

Abstract
The past decade has witnessed the rapid development of radical generation strategies and their applications in C–C bond-forming reactions. Most of these processes require initiators, transition metal catalysts or organometallic reagents. Herein, we report the discovery of a simple organic system (2-azaallyl anions) that enables radical coupling reactions under transition-metal-free conditions. Deprotonation of N-benzyl ketimines generates semi-stabilized 2-azaallyl anions that behave as “super-electron-donors” (SEDs) and reduce aryl iodides and alkyl halides to aryl and alkyl radicals. The SET process converts the 2-azaallyl anions into persistent 2-azaallyl radicals, which capture the aryl and alkyl radicals to form C–C bonds. The radical coupling of aryl and alkyl radicals with 2-azaallyl radicals makes possible the synthesis of functionalized amine derivatives without the use of exogenous radical initiators or transition metal catalysts. Radical clock studies and 2-azaallyl anion coupling studies provide mechanistic insight for this unique reactivity.
TOC image
1. Introduction
Reactions that form C–C bonds have received tremendous attention due to their value in synthetic organic chemistry and drug discovery.1 Of these, the formation of C–C bonds via radical intermediates has witnessed increasing popularity due to the mild nature of many of these processes.1h, 2 Over the past few decades, a series of groundbreaking radical-based coupling strategies have been developed that enable a wide variety of bonds to be formed. Among these, use of visible-light activated transition metal and organo-photocatalysts to generate organic radicals has found tremendous applications in novel C–C bond-forming methods.3 A key feature of such photocatalysts (PC*) is their ability to behave as both reductants and oxidants, as shown in Scheme 1a. Under photocatalytic conditions, a wide variety of reagents undergo oxidative or reductive fragmentation to organic radicals. Another beautiful strategy for radical generation is through redox-active esters, which are converted efficiently to radicals by reductive fragmentation using transition-metals as electron donors (Scheme 1b).4 Finally, a metal-free strategy to generate radicals involves the use of organic super-electron-donors (SEDs).5 SEDs possess reduction potentials as high as −1.50 V (versus SCE)6 and reduce a range of substrates to generate radicals (Scheme 1c). The reducing features of SEDs have been demonstrated in intramolecular radical cyclization experiments.7 In contrast, kinetically slower intermolecular (bimolecular) C–C bond formations enabled by excess amounts of SEDs are far less developed.8
Scheme 1.
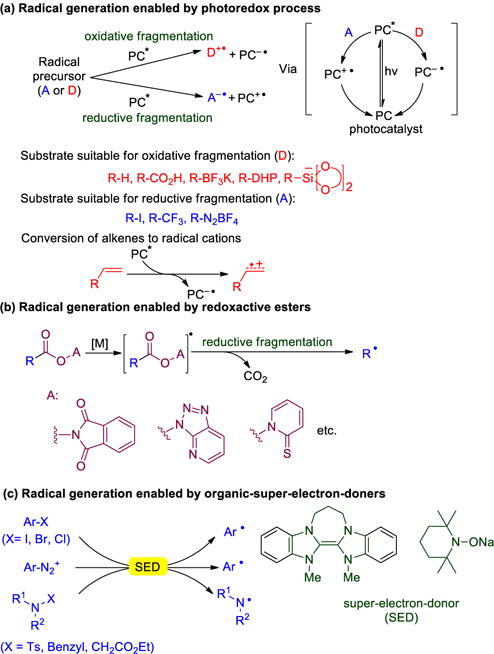
Representative strategies for radical generation.
Along these lines, we recently became interested in radical coupling reactions while investigating a unique transition metal-free reaction of 2-azaallyl species with vinyl halide electrophiles (Scheme 2a). We found that deprotonation of triaryl ketimines to generate semi-stabilized 2-azaallyl anions also resulted in the formation of radical species that were detected by EPR.9 This unexpected observation led us to speculate that the 2-azaallyl anion and neutral ketimine are in equilibrium with the 2-azaallyl radical and ketimine radical anion (Scheme 2b). This hypothesis was supported by DFT computational studies, but experimental evidence for the intermediacy of radicals in the coupling reactions was elusive.
Scheme 2.
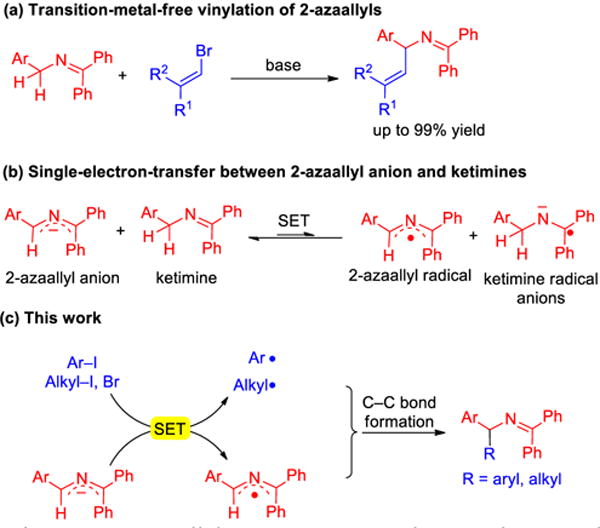
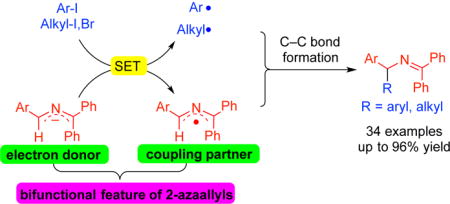
2-Azaallyl anions as super-electron donor and radical coupling partners in transition-metal-free C–C bond-formations.
The proposed reducing nature of 2-azaallyl anions inspired us to ponder their potential to behave as organic super-electron-donors (SEDs) and their possible applications in C–C bond-formation. We hypothesized that 2-azaallyl anions might reduce aryl halides and possibly alkyl halides to generate aryl and alkyl radicals. If the 2-azaallyl anion is indeed sufficiently reducing in nature to undergo SET with aryl and alkyl halides, the resulting 2-azaallyl radical must be relatively stable (long lived). Owing to the persistent radical effect,10 it might behave as a persistent radical and capture the transient radicals which are aryl and alkyl radicals, enabling formation of sp3–sp2 and sp3–sp3 C–C bonds (Scheme 2c). Herein, we report the first transition metal-free arylation and alkylation of 2-azaallyl radicals enabled by the use of 2-azaallyl anions as super-electron donors. A variety of unactivated aryl iodides and alkyl halides were successfully coupled with 2-azaallyl species at room temperature without the use of special initiators, photocatalysts or transition metal catalysts. Notably, the arylation protocol enables the synthesis of diarylmethylamine derivatives, which are of great importance in medicinal and pharmaceutical chemistry.11 Arylation at the benzhydryl carbon (side-reaction) was also observed which afforded pharmaceutically valuable triarylmethylamine derivatives.10c The alkylation protocol overcomes limitations of hindered alkyl halide electrophiles that show little or no reactivity in classic nucleophilic substitution reactions12 and provides efficient synthesis of challenging alkyl amine derivatives.13 In addition, mechanistic studies provide insight into these C–C bond-forming reactions.
2. Result and Discussion
2.1 Transition Metal-free C(sp3)–C(sp2) Coupling to 2-Azaallyl Species
Among organic electron acceptors with C–X bonds, we decided to first examine aryl iodides because their reduction potentials are lower than other aryl halides (Scheme 3a).14 A focused microscale (10 μmol) high-throughput-screen (HTS) was performed for the study of arylation of ketimine 1a with 4-tert-butyliodobenzene (2a) at room temperature for 6 h. Other parameters included six solvents (THF, CPME, DME, MTBE, toluene, and cyclohexane), two bases [LiN(SiMe3)2 and NaN(SiMe3)2] and two concentrations (0.1 M and 0.2 M). The best conditions in this screen involved DME, NaN(SiMe3)2 at 0.2 M concentration. We next monitored the reaction of 4-tert-butyl iodobenzene (2a, 10 μmol) with ketimine 1a and NaN(SiMe3)2 and found that the reaction reached completion in 4 h. (see Figure S1 of Supporting Information for details). A laboratory scale (0.4 mmol) repeat (Scheme 3b) of these optimal conditions gave the C3-arylation product 4aa in 55% isolated yield. To our surprise, the product from arylation at the benzhydryl carbon (C3-arylation, 4aa′) was isolated in 16% yield. We propose that the C3-arylation is derived from the coupling of azaallyl radical with phenyl radical at the more substituted C3 carbon. Steric influences also affect the ratio of C1 vs. C3 arylation, leading to greater arylation at the less substituted carbon in the 2-azaallyl system.
Scheme 3.
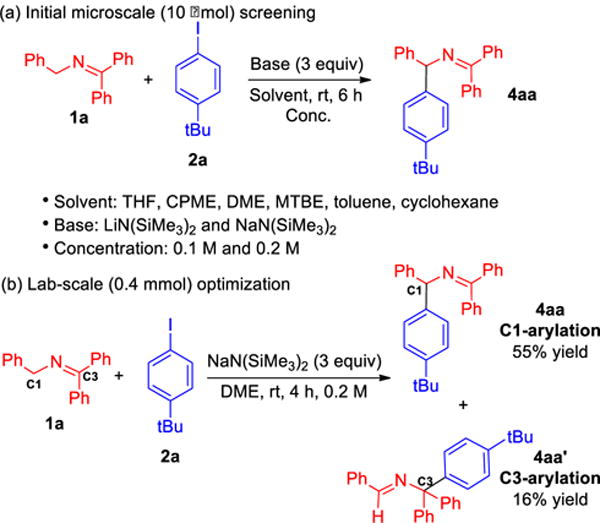
Reaction optimization of arylation. (a) Initial microscale-screening, (b) Lab-scale (0.4 mmol) optimization.
We then turned to the scope of the transition metal-free arylation with different aryl iodides (Table 1). Iodobenzene (2b) furnished C1-arylation product 4ab in 58% yield and C3-arylated 4ab′ in 14% yield. Electron-rich 4-iodoanisole (2c) coupled with 1a in 50% yield at the C1 position (4ac) and 13% yield at the C3 position (4ac′). Using MTBE as solvent, electron-withdrawing 1-bromo-4-iodobenzene (2d) coupled with 1a affording product 4ad (C1) in 43% yield and 4ad′ (C3) in 14% yield. Under the same conditions, coupling between 1-chloro-4-iodobenzene (2e) and 1a led to C1-arylated product 4ae and C3-arylated product 4ae′ in 44% and 15% yield, respectively. We observed that these aryl iodides underwent reaction with high chemoselectivity at the C–I sites, indicating that 2-azaallyl anions preferentially reduce aryl iodides over aryl chlorides or aryl bromides under these reaction conditions. It is noteworthy that aryl iodides 2d and 2e are Bunnett and Crearys’ dihalide mechanistic probes (see the mechanistic study section).15 The fluoro analogue, 4-fluoroiodobenzene (2f), coupled with 1a to give 4af and 4af′ in 48% and 13% yield, respectively. The arylation also preceded with meta-substituted aryl iodides (3-OMe, 2g) to give C1-arylated product 4ag in 47% yield and its C3-arylated counterpart 4ag′ in 17% yield.
Table 1.
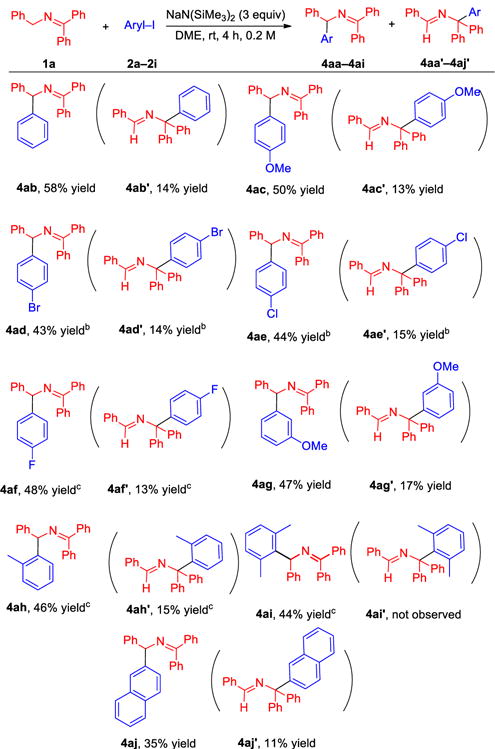
|
Reactions were conducted on a 0.4 mmol scale using 2 equiv. ketimine, 1 equiv. aryl iodide and 3 equiv. NaN(SiMe3)2 at 0.2 M. Isolated yields after chromate-graphic purification.
MTBE.
Reaction time 9 h.
We next wanted to address the possibility of benzyne formation under the basic conditions for the arylation reaction. We therefore examined 2-iodotoluene and 2-iodo-1,3-dimethylbenzene. If benzyne intermediates were involved in the reactions in Table 1, the benzyne generated from 2-iodotoluene would likely undergo reaction at the less hindered position of the benzyne and generate product with a 3-tolyl group. Furthermore, 2-iodo-1,3-dimethylbenzene would be expected to be unreactive if benzyne intermediates are required for the reactions in Table 1. In the event, 2-iodotoluene participated in the arylation to afford the 2-tolyl product, albeit in 21% yield after 4 h. When the reaction time was extended to 9 h, product 4ah was formed in 46% yield. Interestingly, C3 arylated 4ah′ was isolated in 15% yield. More hindered 2-iodo-1,3-dimethylbenzene (2i) furnished 44% yield of the coupling product 4ai. Notably, arylation at the more hindered C3 position was not observed. These results indicate that benzyne intermediates are not necessary in these coupling reactions. Coupling of 2-iodonaphthalene (2j) with ketamine 1a afforded product 4aj in 35% yield and product 4aj′ in 11% yield. From this reaction mixture, we were also able to isolate naphthalene in 20% yield. We hypothesize that naphthalene is derived from hydrogen atom transfer (HAT) to a 2-naphthyl radical intermediate from the DME solvent. This result is significant, because it provides support for the existence of isolated aryl radical intermediates in these transformations.
2.2 Transition Metal-free C(sp3)–C(sp4) Coupling to 2-Azaallyl Species
Given that the 2-azaallyl anion is capable of reacting with aryl iodides, presumably by initial SET, we wanted to probe its reactivity with alkyl halides, which are generally more difficult to reduce.14 We selected neopentyl iodide (3a) as the model alkylation electrophile because its steric hindrance precludes an SN2 coupling pathway.10 The same parameters employed in Scheme 3a were applied to the 10 μmol scale screening of ketimine 1a with neopentyl iodide (3a; rt for 6 h, Scheme 4a). We were pleased and surprised to find that the alkylation reaction proceded. The leading result from the HTS for alkylation was with MTBE and NaN(SiMe3)2 at 0.1 M concentration. In a similar fashion, we next monitored reaction profiles of alkylation at 10 μmol scale and results revealed that coupling of neopentyl iodide 3a with ketimine 1a was completed in 2 h (see Figure S2 of Supporting Information for details). To our delight, lab scale reaction (0.1 mmol) under the optimal conditions gave alkylation product 5aa in 95% isolated yield (Scheme 4b). Notably, C3-alkylation was not observed under these reaction conditions.
Scheme 4.
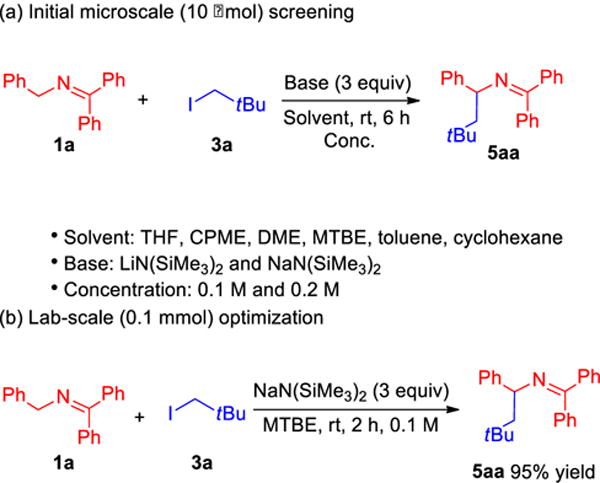
Reaction optimization of arylation. (a) Initial microscale-screening, (b) Lab-scale (0.1 mmol) optimization.
With the optimal conditions, we investigated the scope of the alkylation reaction with respect to unactivated and hindered alkyl halide electrophiles (Table 2). We were concerned that tert-butyl iodide would undergo rapid E2 elimination under the basic reaction conditions. Nevertheless, tert-butyl iodide reacted to afford the alkylation product 5ab in 53% yield. Furthermore, both 1-iodo-1-methylcyclohexane 3c and 1-bromo-1-methylcyclohexane 3d successfully coupled with ketimine 1a, leading to products 5ac in 41% and 37% yield, respectively. Aminoadamantane derivatives are widely present in medications to fight Parkinson’s disease,16 antiviral agents,17 and N-methyl-D-aspartate (NMDA) receptor antagonists.18 To our delight, 1-adamantyl iodide 3e underwent coupling with ketimine 1a in excellent yield (93%). The same product could also be synthesized from 1-adamantyl bromide (3f) in 61% yield with toluene as solvent. Considering the value of aminoadamantane derivatives, we continued surveying the coupling of 1-adamantyl iodide 3e with a few ketimines. tert-Butyl substituted triaryl ketimine 1b underwent coupling with 1-adamantyl iodide 3e in 58% yield. The relatively electron-rich (4-C6H4-OMe) ketimine 1c provided product 5ce in 68% yield. Coupling electron-withdrawing (3-C6H4-CF3) ketimine 1d with 3e proceeded in 80% yield. Heterocyclic 2-furyl (1e) and 2-thiophenyl (1f) ketimines were also examined and afforded products 5ee and 5fe in 60 and 85% yield, respectively. No C3-alkylated products were observed in the coupling of ketimines with 1-adamantyl halides, presumably due to steric effects. Notably, no product deprotonation and/or isomerization to the other ketimine tautomer was detected in the substrates listed in Table 1 and 2.
Table 2.
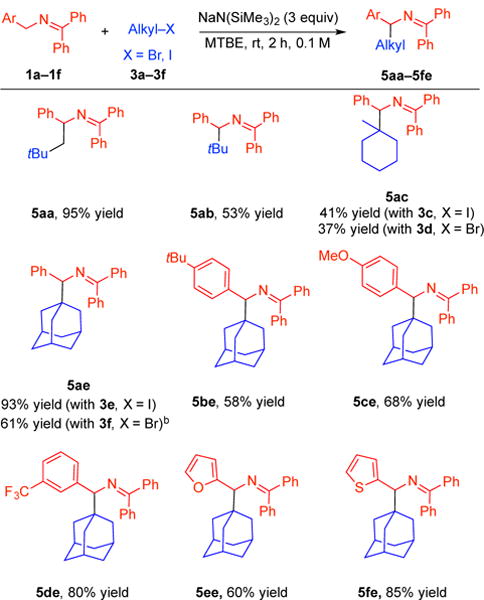
|
Reactions were conducted on a 0.1 mmol scale using 2 equiv. ketimine, 1 equiv. alkyl iodide and 3 equiv. NaN(SiMe3)2 at 0.1 M. Isolated yields after chromatographic purification.
Toluene.
To test the scalability of our method, we conducted the gram-scale synthesis of heterocyclic product 5fe. Thus, ketimine 1f was synthesized from amine and benzophenone imine precursors and dried under vacuum.19 Using the unpurified imine, the alkylation reaction was performed with 1-adamantyl iodide 3e as described above leading to product 5fe in 82% yield (1.69 g, Scheme 5a). Hydrolysis of ketimine 5fe was also performed to afford 1-adamantyl(thiophen-2-yl)methanamine (6fe) in 99% isolated yield (Scheme 5b).
Scheme 5.
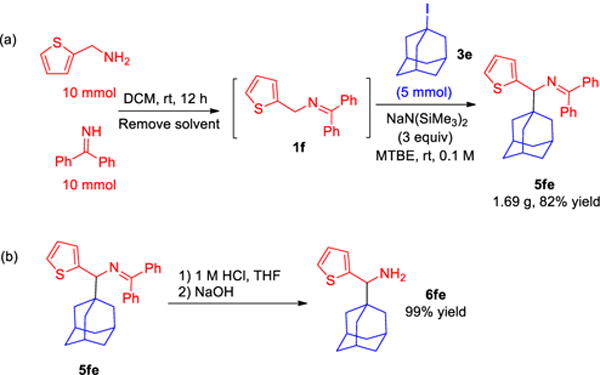
(a) Gram-scale one-pot synthesis of 5fe through a telescoped imine synthesis/alkylation process. (b) Ketimine hydrolysis.
3. Mechanistic Studies
In our initial study of the transition metal-free vinylation of 2-azaallyl species (Scheme 2a),9 our computational work pointed to substrate dependent mechanisms. Reactions were proposed to proceed through either an anionic vinyl substitution pathway with 2-azaallyl anions and β-bromostyrenes or by the addition of 2-azaallyl radicals to the C=C bonds of vinyl halides.20 The reactions presented herein with aryl iodides and alkyl halides, however, must proceed by different pathways and are most easily rationalized by SET from the 2-azaallyl anion (A1, Scheme 6) to either the aryl iodide or alkyl halide to generate radical intermediates. The transient aryl/alkyl radicals can react with the 2-azaallyl radical (A0, proposed to be a persistent radical) to afford the coupled product (Path a, Scheme 6). Or, they might instead react with azaallyl anion (A1) to give a ketiminyl radical anion (A2), which then reduces Ar–I or Alkyl–X to give product and Ar• /Alkyl• in an SRN1 process (Path b, Scheme 6).21
Scheme 6.
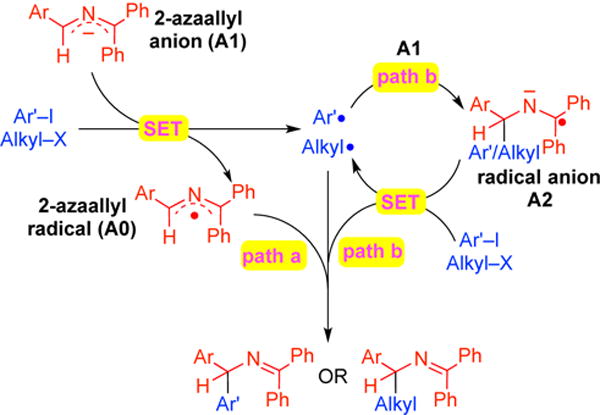
Possible reaction mechanisms. Path a: radical-radical coupling. Path b: SRN1 mechanism.
To investigate the SET process, we undertook a series of experiments with radical clock substrates that undergo rapid cyclization in the presence of radicals. In order to probe for aryl radicals in the coupling of ketimine 1a with aryl iodides, radical clock 2k was employed.22 In the presence of ketimine 1a, 2k was treated with 3 equiv of NaN(SiMe3)2 at rt (Scheme 7). The cyclized product 4ak was obtained in 96% yield as a 1:1 mixture of diastereomers. A second radical trap based on N-allyl-2-iodo-N-methylaniline was subjected to similar conditions and again gave rise to cyclized product in 88% yield. These results confirm the intermediacy of radicals in these reactions.
Scheme 7.
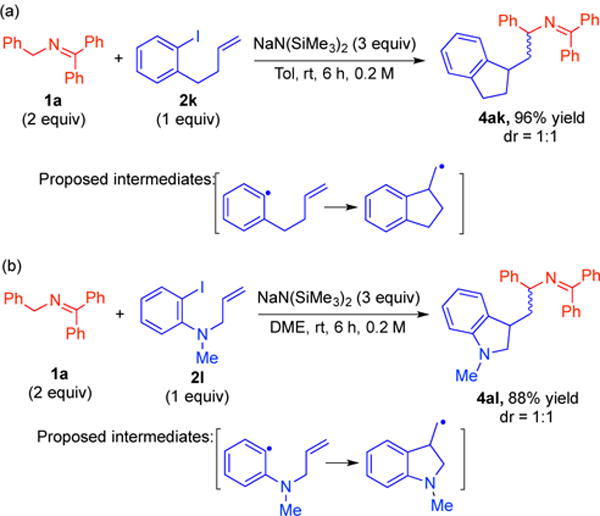
Radical clock study in the arylation.
In the coupling of 1a with N-allylaniline 2l, we observed the formation of rac- and meso-di-ketimines (See Figure S7 of Supporting Information), which we proposed to be derived from dimerization of 2-azaallyl radicals (Scheme 8). NMR spectra of the isolated di-ketimine byproducts matched with the rac- and meso-di-ketimines independently prepared by condensation of the rac- and meso-diamines with benzophenone imine (See Figure S3-S6 of Supporting Information). Such dimers could be observed throughout all of the arylations and alkylations of 2-azaallyl species described above and their formation supports the existence of 2-azaallyl radicals under the reaction conditions.
Scheme 8.
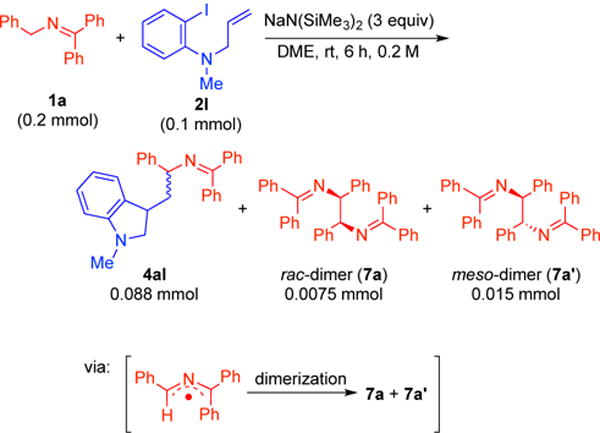
Dimerization of 2-azaallyl radicals.
We next performed parallel mechanistic studies using an alkyl radical clock 3g.22 As outlined in Scheme 9, treatment of ketimine 1a with NaN(SiMe3)2 in the presence of alkyl iodide 3g with a pendent alkene resulted in formation of 5ag in 95% yield as a 1:1 mixture of diastereomers. The formation of the cyclized product confirms that the alkylation reaction, as with the arylation process, proceeds via a radical mechanism.
Scheme 9.
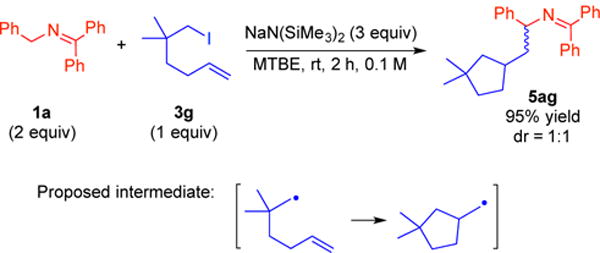
Radical clock study in the alkylation with unactivated alkyl iodides 3g.
In order to further study the role of the 2-azaallyl anion in the SET process, and to shed light on the origin of di-ketimines 7a and 7a′, 2-azaallyl anion stabilized by [K(18-crown-6)]+ cation (8) was synthesized and isolated through recrystallization (Scheme 10a). Mixing salt 8 and aryl iodide 2f under the same conditions used in Table 1 led to product 4af in 12% assay yield (AY, Scheme 10b, i). Moreover, the formation of di-ketimines 7a and 7a′ were observed in 6 and 15% AY, respectively. As there is no neutral ketimine 1a present at the start of the reaction, we propose that di-ketimines are derived from the homo-coupling of 2-azaallyl radicals. To understand the low yields, control experiments using ketimine 1a, KN(SiMe3)2 as base and 18-crwon-6 as additive were conducted. Assay yields of 20% 4af, 4% 7a and 15% 7a′ were observed with KN(SiMe3)2 in the absence of crown ether (Scheme 10b, ii). Addition of 18-crown-6 to the solution of KN(SiMe3)2 and ketimine caused the yield of 4af to drop to 10% with 8% 7a and 22% 7a′ observed (Scheme 10b, iii). These results support our hypothesis that the 2-azaallyl anion behaves as an electron donor in the SET. The synthesis of the sodium salt of 2-azaallyl anion following the procedure employed with the potassium salt in Scheme 10a, but without addition of crown ether to facilitate the recrystallization, was not successful. We are currently probing the origin of the impact of the crown ether on the reactivity of the 2-azaallyl anions, which will be the focus of a subsequent study.
Scheme 10.
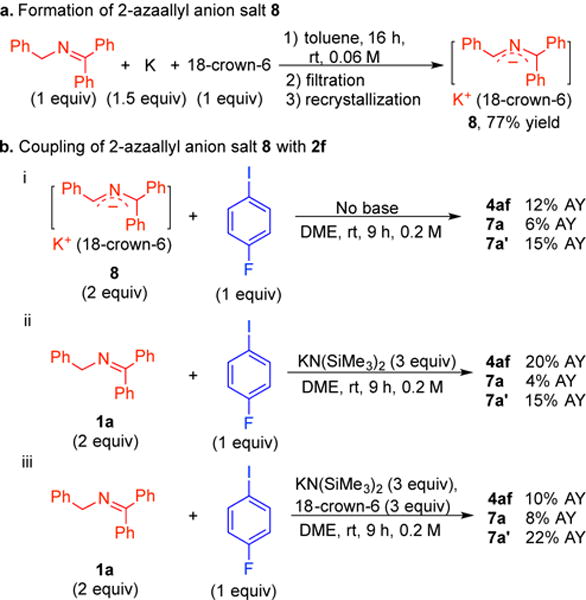
(a) Synthesis of 2-azaallyl anion 8. (b) Study of coupling of 2-azaallyl anion salt 8 with aryl iodide 2f.
The radical clock studies and 2-azaallyl anion coupling studies support the SET process. We next focused on two possible C–C bond forming pathways (radical-radical coupling vs. SRN1, paths a and b in Scheme 6). The goal of this study was to probe the radical anion intermediate (A2, Scheme 6) to determine if the reaction follows the SRN1 mechanism. Hence, we employed Bunnett and Crearys’ dihalides [1-bromo-4-iodobenzene (2d) and 1-chloro-4-iodobenzene (2e) of Table 1], which are widely used in probing SRN1 mechanisms.15, 23 In analogy to Kwong and Lei’s work,23a if the reaction follows the SRN1 mechanism, aryl radical trapping by the 2-azaallyl anion would give radical anion Int 1 or Int 1′ (Scheme 11a). The bromo or chloro group of Int 1 should then dissociate to radical intermediate Int 2, which could undergo HAT to give product 4ab or couple with another equivalent to 2-azaallyl anion to afford bis-coupled products 9 (Scheme 11a). As we reported in Table 1, coupling of 1-bromo-4-iodobenzene (2d) and 1-chloro-4-iodobenzene (2e) with ketimine 1a afforded 4ad and 4ae in 43% and 44% yields with Br and Cl retained in the products. Neither the dehalogenation product nor the bis-ketimine adduct were observed in the reaction. We interpret this observation as evidence against an SRN1 mechanism. Moreover, naphthalene isolated from coupling between 2-iodonaphthalene 2j and ketimine 1a support the existence of radical intermediate Int 4, which favors the radical-radical coupling pathway (Scheme 11b). Overall, the data points to an SET process followed by a radical-radical coupling mechanism as accounting for the unique reactivity of arylation and alkylation of 2-azaallyl species.
Scheme 11.
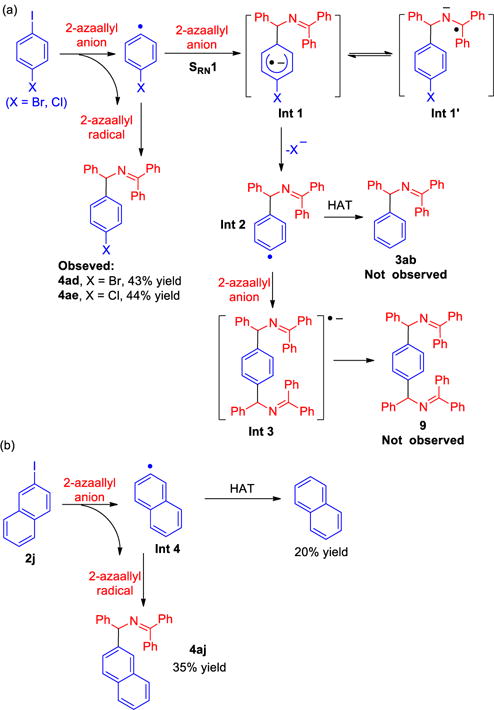
(a) Probe of SRN1 mechanism using Bunnett and Crearys’ 1,4-dihalobenzenes. (b) Reaction of 2-iodonaphthalene.
4. Conclusion
In summary, we report a unique radical generation strategy enabled by simple organic reagents, 2-azaallyl anions, which are a new class of super-electron donors (SEDs). The SED feature enables the reductive fragmentation of unactivated aryl iodides and tertiary alkyl halides to aryl and alkyl radicals. After SET from the 2-azaallyl anion, the resulting 2-azaallyl radical behaves as a persistent radical that then traps both aryl and alkyl radicals forming C–C bonds. Such radical coupling processes occur under mild conditions and do not require special initiators, light or photocatalysts. This radical coupling method enables facile access to diarylmethyl and benzylalkyl amines that are of great importance in medicinal and pharmaceutical chemistry. Diketimine dimer products isolated from the reaction are proposed to arise via homo-coupling of 2-azaallyl radicals. Three radical clock studies, 2-azaallyl anion coupling studies, and Bunnett and Crearys’ dihalide probes confirm the intermediacy of radical intermediates and lend further support to a radical coupling mechanism. Further extension of this unique radical coupling mechanism to suitable reaction partners is underway.
Supplementary Material
Acknowledgments
P. J. W. thanks the National Science Foundation (CHE-1464744) and National Institutes of Health (NIGMS 104349) for financial support. J. J. C. thanks the National Natural Science Foundation of China (NSFC-21372159) for financial support. A. P.-E. thanks MINECO, Spain, for predoctoral and mobility fellowships. G.D. and X. Y. thank the Excellent Young Talents program of Yunnan University.
Footnotes
Supporting Information
Experimental procedures and characterization data for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Diederich F, Stang PJ. Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH. 1998 [Google Scholar]; (b) Magano J, Dunetz JR. Chem Rev. 2011;111:2177. doi: 10.1021/cr100346g. [DOI] [PubMed] [Google Scholar]; (c) Zheng B, Li M, Gao G, He Y, Walsh PJ. Adv Synth Catal. 2016;358:2156. doi: 10.1002/adsc.201600090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gao G, Fu Y, Li M, Wang B, Zheng B, Hou S, Walsh PJ. Adv Synth Catal. 2017;359:2890. doi: 10.1002/adsc.201700438. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wencel-Delord J, Droge T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; (f) Newhouse T, Baran PS. Angew Chem Int Ed. 2011;50:3362. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) McMurray L, O’Hara F, Gaunt MJ. Chem Soc Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (h) Li BJ, Shi ZJ. Chem Soc Rev. 2012;41:5588. doi: 10.1039/c2cs35096c. [DOI] [PubMed] [Google Scholar]; (i) Sun CL, Shi ZJ. Chem Rev. 2014;114:9219. doi: 10.1021/cr400274j. [DOI] [PubMed] [Google Scholar]
- 2.Yan M, Lo JC, Edwards JT, Baran PS. J Am Chem Soc. 2016;138:12692. doi: 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Shaw MH, Twilton J, MacMillan DW. J Org Chem. 2016;81:6898. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Heitz DR, Rizwan K, Molander GA. J Org Chem. 2016;81:7308. doi: 10.1021/acs.joc.6b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tellis JC, Kelly CB, Primer DN, Jouffroy M, Patel NR, Molander GA. Acc Chem Res. 2016;49:1429. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Candish L, Freitag M, Gensch T, Glorius F. Chem Sci. 2017;8:3618. doi: 10.1039/c6sc05533h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Koike T, Akita M. Acc Chem Res. 2016;49:1937. doi: 10.1021/acs.accounts.6b00268. [DOI] [PubMed] [Google Scholar]; (f) Nakajima K, Miyake Y, Nishibayashi Y. Acc Chem Res. 2016;49:1946. doi: 10.1021/acs.accounts.6b00251. [DOI] [PubMed] [Google Scholar]; (g) Kelly CB, Patel NR, Primer DN, Jouffroy M, Tellis JC, Molander GA. Nat Protoc. 2017;12:472. doi: 10.1038/nprot.2016.176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Romero NA, Nicewicz DA. Chem Rev. 2016;116:10075. doi: 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; (i) Skubi KL, Blum TR, Yoon TP. Chem Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;352:801. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan CM, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J Am Chem Soc. 2016;138:2174. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Toriyama F, Cornella J, Wimmer L, Chen TG, Dixon DD, Creech G, Baran PS. J Am Chem Soc. 2016;138:11132. doi: 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Doni E, Murphy JA. Chem Commun. 2014;50:6073. doi: 10.1039/c3cc48969h. [DOI] [PubMed] [Google Scholar]; (b) Murphy JA. J Org Chem. 2014;79:3731. doi: 10.1021/jo500071u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou S, Doni E, Anderson GM, Kane RG, MacDougall SW, Ironmonger VM, Tuttle T, Murphy JA. J Am Chem Soc. 2014;136:17818. doi: 10.1021/ja5101036. [DOI] [PubMed] [Google Scholar]
- 6.Farwaha HS, Bucher G, Murphy JA. Org Biomol Chem. 2013;11:8073. doi: 10.1039/c3ob41701h. [DOI] [PubMed] [Google Scholar]
- 7.Hanson SS, Doni E, Traboulsee KT, Coulthard G, Murphy JA, Dyker CA. Angew Chem Int Ed. 2015;54:11236. doi: 10.1002/anie.201505378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Zard SZ. Chem Soc Rev. 2008;37:1603. doi: 10.1039/b613443m. [DOI] [PubMed] [Google Scholar]; (b) Hartmann M, Li Y, Studer A. J Am Chem Soc. 2012;134:16516. doi: 10.1021/ja307638u. [DOI] [PubMed] [Google Scholar]; (c) Zhang B, Studer A. Org Lett. 2013;15:4548. doi: 10.1021/ol402106x. [DOI] [PubMed] [Google Scholar]; (d) Li Y, Studer A. Angew Chem Int Ed. 2012;51:8221. doi: 10.1002/anie.201202623. [DOI] [PubMed] [Google Scholar]
- 9.Li M, Gutierrez O, Berritt S, Pascual-Escudero A, Yeşilçimen A, Yang X, Adrio J, Huang G, Nakamaru-Ogiso E, Kozlowski MC, Walsh PJ. Nat Chem. 2017;9:997. doi: 10.1038/nchem.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Fischer H. J Am Chem Soc. 1986;108:3925. [Google Scholar]; (b) Fischer H. Chem Rev. 2001;101:3581. doi: 10.1021/cr990124y. [DOI] [PubMed] [Google Scholar]; (c) Studer A. Chem Eur J. 2001;7:1159. doi: 10.1002/1521-3765(20010316)7:6<1159::aid-chem1159>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; (d) Focsaneanu KS, Scaiano JC. Helvetica Chimica Acta. 2006;89:2473. [Google Scholar]
- 11.(a) Li JJ. Contemporary Drug Synthesis; Wiley–Interscience. 2004:221. [Google Scholar]; (b) Grant JA, Riethuisen JM, Moulaert B, DeVos C. Ann Allergy Asthma Immunol. 2002;88:190. doi: 10.1016/S1081-1206(10)61995-3. [DOI] [PubMed] [Google Scholar]; (c) Yang X, Kim BS, Li M, Walsh PJ. Org Lett. 2016;18:2371. doi: 10.1021/acs.orglett.6b00815. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Doggrell SA, Liang LC. N-S Arch Pharmacol. 1998;357:126. doi: 10.1007/pl00005146. [DOI] [PubMed] [Google Scholar]; (e) Plobeck N, Delorme D, Wei ZY, Yang H, Zhou F, Schwarz P, Gawell L, Gagnon H, Pelcman B, Schmidt R, Yue SY, Walpole C, Brown W, Zhou E, Labarre M, Payza K, St-Onge S, Kamassah A, Morin PE, Projean D, Ducharme J, Roberts E. J Med Chem. 2000;43:3878. doi: 10.1021/jm000228x. [DOI] [PubMed] [Google Scholar]; (f) Zhou B, Liu ZF, Deng GG, Chen W, Li MY, Yang LJ, Li Y, Yang XD, Zhang HB. Org Biomol Chem. 2016;14:9423. doi: 10.1039/c6ob01495j. [DOI] [PubMed] [Google Scholar]; (g) Zhou Y, Duan K, Zhu L, Liu Z, Zhang C, Yang L, Li M, Zhang H, Yang X. Bioog Med Chem Lett. 2016;26:460. doi: 10.1016/j.bmcl.2015.11.092. [DOI] [PubMed] [Google Scholar]; (h) Zhang CB, Liu Y, Liu ZF, Duan SZ, Li MY, Chen W, Li Y, Zhang HB, Yang XD. Bioorg Med Chem Lett. 2017;27:1808. doi: 10.1016/j.bmcl.2017.02.053. [DOI] [PubMed] [Google Scholar]
- 12.Fu GC. ACS Cent Sci. 2017;3:692. doi: 10.1021/acscentsci.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Beletskaya I, Averin A, Ranyuk E, Golub S, Buryak A, Savelyev E, Orlinson B, Novakov I. Synthesis. 2007;14:2215. [Google Scholar]; (b) Liu FP, Zhong JC, Zheng B, Li SN, Gao G, Wang ZY, Li MY, Hou SC, Wang M, Bian QH. Tetrahedron: Asymmetry. 2015;26:961. [Google Scholar]; (c) Liu F, Zhong J, Li S, Li M, Wu L, Wang Q, Mao J, Liu S, Zheng B, Wang M, Bian Q. J Nat Prod. 2016;79:244. doi: 10.1021/acs.jnatprod.5b00713. [DOI] [PubMed] [Google Scholar]; (d) Butera JA, Antane MM, Antane SA, Argentieri TM, Freeden C, Graceffa RF, Hirth BH, Jenkins D, Lennox JR, Matelan E, Norton NW, Quagliato D, Sheldon JH, Spinelli W, Warga D, Wojdan A, Woods M. J Med Chem. 2000;43:1187. doi: 10.1021/jm9905099. [DOI] [PubMed] [Google Scholar]; (e) Bugge S, Kaspersen SJ, Larsen S, Nonstad U, Bjorkoy G, Sundby E, Hoff BH. Eur J Med Chem. 2014;75:354. doi: 10.1016/j.ejmech.2014.01.042. [DOI] [PubMed] [Google Scholar]
- 14.Nicewicz D, Roth H, Romero N. Synlett. 2015;27:714. [Google Scholar]
- 15.Studer A, Curran DP. Angew Chem Int Ed. 2011;50:5018. doi: 10.1002/anie.201101597. [DOI] [PubMed] [Google Scholar]
- 16.Danielczyk W. J Neural Transm Suppl. 1995;46:399. [PubMed] [Google Scholar]
- 17.Leophonte P. Bull Acad Natl Med. 2005:341. [PubMed] [Google Scholar]
- 18.Hughes P, Olejnik O, Schiffman R. 2005031652. Patent Application US. 2005
- 19.O’Donnell MJ, Polt RL. J Org Chem. 1982;47:2663. [Google Scholar]
- 20.(a) Li M, Berritt S, Walsh PJ. Org Lett. 2014;16:4312. doi: 10.1021/ol502043j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li M, Gonzalez-Esguevillas M, Berritt S, Yang X, Bellomo A, Walsh PJ. Angew Chem Int Ed. 2016;55:2825. doi: 10.1002/anie.201509757. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li M, Yucel B, Adrio J, Bellomo A, Walsh PJ. Chem Sci. 2014;5:2383. doi: 10.1039/C3SC53526F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cao X, Sha SC, Li M, Kim BS, Morgan C, Huang R, Yang X, Walsh PJ. Chem Sci. 2016;7:611. doi: 10.1039/c5sc03704b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Reddy MVVS, Celalyan-Berthier A, Geoffroy M, Morgantini PY, Weber J, Bernardinelli G. J Am Chem Soc. 1988;110:2748. [Google Scholar]; (f) Gaebert C, Mattay J, Toubartz M, Steenken S, Muller B, Bally T. Chem Eur J. 2005;11:1294. doi: 10.1002/chem.200400557. [DOI] [PubMed] [Google Scholar]; (g) Li M, Yucel B, Jiménez J, Rotella M, Fu Y, Walsh PJ. Adv Synth Catal. 2016;358:1910. doi: 10.1002/adsc.201600075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossi RA, Pierini AB, Penenory AB. Chem Rev. 2003;103:71. doi: 10.1021/cr960134o. [DOI] [PubMed] [Google Scholar]
- 22.Newcomb M. Encyclopedia of Radicals in Chemistry, Biology and Materials. John Wiley & Sons, Ltd; 2012. Radical Kinetics and Clocks. [Google Scholar]
- 23.(a) Liu W, Cao H, Zhang H, Zhang H, Chung KH, He C, Wang H, Kwong FY, Lei A. J Am Chem Soc. 2010;132:16737. doi: 10.1021/ja103050x. [DOI] [PubMed] [Google Scholar]; (b) Liu C, Liu D, Lei A. Acc Chem Res. 2014;47:3459. doi: 10.1021/ar5002044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.