

Abstract
Copy number variants (CNVs) have been strongly implicated in the genetic etiology of schizophrenia (SCZ). However, genome-wide investigation of the contribution of CNV to risk has been hampered by limited sample sizes. We sought to address this obstacle by applying a centralized analysis pipeline to a SCZ cohort of 21,094 cases and 20,227 controls. A global enrichment of CNV burden was observed in cases (OR=1.11, P=5.7×10−15), which persisted after excluding loci implicated in previous studies (OR=1.07, P=1.7 ×10−6). CNV burden was enriched for genes associated with synaptic function (OR = 1.68, P = 2.8 ×10−11) and neurobehavioral phenotypes in mouse (OR = 1.18, P= 7.3 ×10−5). Genome-wide significant evidence was obtained for eight loci, including 1q21.1, 2p16.3 (NRXN1), 3q29, 7q11.2, 15q13.3, distal 16p11.2, proximal 16p11.2 and 22q11.2. Suggestive support was found for eight additional candidate susceptibility and protective loci, which consisted predominantly of CNVs mediated by non-allelic homologous recombination.
Introduction
Studies of genomic copy number variation (CNV) have established a role for rare genetic variants in the etiology of SCZ 1. There are three lines of evidence that CNVs contribute to risk for SCZ: genome-wide enrichment of rare deletions and duplications in SCZ cases relative to controls 2, 3, a higher rate of de novo CNVs in cases relative to controls4–6, and association evidence implicating a small number of specific loci (Supplementary Table 1). All CNVs that have been implicated in SCZ are rare in the population, but confer significant risk (odds ratios 2–60).
To date, CNVs associated with SCZ have largely emerged from mergers of summary data for specific candidate loci 7–9; yet even the largest genome-wide scans (sample sizes typically <10,000) remain under-powered to robustly confirm genetic association for the majority of pathogenic CNVs reported so far, particularly for those with low frequencies (<0.5% in cases) or intermediate effect sizes (odds ratios 2–10). It is important to address the low power of CNV studies with larger samples given that this type of mutation has already proven useful for highlighting some aspects of SCZ related biology 6, 10–13.
The limited statistical power provided by small samples is a significant obstacle in studies of rare and common genetic variation. In response, global collaborations have been formed in order to attain large sample sizes, as exemplified by a study by the Schizophrenia Working Group of the Psychiatric Genomics Consortium (PGC) which identified 108 independent schizophrenia associated loci 14. Recognizing the need for similarly large samples in studies of CNVs for psychiatric disorders, we formed the PGC CNV Analysis Group. Our goal was to enable large-scale analyses of CNVs in psychiatry using centralized and uniform methodologies for CNV calling, quality control, and statistical analysis. Here, we report the largest genome-wide analysis of CNVs for any psychiatric disorder to date, using datasets assembled by the Schizophrenia Working Group of the PGC.
Data processing and meta-analytic methods
Raw intensity data were obtained from 57,577 subjects from 43 separate datasets (Supplementary Table 2). After CNV calling and quality control (QC), 41,321 subjects were retained for analysis. We developed a centralized pipeline for systematic calling of CNVs for Affymetrix and Illumina platforms. (Methods and Supplementary Figure 1). The pipeline included multiple CNV callers run in parallel. Data from Illumina platforms were processed using PennCNV 15 and iPattern 16. Data from Affymetrix platforms were analyzed using PennCNV and Birdsuite 17.Two additional methods, iPattern and C-score 18, were applied to data from the Affymetrix 6.0 platform. In order to ensure proper normalization of the X chromosome, male and female subjects were normalized separately. The CNV calls from each program were converted to a standardized format and a consensus call set was constructed by merging CNV outputs at the sample level. Only CNV segments that were detected by all algorithms were retained. We performed QC at the platform level to exclude samples with poor probe intensity and/or an excessive CNV load (number and length). A final set of rare, high quality CNVs was defined as those >20kb in length, at least 10 probes, and <1% MAF.
Genetic associations were investigated by case-control tests of CNV burden at four levels: (1) genome-wide (2) pathways, (3) genes, and (4) CNV breakpoints. Analyses controlled for SNP-derived principal components, sex, genotyping platform and data quality metrics. Multiple-testing thresholds for genome-wide significance were estimated from family-wise error rates drawn from permutation
Genome wide analysis of CNV burden
An elevated burden of rare CNVs among SCZ cases has been well established 2. We applied our meta-analytic framework to measure the consistency of overall CNV burden across genotyping platforms, and whether a measurable amount of CNV burden persists outside of previously implicated CNV regions. Consistent with previous estimates, the overall CNV burden was significantly greater among SCZ cases when measured as total Kb covered (OR=1.12, p = 5.7×10−15), genes affected (OR=1.21, p = 6.6×10−21), or CNV number (OR=1.03, p = 1×10−3). The burden signal above was driven by CNVs located within genes. Focusing heretofore on the number of genes affected by CNV, the burden metric with the strongest signal of enrichment in our study, the effect size was consistent across all genotyping platforms (Figure 1a). When we split by CNV type, the effect size for copy number losses (OR=1.40, p = 4×10−16) was greater than for gains (OR=1.12, p = 2×10−7) (Supplementary Figures 2 and 3). Partitioning by CNV frequency (based on 50% reciprocal overlap with the full call set, Methods), CNV burden was enriched among cases across a range of frequencies, up to counts of 80 (MAF = 0.4%) in the combined sample (Figure 1b). CNV burden results for individual cohorts are provided in Supplementary Figure 4. We observed no enrichment in CNV burden when considering only variants that did not overlap exons (Supplementary Figure 5)
Figure 1. CNV Burden.

(A) Forest plot of CNV burden (measured here as genes affected by CNV), partitioned by genotyping platform, with the full PGC sample at the bottom. CNV burden is calculated by combining CNV gains and losses. Numbers of case and controls for each platform are listed, and “genes” denotes the mean number of genes affected by a CNV in controls. Burden tests use a logistic regression model predicting SCZ case/control status by CNV burden along with covariates (see methods). The odds ratio is the exponential of the logistic regression coefficient, and odds ratios above one predict increased SCZ risk. (B) CNV burden partitioned by CNV frequency. For reference, for autosomal CNVs, a CNV count of 41 in the sample corresponds to frequency of 0.1% in the full PGC sample. Using the same model as above, each CNV was placed into a single CNV frequency category based on a 50% reciprocal overlap with other CNVs. CNV gene burden with inclusion of all CNVs are shown in green, and burden excluding previously implicated CNV loci are shown in blue.
A primary question in this study is the contribution of novel loci to the excess CNV burden in cases. After removing nine previously implicated CNV loci (where reported p-values exceed our designated multiple testing threshold, Supplementary Table 1), excess CNV burden in SCZ remained significantly enriched (genes affected OR=1.11, p = 1.3×10−7, Figure 1b). CNV burden also remained significantly enriched after removal of all reported loci from Supplementary Table 1, but the effect-size was greatly reduced (OR = 1.08) compared to the enrichment overall (OR = 1.21). When we partitioned CNV burden by frequency, we found that much of the previously unexplained signal was restricted to ultra-rare events (i.e., MAF < 0.1%, Figure 1b).
Gene-set (pathway) burden
We assessed whether CNV burden was concentrated within defined sets of genes involved in neurodevelopment or neurological function. A total of 36 gene-sets were evaluated (for a description see Supplementary Table 3), consisting of gene-sets representing neuronal function, synaptic components and neurological and neurodevelopmental phenotypes in human (19 sets), gene-sets based on brain expression patterns (7 sets), and human orthologs of mouse genes whose disruption causes phenotypic abnormalities, including neurobehavioral and nervous system abnormality (10 sets). Genes not expressed in brain (1 set) or associated with abnormal phenotypes in mouse organ systems unrelated to brain (7 sets) were included as negative controls. We mapped CNVs to genes if they overlapped by at least one exonic basepair.
Gene-set burden was tested using logistic regression deviance test 6. In addition to using the same covariates included in genome-wide burden analysis, we controlled for the total number of genes per subject spanned by rare CNVs to account for signal that merely reflects the global enrichment of CNV burden in cases 19. Multiple-testing correction (Benjamini-Hochberg False Discovery Rate, BH-FDR) was performed separately for each gene-set group and CNV type (gains, losses). After multiple test correction (Benjamini-Hochberg FDR ≤ 10%) 15 gene-sets were enriched for rare loss burden in cases and 4 for rare gains in cases, none of which are negative control sets (Figure 2).
Figure 2. Gene-set Burden.

Gene-set burden test results for rare losses (a, c) and gains (b, d); frames a–b display gene-sets for neuronal function, synaptic components, neurological and neurodevelopmental phenotypes in human; frames c–d display gene-sets for human homologs of mouse genes implicated in abnormal phenotypes (organized by organ systems); both are sorted by –log 10 of the logistic regression deviance test p-value multiplied by the beta coefficient sign, obtained for rare losses when including known loci. Gene-sets passing the 10% BH-FDR threshold are marked with “*”. Gene-sets representing brain expression patterns were omitted from the figure because only a few were significant (losses: 1, gains: 3).
Of the 15 sets significant for losses, the majority consisted of synaptic or other neuronal components (9 sets); in particular, “GO synaptic” (GO:0045202) and the activity-regulated cytoskeleton-associated protein complex, or “ARC complex”, rank first based on statistical significance and effect-size respectively (Figure 2a). Losses in cases were also significantly enriched for genes involved in nervous system or behavioral phenotypes in mouse but not for gene-sets related to other organ system phenotypes (Figure 2c). To account for dependency between synaptic and neuronal gene-sets, we re-tested loss burden following a step-down logistic regression approach, ranking gene-sets based on significance or effect size (Supplementary Table 4). Only GO synaptic and ARC complex were significant in at least one of the two step-down analyses, suggesting that burden enrichment in the other neuronal categories is mostly captured by the overlap with synaptic genes. Following the same approach, the mouse neurological/neurobehavioral phenotype set remained nominally significant, suggesting that a portion of this signal was independent of the synaptic gene set. Pathway enrichment was less pronounced for duplications, consistent with the smaller burden effects for this class of CNV. Among synaptic or other neuronal components, duplication burden was significantly enriched only for NMDA receptor complex; (Figure 2b); none of the mouse phenotype sets passed the significance threshold for duplications (Figure 2d).
Given that synaptic gene sets were robustly enriched for deletions in cases, and with an appreciable contribution from loci that have not been strongly associated with SCZ previously, pathway-level interactions of these sets were further investigated. A protein-interaction network was seeded using the synaptic and ARC complex genes that were intersected by rare deletions in this study (Figure 3). A graph of the network highlights multiple subnetworks of synaptic proteins including pre-synaptic adhesion molecules (NRXN1, NRXN3), post-synaptic scaffolding proteins (DLG1, DLG2, DLGAP1, SHANK1, SHANK2), glutamatergic ionotropic receptors (GRID1, GRID2, GRIN1, GRIA4), and complexes such as Dystrophin and its synaptic interacting proteins (DMD, DTNB, SNTB1, UTRN). A subsequent test of the Dystrophin glycoprotein complex (DGC) revealed that deletion burden of the synaptic DGC proteins (intersection of “GO DGC” GO:0016010 and “GO synapse” GO:0045202) was enriched in cases (Deviance test P = 0.05), but deletion burden of the full DGC was not significant (P = 0.69).
Figure 3. Protein Interaction Network for Synaptic Genes.

Synaptic and ARC-complex genes intersected by a rare loss in at least 4 case or control subjects and with genic burden Benjamini-Hochberg FDR <= 25% (red discs) were used to query GeneMANIA36 and retrieve additional protein interaction neighbors, resulting in a network of 136 synaptic genes. Genes are depicted as disks; disk centers are colored based on rare loss frequency (Freq.SZ and Freq.CT) being prevalent in cases or controls; disk borders are colored to mark (i) gene implication in human dominant or X-linked neurological or neurodevelopmental phenotype, (ii) de novo mutation (DeN) reported by Fromer et al. 28, split between LOF (frameshift, stop-gain, core splice site) and missense or amino acid insertion / deletion, (iii) implication in mouse neurobehavioral abnormality. Pre-synaptic adhesion molecules (NRXN1, NRXN3), post-synaptic scaffolds (DLG1, DLG2, DLGAP1, SHANK1, SHANK2) and glutamatergic ionotropic receptors (GRID1, GRID2, GRIN1, GRIA4) constitute a highly connected subnetwork with more losses in cases than controls.
Gene CNV association
To define specific loci that confer risk for SCZ, we tested CNV association at the level of individual genes, using logistic regression deviance test and the same covariates included in genome-wide burden analysis. To correctly account for large CNVs that affect multiple genes, we aggregated adjacent genes into a single locus if their copy number was highly correlated across subjects (more than 50% subject overlap). CNVs were mapped to genes if they overlapped one or more exons. The criterion for genome-wide significance used the Family-Wise Error Rate (FWER) < 0.05. The criterion for suggestive evidence used a Benjamini-Hochberg False Discovery Rate (BH-FDR) < 0.05.
Of eighteen independent CNV loci with gene-based BH-FDR < 0.05, two were excluded based on CNV calling accuracy or evidence of a batch effect (Supplementary Note). The sixteen loci that remain after these additional QC steps, comprising seventeen separate association signals, are listed in Table 1. P-values for this summary table were obtained by re-running our statistical model across the entire region (Supplementary Note). These sixteen loci represent a set of novel (n=6), previously reported (n=4), and previously implicated (n=7) regions, with 22q11.21 comprising two separate association signals at the same locus. Manhattan plots of the gene association for losses and gains are provided in Figure 4. A permutation-based false discovery rate yielded similar estimates to BH-FDR.
Table 1.
Significant CNV loci from gene-based association test
| CHR | START | END | locus GENE | Status | Putative Mechanism |
CNV test | Direction | FWER | BH-FDR | CAS | CON | Regional P-value |
Odds Ratio [95% CI] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 22 | 17,400,000 | 19,750,000 | 22q11.21 | Previously Implicated | NAHR | loss | risk | yes | 3.54E-15 | 64 | 1 | 5.70E-18 | 67.7 [9.3–492.8] |
| 16 | 29,560,000 | 30,110,000 | 16p11.2 (proximal) | Previously Implicated | NAHR | gain | risk | yes | 5.82E-10 | 70 | 7 | 2.52E-12 | 9.4 [4.2–20.9] |
| 2 | 50,000,992 | 51,113,178 | 2p16.3 NRXN1 | Previously Implicated | NHEJ | loss | risk | yes | 3.52E-07 | 35 | 3 | 4.92E-09 | 14.4 [4.2–46.9] |
| 15 | 28,920,000 | 30,270,000 | 15q13.3 | Previously Implicated | NAHR | loss | risk | yes | 2.22E-05 | 28 | 2 | 2.13E-07 | 15.6 [3.7–66.5] |
| 1 | 144,646,000 | 146,176,000 | 1q21.1 | Previously Implicated | NAHR | loss+gain | risk | yes | 0.00011 | 60 | 14 | 1.50E-06 | 3.8 [2.1–6.9] |
| 3 | 197,230,000 | 198,840,000 | 3q29 | Previously Implicated | NAHR | loss | risk | yes | 0.00024 | 16 | 0 | 1.86E-06 | INF |
| 16 | 28,730,000 | 28,960,000 | 16p11.2 (distal) | Previously Reported | NAHR | loss | risk | yes | 0.0029 | 11 | 1 | 5.52E-05 | 20.6 [2.6–162.2] |
| 7 | 72,380,000 | 73,780,000 | 7q11.23 | Previously Reported | NAHR | gain | risk | yes | 0.0048 | 16 | 1 | 1.68E-04 | 16.1 [3.1–125.7] |
| X | 153,800,000 | 154,225,000 | Xq28 (distal) | Novel | NAHR | gain | risk | no | 0.049 | 18 | 2 | 3.61E-04 | 8.9 [2.0–39.9] |
| 22 | 17,400,000 | 19,750,000 | 22q11.21 | Previously Reported | NAHR | gain | protective | no | 0.024 | 3 | 16 | 4.54E-04 | 0.15 [0.04–0.52] |
| 7 | 64,476,203 | 64,503,433 | 7q11.21 ZNF92 | Novel | NAHR | loss+gain | protective | no | 0.033 | 131 | 180 | 6.71E-04 | 0.66 [0.52–0.84] |
| 13 | 19,309,593 | 19,335,773 | 13q12.11 ZMYM5 | Novel | NHAR | gain | protective | no | 0.024 | 15 | 38 | 7.91E-04 | 0.36 [0.19–0.67] |
| X | 148,575,477 | 148,580,720 | Xq28 MAGEA11 | Novel | NAHR | gain | protective | no | 0.044 | 12 | 36 | 1.06E-03 | 0.35 [0.18–0.68] |
| 15 | 20,350,000 | 20,640,000 | 15q11.2 | Previously Implicated | NAHR | loss | risk | no | 0.044 | 98 | 50 | 1.34E-03 | 1.8 [1.2–2.6] |
| 9 | 831,690 | 959,090 | 9p24.3 DMRT1 | Novel | NHEJ | loss+gain | risk | no | 0.049 | 13 | 1 | 1.35E-03 | 12.4 [1.6–98.1] |
| 8 | 100,094,670 | 100,958,984 | 8q22.2 VPS13B | Novel | NHEJ | loss | risk | no | 0.048 | 7 | 1 | 1.74E-03 | 14.5 [1.7–122.2] |
| 7 | 158,145,959 | 158,664,998 | 7p36.3 VIPR2 WDR60 | Previously Reported | NAHR | loss+gain | risk | no | 0.046 | 20 | 6 | 5.79E-03 | 3.5 [1.3–9.0] |
All seventeen association signals listed contain at least one gene with Benjamini-Hochberg false discovery rate (BH-FDR) < 0.05 in the gene-based test, with eight containing at least one gene surpassing the family-wise error rate (FWER) < 0.05. Genomic positions listed are using hg18 coordinates. For putative CNV mechanisms, non-allelic homologous recombination (NAHR) and non-homologous end joining (NHEJ) are listed as the likely genomic feature driving CNV formation at each locus. Regional p-values and odds ratios listed are from a regional test at each locus, where we combine CNV overlapping the implicated region and run the same test as used for each gene (logistic regression with covariates and deviance test p-value). CNV losses and gains at the 22q11.21 locus are listed as separate association signals, as CNV losses associate with SCZ risk while CNV gains associate with protection from SCZ. For each association we indicate whether it was previously described in the literature (Previously Reported) and if the reported P-value exceeded the multiple testing correction in this study (Previously Implicated).
Figure 4. Gene Based Manhattan Plot.

Manhattan plot displaying the –log10 deviance p-value for (a) CNV losses and (b) CNV gains the gene-based test. P-value cutoffs corresponding to FWER < 0.05 and BH-FDR < 0.05 are highlighted in red and blue, respectively. Loci significant after multiple test correction are labeled.
Eight loci attain genome-wide significance, including copy number losses at 1q21.1, 2p16.3 (NRXN1), 3q29, 15q13.3, 16p11.2 (distal) and 22q11.2 along with gains at 7q11.23 and 16p11.2 (proximal). An additional eight loci meet criterion for suggestive association, including six that have not been reported previously in association with SCZ. Based on our estimation of False Discovery Rates (BH and permutations), we expect to observe less than two associations meeting suggestive criteria by chance. In order to further evaluate the six new candidate loci identified here, we performed experimental validation of CNV calls in a subset of samples by digital droplet PCR (ddPCR, see Methods). Validation rates of 100% were obtained for gains of DMRT1, MAGEA11 and distal Xq28, losses of VPS13B, and gains and losses of ZNF92 (Supplementary Table 5). We obtained a low validation rate at one locus, ZMYM5 (64%), and therefore do not consider the association at this locus convincing.
Breakpoint level CNV association
With our sample size and uniform CNV calling pipeline, many individual CNV loci can be tested with adequate power at the CNV breakpoint level (i.e. the SNP probe defining the start and end of the CNV segment), potentially facilitating discovery at a finer resolution than locus-wide tests. Tests for association were performed at each CNV breakpoint using the residuals of case-control status after controlling for analysis covariates, with significance determined through permutation. Results for losses and gains are shown in Supplementary Figure 6. Four independent CNV loci surpass genome-wide significance, all of which were also identified in the gene-based test, including the 15q13.2–13.3 and 22q11.21 deletions, 16p11.2 duplication, and 1q21.1 deletion and duplication. While these loci represent fewer than half of the previously implicated SCZ loci, we do find support for all loci where the association originally reported meets the criteria for genome-wide correction in this study. We examined association among all previously reported loci showing association to SCZ, including 18 CNV losses and 25 CNV gains (Supplementary Table 6); 8 loci have BH-FDR q-value < 0.05, 13 loci have BH-FDR q-value < 0.1, and 25 of the 42 loci were associated with SCZ at an uncorrected p < .05.
Associations at some loci become better delineated through breakpoint-level analysis. For instance, NRXN1 at 2p16.3 is a CNV hotspot, and exonic deletions of this gene are significantly enriched in SCZ9, 20. In this large sample, we observe a high density of “non-recurrent” deletion breakpoints in cases and controls. A snapshot of the breakpoint association results from the PGC CNV browser (see URLs) reveals a saw-tooth pattern of association. Predominant peaks correspond to exons and transcriptional start sites of NRXN1 isoforms (Figure 5). This example highlights how, with high diversity of alleles at a single locus, the association peak may become more refined, and in some cases converge toward individual functional elements. Similarly, visualization of the previously reported SCZ risk loci on 16p13.2 and 8q11.23 reveals a high density of duplication breakpoints, which better delineate genes in these regions. It is important, however, to note that CNV breakpoints in the current study are estimated from genotyped SNPs around the true breakpoint, and that these breakpoint estimates are limited by the resolution of the genotyping platform, and therefore subject to error.
Figure 5. Manhattan plot of breakpoint-level associations across the Neurexin-1 locus.
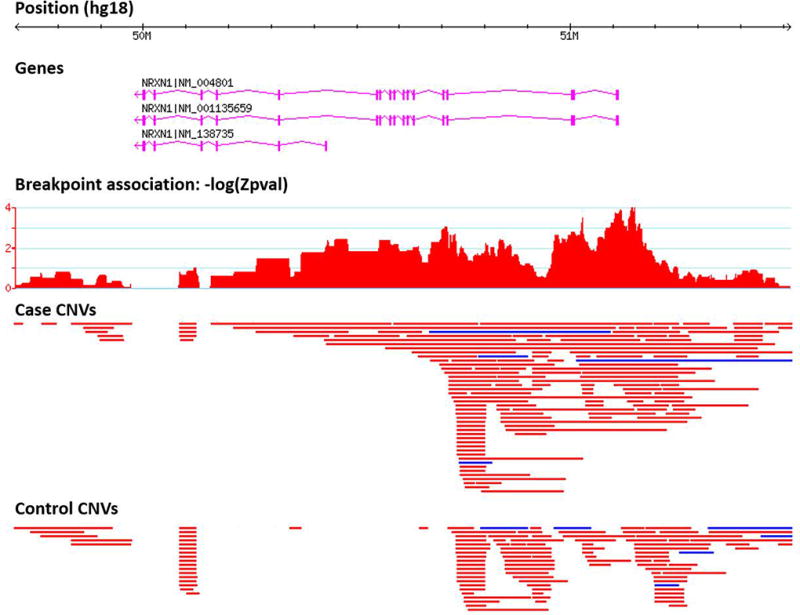
The manhattan plot (for deletions) represents empirical P-values at each deletion breakpoint. CNV tracks display duplications (blue) and deletions (red) detected in cases and controls from the PGC SCZ dataset.
Novel risk alleles are predominantly NAHR-mediated CNVs
Many CNV loci that have been strongly implicated in human disease are hotspots for non-allelic homologous recombination (NAHR), a process which in most cases is mediated by flanking segmental duplications 21. We defined a CNV as “NAHR” when both the start and end breakpoint is located within a segmental duplication. Consistent with the importance of NAHR in generating CNV risk alleles for schizophrenia, most of the loci in Table 1 are flanked by segmental duplications. Moreover, after excluding loci that have been implicated in previous studies, the remaining loci with FDR < 0.05 in the gene-base burden test were NAHR enriched (6.03-fold, P=0.008; Supplementary Figure 7), when compared to a null distribution determined by randomizing the genomic positions of associated genes (Supplemental Note). These findings suggest that the novel SCZ CNVs share similar characteristics to known pathogenic CNVs in that they tend to occur in regions prone to high rates of recurrent mutation.
Discussion
The present study of the PGC SCZ CNV dataset includes the majority of all microarray data that has been generated in genetic studies of SCZ to date. In this, we find definitive evidence for eight loci, surpassing strict genome-wide multiple testing correction. We also find evidence for a contribution of novel CNVs conferring either risk or protection to SCZ, with an FDR < 0.05. The complete results, including CNV calls and statistical evidence at the gene or breakpoint level, can be viewed using the PGC CNV browser (URLs). Our data suggest that the undiscovered novel risk loci that can be detected with current genotyping platforms lie at the ultra-rare end of the frequency spectrum and still larger samples will be needed to identify them at convincing levels of statistical evidence.
Collectively, the eight SCZ risk loci that surpass genome-wide significance are carried by a small fraction (1.4%) of SCZ cases in the PGC sample. We estimate 0.85% of the variance in SCZ liability is explained by carrying a CNV risk allele within these loci (Supplementary Note). As a comparison, 3.4% of the variance in SCZ liability is explained by the 108 genome-wide significant loci identified in the companion PGC GWAS analysis. Combined, the CNV and SNP loci that have been identified to date explain a small proportion (<5%) of heritability. The large dataset here provides an opportunity to evaluate the strength of evidence for a variety of loci where an association with SCZ has been reported previously. Of 44 published findings from the recent literature, we find evidence for 8 loci at a false discovery rate of 5% and nominal support for an additional 17 loci (uncorrected p < 0.05, Supplementary Table 6). Thus, nearly half of the existing candidate loci retain some support in our combined analysis. However we also find a lack of evidence for many of the previously identified loci, underscoring the value of meta-analytic efforts to assess the validity of such reports. A lack of strong evidence in this dataset (which includes samples that overlap with many of the previous studies) may in some cases simply reflect that statistical power is limited for very rare variants, even in large samples. However, it is likely that some of the earlier findings represent chance associations; indeed, the loci that are not supported by our data consist largely of loci for which the original statistical evidence was weak (Supplementary Table 6). Thus, our results help to refine the list of promising candidate CNVs. Continued efforts to evaluate the growing number of candidate variants has considerable value for directing future research efforts focused on specific loci.
The novel candidate loci meeting suggestive criteria in this study include two regions on chromosome X. It has been hypothesized that sex-linked loci contribute to SCZ, based originally on the observation of an increased rate of sex chromosome aneuploidy in cases 22. X-linked loci could not be detected in previous CNV studies of SCZ, because none to date evaluated variants on the sex chromosomes. In the current study, accurate calls were obtained by controlling for sex chromosome ploidy in the normalization and variant calling methods. Notably, duplications of distal Xq28 (regional P = 3.6×10−4, OR = 8.9, Table 1 and Supplementary Figure 8) appear to confer risk for SCZ in both males and females, and the effect size was greatest in males (P = 0.01, OR = ∞). Similar patterns consistent with dominant X-linked effects were observed at other loci (Supplementary Table 7). Duplications of distal Xq28 have been reported in association with developmental delay in both sexes 23, 24. Notably, of 26 subjects that have been described clinically, nearly half (12/26) have behavioral or psychiatric conditions. Of the five reciprocal deletions that were detected in this study, none were observed in males, consistent with hemizygous loss of distal Xq28 being associated with recessive embryonic lethality in males 24. Thus, mounting evidence indicates that increased copy number of distal Xq28 is associated with psychiatric illness. These results also provide a further demonstration that CNV risk factors in schizophrenia overlap with loci that contribute to pediatric developmental disorders 1, 25.
We observed multiple “protective” CNVs that showed a suggestive enrichment in controls, including duplications of 22q11.2, and MAGEA11 along with deletions and duplications of ZNF92. No protective effects were significant after genome-wide correction. Moreover, a rare CNV that confers reduced risk for SCZ may not confer a general protection from neurodevelopmental disorders. For example, microduplications of 22q11.2 appear to confer protection from SCZ 26; however, such duplications have been shown to increase risk for developmental delay and a variety of congenital anomalies in pediatric clinical populations 27. It is probable that some of the undiscovered rare alleles affecting risk for SCZ confer protection but larger sample sizes are needed to determine this unequivocally. If it is true that a proportion of CNVs observed in our control sample represent rare protective alleles, then the heritability of SCZ explained by CNVs may not be fully accounted for by the excess CNV burden in cases.
Our results provide strong evidence that deletions in SCZ are enriched within a highly connected network of synaptic proteins, consistent with previous studies 2, 6, 10, 28. The large CNV dataset here allows a more detailed view of the synaptic network and highlights subsets of genes account for the excess deletion burden in SCZ, including synaptic cell adhesion and scaffolding proteins, glutamatergic ionotropic receptors and protein complexes such as the ARC complex and DGC. Modest CNV evidence implicating Dystrophin (DMD) and its binding partners is intriguing given that the involvement of certain components of the DGC have been postulated 29, 30 and disputed 31 previously. Larger studies of CNV are needed to define a role for this and other synaptic sub-networks in SCZ.
Our current study is well-powered to detect CNVs of large effect that occur in >0.1% of cases, but is underpowered to detect association to variants with modest effect sizes or to ultra-rare variants regardless of effect size. Furthermore, this study did not assess the contribution of common CNVs to SCZ, one instance of which we know: a recent study has demonstrated that the causal variants underlying the strongest common variant association in SCZ include duplications of Complement factor 4A 35. Lastly, we recognize that a majority of structural variants (SVs) are not detectable with current genotyping platforms 32. New technologies for whole genome sequencing will ultimately provide an assessment of the contribution of a wider array of rare variants including balanced rearrangements, small CNVs 33 and short tandem repeats 34.
This study represents a milestone. Large-scale collaborations in psychiatric genetics have greatly advanced discovery through genome-wide association studies. Here we have extended this framework to rare CNVs. Our knowledge of the contribution from lower frequency variants gives us confidence that the application of this framework to large newly acquired datasets has the potential to further the discovery of loci and identification of the relevant genes and functional elements.
Methods
Overview
We assembled a CNV analysis group with the goal of leveraging the extensive expertise within the Psychiatric Genomics Consortium (PGC) to develop a fully automated centralized pipeline for consistent and systematic calling of CNVs for both Affymetrix and Illumina platforms. An overview of the analysis pipeline is shown in Supplementary Figure 1. After an initial data formatting step we constructed batches of samples for processing using four different methods, PennCNV, iPattern, C-score (GADA and HMMSeg) and Birdsuite for Affymetrix 6.0. For Affymetrix 5.0 data we used Birdsuite and PennCNV, for Affymetrix 500 we used PennCNV and C-score, and for all Illumina arrays we used PennCNV and iPattern. We then constructed a consensus CNV call dataset by merging data at the sample level and further filtered calls to make a final dataset Supplementary Table 2. Prior to any filtering, we processed raw genotype calls for a total of 57,577 individuals, including 28,684 SCZ cases and 28,893 controls.
Study Sample
A complete list of datasets that were included in the current study can be found in Supplementary Table 2. A more detailed description of the original studies can be found in a previous publication1
Copy Number Variant Analysis Pipeline Architecture and Sample Processing
All aspects of the CNV analysis pipeline were built on the Genetic Cluster Computer (GCC) in the Netherlands https://userinfo.surfsara.nl/systems/lisa.
Input Acceptance and Preprocessing
For Affymetrix we used the *.CEL files (all converted to the same format) as input, whereas for Illumina we required Genome or Beadstudio exported *.txt files with the following values: Sample ID, SNP Name, Chr, Position, Allele1 – Forward, Allele2 – Forward, X, Y, B Allele Freq and Log R Ratio. Samples were then partitioned into ‘batches’ to be run through each pipeline. For Affymetrix samples we created analysis batches based on the plate ID (if available) or genotyping date. Each batch had approximately 200 samples. Each batch included at least 50 subjects of each sex. Affymetrix Power Tools (APT - apt-copynumber-workflow) was then used to calculate summary statistics about chips analyzed. Gender mismatches identified and excluded as were experiments with MAPD > 0.4. For Illumina data, we first determined the genome build and converted to hg18 if necessary and created analysis batches based on the plate ID or genotyping date.
Composite Pipeline
The composite pipeline comprises CNV callers PennCNV 2, iPattern 3, Birdsuite 4 and C-Score 5 organized into component pipelines. We used all four callers for Affymetrix 6.0 data and we used PennCNV and C-Score for Affymetrix 500. Probe annotation files were preprocessed for each platform. Once the array design files and probe annotation files were pre-processed, each individual pipeline component pipeline was run in two steps: 1) processing the intensity data by the core pipeline process to produce CNV calls, 2) parsing the specific output format of the core pipeline and converting the calls to a standard form designed to capture confidence scores, copy number states and other information computed by each pipeline
Merging of CNV data and Quality control (QC) filtering
Merging of CNV data and Quality control (QC) filtering is described in detail in the supplementary material. Briefly, for each subject CNV calls were made using multiple algorithms. Only CNV calls that were made using multiple algorithms were included in the call set. Sample level QC filtering was performed by removing arrays with excessive probe variance or GC bias and removal of samples with mismatches in gender or ethnicity or chromosomal aneuploidies. The final filtered CNV dataset was annotated with Refseq genes (transcriptions and exons). After this stage of quality control (QC), we had a total of 52,511 individuals, with 27,034 SCZ cases and 25,448 controls. To make our final dataset of rare CNVs for all subsequent analysis we filtered out variants that were present at >= 1% (50% reciprocal overlap) frequency in cases and controls combined. We included in the call set CNVs that were ≥20 kb and ≥10 probes in length and overlapped < 50% with regions tagged as copy number polymorphic on any other platform.
In order to minimize the impact of technical artifacts and potential confounds on CNV association results, we removed from the dataset individuals that did not pass QC filtering from the companion PGC GWAS study of schizophrenia 1 as well as well as case or control samples that could not be matched by array platform or reconciled by using a common set of probes.
Statistics
Regression of potential confounds on case-control ascertainment
The PGC cohorts are a combination of many datasets drawn from the US and Europe, and it is important to ensure that any bias in sample ascertainment does not drive spurious association to SCZ. In order to ensure the robustness of the analysis, burden and gene-set analyses included potential confounding variables as covariates in a logistic regression framework. Due to the number of tests run at breakpoint level association, we employed a step-wise logistic regression approach to allow for the inclusion of covariates in our case-control association, which we term the SCZ residual phenotype.
Covariates included sex, genotyping platform, and ancestry principal components derived from SNP genotypes on the same samples in a previous study1. Control for population stratification is described in the supplementary material. We were unable to control for dataset or genotyping batch, as a subset of the contributing datasets are fully confounded with case/control status. Only principal components that showed a significant association to small CNV burden were used (small CNV being defined as autosomal CNV burden with CNV < 100 kb in size). Among the top 20 principal components, only the 1st, 2nd, 3rd, 4th, and 8th principal component showed association with small CNV burden (with p < 0.01 used as the significance cutoff).
Lastly, in order to control for case-control differences in CNV ascertainment due to data quality we sought to identify data quality metrics that were confounded with case status. Affymetrix (MAPD and waviness-sd) and Illumina (LRRSD, BAFSD, GCWF) QC metrics were re-examined across studies to assess if any additional outliers were present. Only three outliers were removed as their mean B allele (or minor allele) frequency deviated significantly from 0.5. Many CNV metrics are auto-correlated, as they measure similar patterns of variation in the probe intensity. Thus, we focused on the primary measure of probe variance – MAPD and LRRSD. Among Affymetrix 6.0 datasets, MAPD did not differ between in cases and controls (t=1.14, p = 0.25). However, among non-Affymetrix 6.0 datasets, LRRSD showed significant differences between cases and controls (t=−35.3, p < 2e−16), with controls having a higher standardized mean LRRSD (0.227) than cases (−0.199). Thus, to control for any spurious associations driven by CNV calling quality, we included MAPD (for Affymetrix platforms) or LRRSD (for Illumina platforms) as covariates in downstream analysis, which we designate as our “CNV metric” covariate for each individual. Prior to inclusion in the combined dataset, the CNV metric variable was normalized within each respective genotyping platform.
To calculate the SCZ residual phenotype, we first fit a logistic regression model of covariates to affection status, and then extracted the Pearson residual values for use in a quantitative association design for downstream analyses. Residual phenotype values in cases are all above zero, and controls below zero, and are graphed against overall kb burden in Supplementary Figure 9. We removed three individuals with an SCZ residual phenotype greater than three (or negative three in controls). After the post-processing round of QC, we retained a dataset with a total of 41,321 individuals comprising 21,094 SCZ cases and 20,227 controls.
CNV burden analysis
We analyzed the overall CNV burden in a variety of ways to discern which general properties of CNV are contributing to SCZ risk. Overall individual CNV burden was measured in 3 distinct ways – 1) Kb burden of CNVs, 2) Number of genes affected by CNVs, and 3) Number of CNVs. Genes were counted only if the CNV overlapped a coding exon. We also partitioned our analyses by CNV type, size, and frequency. CNV type is defined as copy number losses (or deletions), copy number gains (or duplications), and both copy number losses and gains. To assign a specific allele frequency to a CNV, we used the --cnv-freq-method2 command in PLINK, whereby the frequency is determined as the total number of CNV overlapping the target CNV segment by at least 50%. This method differs from other methods that assign CNV frequencies by genomic region, whereby a single CNV spanning multiple regions may be included in multiple frequency categories.
For Figure 1, and Supplementary Figures 2 and 3, we partitioned CNV burden by genotyping platform, and the abbreviations for each platform are expanded below:
A500: Affymetrix 500
I300: Illumina 300K
I600: Illumina 610K and Illumina 660W
A5.0: Affymetrix 5.0
A6.0: Affymetrix 6.0
omni: OmniExpress and OmniExpress plus Exome
Due to the small sample size of the Omni 2.5 array (28 cases and 10 controls), they were excluded from presentation in the figure, but are included in all burden analyses with the total PGC sample. Using a logistic regression framework with the inclusion of covariates detailed above, we predicted SCZ status using CNV burden as an independent predictor variable, thus allowing us to get an accurate estimate of the contribution of CNV burden. In addition, to determine the proportion of CNV burden risk that is attributable to loci that have not been implicated in previous studies of SCZ, we ran all burden analyses after removing CNVs that overlapped previously implicated CNV boundaries by more than 10%.
CNV breakpoint level association
Association was tested at each respective CNV breakpoint. Three categories of CNV were tested: deletions, duplications, and deletions and duplications combined. All analyses were run using PLINK6.
We ran breakpoint level association using the SCZ residual phenotype as a quantitative variable, with significance determined through permutation of phenotype residual labels. An additional z-scoring correction, explained below, is used to control for any extreme values in the SCZ residual phenotype and efficiently estimate two-sided empirical p-values for highly significant loci. To ensure against the potential loss of power from the inclusion of covariates, we also ran a single degree of freedom Cochran-Mantel-Haenzel (CMH) test stratified by genotyping platform, with a 2 (CNV carrier status) × 2 (phenotype status) × N (genotyping platform) contingency matrix. While the CMH test does not account for more subtle biases that could drive false positive signals, it is robust to signals driven by a single platform and allows for each CNV carrier to be treated equally. Loci the surpassed genome-wide correction in either test was followed up for further evaluation.
Z-score recalibration of empirical testing
Breakpoint level association p-values from the SCZ residual phenotype were initially obtained by performing one million permutations at each CNV position, whereby each permutation shuffles the SCZ residual phenotype among all samples, and retains the SCZ residual mean for CNV carriers and non-carriers. For extremely rare CNV, however, CNV carriers at the extreme ends of the SCZ residual phenotype can produce highly significant p-values. While we understand that such rare events are unable to surpass strict genome-wide correction, we wanted to retain all tests to help delineate the potential fine-scale architecture within a single region of association. To properly account for the increased variance when only a few individuals are tested, we applied an empirical Z-score correction to the CNV carrier mean. In order to get an empirical estimate of the variance for each test, we calculated the standard deviation of residual phenotype mean differences in CNV carriers and non-carriers from 5,000 permutations. Z-scores are calculated as the observed case-control mean difference divided by the empirical standard deviation, with corresponding p-values calculated from the standard normal distribution. Concordance of the initial empirical and Z-score p-values are close to unity for association tests with six or more CNV, whereas Z-score p-values are more conservative among tests with less than six CNV. Furthermore, the Z-score method naturally provides an efficient manner to estimate highly significant empirical p-values that would involve hundreds of millions of permutations to achieve. Genome-wide correction for multiple testing was determined as described in the Supplementary Note
Gene-set burden enrichment analysis: gene-sets
Gene-sets with an a priori expectation of association to neuropsychiatric disorders were compiled and CNV calls were preprocessed as described in the supplementary material.
For each gene-set, we fit the following logistic regression model (as implemented by the R function glm of the stats package), where subjects are statistical sampling units:
y ~ covariates + global + gene-set
Where:
y is the dicotomic outcome variable (schizophrenia = 1, control = 0)
covariates is the set of variables used as covariates also in the genome-wide burden and breakpoint association analysis (sex, genotyping platform, CNV metric, and CNV associated principal components)
global is the measure of global genic CNV burden. This covariate accounts for non-specific association signal that could be merely reflective of an overall difference CNV burden between cases and controls. For the results in the main text, we used the total gene number (abbreviated as U from universe gene-set count); we also calculated results for total length (abbreviated as TL) and variant number plus variant mean length (abbreviated as CNML)
gene-set is the gene-set gene count
The gene-set burden enrichment was assessed by performing a chi-square deviance test (as implemented by the R function anova.glm of the stats package) comparing these two regression models:
y ~ covariates + global
y ~ covariates + global + gene-set
We reported the following statistics:
coefficient beta estimate (abbreviated as Coeff)
t-student distribution-based coefficient significance p-value (as implemented by the R function summary.glm of the stats package, abbreviated as Pvalue_glm)
deviance test p-value (abbreviated as Pvalue_dev)
gene-set size (i.e. number of genes is the gene-set, regardless of CNV data)
BH-FDR (Benjamini-Hochberg False Discovery rate)
percentage of schizophrenia and control subjects with at least 1 gene, 2 genes, etc… impacted by a CNV of the desired type (loss or gain) in the gene-set (abbreviated as SZ_g1n, SZ_g2n, … CT_g1n, …)
Please note that, by performing simple simulation analyses, we realized that Pvalue_glm can be extremely over-conservative in presence of very few gene-set counts different than 0, while Pvalue_dev tends to be slightly under-conservative. While the two p-values tend to agree well for gene-set analysis, Pvalue_glm is systematically over-conservative for gene analysis since smaller counts are typically available for single genes.
Gene association analysis
Subjects were restricted to the ones with at least one rare CNV. Only genes with at least a minimum number of subjects impacted by CNV were tested; this threshold was picked by comparing the BH-FDR to the permutation-based FDR and ensuring limited FDR inflation (permuted FDR < 1.65 * BH-FDR at BH-FDR threshold = 5%) while maximizing power. For gains the threshold was set to 12 counts, while for losses it was set to 8 counts.
For each gene, we fit the following logistic regression model (as implemented by the R function glm of the stats package), where subjects are statistical sampling units:
y ~ covariates + gene
Where:
y is the dichotomous outcome variable (schizophrenia = 1, control = 0)
covariates is the set of variables used as covariates also in the genome-wide burden and breakpoint association analysis (sex, genotyping platform, CNV metric, and CNV associated principal components)
gene is the binary indicator for the subject having or not having a CNV of the desired type (loss or gain) mapped to the gene
The gene burden was assessed by performing a chi-square deviance test (as implemented by the R function anova.glm of the stats package) comparing these two regression models:
y ~ covariates
y ~ covariates + gene
Genome wide correction for multiple testing was determined as described in the supplementary material.
Experimental Validation of CNV calls by digital droplet PCR
For 6 novel candidate loci that were identified in this study, we sought to confirm CNV calling accuracy by experimental validation of CNV calls in a subset of study samples. Within each association peak we a defined a segment was defined that overlapped a majority of calls. Appropriate digital droplet assays were then selected from the BioRad catalog. A single FAM-labeled probe was designed for DMRT1, ZMYM5, ZNF92, MAGEA11 and Distal Xq28. Because some deletions of the VPS13B gene were non-overlapping, two different probes were selected for this locus. CNV calls (up to a maximum of 17) were selected from the core target region. Probe details, CNV calls and validation results can be found in Supplementary Table 5. Study samples were then obtained from two studies (Sweden and CLOZUK) and 4 population control samples were obtained from Coriell Cell repositories (ND00745, ND01936, ND00689, ND01317) to be used as negative controls for ddPCR assays. EcoRI digested samples (10 ng of genomic DNA) were analyzed in triplicate by ddPCR using the Fam-labeled CNV probe and HEX-labeled reference probe M0005 RPP30-HEX (Supplementary Table 5) in the UCSD CFAR Genomics & Sequencing Core. PCR droplets were generated using a Bio-Rad QX100 Droplet Generator, then quantitative PCR was performed using the GeneAmp PCR system 9700 (Applied Biosystems) instrument according to manufacturer’s protocols (40 cycles at 94°C for 30 sec and 60°C for 1 min). PCR droplets were read & analyzed on Bio-Rad QX100 Droplet Reader with QuantaSoft software.
Supplementary Material
Acknowledgments
Core funding for the Psychiatric Genomics Consortium is from the US National Institute of Mental Health (U01 MH094421). We thank Thomas Lehner, Anjene Addington and Geetha Senthil (NIMH). The work of the contributing groups was supported by numerous grants from governmental and charitable bodies as well as philanthropic donation. Details are provided in the Supplementary Notes. Membership of the Wellcome Trust Case Control Consortium and Psychosis Endophenotype International Consortium are provided in the Supplementary Notes.
Footnotes
URLs
PGC CNV browser, http://pgc.tcag.ca/gb2/gbrowse/pgc_hg18.
Visualization 16p13.2: http://bit.ly/1NPgIuq
Visualization of 8q11.23 locus: http://bit.ly/1PwdYTt
Xq28 gene reviews: http://bit.ly/2au9QGb
Genetic Cluster Computer (GCC): https://userinfo.surfsara.nl/systems/lisa
Data Availability
The PGC CNV resource is now publicly available through a custom browser at http://pgc.tcag.ca/gb2/gbrowse/pgc_hg18/ and the rare CNV call set can be obtained from the European Genome-Phenome Archive (Study accession #EGAS00001001960).
Author Contributions
Management of the study, core analyses and content of the manuscript was the responsibility of the CNV Analysis Group chaired by J.S. and jointly supervised by S.W.S. and B.M.N. together with the Schizophrenia Working Group chaired by M.C.O’D. Core analyses were carried out by D.P.H., D.M., and C.R.M. Data Processing pipeline was implemented by C.R.M., B.T., W.W., D.G., M.G., A.S. and W.B. The A custom PGC CNV browser was developed by C.R.M, D.P.H., and B.T. Additional analyses and interpretations were contributed by W.W., D.A. and P.A.H. The individual studies or consortia contributing to the CNV meta-analysis were led by R.A.,O.A.A., D.H.R.B., A.D.B., E. Bramon, J.D.B., A.C., D.A.C., S.C., A.D., E. Domenici, H.E., T.E., P.V.G., M.G., H.G., C.M.H., N.I., A.V.J., E.G.J., K.S.K., G.K., J. Knight, T. Lencz, D.F.L., Q.S.L., J. Liu, A.K.M., S.A.M., A. McQuillin, J.L.M., P.B.M., B.J.M., M.M.N., M.C.O’D., R.A.O., M.J.O., A. Palotie, C.N.P., T.L.P., M.R., B.P.R., D.R., P.C.S, P. Sklar. D.St.C., P.F.S., D.R.W., J.R.W., J.T.R.W. and T.W. The remaining authors contributed to the recruitment, genotyping, or data processing for the contributing components of the meta-analysis. J.S., B.M.N, M.C.O’D, C.R.M, D.P.H., and D.M. drafted the manuscript, which was shaped by the management group. All other authors saw, had the opportunity to comment on, and approved the final draft.
Competing Financial Interest
J.S. is a co-inventor on patents granted (8554488) and pending (20140171371) on genetic methods for the diagnosis of psychiatric disorders. Several of the authors are employees of the following pharmaceutical companies: F. Hoffman-La Roche (E.D., L.E.), Eli Lilly (D.A.C., Y.M., L.N.) and Janssen (A.S., Q.S.L). None of these companies influenced the design of the study, the interpretation of the data, the amount of data reported, or financially profit by publication of the results, which are pre-competitive. The other authors declare no competing interests.
References
- 1.Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–41. doi: 10.1016/j.cell.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–43. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 3.The International Schizophrenia, C. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malhotra D, et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron. 2011;72:951–63. doi: 10.1016/j.neuron.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–5. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 6.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular psychiatry. 2012;17:142–53. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCarthy SE, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–7. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulle JG, et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am J Hum Genet. 2010;87:229–36. doi: 10.1016/j.ajhg.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rujescu D, et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pocklington AJ, et al. Novel Findings from CNVs Implicate Inhibitory and Excitatory Signaling Complexes in Schizophrenia. Neuron. 2015;86:1203–14. doi: 10.1016/j.neuron.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horev G, et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A. 2011;108:17076–81. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Golzio C, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–7. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes AJ, et al. Individual differences in amygdala-medial prefrontal anatomy link negative affect, impaired social functioning, and polygenic depression risk. J Neurosci. 2012;32:18087–100. doi: 10.1523/JNEUROSCI.2531-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang K, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–74. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat.Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vacic V, et al. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raychaudhuri S, et al. Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirov G, et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet. 2008;17:458–65. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 21.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–22. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 22.DeLisi LE, et al. Schizophrenia and sex chromosome anomalies. Schizophr Bull. 1994;20:495–505. doi: 10.1093/schbul/20.3.495. [DOI] [PubMed] [Google Scholar]
- 23.El-Hattab AW, et al. Int22h-1/int22h-2-mediated Xq28 rearrangements: intellectual disability associated with duplications and in utero male lethality with deletions. J Med Genet. 2011;48:840–50. doi: 10.1136/jmedgenet-2011-100125. [DOI] [PubMed] [Google Scholar]
- 24.El-Hattab AW, et al. Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: new cases and literature review. BMC Med Genet. 2015;16:12. doi: 10.1186/s12881-015-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25:528–35. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rees E, et al. Evidence that duplications of 22q11.2 protect against schizophrenia. Mol Psychiatry. 2014;19:37–40. doi: 10.1038/mp.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Campenhout S, et al. Microduplication 22q11.2: a description of the clinical, developmental and behavioral characteristics during childhood. Genet Couns. 2012;23:135–48. [PubMed] [Google Scholar]
- 28.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–84. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zatz M, et al. Cosegregation of schizophrenia with Becker muscular dystrophy: susceptibility locus for schizophrenia at Xp21 or an effect of the dystrophin gene in the brain? J Med Genet. 1993;30:131–4. doi: 10.1136/jmg.30.2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Straub RE, et al. Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet. 2002;71:337–48. doi: 10.1086/341750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mutsuddi M, et al. Analysis of high-resolution HapMap of DTNBP1 (Dysbindin) suggests no consistency between reported common variant associations and schizophrenia. Am J Hum Genet. 2006;79:903–9. doi: 10.1086/508942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sudmant PH, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi: 10.1038/nature15394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brandler WM, et al. Frequency and Complexity of De Novo Structural Mutation in Autism. Am J Hum Genet. 2016;98:667–79. doi: 10.1016/j.ajhg.2016.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gymrek M, et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat Genet. 2016;48:22–9. doi: 10.1038/ng.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekar A, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zuberi K, et al. GeneMANIA prediction server 2013 update. Nucleic Acids Res. 2013;41:W115–22. doi: 10.1093/nar/gkt533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–74. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korn JM, et al. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat.Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarthy SE, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41:1223–7. doi: 10.1038/ng.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Purcell S, et al. PLINK: a toolset for whole-genome association and population-based linkage analysis. American Journal of Human Genetics. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.