

Abstract
Pre-mRNA molecules can form a variety of structures, and both secondary and tertiary structures have important effects on processing, function and stability of these molecules. The prediction of RNA secondary structure is a challenging problem and various algorithms that use minimum free energy, maximum expected accuracy and comparative evolutionary based methods have been developed to predict secondary structures. However, these tools are not perfect, and this remains an active area of research. The secondary structure of pre-mRNA molecules can have an enhancing or inhibitory effect on pre-mRNA splicing. An example of enhancing structure can be found in a novel class of introns in zebrafish. About 10% of zebrafish genes contain a structured intron that forms a bridging hairpin that enforces correct splice site pairing. Negative examples of splicing include local structures around splice sites that decrease splicing efficiency and potentially cause mis-splicing leading to disease. Splicing mutations are a frequent cause of hereditary disease. The transcripts of disease genes are significantly more structured around the splice sites, and point mutations that increase the local structure often cause splicing disruptions. Post-splicing, RNA secondary structure can also affect the stability of the spliced intron and regulatory RNA interference pathway intermediates, such as pre-microRNAs. Additionally, RNA secondary structure has important roles in the innate immune defense against viruses. Finally, tertiary structure can also play a large role in pre-mRNA splicing. One example is the G-quadruplex structure, which, similar to secondary structure, can either enhance or inhibit splicing through mechanisms such as creating or obscuring RNA binding protein sites.
Keywords: Splicing, RNA processing, Secondary Structure, G Quadruplex, Splice Site, ESE, ESS, disease, zebrafish, simple repeats
1. Introduction
1.1 Determining the structure of an RNA molecule
RNAs play a crucial role in many biological processes including regulation of gene expression and catalysis of cellular processes. RNA molecules can form a variety of structures. There is a correlation between the tertiary structure of an RNA molecule and its interactive and functional capacity [1]. It is however very difficult to predict 3D structure of an RNA molecule with high accuracy and precision. Multiple reviews previously covered the experimental tools used to solve RNA secondary structures [2–4]. This review section will focus on the utilization of computational methods for secondary structure predictions.
Many algorithms have been developed to study RNA secondary structure. Generally speaking, folding algorithms can be divided into two categories: single sequence methods and comparative methods, each having their own advantages and disadvantages.
The single sequence category of tools relies on finding the structure with minimum free energy (MFE) for each sequence. Dynamic programming (DP) has been used extensively in such tools. In essence, the algorithm computes the sum of all possible base pairs and their energy parameters or to put it simply, it determines base pairing probability from the partition function calculation. The partition function is used to count the particles of a system that are distributed over the available energy levels (i.e. the particles follow the Boltzmann distribution). In the RNA structure prediction problem, instead of counting particles, possible RNA structures conformations were counted. In accordance with the Boltzmann distribution, the fraction that a secondary structure conformation occupies in the ensemble of structures amounts to , where ΔG is the Gibb’s free energy change of RNA secondary structure conformation S, R is the gas constant, and T is the absolute temperature of the system. Partition function Q is therefore a summation over all possible energy levels of all possible secondary structure conformations . If the partition function is seen as a way to calculate the spread over the energy levels of RNA secondary structures, its reciprocal gives an easy way to calculate the probability of any given secondary structure conformation: . From this equation it follows that to calculate the base pairing probability between nucleotides i and j (Pi,j), we take the sum over the probabilities of all secondary structures k that contain the pair: [5]. Examples of folding algorithms in this category include mfold [6] and RNAfold [7]. Multiple additional assumptions have been used to limit the search space for MFE structures, including limiting base paring to Watson-Crick and wobble GU, as well as exclusion of pseudoknots (features of RNA structure in which base pairings occur between the unpaired bases of a loop and some other bases outside the loop).
Furthermore, the free energy landscape is constantly changing depending on multiple environmental variables like pH, temperature, chaperones and solvent condition. It is therefore not uncommon to consider structures that reside close to the MFE structure on the energy landscape. Programs have been developed to find the suboptimal structures for a given RNA sequence e.g. RNAsubopt [7]. Recently, several methods have been developed to predict secondary structures using maximum expected accuracy (MEA) approach, e.g. CONTRAfold [8], CentroidFold [9], and IPknot [10]. The goal of these algorithms is to find a secondary structure that maximizes the expected base-pair accuracy. In all of the above tools, the accuracy of a prediction is inversely proportional to the sequence length. To counteract this problem, it is possible to predict stable structures using a small window based approach, where base pairs are limited to a particular distance within that window (Figure 1). Algorithms such as Rfold [11] and RNAplfold [7] implement this strategy. However, such approach will not capture the long distance interactions between distant sequences and there is a need for further noise reduction (i.e. exclusion of pairs that exhibit a low thermodynamic probability) after calculation.
Figure 1. Detecting Local Stable Structures.
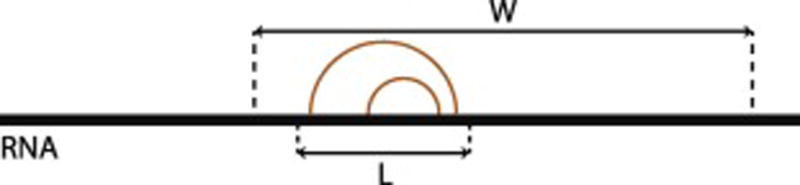
RNA is examined for local stable structures using a sliding window approach. Within window size of W only base pairings within a maximum distance apart (L) are considered. The orange arcs represent possible valid base pairings.
There are many applications where it may not be necessary to accurately predict the entire structure. For example, one may only need to know whether the binding site for a single stranded RNA binding protein (RBP) is accessible. Another frequently used model utilizes secondary structure as a mechanism to bring a distal element near a particular site. To this end simulation studies that insert complimentary k-mers into introns demonstrate a few simple rules of thumb that are a useful means of gaining intuition about the likelihood of distal complementary regions forming a secondary structure. Complimentary k-mers of length 12 can give maximal or near maximal bridging across an intron. Increases in k-mer length, did not result in appreciable increases in pairing (Figure 2) [12].
Figure 2. Complementary dinucleotide repeats at the ends of Zebrafish introns drive secondary structure.
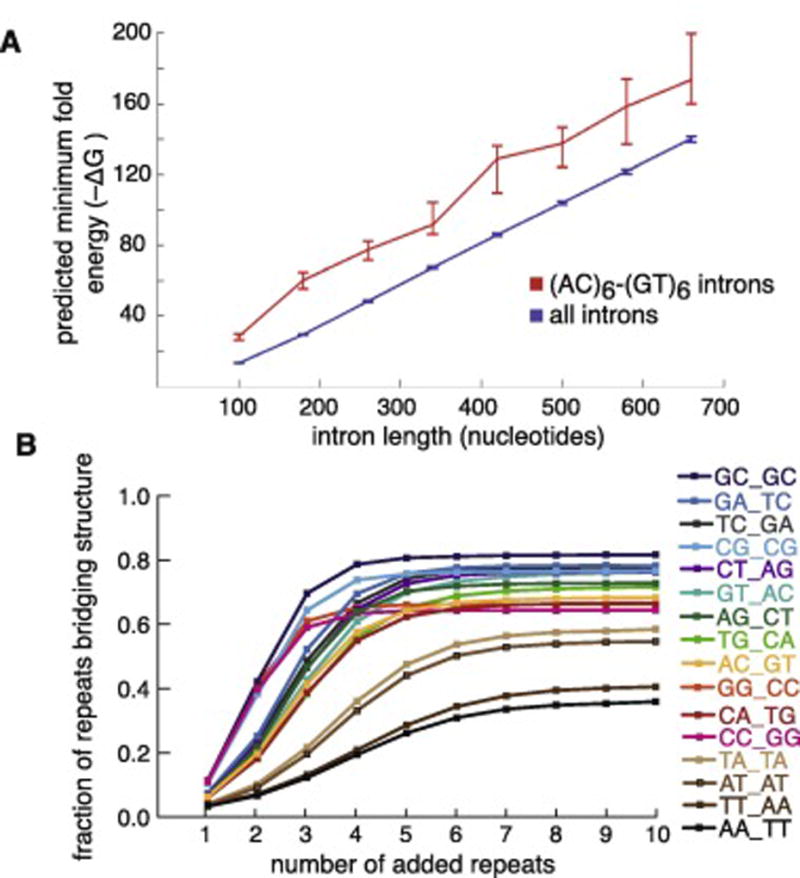
A. Minimum fold energy of Zebrafish (AC)m-(GT)n introns (red) and all Zebrafish introns (blue). Introns are binned by length and minimum fold energy is calculated using RNAfold. B. All possible dinucleotide repeats are added in silico to Zebrafish introns 20 nucleotides downstream of the 5’ss, and the complementary dinucleotide repeat is added 20 nucleotides upstream of the 3’ss. Synthetic introns are folded using RNAfold and the number of introns with predicted repeats base-pairing to form a hairpin structure is counted.
Comparative sequence methods can predict structures with higher accuracy than single sequence models as they take evolutionary information into consideration [13]. The assumption is that multiple molecules that perform similar functions will form similar structures regardless of their differences in sequence. However, it is not always feasible to use this approach, as it requires a priori that a set of homologous sequences be available. RNAalifold [7] and Pfold [14] are some exemplars of commonly used tools for comparative secondary sequence analyses. The combination of conservation and predicted structure is also available in the public annotation of genome in the Evofold track on the UCSC genome browser.
It has been estimated that the current algorithms correctly predict the secondary structure about 80% of the time. Comparative methods surpass single sequence approaches. Within the single sequence analysis category, the MEA methods outperform MFE procedures [13]. Despite the advances in the computational prediction of RNA structure, there are still challenges and thus it remains an active area of research.
2. Secondary structure enforces correct 5' and 3'ss pairing in zebrafish
Pre-mRNA splicing is modulated by RNA secondary structure. RNA secondary structure can assist in productive splicing by bringing distant elements into close proximity for recognition or by creating a binding platform for RBP [15]. On the other hand, RNA structure can also act negatively by preventing RNA-protein interaction or RNA-RNA interactions [16–18]. RNA structure has the potential to play a large role in regulating the splicing outcome.
A positive role of RNA structure for pre-mRNA splicing has been identified using comparative genomics strategies. Using pairwise comparisons of genomic sequences near the intron-exon junctions, lineage-specific k-mers around the splice site were discovered [12]. This unique distribution of k-mers suggests that these discovered sequence motifs regulate splicing in a lineage-specific manner. Using this method, complementary dinucleotide repeats across introns were first discovered in zebrafish and then further confirmed in other teleost species. A k-mer motif consisting of AC repeats at the 5’ end of the intron and GT repeats at the 3’ end of the intron, was found in 2% of zebrafish introns. At least one instance of this novel class of structured intron was found in 10% of all zebrafish genes. Structural analysis including folding energy calculation and biochemical probing showed that the (AC)m-(GT)n introns formed an extensive stem structure to bring the 5’ splice site and the 3’ splice site at two ends of the intron to close proximity to facilitate splice site pairing and accurate splicing.
To explore intronic bridging analogous to the structured (AC)m-(GT)n introns in zebrafish, RNA secondary structure prediction tools such as RNAfold can be used to predict the minimum folding energy and predicted secondary structure [19]. All zebrafish introns less than 740 nucleotides in length were folded using RNAfold (Figure 2A). Predicted minimum fold energy (-ΔG) increases with length of sequence. Zebrafish introns with ACACAC hexamer at the beginning of the intron and GTGTGT hexamer at the end of the intron have significantly higher predicted stability that background introns in the same intron size bins (Figure 2A). (AC)m-(GT)n introns were shuffled and refolded, while preserving dinucleotide composition. The shuffled introns are less structured than their native sequence, demonstrating that their stable predicted structures are not due purely to dinucleotide composition[12]. To determine whether dinucleotide repeats can drive predicted secondary structure, an in silico analysis was performed using all possible dinucleotide repeats. Each dinucleotide was added with different repeat lengths to the beginning 5’ end of the intron, with the complementary sequence added to the 3’ end. These synthetic introns were again folded using RNAfold, and their structures analyzed to determine the percent of introns in which the complementary dinucleotide repeats paired and forced bridging in the structure (Figure 2B). The dinucleotide repeats were able to drive bridging in the predicted structure to varying extents, however, they behaved asymptotically, with all k-mers achieving maximum bridging at a repeat length of six.
In the zebrafish (AC)m-(GT)n introns, the bridged structure-mediated splice site interaction is strong enough to bypass the need of essential splicing factor U2AF2 (U2 Small Nuclear RNA Auxiliary Factor 2), which usually recognize a polypyrimidine track upstream to the 3’ splice site and stabilize the U2 snRNP on the branchsite for the 3’ splice site determination. Knockdown of U2AF2 in vitro and in vivo both lead to defective splicing of the control introns but normal splicing of (AC)m-(GT)n introns. (AC)m-(GT)n introns were also found in lamprey, indicating it was an ancient splicing mechanism evolved before the divergence of Tetrapod and Teleost. The repeats were lost in mammals and other higher vertebrates; instead, exonic splicing enhancer-like sequences were found enriched in the human introns homologous to the (AC)m-(GT)n introns of zebrafish, suggesting a compensatory mechanism evolved to replace the dinucleotide repeats [12].
Although no evidence has been reported for AC and GT repeat-mediated splicing in human transcript processing, multiple copies of complementary GGG and CCC repeats were identified in some human introns [12]. These introns had significant lower folding energy compared to their own shuffled sequence, suggesting that the complementary G/C triplet could stabilize the intron to facilitate splice site interaction like the AC and GT repeats in zebrafish introns. While alternate structure models for G triplets invoking G quadraplex formation are discussed in section 6, the G triplet structures and requirement for splicing factors need to be further explored.
Intronic RNA structure is also critical for splicing the hyper-polymorphic exons. One of the counter-intuitive features of exonic splicing is the co-existence of processing information and coding information in exons. Because of the sequence variations, the sequence information in the exons is mutable, making the exonic splicing enhancers difficult to maintain over evolutionary time. A well-known example of hyper-polymorphic genes is HLA (Human Leukocyte Antigen) family genes. HLA family genes encode proteins recognizing fractions of antigens and representing them on the cell surface to trigger downstream immune responses. Pathogens are under the selective pressure to evolve away from the recognition by the surveillance of the host immune system. The host HLA family genes are then under selective pressure to generate new alleles in order to capture the rapidly evolving and greatly diverged antigen. That is why HLA family genes, especially the exons responsible for interactions with antigens, are among the most variable sequences in the human genome. The splicing of these exons is hence very challenging. Therefore, introns flanking hyper-polymorphic exons evolve into heavy secondary structures to ensure the splice site interactions. It has been shown that there was a positive correlation between exonic SNP (single nucleotide polymorphism) density and the structural stability of the flanking introns [20]. It suggests the secondary structure is used extensively to aid the splicing of highly polymorphic exons.
In conclusion, RNA secondary structure can stabilize the splice site interaction and reduce the need of trans splicing factors that recognize primary sequences in introns or in exons (Figure 3).
Figure 3. Structure-mediate pre-mRNA splicing.
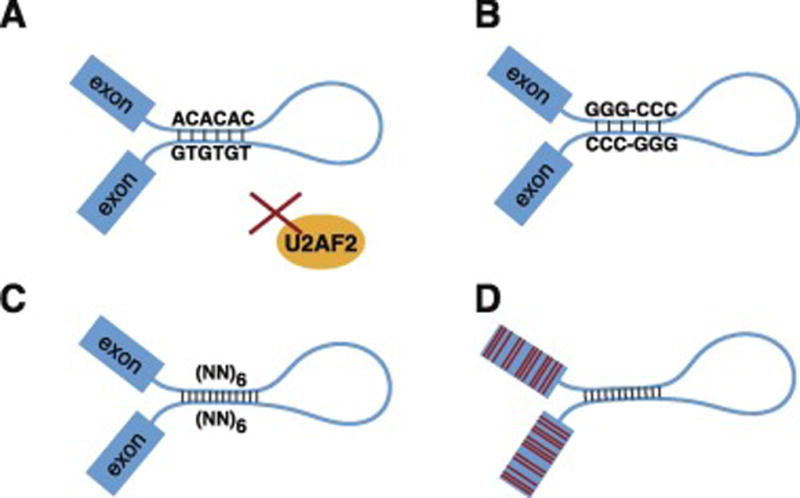
A. In zebrafish, complementary dinucleotide repeats at two ends of the intron bring splice sites to close proximity and facilitate splicing. The structural effect could overcome the absence of the essential splicing factor U2AF2. B. Complementary G/C triplet repeats stabilize human introns structure potentially enhancing splicing similar to A. C. In silico experiments suggest that repeats of complementary dinucleotides can bridge intron ends in the predicted structure of some loci. D. Introns flanking hyper-polymorphic exons are stabilized by secondary structure to ensure splicing.
3. Pre-mRNA Secondary Structure in Human Disease Genes
The importance of RNA secondary structure in splice-site selection has gained interest in numerous studies investigating both individual genes, and with the emergence of a growing achieve of genomic data, at the genome-wide level. When initial studies began experimentally investigating the contribution of pre-mRNA secondary structure to the process of splicing, investigators found that a number of splicing defects were due to an altered pre-mRNA secondary structure near the splice sites [21–26]. Although it was apparent that secondary structure was important to the fidelity of splicing, no explicit trend was discernable from the studies. Instead, it is speculated that a looser structure or set of structures facilitates the correct splicing of a transcript [27].
With the increased capacity to generate genomic data, more studies have taken a global approach to investigating the contribution and importance of pre-mRNA secondary structure to splicing. For example, Patterson et al. found that including pre-mRNA secondary structure information into conventional sequence-based splice-site prediction algorithms improved the programs predictive capabilities [27]. Thus, highlighting the importance of pre-mRNA secondary structure in identifying and predicting appropriate splice-sites. In addition to the significance of pre-mRNA secondary structure in splice-site prediction programs, Hiller et al. found that a) experimentally verified splicing enhancer and silencer motifs were significantly more single-stranded in the local secondary structure of the pre-mRNA near splice-sites and b) this structural context was maintained by purifying selection [28]. More recently, it appears that structures around alternative splice sites are significantly more stable than constitutive and skipped splice sites [29, 30]. This marked differences in stability may mediate splicing by altering splicing regulatory motifs recognition rate to promote or inhibit the binding of splicing silencers or enhancers. Therefore, suggesting an additional structural regulation mechanism in terms of splice-site recognition. As evidenced by these studies, the importance of pre-mRNA secondary structures is an integral component in controlling the fidelity of splicing by prompting the selection of appropriate splice-sites.
Although numerous studies have utilized genomic data to interrogate the importance of pre-mRNA secondary structure in splice-site selection, an exploration of the relationship between pre-mRNA secondary structure of splice-sites in disease has not been investigated. To explore this relationship on a global scale, we have compared the pre-mRNA secondary structure of splice-sites in disease genes reported by the Human Gene Mutation Database (HGMD) to the remaining genes in the genome [31]. Due to the local structure preference of pre-mRNA sequences in vivo [32], a 140 nucleotide window spanning each splice-site (70 nucleotides up- and down-stream of each splice-site) was used to predict the local secondary structure of the splice-sites using a minimum free energy program (MFE), RNAfold [7]. RNA structures with a lower MFE, as calculated by ΔG, are likely to have more base-pairing interactions compared so similar sized sequences with higher ΔG’s. Thus, sequences with lower ΔG’s are generally considered more stably structured. Through our analysis, we found that both the 3’ and 5’ splice sites in disease genes are more stable than splice-sites in the remaining genes of the genome (Figure 4A–B, p = 7.04e-96 and p = 1.61e-219, Mann-Whitney, for 3’ss and 5’ss respectively).
Figure 4. Minimum free energy (ΔG) of disease gene splice sites.
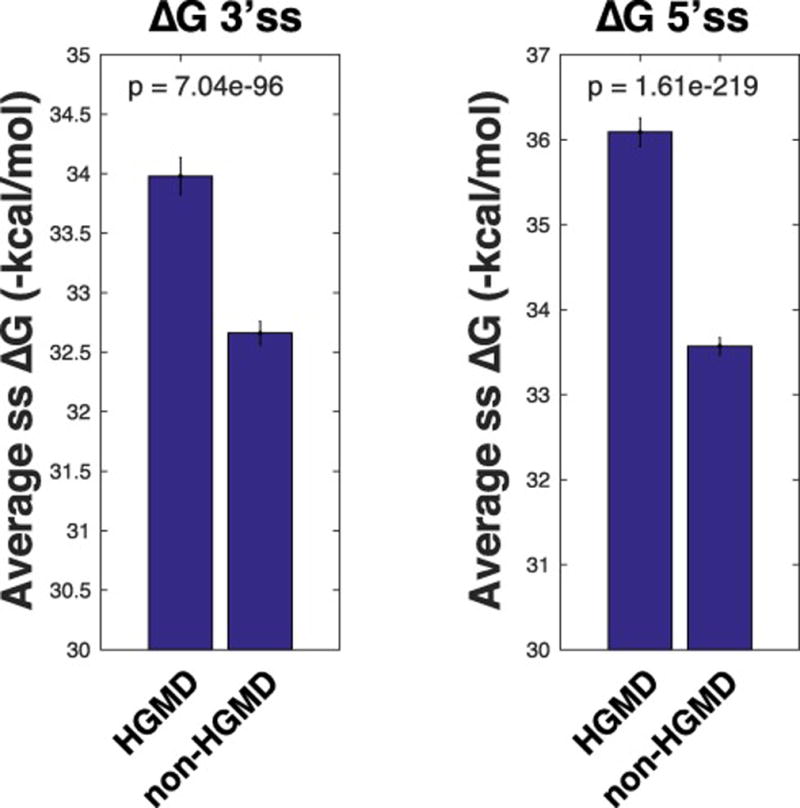
A. RNAfold minimum free energy score (ΔG) of 3’ss in HGMD disease genes (HGMD) and the remaining genes in the genome (non-HGMD). B. RNAfold minimum free energy score (ΔG) of 5’ss in HGMD disease genes (HGMD) and the remaining genes in the genome (non-HGMD).
Sequences with a higher GC content have previously been positively correlated both with structural stability and sites with thermodynamic advantages [30]. To determine if this marked difference in ΔG between disease genes and non-disease genes could be the result of the varying GC content present in the splice-sites, the nucleotide content of the splice-sites were maintained while shuffling the order of the splice-site sequences. When shuffling the arrangement of the splice-site sequences, the same result as in Figure 4 was found. This suggests that the GC content plays a large role in determining the ΔG of the splice-sites. Although it appears that the GC content is an important factor in determining the stability of the splice-sites (in terms of ΔG), it is important to note that this apparent difference in splice-site stability, and GC content, between disease genes and genomic genes highlights the importance of local structures in the recognition and correct splicing of disease genes.
Despite the evident significance of pre-mRNA secondary structure in facilitating splicing and splice-site recognition, it is possible that additional RNA tertiary structures may also be a mechanism regulating splicing [30]. As more technological advancements are made in predicting RNA structures, a clearer trend regarding structure and splicing may become discernible.
4. Disease mutations that alter RNA secondary structures
Mutations that alter RNA secondary structures are known to be deleterious [33, 34]. They may cause structural changes in the coding mRNA transcripts, UTRs, tRNAs or rRNAs, which in turn may affect any of the gene processing steps, including transcription, splicing, translation and decay [33, 35–37]. A recent deep sequencing of RNase-generated fragments of human transcriptomes indicated that as many as 15% of single nucleotide variants alter local RNA secondary structures [38]. There were indications of evolutionary pressure against these variants, termed “riboSNitches”, in RBP binding sites as well as miRNA target sites. They were also shown to be more likely to result in splicing changes, compared to variants that don’t alter the secondary structures [38]. It remains to be determined what fractions of riboSNitches impact the different gene processing steps. Interestingly, antagonistic epistatic interactions involving compensatory mutations that restore fitness by preserving RNA secondary structures have also been described [39]. These findings indicate that variants that alter RNA secondary structures are likely to play a larger role than previously thought in the etiology of human diseases.
We recently developed Massively Parallel Splicing Assay (MaPSy), which enables us to directly compare the splicing performance of thousands of mutant and wildtype substrates [40, 41]. Thermodynamically less stable RNAs (higher free-energy structures) splice more efficiently, particularly those with more open (unpaired) bases in the splice-site regions (Figure 5A,B). Mutations that cause local changes in the RNA secondary structures, particularly those that are in proximity to splice-sites, can significantly alter splicing. For example, several mutations that destabilize the secondary structure of the splice-site donor of exon 10 of the MAPT were found to cause frontotemporal dementia phenotype, a severe neurodegenerative disorder, due to the increased splicing efficiency of exon 10 [24]. The majority of human disease mutations, however, tend to trigger more stable RNA structures, which result in less efficient splicing. Thermodynamically less stable RNAs are more predisposed for having mutations that result in lower free-energy structures (Figure 5C). Interestingly, a subset of Exonic Splicing Mutations (ESM) that was identified with MaPSy substantially changed the predicted folding free energy of the transcripts, mostly to significantly more stable structures (Figure 5D) [7]. Subsequent spliceosome assembly of some of these ESM showed disruptions in all stages of the spliceosome assembly in this subset of ESM, in contrast to the majority of ESM that mainly inhibit one of the stages of the spliceosome assembly. These mutations are likely to be independent of RBP and other trans-acting factors, and thus unlikely to exhibit cell or tissue specific effects.
Figure 5. Role of RNA secondary structures in human diseases.
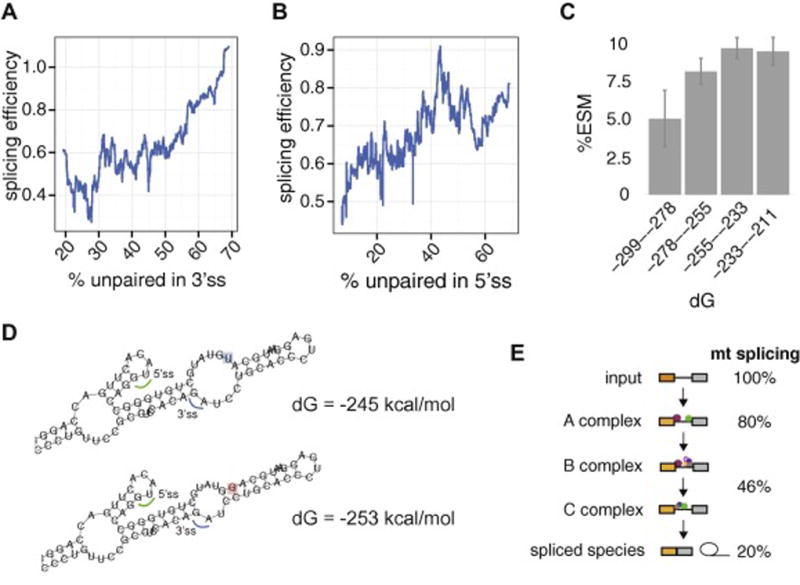
Structural accessibilities around splice sites are important for splicing. Degree of openness (predicted %unpaired bases) in the 3’ splice sites (A) and 5’ splice sites (B) correlates with increased splicing efficiency. (C) Thermodynamically less stable transcripts are more susceptible for exonic splicing mutations (ESM). (D) T to G mutation (blue and red highlight, respectively) in HMBS gene resulted in more stable structure. (E) Mutant splicing relative to wildtype in some mutations that stabilized the RNA structure in the different stages of the spliceosome assembly.
In summary, highly structured substrates splice with lower efficiency than more open substrates. However the splicing of open substrates appear to be more susceptible to ESM, presumably because splicing factors typically recognize ssRNA. The increase of structure is also a mechanism of splicing disruption. Large-scale analysis of splicing mutations identify single point mutations that trigger structural rearrangements that make structures more stable. The effect of this increased stability can be seen as a loss of efficiency at each stage of spliceosome transition.
5. Double Stranded RNA (dsRNA) post splicing and the Innate Immune Response
The immune response is dependent on the recognition of pathogen associated molecular patterns (PAMPs) by pathogen-recognition receptors (PRRs) [42]. Distinguishing between endogenous and exogenous gene products is vital to survival. All organisms from early prokaryotes to vertebrates employ defense strategies to elicit resistance upon detection of foreign molecular signals. Detection of bacterial and fungal PAMPs is well understood. The microbe associated products such as cell wall components, endotoxins, lipidglycans and glycoproteins are generally not found in the host and can be readily discerned [43]. Detection of viruses however depends heavily on the recognition of nucleic acid, in particular dsRNA.
Many viruses exist as dsRNA during the viral life cycle [44]. Endogenous dsRNAs derived from antisense transcription can initiate dynamic posttranslational gene regulation through the regulatory RNA interference (RNAi) pathways. Host dsRNA is cleaved by then enzyme 'Dicer' a ribonuclease III. Dicer recognizes pre-microRNAs, which are short dsRNA hairpins in precursor single stranded RNAs [45]. Pre micro RNAs are commonly derived from primary microRNAs as well as lariat intron intermediates from pre-mRNA splicing. In higher eukaryotes, toll-like receptors (TLRs) function as the primary PRRs for exogenous dsRNA, which generally contain significantly longer regions of complementarity than pre-microRNAs [43, 46]. TLRs are transmembrane proteins characterized by leucine-rich repeats in the extracellular domain. They are differentially expressed in immune cells and are stimulated by a diversity of signals [47]. TLR3 has been shown to specifically act as a receptor for dsRNA by initiating the interferon (IFN) response and activating a number of IFN stimulated genes, such as the apoptosis inducing dsRNA-dependent protein kinase (PKR) and RNA-specific adenosine deaminase (ADAR) which prevents nuclear processing, as well as the inflammatory response [48]. TLR signal transduction is dependent on the E3 ubiquitin ligases TRAF3 and TRAF6 to target molecules for downstream activation [49].
Other PRRs that play an important role in the recognition of exogenous dsRNA and activation of the IFN response are retinoic acid inducible gene-I (RIG-I) like receptors (RLRs). To regulate downstream IFN activity, ubiquitin chains are also necessary for RIG-I to oligomerize and initiate signaling activity, which is thought to be regulated by the E3 ligase tripartite motif protein 25 (TRIM25) [50, 51]. Virus induced host stress responses lead to the localization of RLRs in cytoplasmic stress granules (SGs) with host mRNAs, 40s ribosomes and RBPs. These ribonucleoprotein complexes sequester viral dsRNA, allowing for detection by RLRs although at the same time halt translation of both host and viral mRNAs, performing both pro and antiviral functions simultaneously [43]. While each piece of the cellular dsRNA recognition machinery is highly specialized, they are interdependent in the detection of exogenous RNA. Just as the absence of Dicer triggers the IFN response [52], in the absence of the IFN-I pathway, RNAi is sufficient to prevent viral dsRNA accumulation when viruses contain the homologous sequence [53].
Conservation of RNA secondary structure is a common viral strategy. Stable secondary structures have been reported in the Hepatitis viruses, HIV, SARS, Polio, α and β Influenza viruses, as well as Herpes A and B. They are usually located at or near splice sites and are functional [54–59]. Herpes Simplex Viruses, for example, are well known for their periods of latency and reactivation. In the Herpesvirales family dsRNA has been demonstrated to arise from RNA secondary structure, specifically from pre-mRNA splicing intron lariat intermediates (Figure 6A, B). The latency associate transcript (LAT) is a non-coding RNA with unusually high conservation (~86% identical for a stretch of 500nt at the 3' end) between HSVI, HSVII and, chHSV. In addition to using a non-canonical guanosine branch point nucleotide, the lariat tail of the 2kb LAT intron is thought to form a hairpin [54, 60], protecting the lariat intermediate from being debranched by the debranching enzyme DBR1. The result is the stabilization of the lariat, which presents dsRNA to the host and modulates the immune response.
Figure 6. Structure models of the stable LAT intron lariat intermediate and predicted free energies.
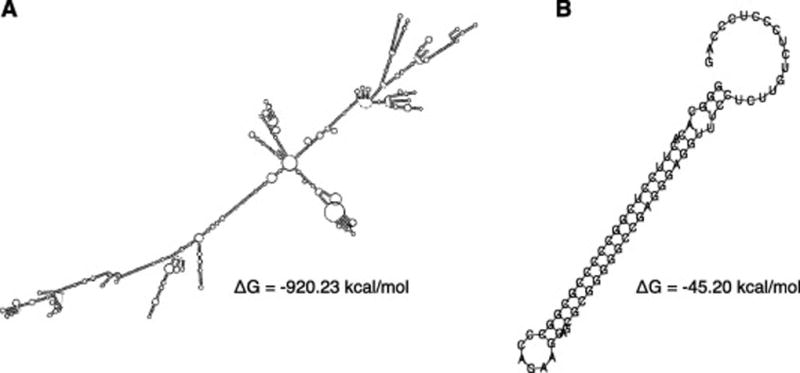
(A) Lariat model structure of the LAT intron from the 5' splice site to the branchpoint nucleotide (loop of lariat). (B) Lariat model structure of the LAT intron from the branch point nucleotide to the 3' splice site (tail of lariat).
The stable LAT intron has been proposed to govern the transition between the latency phase and the lytic phase via the chromatinization of the virus by the host machinery, including the histone chaperones chromatin assembly factor (CAF1) and anti-silencing function 1 (ASF1) [61, 62]. As the chromatin free encapsidated viral genome replicates, it becomes nucleosome occupied in an irregular manner, acquiring euchromatin-associated histone modifications. The induction of latency, however, is coupled with the heterochromatinization and global repression of lytic factors [63]. Deletion of the LAT does not appear to have an effect on establishing latency, but significantly reduces the reactivation of the virus in infected cells [61]. Although the mechanism is not entirely understood, it is clear that this epigenetic switch is directly linked to the expression of the LAT intron, perhaps due to its secondary structure.
6. Moving beyond secondary to tertiary structure: G-Quadruplex
In addition to the numerous roles of RNA secondary structure, RNA tertiary structure may affect alternative splicing. G-quadruplexes (G4) are non-canonical tertiary structures that form in both DNA and RNA. They are composed of stacked G-quartets (i.e. planar tetrads formed through Hoogsteen base-pairing of guanine residues in G-triplet sequences and stabilized by a central monovalent cation) [64] (Figure 7). In DNA, G4s occur frequently in telomeres and promoters regions. They may also form along the lagging strand during DNA replication, which may lead to double-stranded breaks if not processed by helicases [65]. In RNA, G4s are often present at the 5’ end of the first intron, and in both the 5’ and 3’ untranslated regions, but not limited to these regions [66, 67].
Figure 7. Structure of a G-Quadruplex.
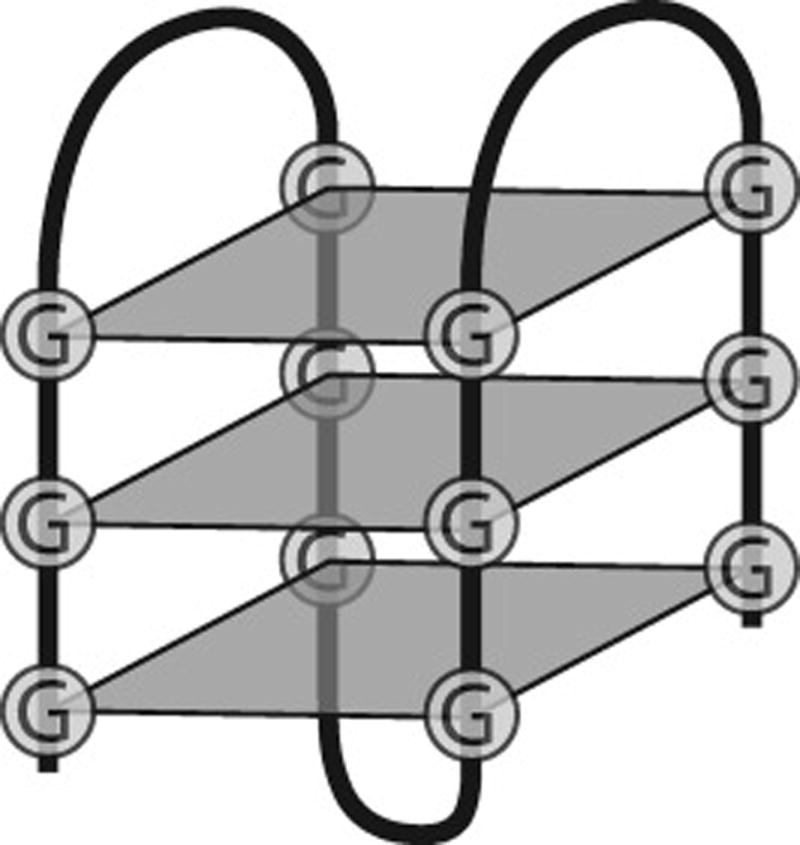
A G-Quadruplex is composed of 4 interspersed G-triplets which fold into stacked G-quartets (gray diamonds). Each guanine residue in a G-quartet is associated with its neighboring guanines through Hoogsteen base-pairing.
G4s are increasingly recognized as important regulators of pre-mRNA processing, including polyadenylation and alternative splicing [68]. In the case of splicing, G4s may act as a binding site for certain RBPs or they may obscure other cis elements that regulate splicing. The Fragile-X Mental Retardation Protein (FMRP), an RBP that is absent in fragile X syndrome, has been shown to bind numerous mRNAs at a G-quartet structural motif. FMRP binds this motif in two locations within its own mRNA transcript, both of which independently lead to ESE activity in exon 15 of the FMR1 transcript, increasing the proportion of the full-length isoform [69]. G4s may also form within introns, and may lead to both enhancement and silencing of splicing in different contexts. The hTERT gene may be alternatively spliced into the inactive hTERT-β isoform through the stabilization of a G4 within intron 6 [70]. Conversely, the tumor suppressor gene TP53 has been shown to form a G4 structure in intron 3, which blocks the retention of intron 2 [71]. Likewise the chicken β-tropomyosin gene contains an intronic G-rich motif that enhances the splicing of exon 6A [72]. Taken together, these examples highlight the ability of G4s to interact with different splicing factors to either enhance or silence splicing in certain cases, either through direct binding to the structural motif itself or through exposure of other cis elements upon formation of a G4.
G-tracts readily assemble into highly-stable G4s in vitro. Their stability is dependent on the monovalent cation they envelope. For example, K+ is positioned halfway in-between 2 G-quartets, yielding a highly stable bi-pyramidal symmetry. However, Li+ has a much larger atomic radius which cannot mimic this symmetry, and thus relatively destabilizes the G4. Although cells have a relatively high concentration of K+ in their cytoplasm compared to other cations, G4s in eukaryotic cells have specifically been shown to be globally unfolded. The particular mechanism is unknown, but eukaryotic cells express several RNA helicases which could unfold G4 structures [73]. Moreover, a large number of G4 stabilizing ligands have been identified, which vary in both their binding location and selectivity for G4 motifs [74]. The use of some of these ligands can affect splicing throughout the transcriptome.
To highlight the potential for some G4 stabilizing ligands to affect splicing, public RNA-seq data of three known G4 ligands (PhenDC3, Daunorubicin and TMPyP4) was processed for differential splicing using the rMATS software package [75, 76]. Compared to amiloride, a compound known to affect splicing [77], the total number or alternative splicing events among the G4 ligands are closer to the baseline level seen in the tetracycline control after correcting for sequencing read counts (unpublished data, Figure 8A). There are 109 events that were shared among all five experimental conditions. The higher number of alternative splicing events seen in TMPyP4 may be attributable to increased preference for RNA G4s over DNA G4s. Using the presence of four or more G-triplets as a proxy for a likely G4, TMPyP4 shows a greater percentage of G-triplets in the flanking introns of its skipped exon events, suggesting that its alternative splicing events are more commonly caused by the stabilization of a G4 (Figure 8B). Taken together, these findings suggest that G4 stabilizing ligands may have a role in causing exon skipping, although the global effect appears to be modest.
Figure 8. Alternative splicing profile for G4 stabilizing ligands.
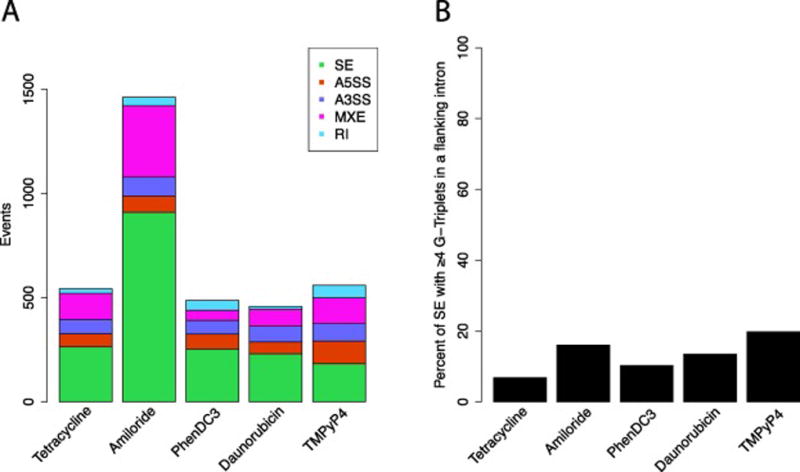
A. Number of alternative splicing events. SE: skipped exon, A5SS: alternative 5’ splice site, A3SS: alternative 3’ splice site, MXE: mutually-exclusive exons, RI: retained intron. B. Percent of SE events in which there are four or more G-triplets in at least one flanking intron.
Highlights.
The prediction of RNA secondary structure is a challenging problem and an evolving field, however many tools have been developed such as mfold, RNAfold, RNAsubopt, CONTRAfold, CentroidFold, IPknot, Rfold, RNAplfold, RNAalifold, and Pfold.
The secondary structure of intronic RNA can assist in pre-mRNA splicing by bridging splice sites together and ensuring proper splice-site pairing.
Disrupting local RNA secondary structures can alter pre-mRNA processing and lead to disease. Disease genes have more structured splice sites than background, and highly structured splice sites globally splice less efficiently than more open RNAs.
Double stranded RNA structures, derived from host introns post-splicing or from viral RNAs, play important roles in gene regulation and the innate immune response.
RNA can also form tertiary structures known as G-quadruplexes, which are comprised of stacked G-quartets and are important regulators of pre-mRNA processing steps, such as polyadenylation and alternative splicing.
Acknowledgments
This work has been supported by the National Institutes of Health [R01GM095612 to W.G.F., R01GM105681 to W.G.F., R21HG007905 to W.G.F.]; and by the Simons Foundation Autism Research Initiative [342705 to W.G.F.].
Abbreviations
- MFE
minimum free energy
- DP
dynamic programming
- MEA
maximum expected accuracy
- RBP
RNA-binding protein
- U2AF2
U2 Small Nuclear RNA Auxiliary Factor 2
- HLA
Human Leukocyte Antigen
- SNP
single nucleotide polymorphism
- HGMD
Human Gene Mutation Database
- ESM
exonic splicing mutations
- ssRNA
single stranded RNA
- dsRNA
double stranded RNA
- PAMP
pathogen associated molecular pattern
- PPR
pathogen-recognition receptor
- RNAi
RNA interference
- TLR
toll-like receptor
- IFN
interferon
- SG
stress granule
- LAT
latency associate transcript
- HSV
herpes simplex viruses
- G4
G-quadruplexes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hajdin CE, Ding F, Dokholyan NV, Weeks KM. On the significance of an RNA tertiary structure prediction. RNA. 2010;16(7):1340–9. doi: 10.1261/rna.1837410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kubota M, Tran C, Spitale RC. Progress and challenges for chemical probing of RNA structure inside living cells. Nat Chem Biol. 2015;11(12):933–41. doi: 10.1038/nchembio.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weeks KM. Advances in RNA structure analysis by chemical probing. Curr Opin Struct Biol. 2010;20(3):295–304. doi: 10.1016/j.sbi.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorenz R, Wolfinger MT, Tanzer A, Hofacker IL. Predicting RNA secondary structures from sequence and probing data. Methods. 2016;103:86–98. doi: 10.1016/j.ymeth.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Mathews DH. Using an RNA secondary structure partition function to determine confidence in base pairs predicted by free energy minimization. RNA. 2004;10(8):1178–90. doi: 10.1261/rna.7650904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Do CB, Woods DA, Batzoglou S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006;22(14):e90–8. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- 9.Sato K, Hamada M, Asai K, Mituyama T. CENTROIDFOLD: a web server for RNA secondary structure prediction. Nucleic Acids Res. 2009;37(Web Server issue):W277–80. doi: 10.1093/nar/gkp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato K, Kato Y, Hamada M, Akutsu T, Asai K. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics. 2011;27(13):i85–93. doi: 10.1093/bioinformatics/btr215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiryu H, Kin T, Asai K. Rfold: an exact algorithm for computing local base pairing probabilities. Bioinformatics. 2008;24(3):367–73. doi: 10.1093/bioinformatics/btm591. [DOI] [PubMed] [Google Scholar]
- 12.Lin CL, Taggart AJ, Lim KH, Cygan KJ, Ferraris L, Creton R, Huang YT, Fairbrother WG. RNA structure replaces the need for U2AF2 in splicing. Genome Res. 2016;26(1):12–23. doi: 10.1101/gr.181008.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puton T, Kozlowski LP, Rother KM, Bujnicki JM. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2013;41(7):4307–23. doi: 10.1093/nar/gkt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knudsen B, Hein J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003;31(13):3423–8. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Mol Cell Biol. 2004;24(24):10505–14. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solnick D. Alternative splicing caused by RNA secondary structure. Cell. 1985;43(3 Pt 2):667–76. doi: 10.1016/0092-8674(85)90239-9. [DOI] [PubMed] [Google Scholar]
- 17.Goguel V, Wang Y, Rosbash M. Short artificial hairpins sequester splicing signals and inhibit yeast pre-mRNA splicing. Mol Cell Biol. 1993;13(11):6841–8. doi: 10.1128/mcb.13.11.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plass M, Codony-Servat C, Ferreira PG, Vilardell J, Eyras E. RNA secondary structure mediates alternative 3'ss selection in Saccharomyces cerevisiae. RNA. 2012;18(6):1103–15. doi: 10.1261/rna.030767.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9(1):133–48. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin CL, Taggart AJ, Fairbrother WG. RNA structure in splicing: An evolutionary perspective. RNA Biol. 2016;13(9):766–71. doi: 10.1080/15476286.2016.1208893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clouet d'Orval B, d'Aubenton-Carafa Y, Brody JM, Brody E. Determination of an RNA structure involved in splicing inhibition of a muscle-specific exon. J Mol Biol. 1991;221(3):837–56. doi: 10.1016/0022-2836(91)80179-x. [DOI] [PubMed] [Google Scholar]
- 22.Deshler JO, Rossi JJ. Unexpected point mutations activate cryptic 3' splice sites by perturbing a natural secondary structure within a yeast intron. Genes Dev. 1991;5(7):1252–63. doi: 10.1101/gad.5.7.1252. [DOI] [PubMed] [Google Scholar]
- 23.Muro AF, Caputi M, Pariyarath R, Pagani F, Buratti E, Baralle FE. Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol Cell Biol. 1999;19(4):2657–71. doi: 10.1128/mcb.19.4.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, Klug A, Goedert M, Varani G. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci U S A. 1999;96(14):8229–34. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35(2):371–89. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gahura O, Hammann C, Valentova A, Puta F, Folk P. Secondary structure is required for 3' splice site recognition in yeast. Nucleic Acids Res. 2011;39(22):9759–67. doi: 10.1093/nar/gkr662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patterson DJ, Yasuhara K, Ruzzo WL. Pre-mRNA secondary structure prediction aids splice site prediction. Pac Symp Biocomput. 2002:223–34. [PubMed] [Google Scholar]
- 28.Hiller M, Zhang Z, Backofen R, Stamm S. Pre-mRNA secondary structures influence exon recognition. PLoS Genet. 2007;3(11):e204. doi: 10.1371/journal.pgen.0030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shepard PJ, Hertel KJ. Conserved RNA secondary structures promote alternative splicing. RNA. 2008;14(8):1463–9. doi: 10.1261/rna.1069408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang J, Kuo CC, Chen L. GC content around splice sites affects splicing through pre-mRNA secondary structures. BMC Genomics. 2011;12:90. doi: 10.1186/1471-2164-12-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003;21(6):577–81. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder R, Grossberger R, Pichler A, Waldsich C. RNA folding in vivo. Curr Opin Struct Biol. 2002;12(3):296–300. doi: 10.1016/s0959-440x(02)00325-1. [DOI] [PubMed] [Google Scholar]
- 33.Siala O, Salem IH, Tlili A, Ammar I, Belguith H, Fakhfakh F. Novel sequence variations in LAMA2 andSGCG genes modulating cis-acting regulatory elements and RNA secondary structure. Genet Mol Biol. 2010;33(1):190–7. doi: 10.1590/S1415-47572010005000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartoszewski RA, Jablonsky M, Bartoszewska S, Stevenson L, Dai Q, Kappes J, Collawn JF, Bebok Z. A synonymous single nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J Biol Chem. 2010;285(37):28741–8. doi: 10.1074/jbc.M110.154575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wittenhagen LM, Kelley SO. Impact of disease-related mitochondrial mutations on tRNA structure and function. Trends Biochem Sci. 2003;28(11):605–11. doi: 10.1016/j.tibs.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 36.Sabarinathan R, Wenzel A, Novotny P, Tang X, Kalari KR, Gorodkin J. Transcriptome-wide analysis of UTRs in non-small cell lung cancer reveals cancer-related genes with SNV-induced changes on RNA secondary structure and miRNA target sites. PloS one. 2014;9(1):e82699. doi: 10.1371/journal.pone.0082699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shabalina SA, Spiridonov NA, Kashina A. Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 2013;41(4):2073–94. doi: 10.1093/nar/gks1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, Chang HY. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505(7485):706–9. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilke CO, Lenski RE, Adami C. Compensatory mutations cause excess of antagonistic epistasis in RNA secondary structure folding. BMC Evol. Biol. 2003;3:3. doi: 10.1186/1471-2148-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soemedi R, Vega H, Belmont JM, Ramachandran S, Fairbrother WG. Genetic variation and RNA binding proteins: tools and techniques to detect functional polymorphisms. Adv. Exp. Med. Biol. 2014;825:227–66. doi: 10.1007/978-1-4939-1221-6_7. [DOI] [PubMed] [Google Scholar]
- 41.Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, Bayrak-Toydemir P, McDonald J, Fairbrother WG. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 2017 doi: 10.1038/ng.3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 43.Yoneyama M, Fujita T. Recognition of viral nucleic acids in innate immunity. Rev Med Virol. 2010;20(1):4–22. doi: 10.1002/rmv.633. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Liu L, Davies DR, Segal DM. Dimerization of Toll-like receptor 3 (TLR3) is required for ligand binding. J Biol Chem. 2010;285(47):36836–41. doi: 10.1074/jbc.M110.167973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yelin R, Dahary D, Sorek R, Levanon EY, Goldstein O, Shoshan A, Diber A, Biton S, Tamir Y, Khosravi R, Nemzer S, Pinner E, Walach S, Bernstein J, Savitsky K, Rotman G. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol. 2003;21(4):379–86. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- 46.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413(6857):732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 47.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91(3):295–8. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 48.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559–68. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang C, Chen T, Zhang J, Yang M, Li N, Xu X, Cao X. The E3 ubiquitin ligase Nrdp1 'preferentially' promotes TLR-mediated production of type I interferon. Nat Immunol. 2009;10(7):744–52. doi: 10.1038/ni.1742. [DOI] [PubMed] [Google Scholar]
- 50.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446(7138):916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 51.Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104(18):7500–5. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White E, Schlackow M, Kamieniarz-Gdula K, Proudfoot NJ, Gullerova M. Human nuclear Dicer restricts the deleterious accumulation of endogenous double-stranded RNA. Nat Struct Mol Biol. 2014;21(6):552–9. doi: 10.1038/nsmb.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maillard PV, Van der Veen AG, Deddouche-Grass S, Rogers NC, Merits A, Reis ESC. Inactivation of the type I interferon pathway reveals long double-stranded RNA-mediated RNA interference in mammalian cells. EMBO J. 2016;35(23):2505–2518. doi: 10.15252/embj.201695086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krummenacher C, Zabolotny JM, Fraser NW. Selection of a nonconsensus branch point is influenced by an RNA stem-loop structure and is important to confer stability to the herpes simplex virus 2-kilobase latency-associated transcript. J Virol. 1997;71(8):5849–60. doi: 10.1128/jvi.71.8.5849-5860.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soszynska-Jozwiak M, Michalak P, Moss WN, Kierzek R, Kierzek E. A Conserved Secondary Structural Element in the Coding Region of the Influenza A Virus Nucleoprotein (NP) mRNA Is Important for the Regulation of Viral Proliferation. PLoS One. 2015;10(10):e0141132. doi: 10.1371/journal.pone.0141132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burrill CP, Westesson O, Schulte MB, Strings VR, Segal M, Andino R. Global RNA structure analysis of poliovirus identifies a conserved RNA structure involved in viral replication and infectivity. J Virol. 2013;87(21):11670–83. doi: 10.1128/JVI.01560-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain N, Morgan CE, Rife BD, Salemi M, Tolbert BS. Solution Structure of the HIV-1 Intron Splicing Silencer and Its Interactions with the UP1 Domain of Heterogeneous Nuclear Ribonucleoprotein (hnRNP) A1. J Biol Chem. 2016;291(5):2331–44. doi: 10.1074/jbc.M115.674564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robertson MP, Igel H, Baertsch R, Haussler D, Ares M, Jr, Scott WG. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2005;3(1):e5. doi: 10.1371/journal.pbio.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stewart H, Bingham RJ, White SJ, Dykeman EC, Zothner C, Tuplin AK, Stockley PG, Twarock R, Harris M. Identification of novel RNA secondary structures within the hepatitis C virus genome reveals a cooperative involvement in genome packaging. Sci Rep. 2016;6:22952. doi: 10.1038/srep22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zabolotny JM, Krummenacher C, Fraser NW. The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J Virol. 1997;71(6):4199–208. doi: 10.1128/jvi.71.6.4199-4208.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol. 1994;68(12):8045–55. doi: 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park YJ, Luger K. Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol. 2008;18(3):282–9. doi: 10.1016/j.sbi.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Paulus C, Nitzsche A, Nevels M. Chromatinisation of herpesvirus genomes. Rev Med Virol. 2010;20(1):34–50. doi: 10.1002/rmv.632. [DOI] [PubMed] [Google Scholar]
- 64.Biffi G, Tannahill D, McCafferty J, Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem. 2013;5(3):182–6. doi: 10.1038/nchem.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rhodes D, Lipps HJ. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015;43(18):8627–37. doi: 10.1093/nar/gkv862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eddy J, Maizels N. Conserved elements with potential to form polymorphic G-quadruplex structures in the first intron of human genes. Nucleic Acids Res. 2008;36(4):1321–33. doi: 10.1093/nar/gkm1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huppert JL, Bugaut A, Kumari S, Balasubramanian S. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res. 2008;36(19):6260–8. doi: 10.1093/nar/gkn511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agarwala P, Pandey S, Maiti S. The tale of RNA G-quadruplex. Org Biomol Chem. 2015;13(20):5570–85. doi: 10.1039/c4ob02681k. [DOI] [PubMed] [Google Scholar]
- 69.Didiot MC, Tian Z, Schaeffer C, Subramanian M, Mandel JL, Moine H. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 2008;36(15):4902–12. doi: 10.1093/nar/gkn472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gomez D, Lemarteleur T, Lacroix L, Mailliet P, Mergny JL, Riou JF. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res. 2004;32(1):371–9. doi: 10.1093/nar/gkh181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marcel V, Tran PL, Sagne C, Martel-Planche G, Vaslin L, Teulade-Fichou MP, Hall J, Mergny JL, Hainaut P, Van Dyck E. G-quadruplex structures in TP53 intron 3: role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis. 2011;32(3):271–8. doi: 10.1093/carcin/bgq253. [DOI] [PubMed] [Google Scholar]
- 72.Expert-Bezancon A, Le Caer JP, Marie J. Heterogeneous nuclear ribonucleoprotein (hnRNP) K is a component of an intronic splicing enhancer complex that activates the splicing of the alternative exon 6A from chicken beta-tropomyosin pre-mRNA. J Biol Chem. 2002;277(19):16614–23. doi: 10.1074/jbc.M201083200. [DOI] [PubMed] [Google Scholar]
- 73.Guo JU, Bartel DP. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science. 2016;353(6306) doi: 10.1126/science.aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monchaud D, Teulade-Fichou MP. A hitchhiker's guide to G-quadruplex ligands. Org Biomol Chem. 2008;6(4):627–36. doi: 10.1039/b714772b. [DOI] [PubMed] [Google Scholar]
- 75.Zheng XH, Nie X, Liu HY, Fang YM, Zhao Y, Xia LX. TMPyP4 promotes cancer cell migration at low doses, but induces cell death at high doses. Sci Rep. 2016;6:26592. doi: 10.1038/srep26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A. 2014;111(51):E5593–601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang JG, Yang DM, Chang WH, Chow LP, Chan WL, Lin HH, Huang HD, Chang YS, Hung CH, Yang WK. Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells. PLoS One. 2011;6(6):e18643. doi: 10.1371/journal.pone.0018643. [DOI] [PMC free article] [PubMed] [Google Scholar]