


Abstract
The RNA-Seq technology has revolutionized transcriptome characterization not only by accurately quantifying gene expression, but also by the identification of novel transcripts like chimeric fusion transcripts. The ‘fusion’ or ‘chimeric’ transcripts have improved the diagnosis and prognosis of several tumors, and have led to the development of novel therapeutic regimen. The fusion transcript detection is currently accomplished by several software packages, primarily relying on sequence alignment algorithms. The alignment of sequencing reads from fusion transcript loci in cancer genomes can be highly challenging due to the incorrect mapping induced by genomic alterations, thereby limiting the performance of alignment-based fusion transcript detection methods. Here, we developed a novel alignment-free method, ChimeRScope that accurately predicts fusion transcripts based on the gene fingerprint (as k-mers) profiles of the RNA-Seq paired-end reads. Results on published datasets and in-house cancer cell line datasets followed by experimental validations demonstrate that ChimeRScope consistently outperforms other popular methods irrespective of the read lengths and sequencing depth. More importantly, results on our in-house datasets show that ChimeRScope is a better tool that is capable of identifying novel fusion transcripts with potential oncogenic functions. ChimeRScope is accessible as a standalone software at (https://github.com/ChimeRScope/ChimeRScope/wiki) or via the Galaxy web-interface at (https://galaxy.unmc.edu/).
INTRODUCTION
A major characteristic of many neoplasias is the presence of chromosomal re-arrangements often leading to higher chances of abnormal fusion of two separate genes (1). These fusion products are considered to be present at DNA level; and are therefore considered unique to cancer. However deep-sequencing technology revealed many more fusion transcripts, without detectable rearrangement at the DNA level and are partly generated via intergenic splicing (2). Over the last decade, an increasing number of fusion transcripts have been identified in major malignancies (3–5). A fusion transcript can contribute to oncogenicity by promoting the expression of a proto-oncogene (6); by deregulating a tumor suppressor gene (7); or by modifying the original structure/function of a protein to form a novel abnormal protein that stimulates tumorigenesis (8). Since oncogenic chimeric transcripts are cancer-specific (9), they offer a unique opportunity to be identified as cancer biomarkers for diagnostic (10,11), prognostic (12) and therapeutic (13,14) purposes to provide targeted cancer treatment. For instance, the first reported fusion gene, known as BCR-ABL1, was discovered in chronic myeloid leukemia (CML) in the early 1970s (15). This fusion gene was found to be associated with the Philadelphia chromosome (16), a recurrent translocation event found in more than 90% of CML patients. BCR-ABL1 encodes a constantly active tyrosine kinase that promotes leukemogenesis (17). Imatinib (18), one of the first drugs that target fusion genes, significantly improves the overall survival rate of CML patients (19) by inhibiting the tyrosine kinase activity of the BCR-ABL1 fusion protein.
High-throughput transcriptome sequencing, or RNA-Seq, has been widely used for fusion transcript detection, where the whole transcriptome from tumor cells, along with the chimeric fusion transcripts, are extracted and sequenced. To date, more than 20 tools have been developed for fusion transcript detection using RNA-Seq data, majority of which are alignment-based (20). Alignment-based methods in general report the optimal alignment results between short reads and the reference transcript sequences, and only achieve better performance when the compared sequences show high levels of homology (21). However, due to the highly perturbed nature of cancer genomes, aligning short reads generated from cancer genomes against the normal reference genome limits these methods from achieving good alignment. This is especially true for the reads derived from complex rearranged regions where fusion events often occur, thereby resulting in low prediction accuracies for these alignment-based methods (20). Moreover, identification of fusion transcripts by the majority of alignment-based methods depends heavily on the number of detected fusion reads. This lead to a bias toward identifying fusion transcripts with moderate to high expression (20) and could potentially leave novel, low expressed fusions undetected.
In order to address issues mentioned above, we developed a novel alignment-free method named ChimeRScope. Alignment-free approaches generally employ a broad collection of methods, including those based on k-mer frequencies or substrings, information theory, graphical representation or sequence clustering. ChimeRScope, interpreted as Chimeric RNA Scope or Chi(k)-meR Scope, predicts fusion transcripts by assessing the gene fingerprint sequences (in the form of k-mers) from RNA-Seq paired-end reads. In this method, reads containing two sets of gene fingerprints from two different genes will be scored based on the quality and the quantity of the fingerprint sequences. Such reads will be marked as fusion event supporting reads (FESRs) for downstream analysis. Using this approach, ChimeRScope eliminates the issues associated with using poorly aligned reads when predicting fusion candidates, thereby avoiding the issues often encountered with alignment-based methods. While the scoring algorithm uses an alignment-free approach, ChimeRScope can take advantage of the discordant reads from the pre-aligned datasets by third-party tools such as TopHat, as it only needs the discordant reads as input for fusion transcript prediction. Also, to identify the exact fusion junctions of the predicted fusion transcripts, toward the end, ChimeRScope also uses a targeted alignment step to visualize the fusion transcripts by aligning the identified FESRs against corresponding fusion partners. The final output from the ChimeRScope pipeline comprises of a list of predicted fusion transcripts with confidence scores and detailed information of the fusion events, fusion orientations and predicted fusion junction sequences, presented as text files and vector-based images.
ChimeRScope, implemented in JAVA, is a platform-independent software that can be installed and configured with minimal effort. It can be run as a command-line application, or integrated into online tools such as Galaxy server (22) for accessing the user-friendly Graphical User Interfaces. For demonstration purpose, we installed ChimeRScope on our local Galaxy server. The ChimeRScope suite, manuals and instructions (including Galaxy server configuration files for ChimeRScope) are available for downloading at ChimeRScope wiki site.
MATERIALS AND METHODS
ChimeRScope method overview
ChimeRScope is a novel alignment-free method that identifies fusion transcript candidates based on k-mer frequencies. It uses each k-mer as an independent unit and a potential gene fingerprint assigned with a weightage score that is negatively correlated with its frequency. ChimeRScope stores all k-mers in the transcriptome into a huge hash table (Gene-Fingerprint library, GF-library or k-mer library; Figure 1 A and B). Each hash key in the table represents a unique k-mer with the corresponding hash value representing the list of transcripts that contain the k-mer. When determining the origins of discordant reads, k-mers with low frequencies across different transcripts are considered as valuable gene fingerprints because they are more discriminative in the scoring function. Using this idea, a paired-end read will be classified as an FESR by ChimeRScope if this read embeds two sets of fingerprint sequences from two different genes with high confidence, which suggests a potential fusion event between these two genes (Figure 1 C–F). ChimeRScope summarizes the confidence scores of all FESRs and outputs a list of high-quality fusion transcripts after processing the entire discordant reads. To further investigate the reported fusion candidates, we also developed a module that performs targeted alignment between FESRs and the associated fusion transcript partners. This module outputs detailed information on fusion partners (e.g. chromosomal locations, orientations, fusion junction sequences and coordinates), all of which aids in downstream experimental validation and functional analysis. This information is also transformed into vector-based (i.e. Scalable Vector Graphics, or SVG) images to understand the fusion events better.
Figure 1.

ChimeRScope strategy for identifying FESRs: an example. A k-mer library is created by first, (A) generating all the k-mer profiles for all the genes. Next, we compare all k-mer profiles so that for each possible k-mer, the list of genes that contains that k-mer can be quickly identified. (B) A snapshot of a hypothetical example of the k-mer library. (C) The Circos map on the right illustrates an example of how ChimeRScope determine an FESR. A discordant paired-end read (100 bp × 2) that fails the stringent alignment against the reference genome is plotted in a circular layout with each nucleobase type represented by a unique color. (D) Four different variations of each k-mer in the read (e.g. highlighted region shows the 11th 17-mer for read 1 from base 11 to 27) will be created and searched against the k-mer library in order to obtain (E) a list of gene IDs that uses the corresponding k-mer as fingerprint. Each block represents a k-mer and each color here represents a unique gene ID. For example, four genes (G1: red, G2: green, G3: yellow and G4: orange) are related to the 11th 17-mer (from the 11th nucleotide to 27th nucleotide, as highlighted in gray region) and two genes (G1 and G4) are associated with the 29th 17-mer (highlighted in light yellow). (F) A complete graph is drawn for all eight matched genes. Each vertex in the complete graph represents a unique gene with the size of the vertex proportional to the overall fingerprint score for that gene. The edge value between two genes is defined by the distance (denoted as d) of two closest fingerprints of the gene pair (only listed several values). Gene pairs with small distance values tend to be false positives due to the similar sequences (See ‘Discussion’ and ‘Materials and Methods’ sections). If we consider only those gene pairs with an overlap of <5 bp (or with the distance more than 17 − 5 = 12) are valid fusion candidates, this read will be classified as a FESR that supports the fusion between G1 and G6 because G1 and G6 are two of the largest vertices (suggesting the most possible origins of the read based on the fingerprint sequences) with the distance value >12.
We designed the ChimeRScope algorithm into four distinct modules (Figure 2). The first module, ChimeRScope Builder, constructs a GF-library that serves as a reference library for searching against the k-mers from the sample reads. This is a one-time step that is carried out prior to processing the sample reads for each reference transcriptome. Next, the ChimeRScope Scanner module identifies an FESR based on the k-mer content of the read using a complete graph network model. This is the most memory and compute-intensive step because of the enormous number of search queries against the GF-library. The third module, ChimeRScope Sweeper, summarizes all the FESRs for each fusion candidate and predicts high quality fusion transcripts. The last module, ChimeRScore Examiner, performs targeted alignment of the FESRs against the reference transcripts of the fusion partners for better visualization of the fusion event to define the fusion junctions. ChimeRScope can use the total raw reads or the unmapped reads from previous alignments as input. However, as it only needs the unmapped reads for fusion transcript prediction, using the unaligned raw reads adds extensive computational burden to the program resulting in slow processing. Hence, we recommend the use of discordantly-aligned or unmapped reads as input for this program for expedited processing. Since the alignment step is a part of the standard RNA-Seq data analysis protocol, the input reads for ChimeRScope Scanner can be extracted directly from the sequence alignment files generated from these standard protocols such as TopHat. Therefore, ChimeRScope pipeline for fusion transcript detection can be integrated with popular RNA-Seq data analysis pipelines like the Tuxedo pipeline (23) for simultaneous analysis of the RNA-Seq datasets for both gene expression and fusion transcript detection.
Figure 2.

The flowchart of the ChimeRScope pipeline for fusion transcript prediction. ChimeRScope can be used completely independently (Option 1) or as an add-on module in the standard RNA-Seq analysis pipeline by using the unmapped reads as input (Option 2) from third-party alignment tools. Four different modules are integrated into the ChimeRScope repository. ChimeRScope builder is a one-time step for GF-library creation and it is often carried out before the analysis. ChimeRScope Scanner identifies FESRs from input reads by using the k-mer library generated by ChimeRScope Builder. ChimeRScope Sweeper then summarizes the FESRs and outputs the list of identified fusion transcripts. Lastly, ChimeRScope Examiner will analyze fusion transcript pairs and the corresponding FESRs for fusion transcript junctions with graphical representations.
ChimeRScope Builder
To generate the gene fingerprint library, we extracted all mRNA sequences (Figure 1A) by combining RefSeq human genome and gene model (GRCh38/hg38) using gtf_to_fasta from Tophat (24) package. For each mRNA sequence, ChimeRScope Builder poses a sliding window of size k, in order to generate the complete k-mer profile for each mRNA sequence. ChimeRScope Builder then compares all the k-mers profiles to obtain a huge hash table that uses each existing k-mer as a hash key, and the list of genes that contains that k-mer as the hash value (Figure 1B). All the hash keys and hash values were coded into binaries to reduce memory usage.
ChimeRScope Scanner
ChimeRScope Scanner takes the discordant paired-end reads (in forward-reverse orientation) in fastq format as inputs. We used the TopHat aligner from the Tuxedo pipeline as the example here since it is one of the most popular pipelines for RNA-Seq data analysis. However, other aligners (e.g. STAR (25) and bowtie (26)) that work well on transcriptome sequencing data can also be used with ChimeRScope for fusion transcript prediction. Samtools (27) was used to combine unmapped.bam (TopHat output file) with all the reads that are not properly mapped as read pairs (27) (SAM flag not equal to 2) from the accepted_hits.bam (TopHat output file). Next, all the discordant reads were extracted from the merged bam file using bamToFastq application from BEDTools (28).
For each discordant paired-end read of length 2L (length L for each end), 2(L − k + 1) k-mers were generated (Figure 1C) and four different k-mer variations (original, reverse, complementary and reverse-complementary k-mer) for each k-mer were created and searched against the k-mer library (Figure 1D and E) in order to cover all possible orientations of a fusion event. Next, for each k-mer, a list of transcripts that are associated with the k-mer and its variations were obtained. ChimeRScope Scanner then assigns a normalized weightage score that is negatively correlated with the size of that transcript list. Additionally, to avoid computational costs on common k-mers like poly-A sequence, k-mers associated with more than 100 transcripts was given zero weightage. The normalized weightage score for each k-mer is calculated using 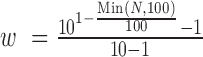 , where w is the weightage score and N is the size of the gene list. After processing the k-mers, ChimeRScope Scanner constructs a complete graph (in the form of 2D matrix) where each vertex represents a unique gene that existed in the k-mer profile of that read (total of n genes). The magnitude of the vertex
, where w is the weightage score and N is the size of the gene list. After processing the k-mers, ChimeRScope Scanner constructs a complete graph (in the form of 2D matrix) where each vertex represents a unique gene that existed in the k-mer profile of that read (total of n genes). The magnitude of the vertex 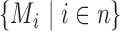 is calculated by summarizing all the weightage scores of the associated k-mers. The edge value
is calculated by summarizing all the weightage scores of the associated k-mers. The edge value 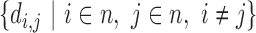 for each gene pair in the graph is calculated by the distance of the two closest gene fingerprints of that pair (Figure 1F). A small distance between two genes indicates that these two genes share high similar gene fingerprint profiles (e.g. distance = 1 means these two genes have k-1 number of nucleotides overlap near the fusion junction) in this read and are more likely to be false positives introduced by point mutations or sequencing errors. If there are several vertices
for each gene pair in the graph is calculated by the distance of the two closest gene fingerprints of that pair (Figure 1F). A small distance between two genes indicates that these two genes share high similar gene fingerprint profiles (e.g. distance = 1 means these two genes have k-1 number of nucleotides overlap near the fusion junction) in this read and are more likely to be false positives introduced by point mutations or sequencing errors. If there are several vertices 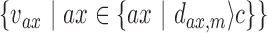 connected with the biggest vertex/vertices
connected with the biggest vertex/vertices 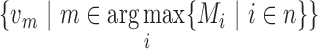 with the edge value more than the distance cut-off c, this read will be classified as a FESR that supports the fusion event between
with the edge value more than the distance cut-off c, this read will be classified as a FESR that supports the fusion event between 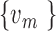 and
and 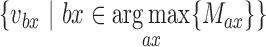 , which should be two of the most evident origins for a FESR (Figure 1F). The weightage score for each FESR is calculated using
, which should be two of the most evident origins for a FESR (Figure 1F). The weightage score for each FESR is calculated using 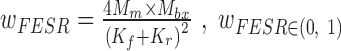 , where Kf and Kr are the maximum number of k-mers each end can have for the read. A FESR will achieve its largest weightage score only when
, where Kf and Kr are the maximum number of k-mers each end can have for the read. A FESR will achieve its largest weightage score only when 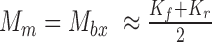 , which shows a perfect fusion transcript event where the k-mer profile for each end of the read is the unique gene fingerprint set for each gene in the pair. Here, FESRs with very low scores (default cut-off is 0.1) are filtered out to prevent false positives.
, which shows a perfect fusion transcript event where the k-mer profile for each end of the read is the unique gene fingerprint set for each gene in the pair. Here, FESRs with very low scores (default cut-off is 0.1) are filtered out to prevent false positives.
ChimeRScope Sweeper
The final confidence score for each fusion transcript is calculated based on the overall distribution of the related FESRs’ weightage scores using an iteration function (Supplementary File 1). This function takes the FESR with the smallest weightage score from the FESR stack and updated it to the confidence score. Therefore, the updated confidence score is always larger than the current FESR weightage score. This guarantees that the final score is always higher than the score of the best FESR. Therefore, fusion transcripts with only a few FESRs can have high confidence scores if they have high quality FESRs. On the other hand, the confidence score accumulates at a slower rate when processing a subset of FESRs with the same level of weightage scores. Using this strategy, it prevents false positives from having high confidence scores when only large amounts of low quality FESRs were present. The final confidence score for a fusion transcript is ranges from 0 to 1. Any fusion transcript with a score of more than 0.5 is considered as a true fusion transcript candidate. The cut-off score indicates that a fusion transcript will be classified as true when it receives, for example, 70% support in average from each fusion partner (0.7*0.7 = 0.49).
Before ChimeRScope reports the final list of fusion transcripts, several filters were applied to remove false positives. These include the homology, sequence similarity, occurrences and genomic-distance filters. ChimeRScope can introduce false positives if the reported fusion transcript pair shares homologous fingerprint profiles (Figure 3A). Therefore, fusion pairs that comes from the same gene family (29) are filtered out. Moreover, ChimeRScope also uses Smith–Waterman algorithm (30) to further exclude those fusion transcripts with similar sequences within the fusion pairs. Adapter contaminations can also introduce false positives (Figure 3B). Although adapter sequences in RNA-Seq are designed not to match any gene sequences, the reversed adapter sequences can be part of the fingerprints for a subset of genes. Reads with adapter contamination sequenced from any expressed genes would exhibit a fusion-like pattern between the expressed gene and genes from that subset. To remove such false positives, ChimeRScope calculates the number of occurrences where one gene has been reported to be fused with other genes among the same dataset. Fusion transcripts involved with such genes with high counts (The default cut-off value is 200. The selection of this value is based on the results from the tested simulated datasets) should be false positives. This is because if the adapter sequences are included in the paired-end reads, they can pair with any associated genes, creating fusion-like patterns between the adapter sequences and these genes. Furthermore, the genomic-distance filter is also used to filter out read-throughs, a class of chimeric transcripts with true fusion-like patterns but are not biologically significant for tumorigenesis (31). At last, for fusion transcript analysis on real RNA-Seq datasets, an alignment search for the fusion sequences against non-coding genes (NCBI BLAST against NR) is recommended for filtering out potential false positives because the regular k-mer library only includes coding mRNAs. This step will filter out false positives caused by non-coding RNAs with fusion-like patterns.
Figure 3.

False positive fusion reads caused by genes with similar sequences and adapter contaminations. (A) An example of the false positives caused by similar sequence region. The reference sequences of G1 and G2 (regions were filled with different patterns) share a similar sequence (illustrated using the same pattern) near the 5΄ region. There are only a few nucleotides that are different (highlighted in red in G2). A paired-end read sequenced from the subject G1 cDNA contains a mutation that changes the fingerprint sequences in the region identical to the sequence of G2. This paired-end read, originated only from G1, will now be classified as a fusion read because it supports the fusion between G1 and G2 due to the above mentioned specific mutation contained in this read. We filtered out this type of false positives by screening for such similar regions in the reported fusion partners. (B) An example of the false positives caused by adapter sequences. Genes and adapters (including the reversed adapter sequence) were filled using different patterns to represent the differences of these sequences. Paired-end reads can have adapter contaminations when the template is smaller than the length of the read or due to other technical issues. ChimeRScope checks four different variations of the k-mers, including the reversed sequence. Although adapter sequences are designed not to significantly match any of the gene sequences, the reverse of the adapter sequences can match to certain genes (G4 matches the reverse adapter sequence in this example). All the paired-end reads sequenced from a cDNA library with adapter contaminations could be classified as fusion reads by ChimeRScope. We filter out this class of false positives by removing fusion transcripts involving partners with high counts because this kind of fusion partners tend to pair with a large number of genes.
ChimeRScope examiner
The Examiner module is designed as a post-analysis module that outputs the models of the fusion transcripts as both text files and SVG images. It performs target alignment using Smith–Waterman algorithm between each fusion transcript reported from the previous module and the corresponding FESRs. ChimeRScope Examiner calculates the estimated fusion junction from each FESR and reports the consensus fusion coordinates for the fusion transcript from the alignment results. In addition, ChimeRScope Examiner also reports the orientations and the strands of the fusion partners, along with the resolved fusion sequences near the fusion junction (+/− 100 bp to the fusion junction). These results were also automatically transformed into vector objects for improved interpretation of the fusion events (Figure 4). The SVG figure is automatically generated for each fusion and can be converted to publication-quality images without changing the aspect ratio. Moreover, most of the elements in the SVG image have their unique name attributes (e.g. transcript ID, exon number, paired-end read name). The name attributes are incorporated at the bottom of the image when users perform mouse-over action on these elements for enhanced understanding of the fusion event.
Figure 4.

A screenshot of the graphical output for fusion transcript DAB2&FRYL. Four different tracks are plotted to illustrate the fusion event. The first track of the SVG image lists the name of the fusion partners with their original orientations and the second track shows the transcripts that might be involved in the fusion event. Each rectangle in the second track represents a coding exon with the size of the box proportional to that exon. The third track, along with the highlighted region between track two and track three, show the amplified region near the fusion junction from each fusion partner. The last track of the image plots a maximum of 71 (determined by the dimension of the forth track) paired-end reads to show the alignment results of selected FESRs against the fusion junction. This figure shows that DAB2 are fused with FRYL near the second exon (5΄ end) of DAB2 and the second exon (3΄ end) of FRYL.
Shannon Index for k-mer size optimization
Shannon Index, also known as Shannon's entropy, was originally described in ecology (32) to quantify the uncertainty in predicting the identity of a species that is taken at random from the dataset. Similarly, k-mer libraries with larger Shannon index values indicate higher uncertainties of predicting the correct origins of randomly sequenced reads (shotgun sequencing). The Shannon index of a k-mer library is calculated using 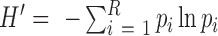 . Here, R is a collection of k-mer classes that have been identified in less than 100 transcripts in a library and pi is the probability of each k-mer class in R. Those k-mers associated with 100 transcripts or more are not included in the k-mer library due to their low specificities; thus they were excluded from the Shannon index calculation.
. Here, R is a collection of k-mer classes that have been identified in less than 100 transcripts in a library and pi is the probability of each k-mer class in R. Those k-mers associated with 100 transcripts or more are not included in the k-mer library due to their low specificities; thus they were excluded from the Shannon index calculation.
Local Galaxy server implementation
Apart from the standalone ChimeRScope package, we also developed a web-based application of ChimeRScope using Galaxy server (22). The Galaxy server instance was installed on our local data analysis server following the instructions provided by Galaxy group (https://wiki.galaxyproject.org/). We changed our local Galaxy server into a production data analysis server using the recommended configurations. We also implemented Galaxy-specific options for ChimeRScope for enhanced compatibility with Galaxy server data management system. All the wrapper scripts used for ChimeRScope applications were implemented in XML (eXtensible Markup Language).
RNA-Seq datasets and data analysis procedures
Simulated datasets
We used two different simulated datasets to test the prediction performance of ChimeRScope against other popular methods. The first simulated datasets, namely 50_pos_set, was included in the FusionMap (33) package. It consists of 50 simulated fusion transcripts with the fusion reads ranging from only 2 read pairs to 1587 read pairs. In total, it contains 57 209 75 bp paired-end reads, with only 4300 reads that spans the fusion junctions. Another simulated dataset we used contains 15 different combinatorial subsets with 3 different read lengths (50, 75 and 100 bp) and 5 different coverage levels (5×, 20×, 50×, 100× and 200×). This dataset, namely comp_sim_set, was obtained from GC. Tseng's group (34). Those 15 datasets contain the same 150 artificial fusion events, all of which were simulated using sequences from Ensembl annotation.
Published RNA-Seq datasets
To evaluate the performance of ChimeRScope in real tumor samples, we tested ChimeRScope on transcriptome data from four breast cancer cell lines (BT-474, MCF-7, KPL-4, skBR-3) (35) with 26 experimentally validated fusion transcripts reported by the original study (The fusion CSE1L-ENSG00000236127 was removed from the list due to the deprecation of ENSG00000236127). The breast cancer cell line datasets were downloaded from NCBI Sequence Read Archive (SRA. http://www.ncbi.nlm.nih.gov/sra) with accession number SRP003186. Additionally, we also tested ChimeRScope on a subset of 272 patient samples from a large glioma study (36) (accession number SRP027383). This validation list consists of 13 RNA-Seq datasets with 31 validated fusion transcripts and it was also used by the JAFFA study (37).
In-house natural killer (NK) cell lines datasets
To further demonstrate the utility of ChimeRScope in vivo, we examined three natural killer (NK) cell lines for experimental validation. Here, four different NK cell line datasets (KHYG1, NKYS, NK92-PMIG, NK92-PRDM1) were downloaded from NCBI SRA database (SRP049695), which can be divided into three different cell lines (KHYG1, NKYS and NK92). The original NK92 cell line samples used for sequencing were transduced with either PMIG, a control vector, or a vector to knock-down PRDM1, a known tumor suppressor. We only used the normal/non-transduced NK92 RNA in the experimental validation step for those fusion transcripts predicted from NK92 samples since the original vector treated NK92 RNA used in the transcriptome sequencing was not available.
Data analysis
We installed ChimeRScope, SOAPfuse (V1.26), FusionCatcher (version 0.99.4d beta), JAFFA (version 1.06), EricScript (version 0.5.5) and MapSplice (version 2.2.1) on a local Linux-based data analysis server (160 cores and 512 GB memory). We processed all simulated datasets and cell lines RNA-Seq datasets mostly using the default settings. Results on NK cell lines were further filtered using awk command. All the settings and the reference database versions can be found in Supplementary File 2.
Experimental validation
The NK cell lines used for fusion detection were KHYG1, NKYS and NK92, which were cultured under standard conditions and total RNA was extracted as per standard protocol. The RNA extracted from these cells lines was reverse transcribed into cDNA using ProtoScript First Strand cDNA Synthesis Kit (New England BioLabs) as per manufacturer's protocol, followed by polymerase chain reaction (PCR) using the oligonucleotide primers designed using Vector NTI Advance (version 11.5.4) and Primer3Plus (38). NCBI primer blast (39) was used to check the off-target amplicons to ensure the binding site specificity of the primers. PCR was performed for each primer set using either the standard Taq PCR kit (New England BioLabs) or the One Taq PCR kit with GC buffer (New England BioLabs) depending on the GC content of the target sequences. All PCR products were analyzed by agarose gel electrophoresis. The PCR products of the approximate expected amplicon size were extracted from the agarose gel using the GeneJET Gel Extraction Kit (ThermoScientific). All the amplicons extracted from the agarose gel were Sanger sequenced using Applied Biosystems (ABI) 3730 DNA Analyzer as per manufacturer's protocol.
RESULTS
Optimization of k-mer size
It is expected that the choice of the k-mer size affects many factors such as the distribution of k-mer frequencies and the computational speed, while it is also restricted by factors like the read length; hence determining the optimal size of k in k-mers is crucial to achieve optimal accuracy and performance of ChimeRScope. Our choice of k-mer size for the ChimeRScope GF-library is based on three criteria. First, k should not be too small, because a small k will always yield less unique gene fingerprint sequences. Second, k should not be too large since sequence variations within a string can affect higher percentage of the k-mers when k is large (e.g. the maximum proportion of k-mers affected by a SNP is calculated by k/(L − k + 1), where L is the length of the read; larger k increases the proportion of the affected k-mers). Last, k should be an odd number because ChimeRScope tracks each k-mer using the index of the middle nucleotide; hence reversing a k-mer (see ‘Materials and Methods’ section) when k is an odd value will not alter the index. To decide the optimal k value for human reference genome GRCh38/hg38 (38 834 mRNA transcripts in RefSeq annotation), we plot the percentages of all k-mer classes (categorized by the occurrences in unique transcripts) for all k-mer libraries using the odd k values ranged from 13 to 29 (Figure 5A). We believe that GF-libraries with a higher level of discriminative k-mers (or lower Shannon Index (40)) are generally more advantageous when evaluating fingerprint sequences (41). Our results (Figure 5 and Supplementary Table S1 in Supplementary File 3) show that k = 17 is the optimal k-mer size for human reference model (GRCh38/hg38) as it is the smallest k size that gives highest levels of discriminative fingerprints (or low Shannon Index) compared with those of the GF-libraries with larger k values.
Figure 5.

k-mer percentages for nine selected k-mer libraries for GRCh38/hg38 (mRNAs only). (A) The k-mer percentages for nine selected k-mer libraries (k = 13, 15, 17, 19, 21, 23, 25, 27, 29) are plotted using 3D line chart, where each line in this chart represents a unique k-mer library. Libraries with larger k values are plotted to the further side of the figure. The x-axis lists all k-mer classes (characterized by the number of transcripts that use the k-mer as a fingerprint) and y-axis shows the corresponding percentages of the k-mer class in the certain k-mer library. For instance, ∼48% of the k-mers in the 15-mer library are unique fingerprint sequences (y = 48% when x = 1 and k = 15). For each k-mer library, the majority of the k-mer classes (∼99%) are discriminative k-mers with <10 associated genes. Overall, larger k often gives better k-mer library because it contains more discriminative k-mers (higher value toward left part of the x-axis). (B) Shannon Indices for all nine GF-libraries. GF-libraries for k = 17 or larger have similar low levels of Shannon Indices (∼1.4, highlighted in the red box). Consequently, k = 17 is the optimal k-mer size for GRCh38/hg38 because k = 17 is the smallest k-mer size that gives highest levels of discriminative k-mers (or low Shannon Index).
In total, the 17-mer GF-library for GRCh38/hg38 consists of ∼62.7 million k-mers (Supplementary Table S2 in Supplementary File 3). Out of which, 52.8% (∼33.1 million) are unique fingerprint sequences (identified in only one transcript) and 23.5% (∼14.5 million) are observed in only two different transcripts (Supplementary Table S3 in Supplementary File 3). Additionally, more than 99% of the k-mer sequences in 17-mer GF-library are associated with no more than eight transcripts indicating the significance of 17-mers across the human transcriptome. Notably, common fingerprints like poly-A sequences can drastically slow down the computational time due to their occurrences in a large number of transcripts. Therefore, we defined that k-mers associated with more than 100 transcripts (531 k-mers. See Supplementary Table S2 in Supplementary File 3) were assigned a weightage score of zero and thus excluded from the scoring step. Furthermore, we also calculated the number of the most discriminative 17-mers in the human transcriptome (Supplementary Table S4 in Supplementary File 4) and ranked all the transcripts by the numbers of the unique 17-mers (Supplementary File 4). Results have shown that 82% of all the transcripts (32001/38834, Supplementary File 4) have at least 10 unique fingerprint sequences. Supplementary Table S4 also shows that more than 99.4% (38587/38834) of the transcripts have at least one of these high-quality k-mers (k-mers found in no more than 10 transcripts). The rest of the transcripts (0.6%) can be grouped into 34 genes (Supplementary Table S4), most of which can be further classified into only four different gene families (Cancer/Testis antigen family, Ubiquitin Specific Peptidase 17-like family, GAGE cancer/testis antigen family and PRoline-Rich protein gene family). Overall, these statistics suggest that the 17-mer GF-library contains sufficient fingerprint sequences for most of the transcripts/genes.
Optimization of time and memory usage
Memory utilization is a common problem often encountered by alignment-free algorithms (42). ChimeRScope Scanner also consumes a lot of computational resources due to the huge hash table (the GF-library containing millions of k-mers) and the large search space (millions of reads). To overcome this issue, we optimized the algorithm by transforming all the k-mers into binaries, reducing the number of iterations and optimizing data structures (Supplementary methods in Supplementary File 5). These modifications led to a 6-fold improvement on the RAM usage for ChimeRScope Builder, while also reducing the run time by 60-fold compared with our first version of ChimeRScope prior to optimization (Table 1). The improved version takes only 6 GB RAM and 12 min for ChimeRScope Scanner to load the GF-library into the memory. The processing speed for ChimeRScope Scanner was enhanced by at least eight times with only 10% of the original memory cost in the improved version. This enables the use of ChimeRScope on projects with a large number of samples. For example, analysis on RNA-Seq datasets with several million discordant reads took more than 10 h using 10 threads with more than 100 GB RAM usage in the original ChimeRScope version. In comparison, the optimized version can analyze the same datasets within 2 h with the same number of threads and 20 GB RAM.
Table 1. Run time for ChimeRScope Scanner, before and after code optimization.
| Memory usage (in GigaByte)* | Time cost/speed | |||
|---|---|---|---|---|
| Before | After | Before | After | |
| Construct 17-mer GF-library | >100 | 17.4 | 25 h | 25 min |
| Loading 17-mer GF-library | 42 | 6 | 1 h | 12 min |
| Parsing reads (per thread) | 10 | 1 | ∼400/min | >5000/min** |
* Memory usage was estimated using TotalMem()-freeMem() functions during the run time. The actual memory usage may vary.
** The processing speed for parsing reads was estimated using the 50_pos_set, simulated with different volumes (10×, 100× and 1000×).
Performance evaluation using simulated datasets
Results from a recently published paper (34) along with other studies (20,37,43–45) suggest that SOAPfuse (43), FusionCatcher (44), JAFFA (37), EricScript (46) and MapSplice (47) are generally the best-performing fusion transcript prediction methods. Therefore, we compared the prediction performance of ChimeRScope with these five methods (results reported either from this study or from previously published studies, if available). We used two different simulated datasets to evaluate the sensitivities and false discovery rates (FDRs). The first simulated dataset was published by FusionMap group with 50 positive fusion events (50_pos_set). The second set of simulated datasets was obtained from a recent study (34) that comprehensively evaluated the performance of 15 different fusion transcript prediction algorithms. These datasets, namely comp_sim_set, are comprised of 15 different combinatorial subsets with 3 different read lengths (50, 75 and 100 bp) and 5 different coverage levels (5×, 20×, 50×, 100× and 200×). Each dataset contains the same 150 artificial fusion events that were simulated based on the Ensembl (48) annotation. We analyzed these datasets and carried out head-to-head comparisons of ChimeRScope against other five methods using the same F-scores (34) for performance assessment. The complete list of fusion transcripts predicted by these six methods on simulated datasets can be found in Supplementary File 6.
The comparative performance of the selected methods on 50_pos_set is shown in Table 2. Because some of the simulated fusion transcripts can be classified as biologically insignificant (e.g. runaway transcripts between neighboring genes) by some filters implemented in ChimeRScope and in other methods like FusionCatcher, we ran our tests both with and without applying these filters on the simulated datasets. We were unable to run JAFFA locally on the 50_pos_set in this study; hence, we used the statistics reported in the original JAFFA publication (37) in Table 2 and we took the best results from the JAFFA hybrid mode output. Overall, ChimeRScope demonstrates the highest F-score (0.97) by predicting 47 true positives out of 50 without reporting any false positives (with all filters disabled). Even with all filters applied, ChimeRScope still reports a higher F-score than other tested methods. The second best-performing method on this dataset, FusionCatcher, also reports 47 true positives but with three false positives in its preliminary result (Supplementary File 6). The filters used by FusionCatcher removed these three false positives at the cost of losing 16 true positives, suggesting that FusionCatcher is very conservative in predicting fusion transcripts.
Table 2. Fusion transcript prediction on the 50_pos_set.
| 50_pos_set | Study | TP* | FP | Precision | Recall | FDR | F-score | Note | Best score |
|---|---|---|---|---|---|---|---|---|---|
| ChimeRScope | Current study | 47 | 0 | 1.00 | 0.94 | 0.00 | 0.97 | Without filters | 0.97 |
| Current study | 45 | 0 | 1.00 | 0.90 | 0.00 | 0.95 | With all filters** | ||
| SOAPfuse | Current study | 38 | 1 | 0.97 | 0.76 | 0.03 | 0.85 | 0.90 | |
| PMID:26862001 | 41 | 0 | 1.00 | 0.82 | 0.00 | 0.90 | |||
| JAFFA | PMID:26019724 | 44 | 0 | 1.00 | 0.88 | 0.00 | 0.94 | JAFFA-hybrid | 0.94 |
| FusionCatcher | Current study | 31 | 0 | 1.00 | 0.62 | 0.00 | 0.77 | Final result | 0.94 |
| Current study | 47 | 3 | 0.94 | 0.94 | 0.06 | 0.94 | Raw result | ||
| EricScript | Current study | 14 | 0 | 1.00 | 0.28 | 0.00 | 0.44 | 0.88 | |
| PMID:26862001 | 39 | 0 | 1.00 | 0.78 | 0.00 | 0.88 | |||
| MapSplice | Current study | 42 | 0 | 1.00 | 0.84 | 0.00 | 0.91 | 0.92 | |
| PMID:26862001 | 43 | 0 | 1.00 | 0.86 | 0.00 | 0.92 |
*TP: true positive; FP: false positive; Recall: sensitivity; FDR: false discovery rate.
**Two fusion transcripts were filtered out by the similarity filter (PRKCA&USP49 score: 384; FKTN&SCAI score: 367). However, the F-score was still higher compared with others methods even with these two fusion transcripts removed.
ChimeRScope predicts 47 true positives (out of 50 true fusion transcripts) without reporting any false positives. Our method achieved the highest F-score with highest sensitivity (recall = 94%) and lowest FDR (0%). In comparison, JAFFA predicts in total 44 true positives and 0 false negatives, reporting the second highest F-score as FusionCatcher (47 true positives and 3 false positives in the raw result).
Next, we analyzed a more extensively simulated dataset, comp_sim_set (with 15 subsets) and compared our results (Supplementary Table S5 in Supplementary File 7) with the original study for this dataset (34). The initial results showed that ChimeRScope was only able to identify a maximum of 135 true fusion transcripts (Supplementary File 6). A closer inspection of these 15 fusion transcripts not predicted by ChimeRScope reveals that they were undetected due to the sequence differences between Ensembl and RefSeq annotation. Specifically, the 15 fusion transcripts in the comp_sim_set are simulated from the Ensembl sequences and fused at non-coding regions defined by RefSeq annotation (Supplementary File 8). Since ChimeRScope GF-library is built entirely based on RefSeq annotation, ChimeRScope could not detect any of these true fusion transcripts. Therefore, we removed these 15 fusion transcripts for all the methods from the final analysis (Table 3). We were unable to run JAFFA successfully on datasets with read lengths of 50 and 75 bps due to the computational issues caused by JAFFA assembly mode; and hence these results were not reported for these datasets in Table 3. Overall, ChimeRScope displays the best F-scores for 13 out of 15 datasets tested. Notably, ChimeRScope has significantly higher F-scores especially on the datasets with low coverage depth (5×), suggesting its unique advantage in detecting fusion transcripts with low expression levels or from datasets with low coverage depth in general. Furthermore, the variation of the F-scores reported by ChimeRScope is subtle (max = 0.957 and min = 0.905) across all the datasets with different read lengths and depth of coverage, indicating that our method is very robust and it generates consistently accurate results irrespective of the read length or sequencing coverage. On the other hand, the F-scores reported by alignment-based methods depend on longer read lengths and larger coverage depths, suggesting that the performances of the alignment-based methods are extremely sensitive to the technical variations in the Next-generation sequencing (NGS) data.
Table 3. F-scores for six major methods on comp_sim_set with 15 incompatible fusion transcripts removed.
| comp_sim_set (TP = 135) | 50 bp | 75bp | 100 bp | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5× | 20× | 50× | 100× | 200× | 5× | 20× | 50× | 100× | 200× | 5× | 20× | 50× | 100× | 200× | |
| ChimeRScope | 0.948 | 0.954 | 0.947 | 0.908 | 0.905 | 0.948 | 0.949 | 0.957 | 0.954 | 0.947 | 0.940 | 0.957 | 0.957 | 0.957 | 0.957 |
| SOAPfuse | 0.807 | 0.922 | 0.935 | 0.913 | 0.943 | 0.836 | 0.921 | 0.921 | 0.929 | 0.925 | 0.840 | 0.942 | 0.928 | 0.932 | 0.929 |
| FusionCatcher | 0.357 | 0.856 | 0.894 | 0.895 | 0.899 | 0.692 | 0.855 | 0.878 | 0.888 | 0.896 | 0.707 | 0.884 | 0.891 | 0.891 | 0.891 |
| JAFFA | 0.609 | 0.849 | 0.856 | 0.856 | 0.856 | ||||||||||
| EricScript | 0.234 | 0.256 | 0.296 | 0.317 | 0.298 | 0.331 | 0.425 | 0.434 | 0.432 | 0.429 | 0.361 | 0.461 | 0.458 | 0.458 | 0.464 |
| MapSplice | 0.235 | 0.514 | 0.548 | 0.579 | 0.594 | 0.381 | 0.525 | 0.586 | 0.591 | 0.598 | 0.383 | 0.541 | 0.568 | 0.583 | 0.577 |
The results were calculated from 135 effective fusion transcripts. ChimerRScope achieved the highest F-scores in 13 out of the 15 datasets. SOAPfuse alone reported higher F-scores in 100×_50 bp and 200×_50 bp, with marginal increase of 0.005 and 0.038 in F-scores, respectively.
Performance evaluation using cancer transcriptome data
To evaluate the performance of ChimeRScope in real tumor samples, we tested ChimeRScope on the transcriptome data from four breast cancer cell lines (Table 4) and 13 glioma patient samples (Table 5). We analyzed these datasets using other five methods for comparative analysis (the complete prediction results for both datasets can be found in Supplementary File 6). Results on four breast cancer cell lines have shown that ChimeRScope identifies the highest number of validated fusion transcripts (22 out of 26). Furthermore, ChimeRScope also reports higher FESRs for fusion transcripts with low coverages or expression levels (Supplementary Table S6 in Supplementary File 9), suggesting that the sensitivity of ChimeRScope to identify such fusion transcripts is higher. Results on the glioma samples have also demonstrated that ChimeRScope performed better than other methods. ChimeRScope reports an F-score of 0.210, which is better than SOAPfuse (0.178), FusionCatcher (0.152), JAFFA (0.106), MapSplice (0.080) and EricScript (0.015). Moreover, ChimeRScope is the only method that identifies FESRs for all 31 fusion transcripts (Three fusion genes, AP2A2&SBF2, CD81&SPAG6 and TPM3&ADAR were filtered out by the similarity filter and the adapter filter. See Supplementary Table S7 in Supplementary File 9). Consequently, these results also demonstrate that ChimeRScope is a better fusion transcript prediction method for cancer transcriptome datasets.
Table 4. Fusion transcript prediction on breast cancer cell lines for selected methods.
| BT474(11) | SKBR3(9)* | MCF7(3) | KPL4(3) | Total TP | Total prediction | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| TP** | Total prediction | TP | Total prediction | TP | Total prediction | TP | Total prediction | |||
| ChimeRScope*** | 10 | 24 | 6 | 24 | 3 | 14 | 3 | 4 | 22 | 66 |
| SOAPfuse | 9 | 35 | 6 | 19 | 2 | 6 | 3 | 8 | 20 | 68 |
| SOAPfuse*** | 7 | 26 | 6 | 11 | 3 | 7 | 3 | 4 | 19 | 48 |
| FusionCatcher | 9 | 31 | 6 | 24 | 2 | 7 | 2 | 5 | 19 | 67 |
| FusionCatcher*** | 4 | 11 | 4 | 5 | 2 | 2 | 2 | 2 | 12 | 20 |
| JAFFA | 8 | 15 | 5 | 9 | 2 | 6 | 2 | 2 | 17 | 32 |
| JAFFA*** | 7 | 16 | 4 | 7 | 3 | 8 | 1 | 2 | 15 | 33 |
| EricScript | 8 | 31 | 4 | 37 | 2 | 10 | 2 | 5 | 16 | 83 |
| EricScript*** | 2 | 21 | 3 | 22 | 0 | 8 | 0 | 7 | 5 | 58 |
| MapSplice | 8 | 27 | 4 | 15 | 2 | 6 | 2 | 5 | 16 | 53 |
| MapSplice*** | 8 | 28 | 5 | 11 | 3 | 8 | 2 | 5 | 18 | 52 |
*CSE1L-ENSG00000236127 was removed because ENSG00000236127 is no longer a valid gene in the latest Ensembl database.
**TP: true positive.
***Marked rows are the results reported by our study. Stats for other rows are reported from Liu et al. (34).
The numbers in parenthesis next to the cell line names (header line) are the total number of validated fusion transcripts in each cell line. ChimeRScope reports the highest number of true positives across four different breast cancer cell lines. Overall, ChimeRScope identified 22 true fusion transcripts out of 26 validated fusion transcripts.
Table 5. Fusion transcript prediction on 13 glioma samples with a total of 31 validated fusion transcripts.
| Total prediction | True positives | Recall (sensitivity) | Precision* | F-score | |
|---|---|---|---|---|---|
| ChimeRScope | 236 | 28 | 0.903 | 0.119 | 0.210 |
| SOAPfuse | 183 | 19 | 0.613 | 0.104 | 0.178 |
| JAFFA | 478 | 27 | 0.871 | 0.056 | 0.106 |
| FusionCatcher | 245 | 21 | 0.677 | 0.086 | 0.152 |
| EricScript | 1464 | 11 | 0.355 | 0.008 | 0.015 |
| MapSplice | 567 | 24 | 0.774 | 0.042 | 0.080 |
* Precision for each method was calculated by dividing the number of true positives by the total prediction (considering that all the untested fusion transcripts as false positives).
ChimeRScope successfully identifies the highest number (28 out of 31) of fusion transcripts, achieving the highest sensitivity, precision and F-score. SOAPfuse reports the second best precision while JAFFA reports the second best sensitivity.
Experimental validation of ChimeRScope predictions
We used paired-end RNA-Seq datasets from three NK lymphoma cell lines that we have published earlier (49). We analyzed these RNA-Seq datasets using all selected methods (Supplementary File 2). In total, ChimeRScope predicted 10 unique fusion transcripts, compared with 25 unique fusion transcripts by SOAPfuse, three by JAFFA and only one by FusionCatcher (Supplementary File 6). On the other hand, EricScript and MapSplice predicted unusually high number (378 and 127 fusion transcripts, respectively) of fusion transcripts compared to the other four methods. Considering that the total number of curated fusion transcripts in all cancers were only around 1000 gene pairs (ChimerKB from ChimerDB 3.0 at http://ercsb.ewha.ac.kr/fusiongene), and the consistently superior performance of the first four methods (ChimeRScope, SOAPfuse, JAFFA and FusionCatcher) against multiple datasets tested, we have limited the experimental validation of the predicted fusion genes to these four methods. Among the four methods, 30 unique fusion transcripts were predicted, including five that were reported by at least two of the four methods (Figure 6). Further analysis on fusion transcripts predicted by SOAPfuse has shown that BOLA2B&SMG1P2, TVP23C&CDRT4 and DSCR4&DSCR4-IT1 are directly associated with well-annotated read-through mRNAs (with NM IDs in NCBI Five Reference Sequence database). This type of predictions is classified as false fusion event by ChimeRScope (see ‘Discussion’ section) and hence is filtered out. In addition, we failed to design primers for ORC6&PLEKG4B and MAPK8&NMU due to low complexity regions. Therefore, these five fusion transcripts predicted by SOAPfuse were also excluded from our experimental validation (Supplementary Table S8 in Supplementary File 10).
Figure 6.
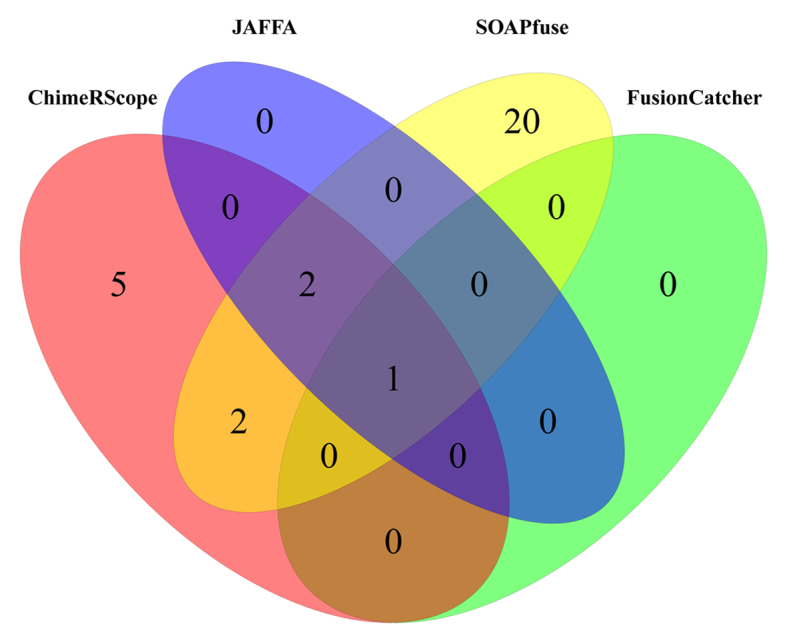
Venn diagram of all fusion transcripts reported by ChimeRScope, SOAPfuse, JAFFA and FusionCatcher on lymphoma cell lines. In total, 30 unique fusion transcripts were reported. ChimeRScope and SOAPfuse predicted the most number of common fusion transcripts (five genes), with two of them exclusively reported by these two methods. The results reported by FusionCatcher have the lowest overlaps with other methods (only one gene).
Of all the fusions predicted by different algorithms, we designed primers for 25 unique fusion transcripts and confirmed 14 fusion transcripts (56%; 14 out of 25 tested fusions) by RT-PCR and Sanger sequencing (Table 6; Supplementary Tables S9 and 10 in Supplementary Files 10 and 11, respectively). Due to space limitations, we chose to show validation results from only four fusion transcripts that are predicted by ChimeRScope with relatively lower number of fusion reads (five to seven FESRs, see Table 6). The Sanger sequencing chromatograms for other validated fusions can be found in Supplementary File 11. Figure 7 highlights the PCR results, primer target regions, Sanger sequencing chromatograms, and the exact fusion junctions marked by red lines (except for RPL14&SRP14 and LRRC37A3&NSF) for these four fusion transcripts. In total, ChimeRScope predicted 10 fusion transcripts from the lymphoma cell lines and we were able to experimentally validate all of these predictions. Thus, there are no false positives predicted by our method (FDR: 0%). However, our method missed four true positives that were predicted by other methods, hence the sensitivity of this method is at 71.4% (10 out of 14), which is the highest among all methods tested. Comparatively, among the 20 tested fusion transcripts from SOAPfuse predictions, only nine fusion transcripts were experimentally confirmed (sensitivity is 64.3% and FDR is 55%). All the fusion transcripts reported by JAFFA (three fusions) and FusionCatcher (one fusion) are also predicted by ChimeRScope and SOAPfuse. Therefore, both JAFFA and FusionCatcher achieve 100% precision rate, but with only 21.4 and 7.1% sensitivities, respectively. We also checked the validated fusion transcripts against those predicted by EricScript and MapSplice. EricScript and MapSplice have correctly predicted only six (sensitivity is 42.9%) and three (sensitivity is 21.4%) of these validated fusion transcripts, respectively. Overall, ChimeRScope reported the best F-score (0.833) for this dataset, compared with 0.529 for SOAPfuse, 0.353 for JAFFA and 0.133 for FusionCatcher (Table 6). We did not include EricScript and MapSplice in the experimental validation; hence, the F-scores and other statistics for these two methods are not available.
Table 6. List of the fusion transcripts confirmed by both PCR and Sanger sequencing in NK cell lines.
| Cell line | Fusion transcript | Fusion read counts* | |||||
|---|---|---|---|---|---|---|---|
| ChimeRScope | JAFFA | FusionCatcher | SOAPfuse | ||||
| KHYG1 | PEX2&YWHAZ | 34 | 22 | (3) | 49 | ||
| KHYG1 | ARIH2&PRKAR2A | (3) | (2) | (2) | 5 | ||
| KHYG1 | CTSC&RAB38 | (2) | (0) | (5) | 10 | ||
| KHYG1 | PRKCH&FLJ22447 | (0) | (0) | (1) | 6 | ||
| NKYS | LRRFIP1&RBM44 | 10 | (0) | (64) | 192 | ||
| NKYS | RPL14&SRP14 | 7 | (0) | (0) | (0) | ||
| NK92 | C15orf57&CBX3 | 6 | (6) | (1) | 10 | ||
| NK92 | DAB2&FRYL | 48 | 33 | 30 | 59 | ||
| NK92 | LEP&SND1 | 92 | 51 | (60) | 121 | ||
| NK92 | LRRC37A3&NSF | 5 | (0) | (0) | (4) | ||
| NK92 | MAST2&METTL21A | 8 | (0) | (0) | (0) | ||
| NK92 | NCOR2&UBC | 5 | (3) | (5) | (4) | ||
| NK92 | NPIPB5&SMG1 | 15 | (0) | (0) | (0) | ||
| NK92 | PTMA&NPM1 | (9) | (0) | (0) | 13 | ||
| Tool | Unique prediction | Test size | TP | FP | Sensitivity | FDR | F-score |
| ChimeRScope | 10 | 10 | 10 | 0 | 0.714 | 0 | 0.833 |
| SOAPfuse** | 25 | 20 | 9 | 11 | 0.643 | 0.55 | 0.529 |
| JAFFA | 3 | 3 | 3 | 0 | 0.214 | 0 | 0.353 |
| FusionCatcher | 1 | 1 | 1 | 0 | 0.071 | 0 | 0.133 |
| EricScript | 378 | 6 | 0.429 | ||||
| MapSplice | 127 | 3 | 0.214 | ||||
*The number of identified fusion reads for each fusion transcript identified by each method is listed in the corresponding cells. Cells with parenthesis indicate that the fusion transcripts were filtered out by the corresponding tools and thus not reported in their final results.
**Five fusion transcripts predicted by SOAPfuse were excluded from the validation list because either the complete fusion sequence were associated with well annotated read-through mRNAs or the specific primer binding sites were not available due to repeated nucleotide sequences.
Fourteen fusion transcripts from the NK cell lines were validated by both PCR and Sanger sequencing. In total, ChimeRScope predicts all 10 positive fusion transcripts with no false positive. Moreover, ChimeRScope reports fusion reads for 13 of the total 14 experimentally validated fusion transcripts, higher than SOAPfuse, JAFFA and FusionCatcher. Overall, ChimeRScope reports the highest F-score (0.833) on the NK cell lines, compared with 0.529, 0.353 and 0.133 for SOAPfuse, JAFFA and FusionCatcher, respectively. Fusion transcripts predicted by EricScript and MapSplice were not included in the experimental validation; thus, the FDRs and F-scores were not calculated for these two methods.
Figure 7.

PCR and Sanger sequencing results for four fusion transcripts with low number of FESRs from ChimeRScope predictions. For each fusion transcript track, the left panel is the PCR panel and the right panel displays the predicted fusion sequence and the primer binding site, along with the Sanger sequencing chromatogram. Specifically, each PCR image has four lanes for a 100-bp ladder marker, the fusion transcript amplicon with the band of the matched product pointed by a red arrow, the positive control (actin beta, or ACTB) and negative control (water). The right panel shows the name of the fusion partners, the predicted fusion junction sequence (100 bp upstream and downstream, separated by the wildcard ‘N’), the binding sites of the primer pair used in the PCR panel, the chromatogram for the highlighted region (mostly the fusion junction, if applicable). The PCR experiments and the Sanger sequencing results confirmed the existences of these four genes in the NK cell lines. We were unable to resolve the fusion junctions for RPL14&SRP14 and LRRC37A3&NSF due to the poor Sanger sequencing data quality. Therefore, the exact fusion junctions of these two fusions were not marked in the chromatograms.
Literature searches for all experimentally validated fusion transcripts have suggested that some of the chimeric transcripts are potentially oncogenic. For instance, a fusion transcript that is predicted by ChimeRScope, NCOR2&UBC, has also been reported previously in CLL patients (50). NCOR2 is a nuclear receptor corepressor that interacts with members of MAPK-signaling (51), Notch and NF-kappa-B pathways (52). The altered expression of this gene is associated with cell cycle progression and apoptosis in multiple cancers (53,54). Figure 8 illustrates the fusion model of NCOR2&UBC with the predicted (55) functional domains in the resulting chimeric protein. This chimeric transcript combines the first exon of NCOR2 and the second exon of UBC, creating a new transcript with the loss of the SANT (named after switching-defective protein 3 or SWI3, adaptor 2 or ADA2, nuclear receptor co-repressor or N-CoR, transcription factor IIIB or TFIIIB) domain that is responsible for chromatin-remodeling and transcription regulation (56,57). Another validated fusion transcript, LRRC37A3&NSF is predicted only by ChimeRScope method. This fusion involves a gene named N-ethylmaleimide sensitive factor (NSF). Studies have shown that NSF directly interacts with CD28 (58), a gene responsible for T-cell activation and survival. Although triggering of human NK cells by CD80 and CD86 (ligands of CD28) seems to be independent of CD28 (59), the absence of CD28 expressions in NK cell lines (59) could be the result of the LRRC37A3&NSF fusion event. Other exclusive ChimeRScope's predictions like MAST2&METTL21A and NPIPB5&SMG1 are kinase fusions (60) that are more likely to have oncogenic functions in cancer because they involve kinases like MAST2 (microtubule-associated serine/threonine kinase 2) and SMG1 (nonsense-mediated mRNA decay associated PI3K related kinase). At last, the only fusion transcript predicted by all four methods, DAB2&FRYL, includes a potential tumor suppressor gene named DAB2 (disabled homolog 2) which has been found to be associated with tumorigenesis in different cancers (61–63). These fusion transcripts mentioned above warrants further investigation to confirm their specific roles in tumorigenesis.
Figure 8.

The fusion model of NCOR2&UBC and the predicted functional domains. This fusion transcript is fused between the first exon of NCOR2 (3΄ end) and the second exon of UBC (5΄ end). The SANT domain from NCOR2 and the ubiquitin domains from UBC are plotted to the approximate position of the corresponding exons. Because the exons with the SANT motif sequence are not included in NCOR2&UBC, the predicted domains of the NCOR2&UBC fusion transcript only contain the ubiquitin domains from UBC.
Computational cost
We compared the computational cost of ChimeRScope against the other methods, in order to check the feasibility of using ChimeRScope on large-scale data analysis. Since each of these methods were developed in different programming languages with different ways of analyzing transcriptome data (Supplementary Table S11 in Supplementary File 12), we chose to run the same four samples from NK cell lines on each method by assigning similar amount of resources and compared the run time of these methods. We executed runs for one tool at a time on our local data analysis server (with 160 cores and 512 GB memory) to prevent potential competition for computational resources if the jobs were run simultaneously. We uniformly assigned 20 threads for each tool with multithreading features. The total run time for each method is listed in Table 7. On average, ChimeRScope pipeline only takes 1.67 h per sample (if integrated with standard RNA-Seq data analysis pipeline) outperforming the other five methods by far with a large margin. EricScript reported the second fastest time (5.21 h per sample). The Scanner and Sweeper modules of ChimeRScope that identify the FESRs and fusion transcript candidates, respectively, took over 90% of the total time due to the compute-intensive nature of these tasks. It is worth mentioning that the search space for ChimeRScope is only around 10% of the total reads (unmapped and discordantly mapped reads), hence our method can be integrated with the standard RNA-Seq data analysis pipeline as an add-on module of post-alignment step using the unaligned read data as input. In contrast, the other five methods are alignment-based methods that independently process the entire read data to predict fusion transcripts.
Table 7. The run time for fusion transcript prediction on NK cell lines using ChimeRScope and other five major methods.
| Total reads | Discordant reads | ChimeRScope | SF* | JF* | FC* | ES* | MS* | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Scanner | Sweeper | Examiner | Total | ||||||||
| SRR1648334 | 39 555 460 | 4 920 296 | 0.60 | 0.78 | 0.07 | 1.45 | 57.03 | 7.67 | 4.02 | 9.87 | |
| SRR1648335 | 64 685 980 | 7 584 282 | 0.63 | 1.17 | 0.10 | 1.90 | 90.75 | 12.28 | 7.40 | 15.07 | |
| SRR1648336 | 48 472 502 | 6 043 845 | 1.02 | 0.57 | 0.13 | 1.72 | 72.74 | 8.37 | 4.40 | 9.92 | |
| SRR1648337 | 49 849 026 | 6 297 921 | 0.98 | 0.53 | 0.10 | 1.61 | 66.63 | 8.67 | 5.00 | 10.05 | |
| Time | 3.23 | 3.05 | 0.40 | 6.68 | 101.75 | 287.15 | 36.99 | 20.82 | 44.91 | ||
| Time per sample | 0.81 | 0.76 | 0.10 | 1.67 | 25.44 | 71.79 | 9.25 | 5.21 | 11.23 | ||
* SF: SOAPfuse; JF: JAFFA; FC: FusionCatcher; ES: EricScript; MS: MapSplice.
The total number of reads for these four samples ranged from 39.6 to 64.7 million. Roughly 12% of the total reads in each sample are defined as discordant reads after TopHat alignment. ChimeRScope only takes >7 h to analyze all discordant reads. The time cost for each step is also listed in this table. Comparatively, the other five methods take significantly longer time to finish, possibly due to the time spent on alignment of the total reads. The run times for SOAPfuse on each individual samples are not available because SOAPfuse analyzes all four different samples in a single run.
Software and web application development
To increase the ease of accessibility to the research community, we developed both the standalone software package and a web-based application of the ChimeRScope using Galaxy server (22). The standalone package is distributed in a single Java Archive (JAR) file and it can be easily integrated with other third-party NGS data analysis software packages. For demonstration purpose and better accessibility, we also installed ChimeRScope on our local Galaxy server. Additionally, we ensured that no differences exist between the web-based Galaxy server version and the command line version of ChimeRScope. The online Galaxy server version of ChimeRScope will benefit the research community, especially for researchers with limited programming experiences. All the scripts and instructions to integrate ChimeRScope into Galaxy server are incorporated in the ChimeRScope wiki page.
DISCUSSION
ChimeRScope method development
ChimeRScope Builder
ChimeRScope predicts fusion transcripts by searching the k-mers from discordant reads against the fingerprint sequences of all known mRNA sequences. These fingerprint sequences are identified by the ChimeRScope Builder module. ChimeRScope Builder takes all the mRNA sequences in fasta format as input, and creates the GF-library with weighted k-mers by comparing the k-mer profiles of these mRNAs. For accurate prediction of the fusion transcripts, we suggest using only curated mRNA sequences because fusion candidates involving mRNAs that are not yet validated can be false positives. For example, the CSE1L-ENSG00000236127 was reported as a true fusion transcript in the breast cancer cell lines (35), but was later deprecated due to the exclusion of ENSG00000236127 in the latest Ensembl database. Therefore, we recommend the use of well curated and non-redundant databases such as the NCBI Reference Sequence (RefSeq) annotation (64,65) for ChimeRScope Builder.
ChimeRScope Scanner
The Scanner module classifies FESRs from unmapped and discordantly mapped reads and it does not allow any mismatches when comparing the fingerprint sequences. Allowing at least one mismatch when comparing k-mers may help improve the sensitivity of the method, however it can increase the processing time of each k-mer up to 3k times for each mismatch and may also introduce more false positives. Nevertheless, hashing algorithm like SimHash (66,67) might be useful to solve this computational issue. SimHash is an efficient algorithm that can be used to find similar fingerprints within a certain Hamming distance. We plan to explore the applicability of using SimHash in the future releases of ChimeRScope (ChimeRScope currently uses HashMap object as the GF-library infrastructure).
ChimeRScope Scanner does not take quality scores into consideration when analyzing the paired-end reads. We believe that low sequencing quality scores near the 3΄ of the reads (often observed in Illumina RNA-Seq datasets) have little impact on the prediction results because only a few of k-mers are affected. Removing reads with overall low quality scores (e.g. average score of all bases lower than 30) is not a requirement for ChimeRScope because such reads are more likely to occur near repetitive regions (GC-rich or AT-rich regions) and polymer regions (68). As such, k-mers generated from these repetitive regions are scored with very low or even zero weightage by ChimeRScope; thus, they are effectively removed by ChimeRScope from further downstream analysis. Besides, low quality reads are often excluded after the quality control step before alignment, as it is a part of the standard NGS data analysis pipeline.
ChimeRScope sweeper
We have implemented several filters in ChimeRScope Sweeper to improve its prediction accuracy when reporting fusion candidates. However, it can still report false positive fusion transcripts, some of which are ChimeRScope-specific. One class of false positives that are often detected by the Sweeper module is the non-coding RNAs that exhibit fusion-like patterns. For instance, the fusion transcript reported in the original SOAPfuse publication (43), GATSL1-GTF2I, can be aligned to several non-coding RNAs (NR_002206.3 and NR_003580.2). ChimeRScope also found a similar fusion transcript named GATSL2-GTF2I in the breast cancer samples (Supplementary File 6). This class of false positives in the preliminary result is later filtered out using BLAST searches (against nucleotide collections). Another group of ChimeRScope-specific false positive fusions include genes with common variations (e.g. SNPs and INDELs). When k-mers generated from reads with common variations are identified as fingerprint sequences of other genes (denoted as GeneX) rather than the original gene (denoted as GeneA), these sequence variations can lead to false fusion events with fusion models of GeneA-GeneX-GeneA. This class of false positives can be identified from ChimeRScope graphical outputs. All the false positives mentioned above tend to be seen repeatedly in real RNA-Seq datasets. We plan to catalogue the common false positive fusion transcripts and programmatically remove them in the future releases.
ChimeRScope examiner
The Examiner module is a post-analysis module that identifies fusion junctions by aligning FESRs against corresponding fusion partner gene sequences. However, for fusion transcripts with only a few FESRs, there might not be enough coverage over the fusion junctions. In such cases, ChimeRScope Examiner will not be able to report the accurate fusion junctions and the SVG figures may not be created.
Performance assessments using published datasets
ChimeRScope performs better compared with five other previously reported alignment-based methods when rigorously tested on two simulated datasets and other cancer transcriptome datasets. Moreover, results also demonstrate that ChimeRScope is more sensitive to detect fusion reads. For instance, ChimeRScope reports the fusion reads for 25 out of 26 validated fusion transcripts from the breast cancer cell lines (Supplementary Table S6 in Supplementary File 9), compared with 23 by SOAPfuse, 16 by FusionCatcher, 17 by JAFFA, 7 by EricScript and 18 by MapSplice, suggesting a better sensitivity of ChimeRScope than these alignment-based methods when searching for fusion reads. Of those validated fusion transcripts, ChimeRScope is the only method that reports the fusion reads (Supplementary Table S6 in Supplementary File 9) of CPNE1-PI3, though CPNE1-PI3 (validated in BT474 cell lines (35)) was further filtered out by ChimeRScope due to the low number of FESRs. A closer inspection of one FESR has shown that an unknown sequence of 23 nt (Supplementary File 13) sits in between the potential fusion junction of CPNE1-PI3 (similar to CPNE1-unknownSeq-PI3). A BLAST search of this sequence against nc/nr collection has shown a 100% match with PI3 gene in Macaca mulatta and Pan troglodytes but the sequence was absent in the human reference transcriptome. In this case, alignment-based methods could not generate reliable alignment results for the related fusion reads using the current transcriptome model and consequently, none of these reads would have been predicted as fusion reads (Supplementary File 13). Comparatively, the analysis on the k-mer content of the same read by ChimeRScope revealed that CPNE1, RBM12 (RBM12 is overlapped with CPNE1 with shared fingerprint sequences) and PI3 are the only possible source of the reads due to the significant fingerprint sequence match. Hence, this read is classified by ChimeRScope as a FESR that supports CPNE1&PI3.
Experimental validation on NK cell lines
To further demonstrate the utility of ChimeRScope in vivo, we examined three NK cell lines for experimental validation. These cell lines, rather than tumor specimens were chosen, due to their derivation from single clones and ease of reproducible validation in cell lines. We have successfully amplified all the 10 predicted fusion products with the exact amplicon size. We also confirmed the exact fusion junctions from the Sanger sequencing for eight out of 10 predictions, but unable to do so for two of the fusion transcripts (RPL14&SRP14 and LRRC37A3&NSF) that have faint PCR bands (Figure 7). However, these two fusions were still considered as true fusion events based on the specific amplicon size and the alignment evidences between the predicted fusion sequences and the Sanger sequencing results (Supplementary File 14). Specifically, the fusion junction of the RPL14&SRP14 fusion contains CAG repeats, which could also affect the Sanger sequencing quality near the 3΄ end of the repeat region. We were unable to resolve the fusion junction due to the CAG repeats, thus the exact fusion junction is not marked for this fusion in Figure 7. For LRRC37A3&NSF, we were only able to design the primer pair with the forward primer spanning the fusion junction (Supplementary Table S9 in Supplementary File 10). Therefore, the Sanger sequencing result generated from the forward primer does not span the fusion junction. Due to the poor Sanger sequencing quality observed in the first 15–40 bases, the chromatogram of LRRC37A3&NSF (Figure 7) only shows the comparison between the predicted fusion sequence roughly 40 bp downstream of the forward primer binding site and the Sanger sequencing result. We were not able to obtain high quality Sanger sequencing result from the reverse primer, thus the Sanger sequencing result that covers the fusion junction was not available for this fusion transcript. Nevertheless, the PCR result shows the band with the exact amplicon size. Additionally, the forward and the reverse primer (Supplementary Table S9 in Supplementary File 10) are very specific to LRRC37A3 and NSF, respectively. Since the Sanger sequencing result shows significant match with the 3΄ gene (NSF), we believe that this fusion transcript is also a true fusion event.
Among the four validated fusion transcripts that were not reported by ChimeRScope, ChimeRScope still identified FESRs for three of those fusions, but filtered them out due to the stringent filters it uses to remove false positives. For example, the preliminary result from ChimeRScope Scanner shows that ChimeRScope identified nine FESRs for PTMA&NPM1. However, some of these FESRs have very low weightage score and was not considered as valid FESRs (see ‘Materials and Methods’ section) due to insufficient fingerprint sequences (possibly caused by evenly distributed sequence variations). Allowing a couple of mismatches when comparing the k-mers could potentially improve the sensitivity of our method to detect such fusion transcripts.
Applications of ChimeRScope
The standalone version of ChimeRScope is easy to configure and install with minimum dependencies (only requires Java Standard Edition Runtime Environment 7), making this accessible to researchers even with minimal programming skills. Additionally, the reference files for ChimeRScope to execute properly can be easily prepared compared with tools such as SOAPfuse, FusionCatcher and JAFFA, which require more than 15 files from various public databases. The GF-library used by ChimeRScope contains only a few files which are automatically generated using ChimeRScope Builder for any well-annotated reference genome. ChimeRScope is distributed as a single JAR file, which is a platform-independent application that can be executed on any operating system (Linux, MacOS or Windows) that supports java with sufficient computational power. Moreover, being a platform-independent tool with minimum dependencies, ChimeRScope can be easily integrated with other third-party genomic research platforms like Galaxy server. As a consequence, ChimeRScope can inherit the advantages (such as the accessibility, reproducibility and transparency of Galaxy server) of these platforms.
CONCLUSION
We present an alignment-free method named ChimeRScope that inspects the k-mer contents of the RNA-Seq paired-end reads for fusion transcript detection. Our results demonstrate that ChimeRScope is suitable for large-scale fusion transcript data analysis with consistently better prediction performance compared with other popular tools, irrespective of read length, sequencing depth and expression levels of the fusion transcripts. The application of this method against large-scale RNA-Seq datasets such as cancer transcriptomes from TCGA resource could lead to the discovery of potentially novel and physiologically relevant drug targets for cancer treatment, or biomarkers for effective diagnosis and prognosis in precision medicine. ChimeRScope is a user-friendly software that can either be set up as a standalone software or installed on genomic research platforms such as Galaxy server. The ChimeRScope software application, detailed manuals, instructions with wrapper scripts for Galaxy server usage, and the pre-built GF-libraries are made available for research community use on our ChimeRScope wikipage at GitHub. An online application of ChimeRScope can also be accessed from a local Galaxy server at https://galaxy.unmc.edu.
Supplementary Material
ACKNOWLEDGEMENTS
We thank George C. Tseng and Silvia Liu from the University of Pittsburgh for sharing the 15 simulated datasets for this study. We also thank the Bioinformatics and System Biology Core at UNMC for providing bioinformatics infrastructure. The authors also thank the University of Nebraska Medical Center's High-Throughput DNA Sequencing and Genotyping Core Facility for performing all Sanger sequencing. We also thank Ms Megan Brown from UNMC for proof reading the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
University of Nebraska Medical Center startup funds (to C.G.); Biomedical Informatics Graduate Fellowship (to L.Y.); Lymphoma Research Foundation [F-263549 to I.J., in part]; Translational Research Program of Leukemia and Lymphoma Society [6129-14 to I.J., in part]; Bioinformatics and System Biology Core at the University of Nebraska Medical Center from the Nebraska Research Initiative (NRI); Nebraska INBRE [2P20GM103427]; CCSG to the Fred and Pamela Buffett Cancer Center [5P30CA036727]. Funding for open access charge: Institutional Start-up Funds (to C.G.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Mitelman F., Johansson B., Mertens F.. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer. 2007; 7:233–245. [DOI] [PubMed] [Google Scholar]
- 2. Jividen K., Li H.. Chimeric RNAs generated by intergenic splicing in normal and cancer cells. Genes Chromosomes Cancer. 2014; 53:963–971. [DOI] [PubMed] [Google Scholar]
- 3. Parker B.C., Zhang W.. Fusion genes in solid tumors: an emerging target for cancer diagnosis and treatment. Chin. J. Cancer. 2013; 32:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bohlander S.K. Fusion genes in leukemia: an emerging network. Cytogenet. Cell Genet. 2000; 91:52–56. [DOI] [PubMed] [Google Scholar]
- 5. Edwards P.A. Fusion genes and chromosome translocations in the common epithelial cancers. J. Pathol. 2010; 220:244–254. [DOI] [PubMed] [Google Scholar]
- 6. Barros-Silva J.D., Paulo P., Bakken A.C., Cerveira N., Lovf M., Henrique R., Jeronimo C., Lothe R.A., Skotheim R.I., Teixeira M.R.. Novel 5΄ fusion partners of ETV1 and ETV4 in prostate cancer. Neoplasia. 2013; 15:720–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Panagopoulos I., Strombeck B., Isaksson M., Heldrup J., Olofsson T., Johansson B.. Fusion of ETV6 with an intronic sequence of the BAZ2A gene in a paediatric pre-B acute lymphoblastic leukaemia with a cryptic chromosome 12 rearrangement. Br. J. Haematol. 2006; 133:270–275. [DOI] [PubMed] [Google Scholar]
- 8. Parker B.C., Annala M.J., Cogdell D.E., Granberg K.J., Sun Y., Ji P., Li X., Gumin J., Zheng H., Hu L. et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J. Clin. Invest. 2013; 123:855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mertens F., Johansson B., Fioretos T., Mitelman F.. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer. 2015; 15:371–381. [DOI] [PubMed] [Google Scholar]
- 10. Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M.. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009; 458:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maher C.A., Palanisamy N., Brenner J.C., Cao X., Kalyana-Sundaram S., Luo S., Khrebtukova I., Barrette T.R., Grasso C., Yu J. et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:12353–12358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moorman A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica. 2016; 101:407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roeder I., Horn M., Glauche I., Hochhaus A., Mueller M.C., Loeffler M.. Dynamic modeling of imatinib-treated chronic myeloid leukemia: functional insights and clinical implications. Nat. Med. 2006; 12:1181–1184. [DOI] [PubMed] [Google Scholar]
- 14. Tang M., Foo J., Gonen M., Guilhot J., Mahon F.X., Michor F.. Selection pressure exerted by imatinib therapy leads to disparate outcomes of imatinib discontinuation trials. Haematologica. 2012; 97:1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rowley J.D. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973; 243:290–293. [DOI] [PubMed] [Google Scholar]
- 16. Nowell P.C. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut. 1962; 8:65–66. [DOI] [PubMed] [Google Scholar]
- 17. Perrotti D., Calabretta B.. Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene. 2002; 21:8577–8583. [DOI] [PubMed] [Google Scholar]
- 18. Carroll M., Ohno-Jones S., Tamura S., Buchdunger E., Zimmermann J., Lydon N.B., Gilliland D.G., Druker B.J.. CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood. 1997; 90:4947–4952. [PubMed] [Google Scholar]
- 19. Kantarjian H., O’Brien S., Jabbour E., Garcia-Manero G., Quintas-Cardama A., Shan J., Rios M.B., Ravandi F., Faderl S., Kadia T. et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood. 2012; 119:1981–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carrara M., Beccuti M., Cavallo F., Donatelli S., Lazzarato F., Cordero F., Calogero R.A.. State of art fusion-finder algorithms are suitable to detect transcription-induced chimeras in normal tissues?. BMC Bioinformatics. 2013; 14(Suppl. 7):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ding L., Wendl M.C., McMichael J.F., Raphael B.J.. Expanding the computational toolbox for mining cancer genomes. Nat. Rev. Genet. 2014; 15:556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giardine B., Riemer C., Hardison R.C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J. et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005; 15:1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. Genome Project Data Processing, S. . The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Povey S., Lovering R., Bruford E., Wright M., Lush M., Wain H.. The HUGO Gene Nomenclature Committee (HGNC). Hum. Genet. 2001; 109:678–680. [DOI] [PubMed] [Google Scholar]
- 30. Smith T.F., Waterman M.S.. Identification of common molecular subsequences. J. Mol. Biol. 1981; 147:195–197. [DOI] [PubMed] [Google Scholar]
- 31. Babiceanu M., Qin F., Xie Z., Jia Y., Lopez K., Janus N., Facemire L., Kumar S., Pang Y., Qi Y. et al. Recurrent chimeric fusion RNAs in non-cancer tissues and cells. Nucleic Acids Res. 2016; 44:2859–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. MacArthur R.H., MacArthur J.W.. On bird species diversity. Ecology. 1961; 42:594–598. [Google Scholar]
- 33. Ge H., Liu K., Juan T., Fang F., Newman M., Hoeck W.. FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011; 27:1922–1928. [DOI] [PubMed] [Google Scholar]
- 34. Liu S., Tsai W.H., Ding Y., Chen R., Fang Z., Huo Z., Kim S., Ma T., Chang T.Y., Priedigkeit N.M. et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016; 44:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Edgren H., Murumagi A., Kangaspeska S., Nicorici D., Hongisto V., Kleivi K., Rye I.H., Nyberg S., Wolf M., Borresen-Dale A.L. et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011; 12:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bao Z.S., Chen H.M., Yang M.Y., Zhang C.B., Yu K., Ye W.L., Hu B.Q., Yan W., Zhang W., Akers J. et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014; 24:1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davidson N.M., Majewski I.J., Oshlack A.. JAFFA: high sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015; 7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3–new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L.. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012; 13:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lancashire L.J., Lemetre C., Ball G.R.. An introduction to artificial neural networks in bioinformatics–application to complex microarray and mass spectrometry datasets in cancer studies. Brief. Bioinform. 2009; 10:315–329. [DOI] [PubMed] [Google Scholar]
- 41. Burge C., Campbell A.M., Karlin S.. Over- and under-representation of short oligonucleotides in DNA sequences. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:1358–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan C.X., Ragan M.A.. Next-generation phylogenomics. Biol. Direct. 2013; 8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jia W., Qiu K., He M., Song P., Zhou Q., Zhou F., Yu Y., Zhu D., Nickerson M.L., Wan S. et al. SOAPfuse: an algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013; 14:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nicorici D., Satalan M., Edgren H., Kangaspeska S., Murumägi A., Kallioniemi O., Virtanen S., Kilkku O.. FusionCatcher–a tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv. 2014; doi:10.1101/011650. [Google Scholar]
- 45. Kumar S., Vo A.D., Qin F., Li H.. Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data. Sci. Rep. 2016; 6:21597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Benelli M., Pescucci C., Marseglia G., Severgnini M., Torricelli F., Magi A.. Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics. 2012; 28:3232–3239. [DOI] [PubMed] [Google Scholar]
- 47. Wang K., Singh D., Zeng Z., Coleman S.J., Huang Y., Savich G.L., He X., Mieczkowski P., Grimm S.A., Perou C.M. et al. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010; 38:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aken B.L., Ayling S., Barrell D., Clarke L., Curwen V., Fairley S., Fernandez Banet J., Billis K., Garcia Giron C., Hourlier T. et al. The Ensembl gene annotation system. Database. 2016; 2016:baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kucuk C., Jiang B., Hu X., Zhang W., Chan J.K., Xiao W., Lack N., Alkan C., Williams J.C., Avery K.N. et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gammadelta-T or NK cells. Nat. Commun. 2015; 6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Obholzer N.D., Haas B.J., Landau D.A., Pochet N., Regev A., Wu C.. Development of a cancer transcriptome analysis toolkit: identification of gene fusions in chronic lymphocytic leukemia. Cancer Res. 2015; 75(Suppl. 15):4859. [Google Scholar]
- 51. Eisold M., Asim M., Eskelinen H., Linke T., Baniahmad A.. Inhibition of MAPK-signaling pathway promotes the interaction of the corepressor SMRT with the human androgen receptor and mediates repression of prostate cancer cell growth in the presence of antiandrogens. J. Mol. Endocrinol. 2009; 42:429–435. [DOI] [PubMed] [Google Scholar]
- 52. Espinosa L., Ingles-Esteve J., Robert-Moreno A., Bigas A.. IkappaBalpha and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NFkappaB pathways. Mol. Biol. Cell. 2003; 14:491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blackmore J.K., Karmakar S., Gu G., Chaubal V., Wang L., Li W., Smith C.L.. The SMRT coregulator enhances growth of estrogen receptor-alpha-positive breast cancer cells by promotion of cell cycle progression and inhibition of apoptosis. Endocrinology. 2014; 155:3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ghoshal P., Nganga A.J., Moran-Giuati J., Szafranek A., Johnson T.R., Bigelow A.J., Houde C.M., Avet-Loiseau H., Smiraglia D.J., Ersing N. et al. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009; 69:4380–4387. [DOI] [PubMed] [Google Scholar]
- 55. Sigrist C.J., de Castro E., Cerutti L., Cuche B.A., Hulo N., Bridge A., Bougueleret L., Xenarios I.. New and continuing developments at PROSITE. Nucleic Acids Res. 2013; 41:D344–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boyer L.A., Latek R.R., Peterson C.L.. The SANT domain: a unique histone-tail-binding module?. Nat. Rev. Mol. Cell Biol. 2004; 5:158–163. [DOI] [PubMed] [Google Scholar]
- 57. Grune T., Brzeski J., Eberharter A., Clapier C.R., Corona D.F., Becker P.B., Muller C.W.. Crystal structure and functional analysis of a nucleosome recognition module of the remodeling factor ISWI. Mol. Cell. 2003; 12:449–460. [DOI] [PubMed] [Google Scholar]
- 58. Heller M., Watts J.D., Aebersold R.. CD28 stimulation regulates its association with N-ethylmaleimide-sensitive fusion protein and other proteins involved in vesicle sorting. Proteomics. 2001; 1:70–78. [DOI] [PubMed] [Google Scholar]
- 59. Wilson J.L., Charo J., Martin-Fontecha A., Dellabona P., Casorati G., Chambers B.J., Kiessling R., Bejarano M.T., Ljunggren H.G.. NK cell triggering by the human costimulatory molecules CD80 and CD86. J. Immunol. 1999; 163:4207–4212. [PubMed] [Google Scholar]
- 60. Stransky N., Cerami E., Schalm S., Kim J.L., Lengauer C.. The landscape of kinase fusions in cancer. Nat. Commun. 2014; 5:4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xie X.M., Zhang Z.Y., Yang L.H., Yang D.L., Tang N., Zhao H.Y., Xu H.T., Li Q.C., Wang E.H.. Aberrant hypermethylation and reduced expression of disabled-2 promote the development of lung cancers. Int. J. Oncol. 2013; 43:1636–1642. [DOI] [PubMed] [Google Scholar]
- 62. Xie Y., Zhang Y., Jiang L., Zhang M., Chen Z., Liu D., Huang Q.. Disabled homolog 2 is required for migration and invasion of prostate cancer cells. Front. Med. 2015; 9:312–321. [DOI] [PubMed] [Google Scholar]
- 63. Tong J.H., Ng D.C., Chau S.L., So K.K., Leung P.P., Lee T.L., Lung R.W., Chan M.W., Chan A.W., Lo K.W. et al. Putative tumour-suppressor gene DAB2 is frequently down regulated by promoter hypermethylation in nasopharyngeal carcinoma. BMC Cancer. 2010; 10:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pruitt K.D., Brown G.R., Hiatt S.M., Thibaud-Nissen F., Astashyn A., Ermolaeva O., Farrell C.M., Hart J., Landrum M.J., McGarvey K.M. et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014; 42:D756–D763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pruitt K.D., Tatusova T., Maglott D.R.. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007; 35:D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sood S., Loguinov D.. Probabilistic near-duplicate detection using simhash. Proceedings of the 20th ACM international conference on Information and knowledge management. 2011; Scotland: ACM, Glasgow; 1117–1126. [Google Scholar]
- 67. Charikar M.S. Similarity estimation techniques from rounding algorithms. Proceedings of the thiry-fourth annual ACM symposium on Theory of computing. 2002; Quebec: ACM, Montreal; 380–388. [Google Scholar]
- 68. Goodwin S., McPherson J.D., McCombie W.R.. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016; 17:333–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.