

Abstract
The ubiquitous molecular chaperone 70-kDa heat shock proteins (Hsp70) play key roles in maintaining protein homeostasis. Hsp70s contain two functional domains: a nucleotide binding domain and a substrate binding domain. The two domains are connected by a highly conserved inter-domain linker, and allosteric coupling between the two domains is critical for chaperone function. The auxiliary chaperone 40-kDa heat shock proteins (Hsp40) facilitate all the biological processes associated with Hsp70s by stimulating the ATPase activity of Hsp70s. Although an overall essential role of the inter-domain linker in both allosteric coupling and Hsp40 interaction has been suggested, the molecular mechanisms remain largely unknown. Previously, we reported a crystal structure of a full-length Hsp70 homolog, in which the inter-domain linker forms a well-ordered β strand. Four highly conserved hydrophobic residues reside on the inter-domain linker. In DnaK, a well-studied Hsp70, these residues are V389, L390, L391, and L392. In this study, we biochemically dissected their roles. The inward-facing side chains of V389 and L391 form extensive hydrophobic contacts with the nucleotide binding domain, suggesting their essential roles in coupling the two functional domains, a hypothesis confirmed by mutational analysis. On the other hand, L390 and L392 face outward on the surface. Mutation of either abolishes DnaK’s in vivo function, yet intrinsic biochemical properties remain largely intact. In contrast, Hsp40 interaction is severely compromised. Thus, for the first time, we separated the two essential roles of the highly conserved Hsp70 inter-domain linker: coupling the two functional domains through V389 and L391 and mediating the interaction with Hsp40 through L390 and L392.
Keywords: molecular chaperone, Hsp70, Hsp40, allosteric coupling, protein folding
Introduction
One of the most fundamental cellular responses to environmental stresses is the expression of molecular chaperones to alleviate protein denaturation damages caused by such stresses. The 70-kDa heat shock proteins (Hsp70s) are one of the major classes of molecular chaperones that are most frequently stress induced. However, under normal conditions, Hsp70s also play essential roles in maintaining cellular protein homeostasis by supporting multiple functions, such as protein folding, assembly, translocation into organelles, and degradation. Recently, more and more evidence has tied Hsp70s with numerous human diseases, such as cancers and neurodegenerative diseases, and aging.1–5
Consistent with their conserved roles in protein homeostasis, Hsp70s are highly conserved.1,5–7 All Hsp70s have two functional domains, corresponding to two intrinsic activities. At the N-terminus is a nucleotide binding domain (NBD), which binds adenine nucleotides ATP and ADP. NBD also has ATPase activity, although relatively low by itself. At the C-terminus is a substrate binding domain (SBD). SBD usually binds unfolded hydrophobic segments of polypeptides in an extended conformation. These two functional domains are connected by a short and highly conserved linker segment, the inter-domain linker. However, these two functional domains have to be tightly coupled upon ATP binding to NBD to make a functional chaperone, which leads to modulation of the two intrinsic activities. When ATP is bound to NBD, Hsp70s have low affinities for peptide substrates, with fast kinetics for both binding and release.8 In contrast, after ATP is hydrolyzed into ADP, both the binding and the release of peptide substrates are dramatically slowed, which results in much higher binding affinities, about 2 orders of magnitude decrease in the dissociation constant Kd. At the same time, peptide substrate binding to SBD stimulates the ATP hydrolysis rate of NBD.9 Thus, the energy from ATP hydrolysis can be used to efficiently regulate peptide substrate binding and release.
It is well accepted that the chaperone activity of Hsp70s is dramatically enhanced by two classes of cofactors: 40-kDa heat shock proteins (Hsp40s) and nucleotide exchange factors (NEFs).1,5–7,10 Hsp40s stimulate the low intrinsic ATPase activity of Hsp70s by specifically enhancing the rate of ATP hydrolysis, whereas NEFs facilitate the nucleotide exchange from ADP to ATP after ATP hydrolysis. While the action of NEFs is relatively clear, little is known about the molecular mechanism of Hsp40 enhancement.
Although the obligate co-chaperone Hsp40 is more diverse than Hsp70, all Hsp40s are characterized by a J domain with a highly conserved tripeptide HPD motif.11–15 The J domain, named after the well-studied Escherichia coli Hsp40 DnaJ, has been shown to be critical for stimulating the ATPase activity of Hsp70s.12 Consistent with their ability to stimulate the ATPase activity of Hsp70s, Hsp40s interact with Hsp70s robustly only in the presence of ATP. However, this association is usually transient, which makes studying the molecular mechanism challenging. A breakthrough in understanding this interaction came from a genetic suppressor screen using DnaK and DnaJ, an Hsp70 and Hsp40 in E. coli, respectively.16 This screen identified a highly conserved residue, R167, in DnaK’s NBD as a contact site for D35 in the HPD motif of DnaJ’s J domain, consistent with an NMR study suggesting that DnaK’s NBD has a binding site for the J domain of DnaJ.17 Subsequent biochemical and structural studies further suggested that Hsp70 has a binding site for the Hsp40 J domain on the bottom cleft of its NBD,16,18–21 where R167 resides. Furthermore, a number of mutations in Hsp70s, including several on the inter-domain linker (as described in more detail below), have been studied and shown to affect Hsp40 interactions; however, almost all exhibited simultaneous domain coupling defects.18,19,21–26 Thus, it seems that allosteric coupling to ATP binding in Hsp70s is required for Hsp40 interaction. Since Hsp40s manifest robust interaction with Hsp70s only in the ATP state, the allosteric active state, it is conceivable that allosteric coupling is essential for this interaction. This fact has made it difficult to tease apart the molecular mechanism of the Hsp40 interaction from the allosteric coupling in Hsp70s.
Previously, we made significant progress in elucidating the molecular mechanism of allosteric coupling in Hsp70s by solving an X-ray crystal structure of a full-length Hsp70 homolog, Sse1, in the ATP-bound state, the allosteric active state.27 This structure explained many previous biochemical and genetic results on Hsp70s. Furthermore, a number of recent studies using both NMR and fluorescence resonance energy transfer have validated that this Sse1-ATP structure represents the elusive ATP state of Hsp70s.28–31 Interestingly, the highly conserved inter-domain linker was seen, for the first time, in a well-ordered conformation. This linker has been shown to be flexible and exposed in ADP or in nucleotide-free states and, thus, was never seen in an ordered form in all other structures except those bound by SBD.18,32–34 In the Sse1-ATP structure, the linker forms a β strand that fits in a bottom cleft on NBD and extends into SBD. Thus, in this allosterically active ATP state, it serves as a bridge to structurally connect the two functional domains. The observed conformation of the linker is consistent with a number of previous studies using NMR and amide hydrogen exchange on Hsp70s,35–37 suggesting that the linker in Hsp70s most likely adopts a similar conformation.
Within the β strand formed by the inter-domain linker, there are four consecutive hydrophobic residues, which are highly conserved in classic Hsp70s. They are V389, L390, L391, and L392 in DnaK, a well-studied model of Hsp70. Several studies mutated multiple residues on the inter-domain linker and constructed a number of truncations to understand the role of the inter-domain linker.18,23,25,36,38 These studies clearly established the involvement of the linker residues 389–393 in allosteric coupling. Moreover, these mutational studies also suggested a potential involvement of these residues in Hsp40 interaction, perhaps indirectly via the defective allosteric coupling. The essential role of the linker in allosteric coupling was further supported by a number of structural studies on the isolated SBD from DnaK39,40 and biochemical studies on other Hsp70s.41,42 More recently, a structural study on bovine Hsc70 (heat shock cognate protein 70 kDa) NBD cross-linked with a J domain proposed that the J domain activates Hsp70’s ATPase activity by directing the linker toward a hydrophobic patch on the NBD surface, although the inter-domain linker is not visible in any of their structures.18 Despite all these extensive studies, the molecular mechanism and the exact role of the contributing residues on the inter-domain linker remain unclear.
In this study, inspired by the specific conformation of these four hydrophobic residues on the inter-domain linker in the Sse1-ATP structure, we hypothesized that these residues have two distinct roles: V389 and L391 mediate allosteric coupling within Hsp70s, while L390 and L392 are directly involved in Hsp40 interaction. Here, we present biochemical analyses to support this hypothesis after mutating each of these residues individually to either Asp or Ala. Thus, Hsp40s may stimulate Hsp70 chaperone activity by directly interacting with the inter-domain linker and then modulating the role of the inter-domain linker in Hsp70 allosteric coupling.
Results
Sse1 shows little interaction with Hsp40 DnaJ
Previous studies suggested that the interaction between Hsp70 and Hsp40 is highly conserved and that Hsp40s also show similar interactions with Hsp70s from different species.12,26,43,44 Sse1 is a distant homolog of Hsp70 from yeast, and its interaction with Hsp40 is still controversial, with one report demonstrating that Hsp40s fail to stimulate its ATPase activity while others report otherwise.44–46 To directly test the putative interaction between Sse1 and Hsp40, we utilized a well-established surface plasmon resonance (SPR) assay with DnaJ, an E. coli Hsp40, immobilized on a sensor chip.16,21 To immobilize DnaJ, we took advantage of a previously characterized DnaJ construct with the biotin carboxyl carrier protein (BCCP) fused at the C-terminus. This DnaJ–BCCP fusion construct remains fully functional and has been used to analyze the interaction with DnaK, the major Hsp70 in E. coli. As a positive control, DnaK was passed through the sensor chip. Consistent with previous observation, a strong signal was observed, indicating a robust interaction (Fig. 1). In contrast, when Sse1 was applied, no obvious signal was obtained, suggesting that there is no interaction with Hsp40.
Fig. 1.
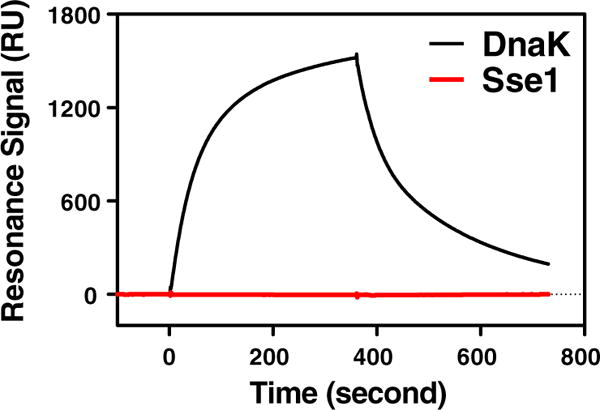
SRP analysis of the interaction with immobilized DnaJ. DnaK (black) and Sse1 (red) at 2 μM were injected through a sensor chip with ∼600 RU of immobilized DnaJ. Relative RU are plotted as a function of time.
Every conserved hydrophobic residue on the inter-domain linker of DnaK is essential
What could account for the lack of Hsp40 interaction with Sse1? One apparent difference between classic Hsp70s and Sse1 can be found within the inter-domain linker, which has been shown to be essential for both allosteric coupling and Hsp40 interaction by truncations and mutations at multiple positions at the same time. In classic Hsp70s, there are four consecutive hydrophobic residues on the inter-domain linker. All of these residues are highly conserved and are usually V, L, and I (Fig. 2a). In contrast, they are F392, K393, F394, and E395 in Sse1, with only the first and third residues being hydrophobic. In our previously published Sse1 structure (Fig. 2b and c), the two conserved hydrophobic residues—F392 and F394— form extensive hydrophobic contacts with NBD. These contacts are highly conserved in all Hsp70s, including Sse1 and the classic Hsp70s, suggesting essential roles of these residues in allosteric coupling. Our previous mutational analysis on the F392 position in two classic Hsp70s has confirmed the crucial role of this residue in in vivo chaperone activities.27 It is intriguing that the two non-conserved residues within the inter-domain linker— K393 and E395—have side chains oriented outward toward the surface, suggesting that they are not involved in domain coupling. Since Sse1 does not show interaction with Hsp40, it is possible that these residues in classic Hsp70s are involved in Hsp40 interaction. Consistent with this hypothesis, a previous suppressor study using DnaK had identified R167 (R170 in Sse1) as a contact site for Hsp40,16 and R170 is proximal to this segment of the inter-domain linker in our Sse1-ATP structure (Fig. 2c). Hence, we hypothesize that K393 and E395 are specifically positioned on the surface of Hsp70s in the ATP state to form the interacting surface for Hsp40s.
Fig. 2.
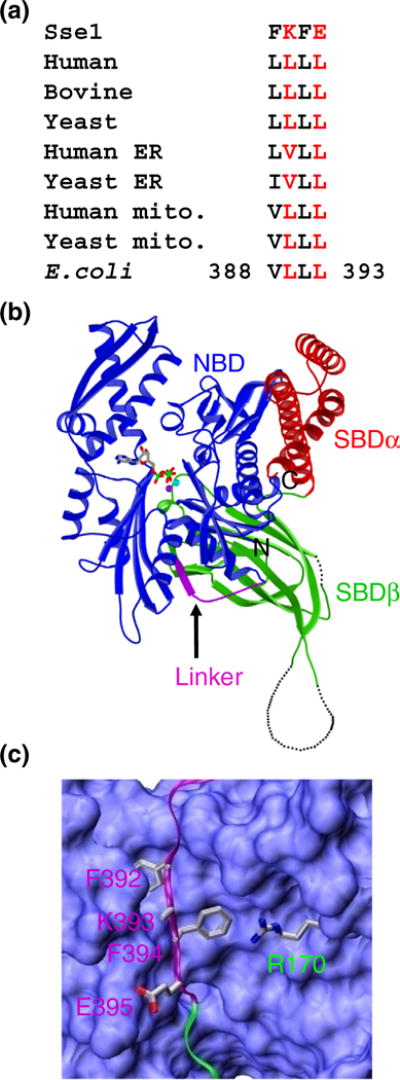
Structural representation and sequence alignment. (a) Sequence alignment of the four highly conserved hydrophobic residues within the Hsp70 inter-domain linker. The Sse1 sequence is on top. The residue numbers for the start and end residues in DnaK are labeled. (b) Ribbon diagram of the Sse1-ATP structure (Protein Data Bank code: 2QXL). NBD is in blue, SBDα is in red, SBDβ is in green, and the inter-domain linker is in purple. The ATP molecule is shown in stick representation, and the two associating ions are shown as purple (Mg2+) and cyan (K+) balls. (c) Close-up view of the inter-domain linker in the Sse1-ATP structure. Color coding is the same as in (b). NBD is shown in surface representation. The inter-domain linker and the SBDβ strand that the linker extends in are shown as ribbons. Side chains for F392, K393, F394, and E395 on the inter-domain linker are shown. These residues correspond to the four highly conserved hydrophobic residues in classic Hsp70s. The side chain for R170 on NBD is shown in stick (instead of surface) representation.
To test the role of these hydrophobic residues on the inter-domain linker, we mutated each in turn within DnaK, a standard model in studying classic Hsp70s. In DnaK, these residues are V389, L390, L391, and L392. As outlined above for the Sse1-ATP structure, we suggest that V389 and L391 are involved in domain coupling, whereas L390 and L392 are essential for Hsp40 interactions. Since Leu and Asp share a similar structure but with different properties, we mutated each residue to Asp to remove the hydrophobic property of Leu. We also mutated each to Ala to simply reduce the side chain. First, we tested whether these mutations affect the in vivo function of DnaK. To do this, we took advantage of the observation that DnaK is required for growth at 37 °C but not at 30 °C. We introduced these mutant DnaK proteins into a dnak deletion E. coli strain and then analyzed the growth at 37 °C using growth at 30 °C as a control. Wild-type (WT) DnaK and empty vector were used as positive and negative controls, respectively. Except for the V389A mutant, all of the mutant DnaKs behaved like the empty vector control (Fig. 3a), indicating a complete loss of in vivo function. Even for the V389A mutant, there is a significant defect in growth. Thus, each of these residues is important for supporting the in vivo function of DnaK. This loss of in vivo function is not due to lack of expression since all the mutant proteins were expressed at levels very similar to those at WT DnaK (Fig. 3b for Asp mutations, and data not shown for the Ala mutations). To better understand the role of these conserved residues, we evaluated each of the mutants biochemically.
Fig. 3.

Growth test on the inter-domain linker mutants in DnaK. (a) Growth test of the linker mutants in DnaK. Serial dilutions of fresh E. coli cultures were spotted on LB agar plates as described in Materials and Methods and incubated at 30 and 37 °C overnight. Empty vector and WT DnaK were used as negative and positive controls, respectively. Dilution factors of cultures are labeled on top. (b) Protein expression levels for the mutant proteins. Equal amounts of E. coli culture carrying each of the mutants of DnaK were loaded onto SDS-PAGE. The top frame is Western blot analysis with anti-DnaK antibody, and the bottom frame is Coomassie-stained SDS-PAGE gel.
Linker mutations have little effect on the two intrinsic activities
Since all four linker residues are far away from both the ATP hydrolysis site and the peptide substrate binding site, mutations should not have any significant influence on either of the two intrinsic activities of Hsp70s: the ATPase and peptide substrate binding activities. To test this hypothesis, we first carried out single-turnover ATPase assays to measure intrinsic ATPase activities. In this assay, a complex between DnaK and ATP was first formed and then the hydrolysis of this prebound ATP was monitored over time, thus allowing us to directly obtain the catalytic constant kcat. For WT DnaK, we obtained a kcat of 0.0101± 0.00029 min−1 at 20 °C (Supplementary Fig. 1a). The observed rate constants for all of the mutant proteins were within 1.5-fold of that for WT, indicating that none of the mutations influenced the intrinsic ATPase activity substantially.
Next, we tested the intrinsic peptide substrate binding affinity by using a previously described fluorescence anisotropy assay21,26,47 and the model peptide NR (sequence NRLLLTG). NR is a well-characterized peptide substrate for DnaK.39,48 The NR peptide was labeled with the fluorescent dye fluorescein at the N-terminus to make the F-NR peptide. The small size of the F-NR peptide has a low fluorescence anisotropy signal, which increases significantly upon DnaK binding. Therefore, following an increase in fluorescence anisotropy can be used to monitor the binding of DnaK to F-NR. To determine binding affinity, we incubated F-NR with increasing concentrations of DnaK protein and recorded fluorescence anisotropy after binding equilibrium was reached. Fitting of the fluorescence anisotropy data to a one-site binding equation gave us the dissociation constant Kd. For the WT protein, we obtained a Kd of 1.471±0.0626 μM (Supplementary Fig. 1b). For the mutant proteins, the Kd values were all within 1.5-fold of that of WT DnaK (Supplementary Fig. 1b). Thus, consistent with our hypothesis, all of the mutant proteins have intrinsic peptide substrate binding affinities similar to that of the WT protein. Taken together, none of the linker mutations affected the intrinsic activities of DnaK appreciably.
Inter-domain coupling is affected differently in the linker mutants
To test the roles of these residues in domain coupling, we first carried out a tryptophan fluorescence assay on the purified proteins, a well-established method to monitor domain coupling in DnaK.49 DnaK has a single tryptophan, Trp102 (in NBD), whose intrinsic fluorescence emission spectrum is sensitive to the nucleotide binding state in DnaK. Relative to the apo form of DnaK and when ADP is bound to DnaK, ATP binding induces a blue shift and a reduced intensity in the fluorescence spectrum only in the context of full-length DnaK when there is allosteric coupling. Thus, the intrinsic fluorescence is a sensitive indicator of ATP-induced allosteric coupling. As shown in Supplementary Fig. 2a, we observed the expected blue shift and reduction in fluorescence intensity for the WT DnaK protein. We first tested the aspartate (D) mutations. Consistent with their expected roles in domain coupling, we did not observe any visible change for either the V389D or the L391D mutant (Supplementary Fig. 2b and d). In contrast, both L390D and L392D mutants showed very similar changes upon ATP binding as was observed for the WT protein (Supplementary Fig. 2c and e). The observation of both blue shift and intensity reduction upon ATP binding indicates that domain coupling was largely intact for these two mutant proteins. In the presence of ADP or in the absence of nucleotide, the tryptophan fluorescence spectra for all of the mutant DnaK proteins behaved like the WT protein (Supplementary Figs. 2f and 3a). Furthermore, addition of acrylamide quenched the intrinsic fluorescence of all the mutant proteins to a similar extent as the WT protein (Supplementary Fig. 3b), indicating that none of the mutations affected the chemical environment of W102 in the undocked state appreciably. For the alanine series, all mutants behaved in ways similar to those of the corresponding aspartate mutations except for L391A, which showed a small degree of blue shift upon ATP binding (data not shown). Thus, as predicted from the Sse1-ATP structure, neither L390 nor L392 plays a significant role in Hsp70 allosteric coupling, while both V389 and L390 are crucial for domain coupling.
To further confirm the above results, we directly tested the inter-domain coupling in DnaK: ATP binding triggers bound peptide substrate release, and at the same time, peptide substrate binding stimulates ATP hydrolysis. First, we examined the bound peptide substrate release triggered by ATP binding, using the fluorescence anisotropy assay described above. F-NR was first bound to DnaK. After binding reached equilibrium, ATP was added to a high concentration, and the fluorescence anisotropy reading was recorded to follow the release of bound F-NR. For WT DnaK, upon ATP binding, the majority of bound F-NR was released as indicated by the decrease in the fluorescence anisotropy readings to values close to the F-NR control levels (Fig. 4a). Consistent with the above tryptophan fluorescence analysis, we observed no obvious release for either V389D or L391D, whereas both L390D and L392D released the bound F-NR, although to different extents. The release for L390D was almost the same as that for the WT protein, while L392D reproducibly released to a less extent, suggesting that L392D may have a mild influence on domain coupling. For all the Ala mutants, we observed similar results, but with fewer defects: both L390A and L392A behaved like the WT, whereas both V389A and L391A released significantly less (Fig. 4b).
Fig. 4.

Allosteric coupling in DnaK. (a and b) ATP-induced bound peptide substrate release for D (a) and A (b) mutants. F-NR was incubated with 5 μM DnaK proteins to allow binding to reach equilibrium. The fluorescence anisotropy values reached steady state (before time 0). At time 0, ATP was added to a final concentration of 2 mM, and then the release of the bound peptide was recorded by anisotropy measurements every 3 s. F-NR in buffer only was used as a negative control to show that addition of ATP does not affect the anisotropy values of F-NR. (c and d) Peptide substrate stimulation on the ATPase activity for D (c) and A (d) mutants. Single-turnover ATPase assays were performed in the presence of increasing concentrations of the model peptide F-NR. Fold of stimulation was calculated by setting the intrinsic ATPase activity rate as 1 and plotted as a function of increasing F-NR concentration. Error bars are shown for ±standard error of the mean with n=3.
Next, we tested the peptide-stimulated ATPase activity using the single-turnover ATPase assay described above. Consistent with previous observations, in the presence of the NR peptide, the ATP hydrolysis rate kcat was dramatically accelerated for WT DnaK (Fig. 4c and d). With increasing concentration of the NR peptide, we observed an increasing ATPase rate. At the highest concentration of the NR peptide used in our assay (400 μM), we obtained about 15-fold stimulation that seemed to approach a plateau. Consistent with both tryptophan fluorescence and ATP-triggered peptide release results shown above, neither V389D nor L391D showed any obvious stimulation over the range of the peptide concentration used in our assay, and there is only about 2.5-fold maximum stimulation for V389A and L391A (Fig. 4c and d). In contrast, all the L390 and L392 mutant proteins were stimulated significantly by the NR peptide. At low concentrations of the NR peptide (such as 5, 10, and 20 μM), the fold of stimulation for the mutant proteins was similar to that for the WT protein. However, at higher concentrations of NR, the fold of stimulation was lower, especially for L392D (about 5-fold stimulation). The lower stimulation in the L392D mutant is not surprising since it also released less bound peptide in the above ATP-triggered peptide release assay.
Taken together, consistent with the prediction from our Sse1-ATP structure, all three domain coupling assays demonstrated that the inter-domain coupling is largely intact for L390 and L392 mutants. In contrast, the inter-domain coupling was dramatically compromised in all of the V389 and L391 mutant proteins, supporting their critical roles in the Hsp70 allosteric coupling.
Mutations at L390, L391, and L392 dramatically reduced the interaction with the Hsp40 co-chaperone
It is well established that DnaJ stimulates DnaK’s ATPase activity by directly accelerating the ATP hydrolysis rate. To test whether L390 and L392 are involved in the interactions with Hsp40, we carried out single-turnover ATPase assays by including DnaJ in our assays. For the WT DnaK protein, in the presence of DnaJ, the ATP hydrolysis rate kcat is dramatically accelerated, about 48.5-fold at 0.8 μM DnaJ (Fig. 5). As expected, none of the L390 and L392 mutants were stimulated significantly by DnaJ. In contrast, DnaJ stimulates the V389A mutant to a similar extent as the WT DnaK, although no obvious stimulation was detected for the other V389 and L391 mutants. The Hsp40 interaction defect in these mutants could be secondary due to defective allosteric coupling.
Fig. 5.
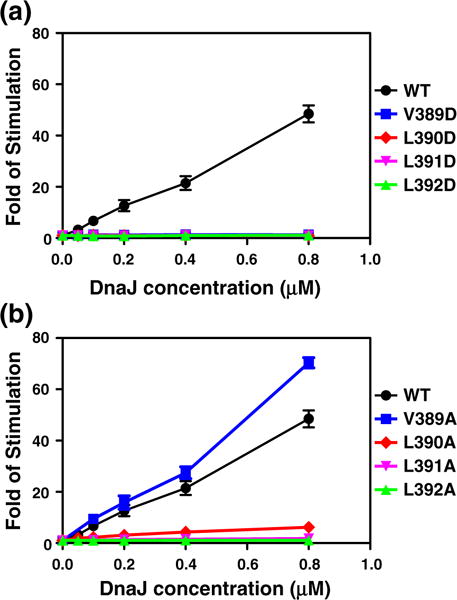
DnaJ stimulation of DnaK’s ATPase activity. Various concentrations of DnaJ were applied to the single-turnover ATPase assays to determine the stimulation by DnaJ. Fold of stimulations were plotted as a function of increasing DnaJ concentration. Error bars are shown for ±standard error of the mean with n=3. (a) D mutants, (b) A mutants.
To further confirm the DnaJ interaction defect, we tested the DnaK mutants using the SRP assay as described above. When we passed the WT DnaK protein through the sensor chip, we observed strong interaction signals in a concentration-dependent manner (Fig. 6a and g). Consistent with the ATPase assays, we observed a drastically reduced signal for all of the DnaK mutants except V389A (Fig. 6). Corresponding to a similar level of ATPase stimulation shown above, a close-to-WT level of interaction was observed with the V389A mutant (Fig. 6h). For the V389D, L390A, and L391A mutants, the SRP signal is about 10% that of the WT DnaK protein (Fig. 6b, i, and j), and for the rest of the mutant proteins, we hardly detected any signal at all even at the concentration of 16 μM, the highest concentration of DnaK used in our assay (Fig. 6c–f, k, and l).
Fig. 6.
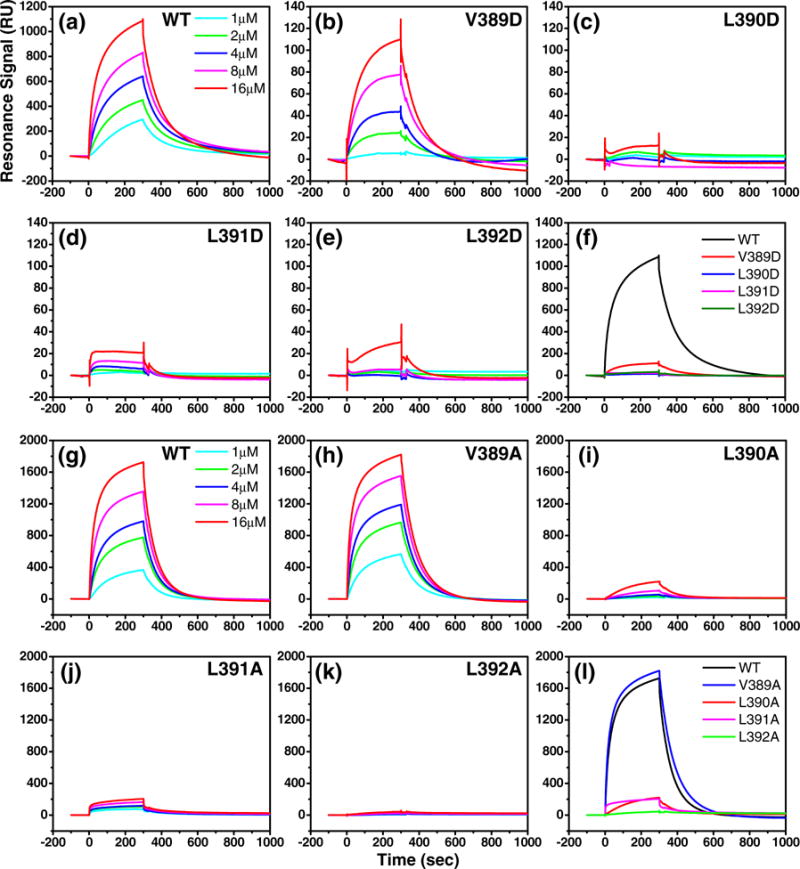
SRP assay on DnaK–DnaJ interaction in the DnaK mutants. After DnaJ was immobilized on an SA sensor chip, serial dilutions of DnaK proteins were injected over the sensor chip at time 0. Responses were recorded over time, and the background binding to a control channel was subtracted from each recording. (a) WT DnaK (performed at the same time with the D mutants), (b) V389D, (c) L390D, (d) L391D, (e) L392D, (g) WT DnaK (performed at the same time with the A mutants), (h) V389A, (i) L390A, (j) L391A, and (k) L392A. The DnaK concentration of each recording was labeled in (a) and (g). (f) and (l) are DnaJ interactions at 16 μM for all the D and A mutants, respectively, in comparison with the WT DnaK.
Although L390 and L392 are not conserved in Sse1, they are highly conversed in classic Hsp70s. Thus, we explored whether these positions in classic Hsp70s can tolerate any changes by mutating each in DnaK to all the other available amino acids. As shown in Fig. 7, except for Ile and Val, all the other changes resulted in dramatic growth defects, demonstrating that the size and hydrophobic properties of the side chains at these two positions are very strict, and any significant change will abolish function.
Fig. 7.

Mutational test at the L390 and L392 positions. L390 and L392 were mutated to all other 19 possible amino acids, and growth test was performed as in Fig. 3. Dilution factors of cultures are labeled on top.
Discussion
In this study, we biochemically dissected the role of the four highly conserved hydrophobic residues on the inter-domain linker. Although earlier studies with truncations and mutations at multiple sites have established the essential role of the whole inter-domain linker in allosteric coupling,18,23,25,36,38 the exact contribution of each residue has been a mystery. Guided by our previously reported Sse1-ATP structure, we hypothesized that these four hydrophobic residues have distinct roles: V389 and L391 are mainly involved in allosteric coupling in Hsp70s, while L390 and L392 function in Hsp40 interactions. As summarized in Table 1, our biochemical data on the linker mutants in DnaK support this hypothesis. None of these residues are involved in the two intrinsic activities. Mutating either V389 or L391 resulted in dramatic allosteric coupling defects. In contrast, allosteric coupling is largely intact when either L390 or L392 is mutated, but Hsp40 interactions are almost completely abolished. Although Sse1 is a distant homolog of Hsp70s, the Sse1-ATP structure has been shown to represent the elusive ATP state of Hsp70s by both our previous mutational studies and a number of recent studies.28–31 Especially for the inter-domain linker, several studies using NMR and amide hydrogen exchange supported that it assumes a similar conformation as seen in the Sse1-ATP structure.35–37 The results presented in this study further support the fact that the Sse1-ATP structure represents the ATP state of classic Hsp70s, especially the conformation of the inter-domain linker, although it is conceivable that the exact conformation of the inter-domain linker in Hsp70s may differ slightly from that in the Sse1-ATP structure.
Table 1.
Summary of the linker mutations
| WT | V389D | L390D | L391D | L392D | V389A | L390A | L391A | L392A | |
|---|---|---|---|---|---|---|---|---|---|
| Allosteric coupling | ++++ | − | +++ | − | ++ | + | ++ | + | +++ |
| Hsp40 interaction | ++++ | − | − | − | − | ++++ | + | − | − |
Our biochemical analysis separated, for the first time, the role of the inter-domain linker in allosteric coupling from that of Hsp40 interaction. Allosteric coupling and interaction with Hsp40 are two crucial activities in understanding the chaperone function of Hsp70s. However, despite extensive efforts, the molecular mechanisms of both are still ill-defined due to the transient nature of both. Since Hsp40s interact with Hsp70s only in the ATP state, the allosteric active state, it is conceivable that Hsp40 interaction depends on the allosteric coupling in Hsp70; however, this has made it difficult for anyone to study Hsp40 interaction independently. Thus, understanding the ATP state of Hsp70s will fundamentally advance our understanding not only of the Hsp70 molecule itself but also of its interaction with the Hsp40 co-chaperones. Before having a structural understanding of this state of Hsp70s, it had been very difficult for us to tease apart the Hsp40 interaction from the allosteric coupling in Hsp70s. Previously, a number of Hsp70 mutations have been studied and shown to have Hsp40 interaction defects,19,21–25 but they all showed simultaneous defects in domain coupling except in one study that used mitochondrial Hsp70.26 These results led to the proposal that the allosteric coupling in Hsp70 is required for the Hsp40 interaction.
The Sse1-ATP structure gave us an unprecedented advantage in understanding both allosteric coupling and Hsp40 interaction. The conserved interaction formed between NBD and two residues from the linker, V389 and L391, explained the long-discovered role of the inter-domain linker in allosteric coupling. Starting from this structure, Smock et al. elegantly combined computational and experimental approaches and identified a structurally contiguous group of residues that form a physical network of linkage between NBD and SBD, which was named a sector.50 This sector forms the physical bases for allosteric coupling in Hsp70s. Interestingly, the inter-domain linker seems to be at the center of this sector. Thus, it is tempting to propose that allosteric coupling is achieved through coordinated action of the inter-domain linker and the other residues in the sector from both NBD and SBD, such as the newly identified D326 and N415 in this study.
On the other hand, we found that the V389A mutant has a normal interaction with Hsp40, although allosteric coupling is defective, suggesting that Hsp40 interaction does not strictly depend on allosteric coupling as proposed before. Since the V389A mutant showed partial rescue of the lack of DnaK, it is possible that its normal Hsp40 interaction helps to alleviate some of the in vivo functional defect caused by the severe allosteric coupling defects. Furthermore, mutations at L390 and L392 positions, especially L390A and L392A, lost their interaction with Hsp40, but allosteric coupling is largely intact, indicating their specific roles in Hsp40 interaction. These two residues may directly mediate the interaction with Hsp40; thus, the Hsp70–Hsp40 interaction may also involve hydrophobic contacts. Previously, an NMR study suggested that the Hsp70–Hsp40 interaction is dominated by electrostatic interactions,17 which is consistent with the identification of R167 on DnaK as a contact site for D35 from DnaJ.16 However, this same NMR study and a mutagenesis study identified several hydrophobic residues in DnaJ that are on the surface of the J domain structure and may be involved in contacting Hsp70: Y25, M30, and F47 (Fig. 8a).51 Consistent with the high conservation of the Hsp70–Hsp40 interaction, all of these residues are highly conserved (Fig. 8b). These hydrophobic residues may contact either L390 or L392 on the Hsp70 inter-domain linker. The J domain structure for DnaJ has been solved by way of NMR.52,53 It is composed of four α helices, with the highly conserved HPD motif on the loop between helix II and helix III. D35 in DnaJ is the last residue in the HPD motif. With D35 on DnaJ as a contact site for R167 on DnaK, the positions of L390 and L392 on the linker are in a good range to interact with these hydrophobic residues on DnaJ. However, in the crystal structure of the bovine Hsc70 NBD cross-linked with a J domain, only the corresponding residue of F47 contacts two hydrophobic residues from Hsc70 NBD: L380 and I179.18 It is possible that this J domain is not from a canonical Hsp40, which has been shown to interact differently with Hsp70s.17,54 More importantly, the inter-domain linker is missing in their structures. Further biochemical and structural studies are needed to clarify this issue.
Fig. 8.

The structure representation and sequence alignment of the J domain. (a) Ribbon diagram of the NMR structure of DnaJ J domain (Protein Data Bank code: 1XBL). Residues D35, Y25, M30, and F47 are in bonds. (b) Sequence alignment of the J domain (from helix II to helix III) from a number of Hsp40s. Ydj1 and Sis1 are two major Hsp40s from yeast, and Hdj1 and Hdj2 are from human. Residues Y25, M30, and F47 are highlighted in red, and the HPD motif is shaded in blue.
The highly conserved inter-domain linker in Hsp70s is very special, although short. During the Hsp70 chaperone cycle, this linker undergoes unique conformational changes. All available evidence suggested that it is only ordered in the ATP state, in a conformation possibly as seen in the Sse1-ATP structure, but very flexible in other states. Thus, it seems that only in the ATP state are L390 and L392 specifically positioned in the suitable orientation to interact with the Hsp40 co-chaperone. This may explain the ATP requirement for the Hsp70–Hsp40 interaction. Although the Hsp40 interaction is also dramatically affected in L389D, L391D, and L391A mutations, these Hsp40 interaction defects are most likely secondary since V389A has a normal Hsp40 interaction. These mutations are likely to have changed the linker conformation or position; thus, either L390 or L392 would no longer be in the correct position for Hsp40 interaction. Alternatively, Hsp40 stimulation may require allosteric coupling through these sites, especially L391. It is possible that Hsp40 stimulates the ATP hydrolysis rate of Hsp70 through further manipulation of the linker conformation, which could change the linker’s contact with NBD or SBD and, thus, its role in allosteric coupling. Therefore, it seems that the highly conserved inter-domain linker behaves like a functional switch mechanism on the Hsp70 molecules. It is rather intriguing how much information this small inter-domain linker is transmitting in the function and regulation of Hsp70s. Further structural and biochemical studies will help us define the exact mechanism.
Various NEFs have been discovered to accelerate the nucleotide exchange in Hsp70s. Intensive studies suggested that Hsp70s’ NBD itself is sufficient to interact with NEF.55–60 Up to now, there is no evidence indicating that the inter-domain linker is involved in the interaction with any NEF. Moreover, NEFs interact with the ADP-bound and nucleotide-free states of Hsp70s, where the linker is in a disordered conformation, with an affinity much higher than that of the ATP-bound state. Taken together, it is most likely that the role of the inter-domain linker in NEF interaction is minimal.
Our results also explain why Sse1 does not interact with Hsp40s. Sse1 belongs to the Hsp110 family, a distant homolog of classic Hsp70s. Sequence alignment suggested that although V389 and L391 are highly conserved in both classic Hsp70s and their Hsp110 homologs, L390 and L392 are not. In Sse1, L390 and L392 are K and E, respectively. When we mutated either L390 or L392 in DnaK, we did not observe any Hsp40 interaction. Since Sse1 has a much slower ATP hydrolysis rate,27,46,56 most of the time, it is in the ATP state, which is the conformation that Hsp40s would bind. If Sse1 were to have the same linker as classic Hsp70s, it could bind Hsp40s so tightly and sequester Hsp40s from productively interacting with the classic Hsp70s, which may have an adverse effect. As a distant homolog and major NEF for the cytosolic Hsp70,61 Sse1 most likely does not need to interact with Hsp40 for its function.
Materials and Methods
Site-directed mutagenesis and growth test
As described previously, we used a dnaK expression plasmid pBB46 (ampR) to test the in vivo function of mutant DnaK.27,62 The four hydrophobic residues on the inter-domain linker of DnaK (V389, L390, L391, and L392) were mutated to aspartic acid or alanine one by one using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The pBB46 plasmid carrying either the WT or mutant DnaK was transformed into a dnak deletion strain BB205 (camR kanR). Transformants were selected at 30 °C on LB plates containing 50 μg/ml ampicillin, 25 μg/ml kanamycin, and 25 μg/ml chloramphenicol. A fresh overnight culture was grown from a single colony in LB containing all three abovementioned antibiotics. Tenfold serial dilutions of these overnight cultures were spotted on LB plates containing 20 μM IPTG and all three antibiotics and then incubated at either 30 or 37 °C overnight to evaluate growth.
Protein purification
The Sse1 protein was purified as described before.27
To facilitate the purification of DnaK, we constructed a plasmid, pBB46-His, from pBB46 by inserting a hexahistidine tag at the very C-terminus of DnaK. Since this hexahistidine tag has little influence on the growth phenotype of the WT and the mutant DnaK, we could use it to analyze the difference between the WT and the mutant proteins. All the DnaK proteins were expressed in the dnak deletion strain BB205 (camR kanR) at 30 °C with 1 mM IPTG in YT medium supplemented with 50 μg/ml ampicillin, 25 μg/ml kanamycin, and 25 μg/ml chloramphenicol.
After a 5-h induction, cells were harvested by centrifu-gation at 5000g. Cell pellet was kept cold on ice and resuspended in cold 2× phosphate-buffered saline (20 mM Na2HPO4, 1.76 mM KH2PO4, 274 mM NaCl, and 5.4 mM KCl). After a 1-h incubation with lysozyme (final concentration, 0.01 mg/ml), sonication was performed on ice for 3 min. The cell lysate was cleared by centrifugation at 20,000g for 1 h and then loaded onto a HisTrap column (GE Healthcare). DnaK proteins eluted from the HisTrap column were further purified with a HiTrap Q column (GE Healthcare). Before being flash frozen in liquid nitrogen, fractions containing DnaK protein from the HiTrap Q were pooled and concentrated to N20 mg/ml in a buffer containing 10 mM Hepes–KOH, pH 7.5, and 50 mM KCl.
For the purification of DnaJ protein, the DnaJ open reading frame was amplified from E. coli genomic DNA and cloned into pSMT3 vector as an Smt3 fusion protein with an N-terminal hexahistidine tag. Induction was performed at 30 °C with 1 mM IPTG in YT medium for 3 h. Cells were harvested by centrifugation and resus-pended in a phosphate-buffered saline–KCl buffer (20 mM Na2HPO4, 1.76 mM KH2PO4, 280 mM KCl, and 10% glycerol). After sonication, cell lysate was cleared by centrifugation at 20,000g and then loaded onto a HisTrap column (GE Healthcare). After the removal of the Smt3 tag with Ulp1, the DnaJ protein was further purified on a HisTrap column and a Superdex 75 16/60 size-exclusion column (GE Healthcare). The pSMT3 vector was a generous gift from Dr. Lima.
SPR analysis
To measure the interaction between Hsp70 and Hsp40, we performed SRP experiments with the help of a Biacore T100 system (GE Healthcare). A fusion construct of DnaJ with the BCCP fused at the C-terminus was immobilized on a streptavidin (SA) sensor chip surface (GE Health-care), as described previously.16,21 About 200 to 250 response units (RU) of DnaJ fusion protein was immobilized. All SPR experiments were conducted at 24 °C in Biacore buffer [25 mM Hepes–KOH, pH 7.5, 100 mM KCl, 10 mM Mg(OAc)2, 0.003% P20 surfactant, and 1 mM ATP] at a flow rate of 30 μl/min according to the standard protocol provided by the manufacturer. Right before injection, DnaK or Sse1 proteins were diluted to the desired concentrations with Biacore buffer and incubated at room temperature for 2 min. Each sample was injected for 5 min, and after each injection, Biacore buffer was run through the sensor chip until complete dissociation. The first channel on the SA sensor chip was used as background control, and this background binding was subtracted from each recording.
Fluorescence anisotropy peptide binding assay
To determine peptide substrate binding, we used a model peptide, F-NR. F-NR is an N-terminally fluorescein-labeled peptide with amino acid sequence NRLLLTG39,48 (synthesized by NEO Bioscience). First, we prepared a serial dilution of DnaK proteins in buffer A [25 mM Hepes–KOH, pH 7.5, 100 mM KCl, and 10 mM Mg(OAc)2] with 100 μM ADP. Then, F-NR was added to a final concentration of 10 nM and incubated for several hours at room temperature to reach equilibrium. Fluorescence anisotropy measurements were made using the Beacon 2000 Fluorescence Polarization System (Invitrogen) at 25 °C with excitation at 490 nm and emission at 535 nm. The dissociation constant Kd was calculated by fitting the fluorescence anisotropy data to a one-site binding equation by nonlinear regression analysis using Prism (GraphPad).
To assay peptide release upon ATP binding, we incubated 5 μM DnaK protein with 10 nM F-NR at room temperature in the presence of 100 μM ADP in buffer A. After several hours, binding reached equilibrium. Then, ATP was added to a final concentration of 2 mM, without changing the volume significantly. Right before and after ATP was added, fluorescence anisotropy was measured continuously every 3 s for 150 s.
Single-turnover ATPase assay
Single-turnover ATPase assays with DnaK proteins were performed as previously described, with modifications.21,26 To form a DnaK–ATP complex, we incubated the DnaK protein (20 μg) with 25 μCi of [α-32P] ATP (NEG503H250UC, 3000 Ci/mmol; Perkin Elmer) in buffer A containing 20 μM unlabeled ATP for 2 min at 4 °C. The complex was separated from free ATP on a spin column pre-equilibrated with spin column buffer [25 mM Hepes–KOH, pH 7.5, 100 mM KCl, 10 mM Mg(OAc)2, and 10% glycerol]. The ATPase assay was started by mixing equal volumes of the DnaK–ATP complex with the NR peptide or the DnaJ protein in buffer A at various concentrations. At specific time points, reactions were stopped, and the ATP was separated from ADP using thin-layer chromatography. [α-32P]ATP and [α-32P]ADP in the reactions were visualized and quantified with a Typhoon phosphoimaging system (GE Healthcare) to calculate the percentage of hydrolyzed ATP. We determined the rate of ATP hydrolysis (kcat) by fitting the ATP hydrolysis data to a first-order rate equation by nonlinear regression analysis using Prism (GraphPad).
Supplementary Material
Acknowledgments
We thank Dr. Elizabeth A. Craig for providing the DnaJ–BCCP construct used in the SPR assay and Drs. Wayne A. Hendrickson, Elizabeth A. Craig, Diomedes E. Logothetis, Louis J. De Felice, Young-Jia You, Jason Rife, Carlos Escalante, and Lei Zhou for critically reading the manuscript and providing insightful suggestions. We also thank Dr. Carlos Escalante for the PC1 Photon Counting Spectrofluorimeter, Dr. Daniel H. Conrad for the Biacore T100 system and for discussion, Dilhara Dewasinghe for technical support, and Dr. Xinping Xu for discussion. This work was supported by startup funds from the School of Medicine, Virginia Commonwealth University (Q.L.), and a New Scholar Award in Aging from the Ellison Medical Foundation (Q.L.).
Abbreviations
- Hsp70
70-kDa heat shock protein
- Hsp40
40-kDa heat shock protein
- NBD
nucleotide binding domain
- SBD
substrate binding domain
- NEF
nucleotide exchange factor
- SPR
surface plasmon resonance
- BCCP
biotin carboxyl carrier protein
- WT
wild type
- SA
streptavidin
- RU
response units
Footnotes
Edited by J. E. Ladbury
Supplementary Data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2011.07.001
References
- 1.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev, Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 4.Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 6.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 7.Young JC. Mechanisms of the Hsp70 chaperone system. Biochem Cell Biol. 2010;88:291–300. doi: 10.1139/o09-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmid D, Baici A, Gehring H, Christen P. Kinetics of molecular chaperone action. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 9.Flynn GC, Chappell TG, Rothman JE. Peptide binding and release by proteins implicated as catalysts of protein assembly. Science. 1989;245:385–390. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- 10.Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc Natl Acad Sci USA. 1991;88:2874–2878. doi: 10.1073/pnas.88.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev, Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan CY, Lee S, Cyr DM. Mechanisms for regulation of Hsp70 function by Hsp40. Cell Stress Chaperones. 2003;8:309–316. doi: 10.1379/1466-1268(2003)008<0309:mfrohf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Qian X, Sha B. Heat shock protein 40: structural studies and their functional implications. Protein Pept Lett. 2009;16:606–612. doi: 10.2174/092986609788490159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hennessy F, Nicoll WS, Zimmermann R, Cheetham ME, Blatch GL. Not all J domains are created equal: implications for the specificity of Hsp40–Hsp70 interactions. Protein Sci. 2005;14:1697–1709. doi: 10.1110/ps.051406805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63:2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suh WC, Burkholder WF, Lu CZ, Zhao X, Gottesman ME, Gross CA. Interaction of the Hsp70 molecular chaperone, DnaK, with its cochaperone DnaJ. Proc Natl Acad Sci USA. 1998;95:15223–15228. doi: 10.1073/pnas.95.26.15223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greene MK, Maskos K, Landry SJ. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc Natl Acad Sci USA. 1998;95:6108–6113. doi: 10.1073/pnas.95.11.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang J, Maes EG, Taylor AB, Wang L, Hinck AP, Lafer EM, Sousa R. Structural basis of J cochaperone binding and regulation of Hsp70. Mol Cell. 2007;28:422–433. doi: 10.1016/j.molcel.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gassler CS, Buchberger A, Laufen T, Mayer MP, Schröder H, Valencia A, Bukau B. Mutations in the DnaK chaperone affecting interaction with the DnaJ cochaperone. Proc Natl Acad Sci USA. 1998;95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wisen S, Bertelsen EB, Thompson AD, Patury S, Ung P, Chang L, et al. Binding of a small molecule at a protein–protein interface regulates the chaperone activity of hsp70–hsp40. ACS Chem Biol. 2010;5:611–622. doi: 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis JE, Voisine C, Craig EA. Intragenic suppressors of Hsp70 mutants: interplay between the ATPase- and peptide-binding domains. Proc Natl Acad Sci USA. 1999;96:9269–9276. doi: 10.1073/pnas.96.16.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suh WC, Lu CZ, Gross CA. Structural features required for the interaction of the Hsp70 molecular chaperone DnaK with its cochaperone DnaJ. J Biol Chem. 1999;274:30534–30539. doi: 10.1074/jbc.274.43.30534. [DOI] [PubMed] [Google Scholar]
- 23.Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of hsp70 chaperones by DnaJ cochaper-ones. Proc Natl Acad Sci USA. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Awad W, Estrada I, Shen Y, Hendershot LM. BiP mutants that are unable to interact with endoplasmic reticulum DnaJ proteins provide insights into interdomain interactions in BiP. Proc Natl Acad Sci USA. 2008;105:1164–1169. doi: 10.1073/pnas.0702132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han W, Christen P. Mutations in the interdomain linker region of DnaK abolish the chaperone action of the DnaK/DnaJ/GrpE system. FEBS Lett. 2001;497:55–58. doi: 10.1016/s0014-5793(01)02435-8. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q, Krzewska J, Liberek K, Craig EA. Mitochondrial Hsp70 Ssc1: role in protein folding. J Biol Chem. 2001;276:6112–6118. doi: 10.1074/jbc.M009519200. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Hendrickson WA. Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell. 2007;131:106–120. doi: 10.1016/j.cell.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlecht R, Erbse AH, Bukau B, Mayer MP. Mechanics of Hsp70 chaperones enables differential interaction with client proteins. Nat Struct Mol Biol. 2011;18:345–351. doi: 10.1038/nsmb.2006. [DOI] [PubMed] [Google Scholar]
- 29.Marcinowski M, Höller M, Feige MJ, Baerend D, Lamb DC, Buchner J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat Struct Mol Biol. 2011;18:150–158. doi: 10.1038/nsmb.1970. [DOI] [PubMed] [Google Scholar]
- 30.Mapa K, Sikor M, Kudryavtsev V, Waegemann K, Kalinin S, Seidel CA, et al. The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol Cell. 2011;38:89–100. doi: 10.1016/j.molcel.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharya A, Kurochkin AV, Yip GN, Zhang Y, Bertelsen EB, Zuiderweg ER. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J Mol Biol. 2009;388:475–490. doi: 10.1016/j.jmb.2009.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang J, Prasad K, Lafer EM, Sousa R. Structural basis of interdomain communication in the Hsc70 chaperone. Mol Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang YW, Sun YJ, Wang C, Hsiao CD. Crystal structures of the 70-kDa heat shock proteins in domain disjoining conformation. J Biol Chem. 2008;283:15502–15511. doi: 10.1074/jbc.M708992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertelsen EB, Chang L, Gestwicki JE, Zuiderweg ER. Solution conformation of wild-type E. coli Hsp70 (DnaK) chaperone complexed with ADP and substrate. Proc Natl Acad Sci USA. 2009;106:8471–8476. doi: 10.1073/pnas.0903503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J Biol Chem. 2006;281:16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- 36.Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Revington M, Zhang Y, Yip GN, Kurochkin AV, Zuiderweg ER. NMR investigations of allosteric processes in a two-domain Thermus thermophilus Hsp70 molecular chaperone. J Mol Biol. 2005;349:163–183. doi: 10.1016/j.jmb.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 38.Vogel M, Mayer MP, Bukau B. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem. 2006;281:38705–38711. doi: 10.1074/jbc.M609020200. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–1614. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liebscher M, Roujeinikova A. Allosteric coupling between the lid and interdomain linker in DnaK revealed by inhibitor binding studies. J Bacteriol. 2009;191:1456–1462. doi: 10.1128/JB.01131-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blamowska M, Sichting M, Mapa K, Mokranjac D, Neupert W, Hell K. ATPase domain and interdomain linker play a key role in aggregation of mitochondrial Hsp70 chaperone Ssc1. J Biol Chem. 2010;285:4423–4431. doi: 10.1074/jbc.M109.061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amor-Mahjoub M, Gomez-Vrielyunck N, Suppini JP, Fouchaq B, Benarouj N, Ladjimi M. Involvement of the interdomain hydrophobic linker and the C-terminal helices in self-association of the molecular chaperone HSC70. Arch Inst Pasteur Tunis. 2006;83:53–62. [PubMed] [Google Scholar]
- 43.Craig EA, Huang P, Aron R, Andrew A. The diverse roles of J-proteins, the obligate Hsp70 co-chaperone. Rev Physiol, Biochem Pharmacol. 2006;156:1–21. doi: 10.1007/s10254-005-0001-0. [DOI] [PubMed] [Google Scholar]
- 44.Goeckeler JL, Petruso AP, Aguirre J, Clement CC, Chiosis G, Brodsky JL. The yeast Hsp110, Sse1p, exhibits high-affinity peptide binding. FEBS Lett. 2008;582:2393–2396. doi: 10.1016/j.febslet.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raviol H, Sadlish H, Rodriguez F, Mayer MP, Bukau B. Chaperone network in the yeast cytosol: Hsp110 is revealed as an Hsp70 nucleotide exchange factor. EMBO J. 2006;25:2510–2518. doi: 10.1038/sj.emboj.7601139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raviol H, Bukau B, Mayer MP. Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 2006;580:168–174. doi: 10.1016/j.febslet.2005.11.069. [DOI] [PubMed] [Google Scholar]
- 47.Montgomery DL, Morimoto RI, Gierasch LM. Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J Mol Biol. 1999;286:915–932. doi: 10.1006/jmbi.1998.2514. [DOI] [PubMed] [Google Scholar]
- 48.Gragerov A, Zeng L, Zhao X, Burkholder W, Gottesman ME. Specificity of DnaK–peptide binding. J Mol Biol. 1994;235:848–854. doi: 10.1006/jmbi.1994.1043. [DOI] [PubMed] [Google Scholar]
- 49.Buchberger A, Theyssen H, Schröder H, McCarty JS, Virgallita G, Milkereit P, et al. Nucleotide-induced conformational changes in the ATPase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- 50.Smock RG, Rivoire O, Russ WP, Swain JF, Leibler S, Ranganathan R, Gierasch LM. An interdomain sector mediating allostery in Hsp70 molecular chaperones. Mol Syst Biol. 2010;6:1–9. doi: 10.1038/msb.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Genevaux P, Schwager F, Georgopoulos C, Kelley WL. Scanning mutagenesis identifies amino acid residues essential for the in vivo activity of the Escherichia coli DnaJ (Hsp40) J-domain. Genetics. 2002;162:1045–1053. doi: 10.1093/genetics/162.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szyperski T, Pellecchia M, Wall D, Georgopoulos C, Wuthrich K. NMR structure determination of the Escherichia coli DnaJ molecular chaperone: secondary structure and backbone fold of the N-terminal region (residues 2–108) containing the highly conserved J domain. Proc Natl Acad Sci USA. 1994;91:11343–11347. doi: 10.1073/pnas.91.24.11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill RB, Flanagan JM, Prestegard JH. 1H and 15N magnetic resonance assignments, secondary structure, and tertiary fold of Escherichia coli DnaJ(1–78) Biochemistry. 1995;34:5587–5596. doi: 10.1021/bi00016a033. [DOI] [PubMed] [Google Scholar]
- 54.Garimella R, Liu X, Qiao W, Liang X, Zuiderweg ER, Riley MI, Van Doren SR. Hsc70 contacts helix III of the J domain from polyomavirus T antigens: addressing a dilemma in the chaperone hypothesis of how they release E2F from pRb. Biochemistry. 2006;45:6917–6929. doi: 10.1021/bi060411d. [DOI] [PubMed] [Google Scholar]
- 55.Hendrickson WA, Liu Q. Exchange we can believe in. Structure. 2008;16:1153–1155. doi: 10.1016/j.str.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 56.Polier S, Dragovic Z, Hartl FU, Bracher A. Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell. 2008;133:1068–1079. doi: 10.1016/j.cell.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 57.Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor GrpE bound to the ATPase domain of the molecular chaperone DnaK. Science. 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- 58.Sondermann H, Scheufler C, Schneider C, Hohfeld J, Hartl FU, Moarefi I. Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science. 2001;291:1553–1557. doi: 10.1126/science.1057268. [DOI] [PubMed] [Google Scholar]
- 59.Cyr DM. Swapping nucleotides, tuning Hsp70. Cell. 2008;133:945–947. doi: 10.1016/j.cell.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shomura Y, Dragovic Z, Chang HC, Tzvetkov N, Young JC, Brodsky JL, et al. Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol Cell. 2005;17:367–379. doi: 10.1016/j.molcel.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 61.Morano KA. New tricks for an old dog: the evolving world of Hsp70. Ann N Y Acad Sci. 2007;1113:1–14. doi: 10.1196/annals.1391.018. [DOI] [PubMed] [Google Scholar]
- 62.Burkholder WF, Zhao X, Zhu X, Hendrickson WA, Gragerov A, Gottesman ME. Mutations in the C-terminal fragment of DnaK affecting peptide binding. Proc Natl Acad Sci USA. 1996;93:10632–10637. doi: 10.1073/pnas.93.20.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.