

Abstract
Radiologic imaging of disease sites plays a pivotal role in the management of patients with cancer. Response Evaluation Criteria in Solid Tumours (RECIST), introduced in 2000, and modified in 2009, has become the de facto standard for assessment of response in solid tumours in patients on clinical trials.
The RECIST Working Group considers the ability of the global oncology community to implement and adopt updates to RECIST in a timely manner to be critical. Updates to RECIST must be tested, validated and implemented in a standardised, methodical manner in response to therapeutic and imaging technology advances as well as experience gained by users. This was the case with the development of RECIST 1.1, where an expanded data warehouse was developed to test and validate modifications. Similar initiatives are ongoing, testing RECIST in the evaluation of response to non-cytotoxic agents, immunotherapies, as well as in specific diseases.
The RECIST Working Group has previously outlined the level of evidence considered necessary to formally and fully validate new imaging markers as an appropriate end-point for clinical trials. Achieving the optimal level of evidence desired is a difficult feat for phase III trials; this involves a meta-analysis of multiple prospective, randomised multicentre clinical trials. The rationale for modifications should also be considered; the modifications may be proposed to improve surrogacy, to provide a more mechanistic imaging technique, or be designed to improve reproducibility of the imaging biomarker.
Here, we present the commonly described modifications of RECIST, each of which is associated with different levels of evidence and validation.
Keywords: RECIST, Modifications, Tumour response
1. Introduction
Radiologic imaging of disease sites plays a pivotal role in the management of patients with cancer. It has been used to estimate disease burden to plan optimal treatment, as well as to evaluate response to treatment to inform medical decisions such as discontinuation and or switching to other therapies. These same clinical tools have been used to measure end-points in cancer drug trials to quantify efficacy in candidate compounds. This quantification assists in the decisions of moving an agent from phase I toward registration. The value of imaging in early clinical trials of potential new cancer therapeutics rests on the validation and reproducibility of the imaging biomarkers to serially and non-invasively assess the extent of tumour change during therapy as surrogates of the previously accepted gold standards of patient benefit of prolongation of survival or improvement in quality of life.
Response Evaluation Criteria in Solid Tumours (RECIST), introduced in 2000, and modified in 2009, has become the de facto standard for assessment of response in solid tumours. The standardisation and ubiquitous use of RECIST allows for easy historical comparisons, especially in phase II trials. While RECIST has served a critical role in drug development, the emergence of new classes of drugs, new treatment paradigms and new imaging modalities and techniques requires continued re-evaluation of RECIST as a response assessment tool.
The RECIST Working Group considers the ability of the global oncology community to implement and adopt any updates to RECIST in a timely manner to be critical. Updates to RECIST must be tested, validated and implemented in a standardised, methodical manner in response to therapeutic and imaging technology advances as well as experience gained by users. This was the case with the development of RECIST 1.1, where an expanded data warehouse was developed to test and validate modifications to the criteria. Similar initiatives are ongoing, testing RECIST in the evaluation of response to non-cytotoxic agents, immunotherapies, as well as specific diseases.
The RECIST Working Group has previously outlined the level of evidence considered necessary to formally and fully validate new imaging markers as an appropriate end-point for clinical trials [1]. The ideal level of evidence would require a series of prospective, randomised multicentre clinical trials, which is not feasible. The rationale for modifications should also be considered; the modifications may be proposed to improve surrogacy, to provide a more mechanistic imaging technique, or be designed to improve reproduc-ibility of the imaging biomarker.
As reported by the RECIST survey in 2014, 60% of the responders also used other criteria or modified RECIST criteria, when RECIST was not applicable in certain disease types or novel therapies were involved [2]. In this paper, we summarise several of the commonly described modifications of RECIST, each of which is associated with different levels of evidence and validation. We used the Oxford Centre for Evidence-Based Medicine approach for categorising levels of evidence (level 1 consisting of systematic reviews of randomised clinical trials or individual clinical trials with narrow confidence intervals. Level 2 in general consists of systematic reviews of cohort studies, lower quality randomised trials and outcomes type research, while level 3 is case control type studies and level 4 is case series. Level 5 is expert opinion and consensus statements) [3].
In addition to being a useful summary of this field, the summary will assist the RECIST Working Group decisions regarding which future techniques and modifications may be relevant to incorporate into RECIST after appropriate validation.
2. Disease and therapy-specific modifications to RECIST
2.1. Prostate cancer
In 2008, the Prostate Cancer Working Group (PCWG2) outlined principles for trial eligibility, design, and conduct in progressive metastatic castration resistant prostate cancer (CRPC) [4]. PCWG2 built upon the standards for baseline evaluations recommended by RECIST and provides guidelines for imaging and symptom assessment specific to prostate cancer. The requirement for target lesions for trial entry, as defined by RECIST, was not recommended by PCWG2 because it would shift the focus of assessment from bone metastases (the most common and relevant disease site) to other disease sites (such as lymph nodes). Bone metastases develop in approximately 90% of patients, while lymph node metastases occur in only 20% of patients and more important cause less morbidity than bone metastases. PCWG2 proposed a composite end-point, including PSA, focused on progression. A major modification of the RECIST criteria in PCWG2 is to account for the healing of lesions on Tc-99m MDP bone scan, causing an osteoblastic response that may falsely appear as new lesions, known as flare phenomena (Fig. 1) [5].
Fig. 1.
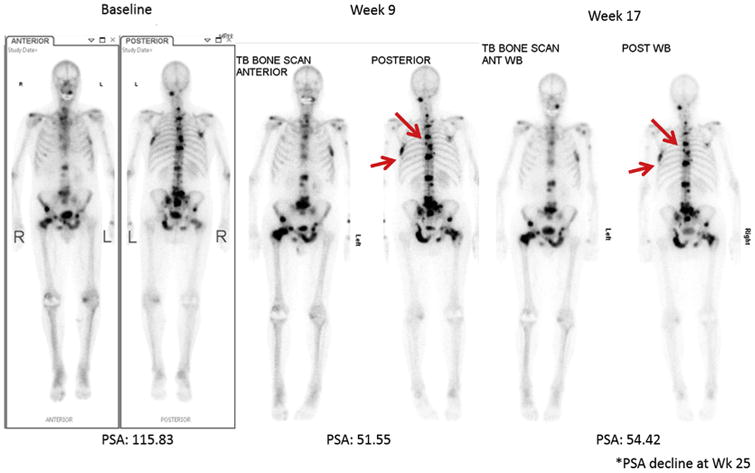
Tc-99m MDP bone scan in a patient with metastatic prostate cancer. The patient with >20 bone lesions at baseline scan. At the week 9 visit, patient presented with two new bone lesions. At week 17 and 25, patient did not have new lesions compared to the week 9 bone scan. Image courtesy of Michael Morris, MD, Memorial Sloan Kettering Cancer Center.
To validate time to radiographic progression criteria, the PCWG2 criteria have been incorporated into three prospective randomised placebo-controlled studies of metastatic CRPC. One of these tested abiraterone plus prednisone compared to placebo plus prednisone in 1088 patients. Radiographic progression-free survival (rPFS) (using PCWG2 criteria) was the co-primary end-point, with overall survival. Patients in the abiraterone arm had a 25% reduction in the risk of death (hazard ratio [HR] 0.75, P = 0.0097) and a 57% relative improvement in rPFS (HR 0.43, P < 0.0001) [6]. Together with the other two prospective clinical trials [7,8], these modifications would support a level of evidence of Ib.
Recently, updates to PCWG2 have been introduced as PCWG3 to update the validation of measurements of tumour burden in prostate cancer, both in bone and soft tissue [9].
2.2. Mesothelioma
Malignant pleural mesothelioma (MPM) grows in a pattern more consistent with a complex rind rather than a sphere (Fig. 2). As a result, uni- or bidimensional measurement tools and response criteria have long been acknowledged as being poorly suited to assess response in MPM. Recognising that the major problem in using RECIST in MPM is the reproducible placement of the ‘longest unidimensional diameter’ of the target tumour mass, a modification to RECIST incorporating consistent anatomic landmarks has been proposed [10]. Tumour thickness perpendicular to the chest wall or mediastinum is measured in two positions at three separate levels on chest computed tomography (CT) scans. Rather than ‘increase’ the number of target lesions per organ, the measurements are consolidated into a single sum of the pleural disease. With these modifications, the RECIST criteria correlated with both survival and lung function in small studies [10]. These modifications support a level of evidence of IIb.
Fig. 2.
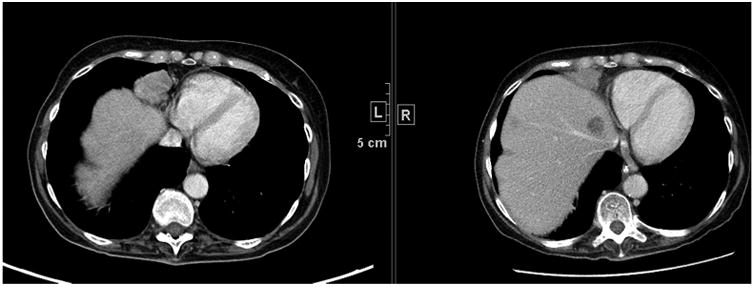
Computed tomography scan in a patient with malignant pleural mesothelioma in the right lung at baseline and follow-up. Note the difficulty in finding a unidimensional measurement that captures the change in tumour size from baseline to follow-up.
2.3. Lymphoma
In 1999, the National Cancer Institute (NCI) Working Group published the first widely accepted response assessment criteria for non-Hodgkin lymphoma, but used as well for Hodgkin lymphoma [11]. These criteria were revised in 2007 by the International Working Group (IWG) to incorporate fluorodeoxyglucose positron emission tomography (FDG-PET), bone marrow immunohistochemistry and flow cytometry in response assessment [12].
FDG-PET has become a well utilised standard for staging, restaging, and response assessment in lymphomas. Compared to conventional anatomic imaging techniques such as CT, FDG-PET can differentiate viable tumour from necrosis or fibrosis in remaining soft tissue or lymph node masses. There are a number of technical considerations for both the acquisition and interpretation of PET scans. FDG-PET is associated with false-positive findings due to infection, inflammation, sarcoidosis, and the misinterpretation of entities such as brown fat. Furthermore, not all lymphoma subtypes are FDG avid and variability exists in FDG avidity among histologic subtypes of lymphoma. Despite these limitations, FDG-PET has become the clinical imaging standard for assessment of lymphoma. Most studies do not find value in routine surveillance with FDG-PET scans.
The incorporation of FDG-PET and anatomic CT imaging (Fig. 3a and b) in the IWG criteria maintains the 4-point categorical responses of complete response, partial response, stable disease and progressive disease (PD). In addition to tumour measurements on CT and assessment of FDG uptake on PET, evaluation of the spleen and liver size as well as bone marrow is necessary to accurately categorise a patients' response at each time point.
Fig. 3.
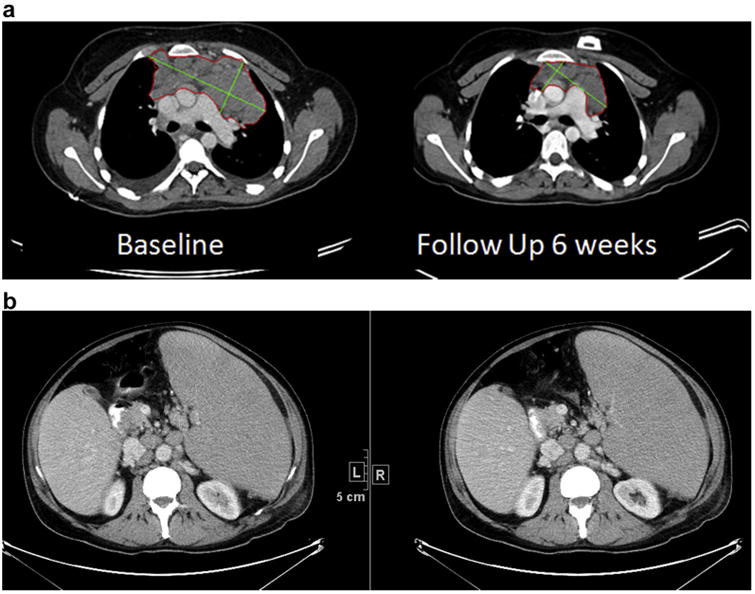
a. Computed tomography (CT) scan in a patient with diagnosis tumour type confluent mediastinal lymph node mass measured bidimensionally at baseline and 6-week follow-up. b. CT scan in a patient with massive splenomegally and multiple pathologically enlarged retroperitoneal lymph nodes.
Further updates to these response criteria were presented at the 4th International Workshop on PET in Lymphoma in Menton, France, and the 12th International Congress on Malignant Lymphonea (ICML) in Lugano, Switzerland, to update the International Harmonisation Project guidance regarding PET [13]. These updates recommended the use of FDG-PET/CT in PET avid lymphomas and grading the uptake in a 5-point scale which has been shown to be robust in multi-centre trials [14]. Mid-cycle PET/CT was also recommended over PET alone. The group further outlined other elements, including residual mass evaluation, volumetric measurements, and PET/CT in certain subtypes of lymphoma as areas warranting further evaluation [15]. In summary, these modifications would support a level of evidence of IIa.
2.4. Immune-related response criteria
Response patterns to immunotherapies may include an initial increase in tumour burden or the appearance of new lesions prior to subsequent reduction in tumour size followed by regression or long-standing or durable stable disease [16]. While this pattern was first described in melanoma with CTLA-4 blockade, it has been reported in other diseases and with other immunoncology agents (Fig. 4). The apparent increase in size of detectable lesions or development of new lesions may be due to T-cell infiltration tumour deposits, and at least some patients with this ‘pseudo-progression’ go on to durable complete or partial responses. Following initial observations in clinical trials with ipilimumab, modifications to World Health Organisation (WHO) response assessment criteria, which are based upon bidimensional tumour measurements, were proposed [17].
Fig. 4.
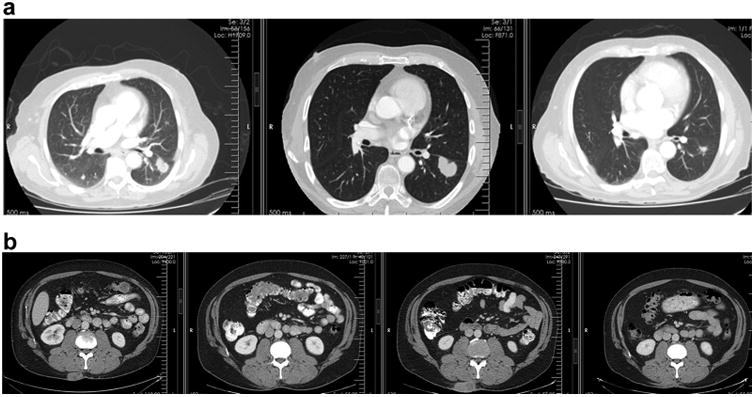
a. Computed tomography (CT) scan of patient with non-small-cell lung cancer and evidence of pseudo-progression at the first follow-up time point with subsequent near resolution of the mass. b. CT scan with a subcutaneous metastasis in the right paraspinal region. There is transient increase in size at the second follow-up time point indicative of pseudo-progression, which subsequently resolves.
The principal modification to the criteria is how ‘new’ lesions are assessed. At the baseline tumour assessment, the sum of the products of the two largest perpendicular diameters (SPD) of all index lesions (five lesions per organ, up to ten visceral lesions and five cutaneous index lesions) is calculated. At each subsequent tumour assessment, the SPD of the index lesions and of any new, measurable lesions (≥5 × 5 mm; up to five new lesions per organ: five new cutaneous lesions and ten visceral lesions) are added together to provide the total tumour burden. Declaration of PD requires at least 25% increase in tumour burden in two consecutive observations at least 4 weeks apart compared with nadir (at any single time point). Four response patterns were discriminated, including response after an increase in total tumour burden and response in the presence of new lesions. All patterns were associated with favourable trends toward longer overall survival. In clinical trials, this has allowed improved characterisation of a small but significant group of patients who would have been characterised as experiencing PD by WHO criteria but actually had clinical benefit. These patients were characterised as responders by immune response criteria [17]. These modifications are supported by a level of evidence of Ib.
The best method to determine treatment response for immune modulating agents is still unknown. Often, both the RECIST 1.1 criteria or immune-related response criteria (irRC) are used in the same trial, or a modified RECIST 1.1 is used, incorporating the principles of irRC. The RECIST Working Group and authors of the irRC are collaborating to harmonise and validate the criteria so that clinical trials involving both immune therapy and other therapies may be evaluated uniformly and easily.
2.5. Response assessment in neuro-oncology of high-grade gliomas
Over the past several years, the response assessment in neuro-oncology (RANO) Working Group has proposed new criteria for the assessment of high-grade glioma to replace the older Macdonald criteria [18]. Macdonald criteria use bidimensional tumour measurements of the enhancing component of brain tumours (Fig. 5), but there has been recognition that changes in contrast enhancement may not be indicative of tumour response. For example, patients receiving anti-angiogenic treatment may have a decrease in contrast enhancement of the tumour due to changes in vascular permeability rather than to antitumour effect. The RANO Working Group defined measurable and non-measurable disease as well as criteria for determining first progression after initial chemoradiotherapy due to the difficulty of differentiating pseudo-progression from actual tumour progression. Definitions of response were also modified to account for non-enhancing tumour by utilising T2-weighted and fluid-attenuated inversion recovery magnetic resonance imaging (MRI). The use of the RANO criteria for high-grade glioma in subsequent trials would support a level of evidence of IIa [19].
Fig. 5.
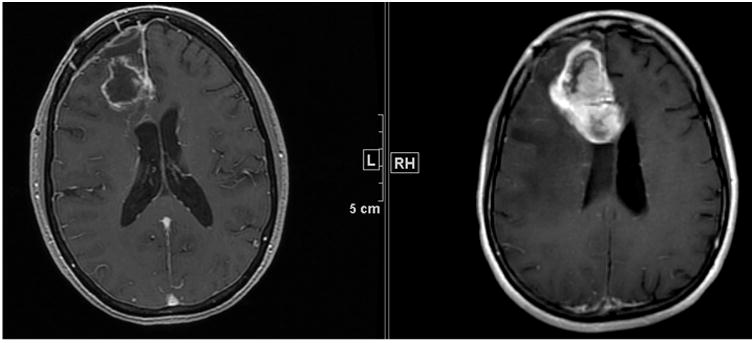
Post-contrast T1-weighted MRI in a patient with a glioma – note the degree of enhancement has increased greatly over the 6-week interval between the two scans.
2.6. Response assessment in neuro-oncology of brain metastases
The response assessment in neuro-oncology brain metastases (RANO-BM) Working Group is an international, multidisciplinary effort to develop standard response and progression criteria for use in clinical trials of treatment for brain metastases. They recently published their recommendations for standard response and progression criteria for the assessment of brain metas-tases in clinical trials [20]. The use of the RANO-BM criteria in subsequent trials would support a level of evidence of V.
2.7. Modified RECIST for hepatocellular carcinoma
The European Association for the Study of the Liver (EASL) proposed amending the conventional response criteria for hepatocellular carcinoma (HCC) to incorporate visible tumour necrosis commonly reported after loco-regional therapy of HCC and with some targeted drugs [21]. To incorporate tumour necrosis, the residual viable tumour must be quantified, and was defined as uptake of contrast agent in the arterial phase of dynamic CT or MRI (Fig. 6). The concept of viable tumour proposed by the EASL panel has also been sanctioned by the American Association for the Study of Liver Diseases (AASLD). The AASLD practice guideline on the management of HCC issued in 2005 stated that the evaluation of the treatment response should ‘take into account the induction of intratumoural necrotic areas in estimating the decrease in tumour load, and not just an overall reduction in tumour size’ [22]. Special considerations in HCC response assessments have also been proposed on the basis of HCC patterns of tumour spread, including portal vein thrombosis, porta hepatis lymph nodes and pleural effusion and ascites. Similar to the immune therapy modification of new disease, a newly detected hepatic nodule is a diagnostic dilemma in HCC response assessment, as this nodule may represent HCC or a non-malignant entity such as a dysplastic nodule or perfusion abnormality, and must be either strongly indicative of HCC or proven to be HCC by biopsy. These modifications would support a level of evidence of IV.
Fig. 6.
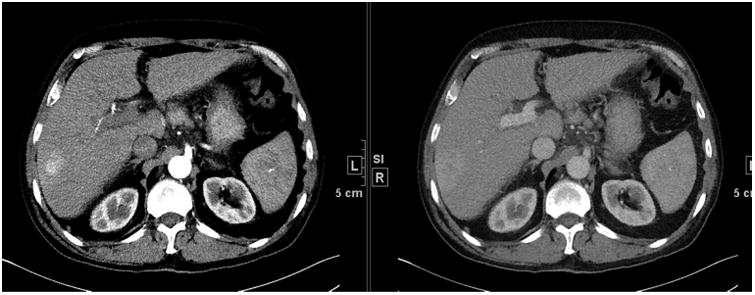
Small foci of hepatocellular carcinoma in the right lobe of the liver on the arterial phase and delayed portal venous phase. Note the potential difference in measurement between the phases.
3. Conclusions
As previously noted, the level of evidence required to validate a new imaging biomarker as an end-point for a specific disease or in phase II trials is considerable, and is usually not accomplished. In several of the examples above, validation could only be achieved by evaluating retrospectively clinical trials conducted with novel therapeutics or in specific diseases. Given the increasing diversity of therapeutic agents, the increasing diversity of mechanisms of actions of these agents and the potential for combination therapies, the need for disease specific modifications of RECIST will continue to expand. However, the need for a common approach and set of rules for response assessment, to be able to compare novel therapies with historical controls, is paramount. Perhaps, equally important is the need to better define disease-specific states of response and progression. Careful attention to both the presumed mechanism of drug action and reproducibility of the biomarker, in this case imaging, is critical.
The RECIST Working Group maintains an active website sponsored by the European Organisation for Research and Treatment of Cancer (EORTC) and is located at http://www.eortc.org/recist/. There are a number of active projects underway. Among the most advanced project is the collection of individual participant data from clinical trials with targeted agents, approximately 50 clinical trials from academic partners (EORTC, CCTG [formerly NCIC CTG], Eastern Cooperative Oncology Group, SWOG, Dutch Colorectal Cancer Group) as well as partners from industry (Amgen, Pfizer, Genentech/Roche, Astra Zeneca, GSK, Sanofi and Merck Serono). The mode of action of targeted agents is often perceived to be different from that of cytotoxic agents and raises questions as to whether a modification to the response criteria is required to robustly evaluate the activity of targeted therapies.
The RECIST Working Group supports the evaluation and validation of novel biomarkers in cancer therapy and will continue to work to incorporate these criteria into RECIST as the techniques and therapeutics become widely used and globally available.
Acknowledgments
Canadian Cancer Trials Group participation was supported by the Canadian Cancer Society Research Institute (grant #021039). This publication was supported by the EORTC Cancer Research Fund and by the National Cancer Institute grant number 5U10-CA11488-45.
E. de Vries: Research grants from Roche/Genentech, Amgen, Novartis, Pieris and Servier to the institute, data monitoring committee Biomarin, advisory board Synthon. R. Ford: Dr. Ford is not a stockholder in any pharmaceutical company and holds no options, grants or patents. He is currently or was previously a consultant for the following companies (either directly through Clinical Trials Imaging Consulting, LLC., or indirectly through other pharmaceutical service companies for which he consults) including ACR Image Metrix, AbbVie, Amgen, Aptiv, Aragon, Bioclinica, Biomedical Systems, Bristol Myers Squibb, Celldex, Celgene, Celsion, Covance, DNAtrix, Eisai, Exelixis, Genentech/Roche, ICON Medical Imaging, Image Endpoints, Janssen, Kyowa, Merck, Mirati Therapeutics, Novartis, Novocure, Oncothyreon, Ono, Optimer, OrbiMed Advisors, Pfizer, Radiant Sage, Quintiles,Red Hill Pharma, Sun Advanced Pharma Research Company, Tokai Pharmaceuticals, Vascular Biogenics and Virtualscopics. S. Hodi: Consultant for Merck, Novartis, Genentech, Synta; research support to institution from Bristol-Myers Squibb. N. Lin: Research funding for clinical trials from (no direct salary support): Genentech, GSK, Novartis, Synta, Array Biopharma, Kadmon, and Puma. J.D. Wolchok: Research grants from Bristol-Myers Squibb; advisory board member for Brisol-Myers Squibb, Merck, Medimmune, Genentech. LH Schwartz: Research grants from Novartis, Astellas, Eli Lilly, Merck, Pfizer, Consultant to GSK, Novartis.
Footnotes
Conflict of interest statement: All authors have no conflicts relevant to the study.
References
- 1.Sargent DJ, Rubinstein L, Schwartz L, Dancey JE, Gatsonis C, Dodd LE, et al. Validation of novel imaging methodologies for use as cancer clinical trial end-points. Eur J Cancer. 2009;45:290–9. doi: 10.1016/j.ejca.2008.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Y, Litière S, de Vries EG, Sargent D, Shankar L, Bogaerts J, et al. The role of response evaluation criteria in solid tumour in anticancer treatment evaluation: results of a survey in the oncology community. Eur J Cancer. 2014;50:260–6. doi: 10.1016/j.ejca.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 3.Oxford Centre for Evidence-based Medicine – Levels of evidence (March 2009) [accessed 14.03.16]; http://www.cebm.net/oxford-centre-evidence-based-medicine-levels-evidence-march-2009/
- 4.Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26:1148–59. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollen JJ, Witztum KF, Ashburn WL. The flare phenomenon on radionuclide bone scan in metastatic prostate cancer. AJR Am J Roentgenol. 1984;142:773–6. doi: 10.2214/ajr.142.4.773. [DOI] [PubMed] [Google Scholar]
- 6.Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 8.Parker C, Nilsson S, Heinrich D, Helle SI, O'Sullivan JM, Fossa SD, et al. ALSYMPCA Investigators. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369(3):213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 9.Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016 Feb 22; doi: 10.1200/JCO.2015.64.2702. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byrne MJ, Nowak AK. Modified RECIST criteria for assessment of response in malignant pleural mesothelioma. Ann Oncol. 2004;15:257–60. doi: 10.1093/annonc/mdh059. [DOI] [PubMed] [Google Scholar]
- 11.Cheson BD, Horning SJ, Coiffier B, Shipp MA, Fisher RI, Connors JM, et al. Report of an international workshop to standardize response criteria for non-Hodgkin's lymphomas. J Clin Oncol. 1999;17:1244–53. doi: 10.1200/JCO.1999.17.4.1244. [DOI] [PubMed] [Google Scholar]
- 12.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 13.Meignan M, Barrington S, Itti E, Gallamini A, Haioun C, Polliack A. Report on the 4th International Workshop on Positron Emission Tomography in Lymphoma held in Menton, France, 3-5 October 2012. Leuk Lymphoma. 2014;55:31–7. doi: 10.3109/10428194.2013.802784. [DOI] [PubMed] [Google Scholar]
- 14.Barrington SF, Qian W, Somer EJ, Franceschetto A, Bagni B, Brun E, et al. Concordance between four European centres of PET reporting criteria designed for use in multicentre trials in Hodgkin lymphoma. Eur J Nucl Med Mol Imaging. 2010;37:1824–33. doi: 10.1007/s00259-010-1490-5. [DOI] [PubMed] [Google Scholar]
- 15.Barrington SF, Mikhaeel NG, Kostakoglu L, Meignan M, Hutchings M, Müeller SP, et al. Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol. 2014;32:3048–58. doi: 10.1200/JCO.2013.53.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoos A, Parmiani G, Hege K, Sznol M, Loibner H, Eggermont A, et al. A clinical development paradigm for cancer vaccines and related biologics. J Immunother. 2007;30:1–15. doi: 10.1097/01.cji.0000211341.88835.ae. [DOI] [PubMed] [Google Scholar]
- 17.Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe´ C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 18.Macdonald DR, Cascino TL, Schold SC, Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8:1277–80. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 19.Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28:1963–72. doi: 10.1200/JCO.2009.26.3541. [DOI] [PubMed] [Google Scholar]
- 20.Lin NU, Lee EQ, Aoyama H, Barani IJ, Barboriak DP, Baumert BG, et al. Proposed response assessment criteria for brain metastases: Response Assessment in Neuro-Oncology (RANO) Working Group. Lancet Oncol. 2015;16:e270–8. doi: 10.1016/S1470-2045(15)70057-4. [DOI] [PubMed] [Google Scholar]
- 21.Bruix J, Sherman M, Llovet JM, Beaugrand M, Lencioni R, Burroughs AK, et al. EASL Panel of Experts on HCC. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. J Hepatol. 2001;35:421–30. doi: 10.1016/s0168-8278(01)00130-1. [DOI] [PubMed] [Google Scholar]
- 22.Bruix J, Sherman M Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–36. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]