from three independent experiments using three (G) and two different pMEFs littermate pairs (J). Asterisks (*), color-coded in (A, B, C, E), indicate the significance. *P <0.05; **P <0.01; ***P <0.001, as determined by a two-way ANOVA with Bonferroni′s post-tests (A, B, C) and with Tukey's post-tests (E, G, J).
PARP1D993A/D993A pMEFs undergo senescence in response to low doses of alkylation
The reduced colony formation was not due to a proliferation defect, because PARP1D993A/D993A pMEFs proliferated normally during long-term passaging in culture (Supplementary Figure S6A and B). However, it could be attributed to a combination effect of cell death and cytostasis. We next tested whether senescence in PARP1D993A/D993A and PARP1−/− pMEFs could have contributed to the reduced colony forming ability. To this, we treated cells with 10 μM MNNG. PARP1−/− and PARP1D993A/D993A pMEFs gradually lost the Ki67 positive cells (cycling cells) as well as the EdU incorporation (Supplementary Figure S7A and B). We also analysed the senescence-associated β-Galactosidase (SA-β-Gal) activity in pMEFs at 3 days post-MNNG and found a higher number of SA-β-Gal positive cells in PARP1D993A/D993A genotypes as compared to WT (Figure 5F and G). Interestingly, MNNG also induced more senescence in PARP1−/− cells compared to WT cells, but much less compared to PARP1D993A/D993A cells (Figure 5G). To complete the analysis of cellular senescence, we measured the cell cycle exit index of pMEFs after MNNG exposure (Figure 5H). To this end, we labelled cells with EdU for 24 h after 2 days of recovery, and analysed the cells positive for both EdU and Ki67 among all EdU+ cells (Figure 5H). In line with the increased SA-β-Gal activity, PARP1−/− and PARP1D993A/D993A exhibited a higher cell cycle exit after alkylation as compared to WT (Figure 5I, J and Supplementary Figure S7A and B). Consistently, the downregulation of the S-phase Cyclin A was stronger in PARP1D993A/D993A and PARP1−/− pMEFs compared to WT controls (Supplementary Figure S7C). These data indicate that hypo-PARylation renders cells very susceptible to senesce whereas the absence of PARylation in PARP1 KO cells may rather induce cell death.
PARP1D993A mutation sensitizes mice to DNA alkylation
The poor long-term recovery of PARP1D993A/D993A and PARP1−/− pMEFs from base lesions prompted us to assess the impact of the PARP1D993A mutation on the tissue homeostasis of mice upon acute genotoxic stress. We monitored the survival of PARP1D993A/D993A and control cohorts (WT and PARP1+/−) after a single intraperitoneal (i.p.) injection of the alkylating agent MNU. Homozygous PARP1−/− mice, which are known to be hypersensitive to MNU (15,20), were included as a control. While 80% of the MNU-injected control mice survived for several weeks without any signs of sickness, like solvent-treated (sham) animals, strikingly, all PARP1D993A/D993A mice died after six days, similar to PARP1−/− mice (Figure 6A). We noted that PARP1−/− and PARP1D993A/D993A mice exhibited signs of colitis, such as diarrhoea. Histological analyses of the small intestine of these mice revealed that at 2 days after the MNU injection, the villi-crypt structure of the intestine in all genotypes appeared to be dispersed (Figure 6B). In contrast to control mice, which had fully restored the villi after 5 days, the PARP1−/− and PARP1D993A/D993A mice displayed a complete loss of the villi (Figure 6B). Notably, this was accompanied by the loss of crypts in PARP1−/− and PARP1D993A/D993A mice. These results indicate that hypo-PARylation caused an exhaustion of intestinal stem cell pools and the loss of differentiated cells. These effects indicate that the intestinal atrophy in PARP1−/− and PARP1D993A/D993A mice was probably among the primary causes of lethality in response to DNA alkylation. These data demonstrate that in contrast to a modest compromise of cell survival after DNA damage under culture conditions, hypo-PARylation completely abrogates tissue integrity in response to acute alkylating DNA damage. Strikingly, a disturbance of the PARylation dynamics and complexity apparently equals a complete elimination of PARP1 in vivo.
Figure 6.

Parp1D993A mutation sensitizes mice to alkylation and the working model. (A) Survival of male mice (age: 2–3 months) with the indicated PARP1 genotypes (control (WT and PARP1+/D993A); PARP1D993A/D993A (D993A) and PARP1 knockout (−/−)) after receiving a single dose of the alkylating agent MNU (150 mg/kg of body weight) or solvent (Sham) by intraperitoneal (i.p.) injection. The control (Co.) group is PARP1+/+ and PARP1+/− mice. (B) Hematoxylin and eosin stained sections of small intestines of mice with the indicated PARP1 genotype sacrificed at 5 days after i.p. injection of the solvent (sham) or at the indicated time points after MNU (100 mg/kg of body weight). C: crypts, V: vili. (C) The working model on PARylation-dependent cell fate decision. In response to acute base damage (step 1), a normal PARylation response, like in PARP1-WT cells, is critical for DNA repair and the clearance of potential obstacles for replication forks to prevent cell death. When PAR formation in response to genotoxic stress is compromised (hypo-PARylation) with regards to the timing of PARylation, a total level or the complexity of PARs (step 2), elevated replication stress due to compromised clearance of base lesions by BER in S-phase occurs (step 3). Null-PARylation of cells by either PARP1 deletion (−/−) or PARP inhibitors (PARPi) (step 2) additionally accumulate replication-independent DNA breaks as well as replication stress (step 3), leading to excessive DNA damage progression. As a consequence of the elevated genotoxicity in S-phase (step 3), cells with hypo-PARylation preferentially undergo senescence (step 4). In contrast, PARP1 deletion (−/−) or PARP inhibitors (PARPi) cause excessive cell death due to DNA damage overload (step 4). Eventually, both cellular phenotypes, i.e. senescence and cell death, dictate a striking organismal recovery defect (step 5). Notably, the PARylation acceptors, such as PARP1 (as a major target), and the binding partners of PAR would probably participate in the cell fate determination.
DISCUSSION
In the current study, we engineered a novel PARP1 mutant mouse model, in which the PARP1 protein is intact, but its catalytic capacity is impaired. Similar to the PARP1−/− knockout mice (15,50,58), PARP1D993A/D993A mice are viable and phenotypically normal. We showed that a moderate modulation of the kinetics of the PARP1 activity and a reduction of the PAR chain complexity is sufficient to sensitize mice to alkylating agents, but does not affect normal tissue development and homeostasis. Neither the PARP1 protein, nor its full activity, is essential for the development and adult life under physiological conditions. Thus, a delicate PARylation is a molecular stress sensor in vivo, which is essential for the maintenance of tissue homeostasis under conditions of genotoxic stress.
The kinetics of the PAR formation by the D993A mutant PARP1 is strongly compromised, characterized by a substantial delay of PARylation in response to DNA damage in vitro (at 2 min, Figure 1D and E) and in vivo (Figure 1C, Supplementary Figure S2B–D). Although the PARP1D993A mutant can catch up the WT PARylation capacity in the in vitro assay (Figure 1D and E), the maximal PARylation of mutant PARP1 is compromised in vivo (Figure 1C, Supplementary Figure S2B–D). Importantly, the structure of the PAR chains (i.e. length, branching, complexity) formed by PARP1D993A is also affected. Notably, these defects are associated with an impairment of BER and result in DNA damage accumulation in S-phase.
Given a massive PARylation by wildtype PARP1 in response to DNA damage, it might be surprising to note that a hypo-PARylation and delayed PAR formation does not affect XRCC1 recruitment kinetics. This might have been expected because XRCC1 can bind low levels of PAR, which is apparently sufficient for the XRCC1 recruitment to damage sites (59). The precise function of PARP1 or PAR in BER is still debatable: While PARP1 knockout cells show proficient BER (50,60,61), others demonstrate a PARP1/PARylation dependence in BER (59,62,63). It is possible that the contribution of PARP1/PARylation in BER may involve an interconnectivity of repair proteins, e.g. of PARP1 and PARP2, in a feedback loop (64,65). Interestingly, when compared to WT cells, the retention of XRCC1 is impaired in PARP1D993A/D993A cells (Figure 2D). Although the recruitment of XRCC1 to the DNA lesion has been well studied, the meaning of its retention is poorly understood. Our data suggest that a high degree of the PAR chain complexity likely prevents the premature fall of XRCC1 from the damaged site, which is required for a full efficiency of BER/SSB repair. It seems that the full speed and the complexity of the PAR formation are important for BER efficiency, which probably affects the ensuing coordination of the DDR with the DNA replication machinery. Indeed, XRCC1-deficient cells and PARP inhibitor-treated cells display a higher level of replication-associated γH2AX foci when exposed to the alkylating agent MMS (54). In line with this, XRCC1 has been reported to be recruited to stalled replication forks by PARylation to conduct BER and, importantly, an effective replication restart (66).
Consistent with a previous study showing that PARP1-deleted cells acquire S-phase γH2AX foci following alkylation-induced replication stress (54), PARP1D993A/D993A pMEFs exhibit an increased DNA damage in S-phase when challenged with oxidative and alkylating damage, although not as high as in PARP1−/− cells (Figure 4B). These are in line with the requirement of PARP1 and the PARP activity for intra-S checkpoint activation (6,10,12,13). The oxidized bases and alkylating damage are primarily repaired by BER, and can stall replication forks, if remain unrepaired. Notably, the BER defect is relatively mild in the PARP1D993A/D993A pMEFs, which nevertheless correlates well with the increased γH2AX foci in the S-phase. The high level of S-phase-associated DNA damage could be due to a deficient BER in these hypo-PARylation mutant cells. BER components are known to physically and functionally interact with replication forks, being engaged in their repair during replication stress by HU or DNA base lesions (66–68). It is conceivable that the impaired assembly of the BER complex by slow or low PAR formation delays the repair of replication forks, which hinder their stabilization and subsequent restart. Although the basal BER defect is rather mild, the rapid repair of oxidative lesions and alkylation damage is critical specifically during S-phase to avoid that a replication fork meets a DNA lesion or an active repair site, causing fork stalling and collapses. Indeed, we find the emergence of highly toxic DSBs during the S-phase under oxidative stress. The replication-coupled DNA damage (γH2AX foci) and DSBs (53BP1 foci) were severely aggravated in the absence of the PARP1 protein (PARP1−/−; Figure 4B and E).
Consistent with the notion that a disturbed restart of DNA synthesis following base lesions could entail DSBs (69), PARP1−/− and PARP1D993A/D993A cells showed a moderate, but a significant increase in DSBs, coinciding with replication restart (Figure 4B and E). It is perhaps unexpected that just a modest reduction of PAR formation is sufficient for accumulating DNA damage and DSBs, albeit at a relatively lower level compared to complete PARP inhibition by inhibitors. Thus, the full speed of PARylation by PARP1 and the PAR structures (length and branching) are critical for preventing a replication fork collapse and for mitigating the rise of replication-restart-associated DSBs during the recovery from DNA base lesions. Indeed, PARP1 and PARylation are involved in replication restart at various levels. Firstly, PARP1 can promote replication restart by recruiting MRE11 to facilitate the end-resection of stalled replication forks for a homology-directed repair (10). Secondly, PARP activity inhibits the replication fork restart by stabilizing them in their regressed state via inhibition of their RECQ1-mediated reactivation (70). Taken together, it seems that the rapid activity of PARP1 upon acute DNA damage is most likely less critical in other cell cycle phases, but it is essential for the repair of DNA base lesions in the S-phase.
As a major outcome of the defective BER and increased replication-associated DSBs, the PARP1D993A/D993A mutation changes the cells’ fate following an exposure to genotoxic stress. In this regard, PARP1−/− and PARP1D993A/D993A cells displayed a poor survival in cellular survival assays (Figure 5). Nevertheless, the survival of PARP1D993A/D993A cells always displays an intermediated degree in between WT and PARP1−/− cells, correlating with the extent of the PARylation impairment of D993A mutant PARP1 in DDR. Interestingly, a low dose of MNNG induces senescence particularly in PARP1D993A/D993A and, to a lesser extent, in PARP1−/− pMEFs (Figure 5G). Using different doses of MNNG allows visualizing that while the absence of PARP1 and its PAR formation (e.g. in PARP1−/−) aggravates cell death and does not strongly drive senescence in response to alkylation, hypo-PARylation rather renders cells exquisitely prone to senesce but less to undergo cell death.
We propose that a timely PARylation on target proteins (including PARP1) in response to DNA damage primes cellular and organismal outcome by safeguarding cell fate choice. Intriguingly, the slowing of DNA damage-induced PAR formation and a moderate alteration of the PAR structures are sufficient to change cell fate (Figure 6C). A deficiency in PARylation impairs BER, which impedes the repair and restart of replication forks eventually leading to SSBs and DSBs and, causing cellular senescence under hypo-PARylation, or cell death under null-PARylation. It is conceivable that PAR binders may participate in this response by being orchestrated by the accurate kinetics and the level of the PAR complexity, which provide the spatiotemporal context for PAR binders and downstream responses, for example, the faithful repair of damaged replication forks (Figure 6C). Altogether, we conclude that both the dynamics of PARylation and the complexity level of PARs are decisive for cell fate in response to DNA damage.
Despite a relative protective action of PARP1D993A/D993A cells on cell death compared to the PARP1−/− counterpart, the alkylating agent MNU inflicts gastrointestinal cell death resulting in colitis in PARP1D993A/D993A animals, completely phenocopying PARP1−/− mice (Figure 6A; (15,71)). Of note, similar to PARP1−/− mice (15,17,20) PARP1D993A/D993A mice are also sensitive to whole body ionizing radiation (2). These in vivo results are striking, given the modest reduction of the PAR complexity and the slow PARylation in biochemical and cellular assays. Several possibilities can account for this striking phenotype: (i) The effect in MEFs and in culture conditions can be compensated for by other pathways. (ii) In vivo, there are mixtures of different cell types, which each have different responses and interactions with each other. (iii) A compromised complexity of PARs may engage different PAR binders, which in turn trigger different signalling cascades in tissues, which cannot be assessed in cell cultures. Thus, it is plausible that a tissues response may reflect a synergistic effect of various cellular responses, which may amplify a DDR from only a modest compromise of PARylation leading to a severe biological outcome after DNA damage. It is interesting to note that the polymorphism of PARP1 at V762A imposes a very mild difference on the PARP1 activity in human tissues and cells, which, however, shows an association with cancer risks (72–75), indicating that a delicate modulation of the PARP1 activity would have a biological impact in organisms. However, the importance of the PARP1 protein and PAR homeostasis in other biological pathways, for example in chromatin remodelling, or as a cofactor to control transcription (23,26–29) have not been investigated in the current study.
The present study documents that a minor disturbance in the homeostasis of PAR, exemplified here by a mutation causing hypo-PARylation, can amplify the mild survival or senescence response at the cellular level up to a detrimental lethality of the whole organism. The dynamics of PAR formation is hence critical for cells and organisms to cope with acute DNA damage. Collectively, we conclude that PARP1’s full activity is dispensable under unperturbed physiological conditions, whereas the proper PARylation response is essential for organismal survival upon genotoxic stress.
Supplementary Material
ACKNOWLEDGEMENTS
We cordially thank T. Joerss for performing the blastocyst injections. We are also grateful for D. Galendo, C. Birch-Hirschfeld, S. Hoppe for their excellent assistance in the maintenance of the mouse colonies. We thank Drs A. Gompf and N. Andreas for technical assistance in flow cytometry. H.S. was a member of the DFG-funded RTG1715. We thank E. Stockel for editing the manuscript. We thank all members of Wang Lab for critical discussion of the project.
Author contributions: H.S. designed and performed the majority of the experiments, analysed data and wrote the manuscript; C.B. performed gene targeting, contributed to the PAR-IF and HiMAC data analysis; K.S. contributed to MEF immortalization and cell survival assay; K.K. and S.E. contributed to the gene targeting in ES cells; WK.M. performed part of the cell death assays; P.G. performed XRCC1-recruitment; A.K. and T.Z. performed PAR chain analysis; E.F. characterized PARylation; D.L.P. contributed to preparation of histology; T.K. contributed to high-content microscopy and analysis; A.B. and A.M. contributed to the analysis of PARylation and discussion; M.O.H. contributed to the project design and discussion; Z.-Q.W. designed the experiments and wrote the manuscript.
Footnotes
Present addresses:
Harald Schuhwerk, Department of Experimental Medicine 1, FAU University Erlangen-Nürnberg, Erlangen, Germany.
Christopher Bruhn, The FIRC Institute of Molecular Oncology, Milan, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
C.B. was a recipient of a student fellowship from Studienstiftung des Deutschen Volkes, Germany. A.K. and T.Z. are members of the DFG-funded Konstanz Research School Chemical Biology (KoRSCB). Mass spectrometric analysis at University of Konstanz was supported by a DFG grant for scientific instrumentation [INST 38/537-1]; ADP-ribosylation research in the laboratory of M.O. Hottiger is funded by the Kanton of Zurich and the Swiss National Science Foundation [310030B_138667 and 310030_157019]. This work was supported in part by the Deutsche Forschungsgemeinschaft (DFG), the Deutsche Krebshilfe (DKH) and the Leibniz Gemeinschaft. Funding for open access charge: Institutional funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bai P. Biology of poly(ADP-ribose) polymerases: the factotums of cell maintenance. Mol. Cell. 2015; 58:947–958. [DOI] [PubMed] [Google Scholar]
- 2. Schuhwerk H., Atteya R., Siniuk K., Wang Z.Q.. PARPing for balance in the homeostasis of poly(ADP-ribosyl)ation. Semin. Cell Dev. Biol. 2016; 63:81–91. [DOI] [PubMed] [Google Scholar]
- 3. Abplanalp J., Hottiger M.O.. Cell fate regulation by chromatin ADP-ribosylation. Semin. Cell Dev. Biol. 2016; 63:114–122. [DOI] [PubMed] [Google Scholar]
- 4. Gibson B.A., Kraus W.L.. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012; 13:411–424. [DOI] [PubMed] [Google Scholar]
- 5. Hottiger M.O., Hassa P.O., Luscher B., Schuler H., Koch-Nolte F.. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010; 35:208–219. [DOI] [PubMed] [Google Scholar]
- 6. Min W., Bruhn C., Grigaravicius P., Zhou Z.W., Li F., Kruger A., Siddeek B., Greulich K.O., Popp O., Meisezahl C. et al. Poly(ADP-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat. Commun. 2013; 4:2993. [DOI] [PubMed] [Google Scholar]
- 7. Teloni F., Altmeyer M.. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res. 2016; 44:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Min W., Wang Z.Q.. Poly (ADP-ribose) glycohydrolase (PARG) and its therapeutic potential. Front. Biosci. (Landmark Ed.). 2009; 14:1619–1626. [DOI] [PubMed] [Google Scholar]
- 9. Martin-Hernandez K., Rodriguez-Vargas J.M., Schreiber V., Dantzer F.. Expanding functions of ADP-ribosylation in the maintenance of genome integrity. Semin. Cell Dev. Biol. 2016; 63:92–101. [DOI] [PubMed] [Google Scholar]
- 10. Bryant H.E., Petermann E., Schultz N., Jemth A.S., Loseva O., Issaeva N., Johansson F., Fernandez S., McGlynn P., Helleday T.. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009; 28:2601–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sugimura K., Takebayashi S., Taguchi H., Takeda S., Okumura K.. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008; 183:1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horton J.K., Stefanick D.F., Kedar P.S., Wilson S.H.. ATR signaling mediates an S-phase checkpoint after inhibition of poly(ADP-ribose) polymerase activity. DNA Repair (Amst.). 2007; 6:742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kedar P.S., Stefanick D.F., Horton J.K., Wilson S.H.. Interaction between PARP-1 and ATR in mouse fibroblasts is blocked by PARP inhibition. DNA Repair. 2008; 7:1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M.. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012; 19:417–423. [DOI] [PubMed] [Google Scholar]
- 15. de Murcia J.M., Niedergang C., Trucco C., Ricoul M., Dutrillaux B., Mark M., Oliver F.J., Masson M., Dierich A., LeMeur M. et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trucco C., Oliver F.J., de Murcia G., Menissier-de Murcia J.. DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 1998; 26:2644–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Z.Q., Stingl L., Morrison C., Jantsch M., Los M., Schulze-Osthoff K., Wagner E.F.. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997; 11:2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Y.G., Cortes U., Patnaik S., Jasin M., Wang Z.Q.. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 2004; 23:3872–3882. [DOI] [PubMed] [Google Scholar]
- 19. Durkacz B.W., Omidiji O., Gray D.A., Shall S.. (ADP-ribose)n participates in DNA excision repair. Nature. 1980; 283:593–596. [DOI] [PubMed] [Google Scholar]
- 20. Cortes U., Tong W.M., Coyle D.L., Meyer-Ficca M.L., Meyer R.G., Petrilli V., Herceg Z., Jacobson E.L., Jacobson M.K., Wang Z.Q.. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell. Biol. 2004; 24:7163–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Min W., Cortes U., Herceg Z., Tong W.M., Wang Z.Q.. Deletion of the nuclear isoform of poly(ADP-ribose) glycohydrolase (PARG) reveals its function in DNA repair, genomic stability and tumorigenesis. Carcinogenesis. 2010; 31:2058–2065. [DOI] [PubMed] [Google Scholar]
- 22. Gao H., Coyle D.L., Meyer-Ficca M.L., Meyer R.G., Jacobson E.L., Wang Z.Q., Jacobson M.K.. Altered poly(ADP-ribose) metabolism impairs cellular responses to genotoxic stress in a hypomorphic mutant of poly(ADP-ribose) glycohydrolase. Exp. Cell Res. 2007; 313:984–996. [DOI] [PubMed] [Google Scholar]
- 23. Posavec Marjanovic M., Crawford K., Ahel I.. PARP, transcription and chromatin modeling. Semin. Cell Dev. Biol. 2016; 63:102–113. [DOI] [PubMed] [Google Scholar]
- 24. Tulin A., Spradling A.. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science. 2003; 299:560–562. [DOI] [PubMed] [Google Scholar]
- 25. Kim M.Y., Mauro S., Gevry N., Lis J.T., Kraus W.L.. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004; 119:803–814. [DOI] [PubMed] [Google Scholar]
- 26. Chang W.J., Alvarez-Gonzalez R.. The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J. Biol. Chem. 2001; 276:47664–47670. [DOI] [PubMed] [Google Scholar]
- 27. Hassa P.O., Covic M., Hasan S., Imhof R., Hottiger M.O.. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001; 276:45588–45597. [DOI] [PubMed] [Google Scholar]
- 28. Nakajima H., Nagaso H., Kakui N., Ishikawa M., Hiranuma T., Hoshiko S.. Critical role of the automodification of poly(ADP-ribose) polymerase-1 in nuclear factor-kappaB-dependent gene expression in primary cultured mouse glial cells. J. Biol. Chem. 2004; 279:42774–42786. [DOI] [PubMed] [Google Scholar]
- 29. Zerfaoui M., Errami Y., Naura A.S., Suzuki Y., Kim H., Ju J., Liu T., Hans C.P., Kim J.G., Abd Elmageed Z.Y. et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J. Immunol. 2010; 185:1894–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haince J.F., McDonald D., Rodrigue A., Dery U., Masson J.Y., Hendzel M.J., Poirier G.G.. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008; 283:1197–1208. [DOI] [PubMed] [Google Scholar]
- 31. Hochegger H., Dejsuphong D., Fukushima T., Morrison C., Sonoda E., Schreiber V., Zhao G.Y., Saberi A., Masutani M., Adachi N. et al. Parp-1 protects homologous recombination from interference by Ku and ligase IV in vertebrate cells. EMBO J. 2006; 25:1305–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang M., Wu W., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G.. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Claybon A., Karia B., Bruce C., Bishop A.J.. PARP1 suppresses homologous recombination events in mice in vivo. Nucleic Acids Res. 2010; 38:7538–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schultz N., Lopez E., Saleh-Gohari N., Helleday T.. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003; 31:4959–4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orsburn B., Escudero B., Prakash M., Gesheva S., Liu G., Huso D.L., Franco S.. Differential requirement for H2AX and 53BP1 in organismal development and genome maintenance in the absence of poly(ADP)ribosyl polymerase 1. Mol. Cell. Biol. 2010; 30:2341–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang F., Shi J.Z., Bian C.J., Yu X.C.. Poly(ADP-ribose) mediates the BRCA2-dependent early DNA damage response. Cell Rep. 2015; 13:678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ying S., Hamdy F.C., Helleday T.. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012; 72:2814–2821. [DOI] [PubMed] [Google Scholar]
- 38. Morrison C., Smith G.C., Stingl L., Jackson S.P., Wagner E.F., Wang Z.Q.. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat. Genet. 1997; 17:479–482. [DOI] [PubMed] [Google Scholar]
- 39. Luo X., Kraus W.L.. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012; 26:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burkle A., Virag L.. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 2013; 34:1046–1065. [DOI] [PubMed] [Google Scholar]
- 41. Shieh W.M., Ame J.C., Wilson M.V., Wang Z.Q., Koh D.W., Jacobson M.K., Jacobson E.L.. Poly(ADP-ribose) polymerase null mouse cells synthesize ADP-ribose polymers. J. Biol. Chem. 1998; 273:30069–30072. [DOI] [PubMed] [Google Scholar]
- 42. Liscio P., Camaioni E., Carotti A., Pellicciari R., Macchiarulo A.. From polypharmacology to target specificity: the case of PARP inhibitors. Curr. Top. Med. Chem. 2013; 13:2939–2954. [DOI] [PubMed] [Google Scholar]
- 43. Tong W.M., Cortes U., Wang Z.Q.. Poly(ADP-ribose) polymerase: a guardian angel protecting the genome and suppressing tumorigenesis. Biochim. Biophys. Acta. 2001; 1552:27–37. [DOI] [PubMed] [Google Scholar]
- 44. Zhou Z.W., Liu C., Li T.L., Bruhn C., Krueger A., Min W., Wang Z.Q., Carr A.M.. An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet. 2013; 9:e1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hassa P.O., Buerki C., Lombardi C., Imhof R., Hottiger M.O.. Transcriptional coactivation of nuclear factor-kappaB-dependent gene expression by p300 is regulated by poly(ADP)-ribose polymerase-1. J. Biol. Chem. 2003; 278:45145–45153. [DOI] [PubMed] [Google Scholar]
- 46. Hassa P.O., Haenni S.S., Buerki C., Meier N.I., Lane W.S., Owen H., Gersbach M., Imhof R., Hottiger M.O.. Acetylation of Poly(ADP-ribose) Polymerase-1 by p300/CREB-binding Protein Regulates Coactivation of NF-Œ∫B-dependent Transcription. J. Biol. Chem. 2005; 280:40450–40464. [DOI] [PubMed] [Google Scholar]
- 47. Fahrer J., Kranaster R., Altmeyer M., Marx A., Burkle A.. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007; 35:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martello R., Mangerich A., Sass S., Dedon P.C., Burkle A.. Quantification of cellular poly(ADP-ribosyl)ation by stable isotope dilution mass spectrometry reveals tissue- and drug-dependent stress response dynamics. ACS Chem. Biol. 2013; 8:1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zubel T., Martello R., Burkle A., Mangerich A.. Quantitation of poly(ADP-ribose) by isotope dilution mass spectrometry. Methods Mol. Biol. (Clifton, N.J.). 2017; 1608:3–18. [DOI] [PubMed] [Google Scholar]
- 50. Wang Z.Q., Auer B., Stingl L., Berghammer H., Haidacher D., Schweiger M., Wagner E.F.. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995; 9:509–520. [DOI] [PubMed] [Google Scholar]
- 51. Bruhn C., Kroll T., Wang Z.Q.. Systematic characterization of cell cycle phase-dependent protein dynamics and pathway activities by high-content microscopy-assisted cell cycle phenotyping. Genomics Proteomics Bioinformatics. 2014; 12:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kamentsky L., Jones T.R., Fraser A., Bray M.A., Logan D.J., Madden K.L., Ljosa V., Rueden C., Eliceiri K.W., Carpenter A.E.. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011; 27:1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grigaravicius P., Rapp A., Greulich K.O.. A direct view by immunofluorescent comet assay (IFCA) of DNA damage induced by nicking and cutting enzymes, ionizing (137)Cs radiation, UV-A laser microbeam irradiation and the radiomimetic drug bleomycin. Mutagenesis. 2009; 24:191–197. [DOI] [PubMed] [Google Scholar]
- 54. Ensminger M., Iloff L., Ebel C., Nikolova T., Kaina B., Lbrich M.. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014; 206:29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Simonin F., Poch O., Delarue M., de Murcia G.. Identification of potential active-site residues in the human poly(ADP-ribose) polymerase. J. Biol. Chem. 1993; 268:8529–8535. [PubMed] [Google Scholar]
- 56. El-Khamisy S.F., Masutani M., Suzuki H., Caldecott K.W.. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003; 31:5526–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bruhn C., Zhou Z.W., Ai H., Wang Z.Q.. The essential function of the MRN complex in the resolution of endogenous replication intermediates. Cell Rep. 2014; 6:182–195. [DOI] [PubMed] [Google Scholar]
- 58. Masutani M., Suzuki H., Kamada N., Watanabe M., Ueda O., Nozaki T., Jishage K., Watanabe T., Sugimoto T., Nakagama H. et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:2301–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Breslin C., Hornyak P., Ridley A., Rulten S.L., Hanzlikova H., Oliver A.W., Caldecott K.W.. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015; 43:6934–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Strom C.E., Johansson F., Uhlen M., Szigyarto C.A., Erixon K., Helleday T.. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011; 39:3166–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vodenicharov M.D., Sallmann F.R., Satoh M.S., Poirier G.G.. Base excision repair is efficient in cells lacking poly(ADP-ribose) polymerase 1. Nucleic Acids Res. 2000; 28:3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Campalans A., Kortulewski T., Amouroux R., Menoni H., Vermeulen W., Radicella J.P.. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res. 2013; 41:3115–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hanssen-Bauer A., Solvang-Garten K., Sundheim O., Pena-Diaz J., Andersen S., Slupphaug G., Krokan H.E., Wilson D.M. 3rd, Akbari M., Otterlei M.. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ. Mol. Mutagen. 2011; 52:623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mortusewicz O., Ame J.C., Schreiber V., Leonhardt H.. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007; 35:7665–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hanzlikova H., Gittens W., Krejcikova K., Zeng Z., Caldecott K.W.. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 2017; 45:2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ying S., Chen Z., Medhurst A.L., Neal J.A., Bao Z., Mortusewicz O., McGouran J., Song X., Shen H., Hamdy F.C. et al. DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2015; 76:1078–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Parlanti E., Locatelli G., Maga G., Dogliotti E.. Human base excision repair complex is physically associated to DNA replication and cell cycle regulatory proteins. Nucleic Acids Res. 2007; 35:1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Levy N., Oehlmann M., Delalande F., Nasheuer H.P., Van Dorsselaer A., Schreiber V., de Murcia G., Menissier-de Murcia J., Maiorano D., Bresson A.. XRCC1 interacts with the p58 subunit of DNA Pol alpha-primase and may coordinate DNA repair and replication during S phase. Nucleic Acids Res. 2009; 37:3177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Petermann E., Helleday T.. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010; 11:683–687. [DOI] [PubMed] [Google Scholar]
- 70. Berti M., Ray Chaudhuri A., Thangavel S., Gomathinayagam S., Kenig S., Vujanovic M., Odreman F., Glatter T., Graziano S., Mendoza-Maldonado R. et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013; 20:347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Masutani M., Nozaki T., Nakamoto K., Nakagama H., Suzuki H., Kusuoka O., Tsutsumi M., Sugimura T.. The response of Parp knockout mice against DNA damaging agents. Mutat. Res. 2000; 462:159–166. [DOI] [PubMed] [Google Scholar]
- 72. Yu H., Ma H., Yin M., Wei Q.. Association between PARP-1 V762A polymorphism and cancer susceptibility: a meta-analysis. Genet. Epidemiol. 2012; 36:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lockett K.L., Hall M.C., Xu J., Zheng S.L., Berwick M., Chuang S.C., Clark P.E., Cramer S.D., Lohman K., Hu J.J.. The ADPRT V762A genetic variant contributes to prostate cancer susceptibility and deficient enzyme function. Cancer Res. 2004; 64:6344–6348. [DOI] [PubMed] [Google Scholar]
- 74. Alanazi M., Pathan A.A., Abduljaleel Z., Shaik J.P., Alabdulkarim H.A., Semlali A., Bazzi M.D., Parine N.R.. Association between PARP-1 V762A polymorphism and breast cancer susceptibility in Saudi population. PLoS One. 2013; 8:e85541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rank L., Veith S., Gwosch E.C., Demgenski J., Ganz M., Jongmans M.C., Vogel C., Fischbach A., Buerger S., Fischer J.M. et al. Analyzing structure-function relationships of artificial and cancer-associated PARP1 variants by reconstituting TALEN-generated HeLa PARP1 knock-out cells. Nucleic Acids Res. 2016; 44:10386–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.