

Abstract
Boronic acids and esters have crucial roles in the areas of synthetic organic chemistry, molecular sensors, materials science, drug discovery, and catalysis. Many of the current applications of boronic acids and esters require materials with very low levels of transition metal contamination. Most of the current methods for the synthesis of boronic acids require, however, transition metal catalysts and ligands that have to be removed via additional purification procedures. This protocol describes a simple, metal- and additive-free method of conversion of haloarenes directly to boronic acids and esters. This photoinduced borylation protocol does not require expensive and toxic metal catalysts and ligands and produces innocuous and easy-to-remove by-products. Furthermore, the reaction can be carried out on multigram scales in common grade solvents without the need for reaction mixtures to be deoxygenated. The setup and purification steps are typically accomplished within 1–3 h. The reactions can be run overnight, and the protocol can be completed within 13–16 h. Two representative procedures that are described in this protocol provide details for preparation of a boronic acid (3-cyanopheylboronic acid) and a boronic ester (1,-benzenediboronic acid bis(pinacol)ester). We also discuss additional details of the method that will be helpful in the application of the protocol to other haloarene substrates.
Introduction
Boronic acids and esters are key materials in the areas of organic synthesis1-3, catalysis4-6, materials science7,8, drug discovery9-11, molecular self-assembly12,13, carbohydrate analysis14-16, and molecular sensing17-20. Photoinduced dissociation of Ar–X bonds has enabled a number of synthetically useful carbon–carbon and carbon–heteroatom bond-forming transformations, e.g. photoinduced nucleophilic substitution21-24, alkylation25, arylation26, and photocyclization27,28 reactions. We recently reported that haloarenes, including electron-rich fluoroarenes, as well as quaternary arylammonium salts readily react with diboron compounds to produce aromatic boronic acids and esters (Fig. 1a-c) under photochemical activation conditions29. A similar photoinduced borylation method was also developed by Li30,31. We also showed that a 1,2- and 1,3-regioselective dual C–H/C–X borylation of haloarenes can be achieved on preparative scale32 (Fig. 1d).
Figure 1.
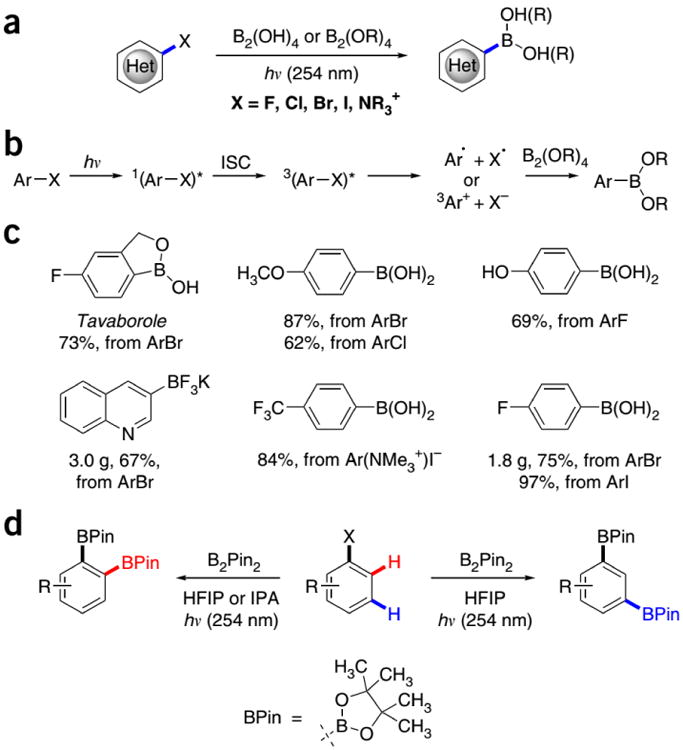
Photoinduced borylation of haloarenes. (a) General borylation scheme. (b) Reaction mechanism. (c) Typical products of the photoinduced borylation. (d) Regiodivergent dual 1,2- and 1,3-C–H/C–X borylation of haloarenes. ISC = intersystem crossing, HFIP = hexafluoroisopropanol, IPA = isopropyl alcohol.
The photochemical method of conversion of haloarenes directly to boronic acids and esters has a broad functional group tolerance and a scope that includes iodo-, bromo- and chloroarenes, as well as electron-rich fluoroarenes and arylammonium salts. The relevant reactions do not require heating, which is an advantage in the preparation of temperature-sensitive compounds. The present approach to borylation does not require expensive and toxic metal catalysts and ligands and produces innocuous and easy-to-remove by-products. Furthermore, the reaction can be carried out in common grade solvents without the need to deoxygenate the reaction mixtures.
The purification procedure is very simple, and, in the case of boronic acids, typically does not require chromatography. Since no metal catalysts are used, the products do not contain metal contaminants, making this method particularly suitable for the preparation of intermediates in pharmaceutical, nanotechnology, advanced materials, and analytical sensors industries that use organoboron compounds and have stringent requirements for trace metal impurities33,34.
By contrast, transition metal–catalyzed borylation methods35-41 suffer from several limitations, including low functional group compatibility and the requirement for toxic and expensive precious metal catalysts and expensive ligands, high reaction temperatures, inert atmosphere, and difficult purification procedures that typically involve chromatography and metal scavengers.
We describe herein a detailed protocol that includes a step by step description of the reaction setup and purification procedures, as well as troubleshooting techniques. The protocol opens new avenues for the synthesis of organoboron compounds on preparative scales and reduces dependence on the transition metal–catalyzed borylation methods.
In the Procedure, we provide instructions for two exemplary syntheses of organoboron compounds using this new method. In the first part of the Procedure (steps 1-17), preparation of boronic acids is exemplified by the grams-scale synthesis of 3-cyanophenylboronic acid (1) from 3-bromobenzonitrile (2) (Fig. 2a). In the second part (steps 18-26), preparation of boronic esters is exemplified by the synthesis of 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (3) from bromoarene 4 (Fig. 2b). Naturally, the two synthetic procedures just alluded to are inherently independent of each other, and readers may decide to implement only one of them, depending on whether they plan to synthesize a boronic acid (adapting the instructions in steps 1-17) or a boronic ester (adapting the instructions in steps 18-26). Similarly, should readers decide to implement both synthetic procedures to obtain a boronic acid and a boronic ester, the relative order in which the two synthetic procedures are implemented is irrelevant.
Figure 2.

Photoinduced borylation reactions described in this protocol. (a) preparation of 3-cyanophenylboronic acid (1) from 3-bromobenzonitrile (2). (b) preparation of boronic ester 3 from bromoarene 4. (c) typical reaction set-up in a Rayonet RPR-100 chamber.
Experimental Design
The reactions are typically carried out in quartz test-tubes. Although substantial amounts of water (>20 vol%) generally slow down the borylation reaction, the presence of trace amounts of moisture does not affect it. Hence, commercial-grade solvents can be used without further purification. The reaction is not air-sensitive and no deoxygenation of the reaction mixture is necessary. We routinely use the photochemical reactor Rayonet RPR-100 equipped with 16 UV-C (254 nm) low-pressure lamps. However, other reactor designs can also be used (e.g. a photochemical immersion well reactor), keeping in mind that UV lamps with wavelengths other than 254 nm (e.g. 300 or 350 nm) are not effective. The yields are generally higher if the reaction mixture is placed in the narrowest possible test-tube that can accommodate the desired reaction mixture volume, to maximize exposure to light from the vertically arranged tubular UV lamps (Fig. 2c). It is also advisable to place the test-tube 1-2 cm from one of the UV lamps. The fan should be on throughout the synthesis to maintain a temperature of 26 °C in the photochemical chamber. The borylation generally proceeds smoothly at 10-30 °C. At higher temperatures, however, formation of the dehalogenated arene by-product (for example, benzonitrile from 3-bromobenzonitrile) may compete with borylation. The solvent of choice for the borylation with tetrahydroxydiboron is methanol. The borylations with diboron esters (e.g., B2pin2) can be carried out in methanol, acetonitrile, isopropanol, and trifluoroethanol.
For practical reasons, we typically maintain substrate concentration at 0.3–1.2 mol·L−1, although in the case of poorly soluble substrates, more dilute reaction mixtures can also be used. Please be aware, however, that at substantially lower concentrations than those of the typical reaction (e.g., below 0.05 mol·L−1), formation of the dehalogenated arene by-product may compete with borylation. The reactions typically do not require stirring, provided that the substrate is dissolved in the reaction mixture. The progress of the reaction, as well as formation of the dehalogenation by-product and boroxines can be monitored by means of 1H NMR spectroscopy of an aliquot of the reaction mixture dissolved in d4-methanol. We have successfully carried out borylations on 0.6-20 mmol scales with haloarenes as limiting reagents. Boronic esters that cannot be isolated by crystallization can be purified by column chromatography on silica gel using a mixture of hexane and ethyl acetate as an eluent (typically 0-50% EtOAc in hexane).
Materials
Reagents
! CAUTION All chemicals used in this protocol should be handled with care. Standard protective equipment should be worn (lab coat, gloves, safety goggles), and all manipulations should be performed in a ventilated laboratory fume hood.
Bis(pinacolato)diboron (B2Pin2) (Synthonix, cat. no. B1760G100)
3-Bromobenzonitrile (Matrix Scientific, cat. no. 075372)
Deionized water
Deuterated chloroform (Cambridge Isotope Laboratories)
Deuterated dimethyl sulfoxide (Cambridge Isotope Laboratories)
Deuterated methanol (Cambridge Isotope Laboratories)
Ethyl acetate (Fisher Scientific, cat. no. E145-20, ACS grade)
Hexane (Fisher Scientific, cat. no. H292-20, ACS grade)
12M Hydrochloric acid (Sigma-Aldrich, cat. no. H1758)
Methanol (Fisher Scientific, cat. no. A412-20, ACS grade)
Sodium hydrogen carbonate (Chem-Impex, cat. no. 00084)
Sodium sulfate (Chem-Impex, cat. no. 00274)
Tetrahydroxydiboron (Boron Molecular, cat. no. BM212)
Equipment
! CAUTION UV protective safety goggles, lab coat and gloves should be worn while working with sources of ultraviolet light.
Access to NMR and IR spectrometry instruments
2-(4-Bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4) (TCI America, cat. no. B4771)
Cotton pad
Disposable syringes and injection needles
Erlenmeyer flasks
Fritted glass Buchner funnel (medium porosity)
Glass pipettes
Magnetic Stirplate
Melting point apparatus
NMR tubes (5 mm width)
pH Indicator paper (pH 0-14)
Plastic funnel
Polyethylene stopper (Chemglass, cat. no. CG-3021-03)
Quartz test-tube (10 cm length × 1.2 cm internal diameter, Quartz Scientific, cat. no. 315010)
-
Quartz test-tube (30 cm length × 1.5 cm internal diameter, Quartz Scientific, cat. no. 317010)
▲ CRITICAL Quartz test-tubes should be used, since borosilicate glass absorbs UV light.
-
Rayonet RPR-100 photoreactor equipped with 16 Ushio 8W T5 UV-C lamps (254 nm).
▲ CRITICAL Photoreactor should be equipped with UV lamps emitting 254 nm light. Use of lamps with other maximum emittance wavelengths may result in lower yields.
Rotary evaporator
Round bottom flasks
Rubber bulb
Separatory funnel
Spatulas
Stirbar (12 mm length × 4 mm diameter, Fisher Scientific, cat. no. 14-513-93)
Ultrasonic bath
Weighing balance
Weighing paper
Reagent Setup
Cold 12 M hydrochloric acid
Cool this reagent to 0-5 °C right before use in step 12 of the Procedure.
Cold hexane
Cool this reagent to 0-5 °C right before use in step 21 of the Procedure.
Procedure
Synthesis of 3-cyanophenylboronic acid (1)-reaction setup. •TIMING 15 min
1| Weigh out 1.82 g (10.0 mmol) of 3-bromobenzonitrile (2) and 1.34 g (15 mmol) of tetrahydroxydiboron. Place the reagents into a quartz test-tube (30 cm length × 1.5 cm internal diameter) equipped with a stirbar. Add 10 ml of methanol.
2| Stir the mixture alternately at 40 °C and room temperature (23 °C) until the solids completely dissolve and a clear solution is formed. Cap the test-tube with a polyethylene stopper, which is vented with a needle.
Synthesis of 3-cyanophenylboronic acid (1)-borylation reaction. •TIMING 12 h
-
3| Place the quartz test-tube containing the reaction mixture into the photoreactor and ensure that the distance between the quartz test-tube and one of the lamps is 1-2 cm. Turn on the lamps and the fan in the photoreactor. Irradiate the reaction mixture without stirring for 12 h. Please note that leaving the reaction overnight (12-17 h) typically does not affect the results.
▲ CRITICAL The fan should remain on for the duration of the reaction to maintain the temperature within the optimal range. Carrying out the experiment with the fan off may result in formation of the dehalogenation by-product, due to the increased temperature in the reactor chamber.
? Troubleshooting
Synthesis of 3-cyanophenylboronic acid (1)-purification. •TIMING 1 h
4| Pour the reaction mixture into a 50-ml round bottom flask. Add 1 g of sodium hydrogen carbonate and concentrate the mixture to dryness on a rotary evaporator (water bath temperature 30–35 °C).
5| Add to the flask 20 mL of hexane, swirl to mix the components and concentrate the mixture to dryness using a rotary evaporator (water bath temperature 30–35 °C).
-
6| Repeat step 5 three more times.
■CRITICAL STEP Evaporation of hexane ensures that residual methanol is removed from the crude product. Failure to remove residual methanol may prevent complete extraction of impurities and by-products (e.g. unreacted haloarene and the dehalogenation product) from the crude material in the following step.
7| Add to the residue obtained after step 6 20 mL of hexane, sonicate the resulting mixture for 30 sec then decant the hexane layer. The hexane layer may at this point be discarded.
8| Dry the residue under reduced pressure on a rotary evaporator (water bath temperature 30–35 °C).
-
9| Add to the residue 10 ml of methanol and concentrate under reduced pressure on a rotary evaporator (water bath temperature 30-35 °C)
■CRITICAL STEP Methanol needs to be added at this point to prevent the formation of boroxines.
10| Add to the residue obtained in the previous step 50 mL of ethyl acetate, swirl, and decant the ethyl acetate layer into a 250 mL separatory funnel.
11| Add 10 mL of deionized water into the round bottom flask with the remaining solid material.
12| Add 5 mL of cold 12 M hydrochloric acid dropwise to the mixture that remained in the round bottom flask to acidify the solution to pH 1, then transfer the mixture into a separatory funnel.
13| Shake the mixture in the separatory funnel vigorously. Decant the top (organic) layer into a 250-mL Erlenmeyer flask.
14| Extract the aqueous phase left in the separatory funnel with 30 mL of ethyl acetate.
15| Repeat the extraction described in the previous step.
16| Combine the ethyl acetate solutions obtained in steps 13, 14, and 15 in a 250-mL Erlenmeyer flask, and add to it 10 g of sodium sulfate and 1 g of sodium hydrogen carbonate. Stir the resulting mixture for 5 min. Filter off the solids using a plastic funnel with a cotton pad, and wash the solids three times with 10 mL of ethyl acetate each time.
-
17| Concentrate the filtrate obtained in the previous step on a rotary evaporator (water bath temperature 30–35 °C). Add to the resulting residue 5 mL of ethyl acetate and 40 mL of hexane. Concentrate the mixture obtained to dryness on a rotary evaporator (water bath temperature 30–35 °C). The product can now be weighed to establish the yield, and the purity of the product can be determined by means of 1H, 13C, and 11B NMR spectroscopy using either CD3OD or (CD3)2SO as solvents.
■PAUSE POINT Boronic acids like 1 may be stored at room temperature for several months or even years. However, such compounds may undergo dehydration to form boroxines in the mentioned conditions over time. Although such formation of boroxines does not usually affect the performance of the material in most of typical cross-coupling reactions, we recommend storing boronic acids at lower temperatures (e.g. 0 °C) to slow down the formation of boroxines.
? Troubleshooting
Synthesis of 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (3)– reaction setup •TIMING 15 min
18| Weigh out 0.7 g (2.5 mmol) of 2-(4-bromophenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (4) and 1.3 g (5.0 mmol) of B2Pin2. Place the reagents into a quartz test-tube (10 cm length × 1.2 cm internal diameter) equipped with a stirbar and add to it 6 ml of methanol.
19| Stir the mixture at room temperature until the solids completely dissolve and a clear solution is formed. Cap the test-tube with a polyethylene stopper that is vented with a needle.
Synthesis of 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (3)-borylation reaction. •TIMING 12 h
-
20| Place the quartz tube containing the reaction mixture into the photoreactor and ensure that the distance between the quartz tube and the lamp is 1-2 cm. Turn on the lamps and the fan in the photoreactor. Irradiate the reaction mixture without stirring for 12 h. Please note that leaving the reaction overnight (12–17 h) typically does not affect the results. The borylation product will partially precipitate from the reaction mixture.
▲ CRITICAL The fan should remain on for the duration of the reaction to maintain the temperature within the optimal range. Carrying out the experiment with the fan off may result in formation of the dehalogenation by-product, due to the increased temperature in the reactor chamber.
? Troubleshooting
Synthesis of 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (3)-purification •TIMING 2-3 h
21| Filter the solid that precipitated from the reaction mixture using a fritted glass Buchner funnel. Wash the solid left in the funnel with 10 mL of cold hexane.
22| Combine the filtrate and the hexane washings and concentrate the combined solution to dryness on a rotary evaporator (water bath temperature 30-35 °C).
23| Add 10 mL of hexane to the residue obtained in the previous step and place the flask in a refrigerator at 4 °C for 1 h, until a solid precipitate has formed.
24| Filter the precipitate and wash it with 5 mL of hexane.
25| Repeat steps 22-24 once more.
-
26| Combine the isolated solid product fractions obtained in steps 21-25 and dry the material in the air. The product can now be weighed to establish the yield and, the purity of the product can be determined by means of 1H, 13C, and 11B NMR spectroscopy using CDCl3 as a solvent.
■PAUSE POINT Pinacol esters of boronic acids can be stored for extended periods of time (e.g. >6 months) without noticeable decomposition at room temperature in capped storage containers.
? TROUBLESHOOTING Troubleshooting advice can be found in Table 1.
Table 1. Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 3, 20 | Incomplete reaction | The lamp wavelength is not 254 nm or the lamp intensity is insufficient | Use lamps emitting 254 nm light or use higher intensity UV sources |
| The substrates utilized by the experimenter are less reactive than bromoarenes 2 and 4 | Allow the reaction to proceed for a longer time until it is complete | ||
|
| |||
| 3, 20 | A substantial amount of dehalogenation by-product is formed | The temperature in the reactor is too high or the reaction mixture is too dilute | Turn on the fan or provide a cool air supply to cool the reaction mixture down to the optimal temperature (10-30 °C) |
| Reduce the solvent volume | |||
|
| |||
| 17 | Boroxine is formed as by-product | The product was dried at >40 °C, causing its dehydration | Add 10 mL of methanol and evaporate the resulting solution at <40 °C |
|
| |||
| 17 | Impure product is obtained | The reaction did not go to completion | Allow the reaction to proceed to completion over a longer a period of time |
| Make sure that the lamp emits 254-nm light | |||
|
| |||
| 17 | Boronic acid is obtained in low yield | Too much water is added in step 11, boronic acid is soluble in water, or an insoluble boroxine is formed | Use the specified amount of water in step 11. To prevent formation of boroxines, dissolve the solid obtained after the hexane wash in step 7 in methanol and evaporate the solution. Repeat the process |
•TIMING
Preparation of boronic acid 1
Steps 1 and 2: 15 min
Step 3: 12 h
Steps 4–17: 1 h
Preparation of boronic ester 3
Steps 18 and 19: 15 min
Step 20: 12 h
Steps 21–26: 2–3 h
Anticipated Results
Following the procedure described above in steps 1-17, 3-cyanophenylboronic acid (1) is obtained as a colorless solid in 88% yield (1.3 g).
Following the procedure described above in steps 18-26, 1,4-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (3) is obtained as a colorless crystalline solid in 75% yield (0.6 g). The products are characterized by 1H, 13C, 11B NMR, and IR spectroscopy. For typical applications characterization by 1H, 13C, 11B NMR is sufficient to establish the identity and purity of the isolated products.
Analytical data
3-Cyanophenylboronic acid (1): m.p. > 260 °C. – 1H NMR (300 MHz, (CD3)2SO): 8.43 (2 H, br s), 8.14 (1 H, s), 8.09 (1 H, dd, J = 7.5, 1.5 Hz), 7.89 (1 H, dd, J = 7.5, 1.2 Hz), 7.59 (1 H, t, J = 7.5 Hz) ppm. – 13C NMR (75 MHz, (CD3)2SO):138.7, 137.6, 133.6, 128.7, 119.3, 110.6 ppm. – 1H NMR (300 MHz, (CD3OD): 7.97 (2 H, br m), 7.74 (1 H, dd, J = 7.5, 1.5 Hz), 7.53 (1 H, t, J = 7.5 Hz) ppm. – 13C NMR (75 MHz, (CD3OD): 139.1, 138.3, 134.2, 129.5, 120, 112.8 ppm. – 11B NMR (160 MHz, CDCl3): 27.5 ppm. – IR: 3358, 2232, 1599, 1575, 1429, 1339, 1339, 1201, 1039, 907, 850, 799 cm−1. – Calcd. for C7H6BNO2: C, 57.22; H, 4.12; N, 9.53. Found C, 57.36; H, 4.12; N, 9.39.
1,4-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (3): m.p. 241– 243 °C. – 1H NMR (300 MHz, CDCl3): 7.80 (4 H, s), 1.34 (24 H, s) ppm. – 13C NMR (75 MHz, CDCl3): 133.8, 83.8, 24.8 ppm. – 11B NMR (160 MHz, CDCl3): 30.8 ppm. – IR: 2975, 1522, 1458, 1392, 1371, 1346, 1324, 1257, 1214, 1167, 1140, 1097, 1019, 961, 856, 844 cm−1. – Calcd. for C18H28B2O4: C, 65.51; H, 8.55. Found C, 65.63; H, 8.50.
Supplementary Material
Acknowledgments
Financial support by the Welch Foundation (AX-1788), the NSF (CHE-1455061), NIGMS (SC3GM105579, 4T34GM008073-32), and UTSA is gratefully acknowledged.
Footnotes
Note: Supporting Information with the NMR spectra of products 1 and 3 is available in the online version of the paper.
Author Contributions A.M.M., B.D.S. and W.C. carried out the experiments. O. V. L. and A.M.M. designed the experiments and analyzed the data. O.V.L. directed the research. O.V.L. and A.M.M. wrote the manuscript.
Competing Financial Interests The authors declare no competing financial interests.
References
- 1.Suzuki A, Brown HC. Organic Syntheses Via Boranes. Vol. 3. Aldrich Chemical Company; 2003. [Google Scholar]
- 2.Hall DG, editor. Boronic Acids. 2nd. Wiley-VCH; 2011. [Google Scholar]
- 3.Gutekunst WR, Baran PS. C-H functionalization logic in total synthesis. Chem Soc Rev. 2011;40:1976–1991. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- 4.Corey EJ. Catalytic enantioselective Diels-Alder reactions: methods, mechanistic fundamentals, pathways, and applications. Angew Chem, Int Ed. 2002;41:1650–1667. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 5.Dimitrijevic E, Taylor MS. Organoboron acids and their derivatives as catalysts for organic synthesis. ACS Catal. 2013;3:945–962. [Google Scholar]
- 6.Ishihara K. Synthesis and application of organoboron compounds. Top Organomet Chem. 2015;49:243–270. [Google Scholar]
- 7.Lorbach A, Huebner A, Wagner M. Aryl(hydro)boranes: versatile building blocks for boron-doped π-electron materials. Dalton Trans. 2012;41:6048–6063. doi: 10.1039/c2dt30118k. [DOI] [PubMed] [Google Scholar]
- 8.Jäkle F. Recent advances in the synthesis and applications of organoborane polymers. Top Organomet Chem. 2015;49:297–325. [Google Scholar]
- 9.Trippier PC, McGuigan C. Boronic acids in medicinal chemistry: anticancer, antibacterial and antiviral applications. Med Chem Commun. 2010;1:183–198. [Google Scholar]
- 10.Yang W, Gao W, Wang B. Applications of boronic acids in chemical biology andmedicinal chemistry. In: Hall DG, editor. Boronic Acids. 2nd. Wiley-VCH; 2011. pp. 591–619. [Google Scholar]
- 11.Ban HS, Nakamura H. Boron-based drug design. Chem Rec. 2015;15:616–635. doi: 10.1002/tcr.201402100. [DOI] [PubMed] [Google Scholar]
- 12.Fujita N, Shinkai S, James TD. Boronic acids in molecular self-assembly. Chem Asian J. 2008;3:1076–1091. doi: 10.1002/asia.200800069. [DOI] [PubMed] [Google Scholar]
- 13.Mastalerz M. Shape-persistent organic cage compounds by dynamic covalent bond formation. Angew Chem, Int Ed. 2010;49:5042–5053. doi: 10.1002/anie.201000443. [DOI] [PubMed] [Google Scholar]
- 14.James TD, Shinkai S. Artificial receptors as chemosensors for carbohydrates. Top Curr Chem. 2002;218:159–200. [Google Scholar]
- 15.Jelinek R, Kolusheva S. Carbohydrate biosensors. Chem Rev. 2004;104:5987–6015. doi: 10.1021/cr0300284. [DOI] [PubMed] [Google Scholar]
- 16.Pal A, Berube M, Hall DG. Design, synthesis, and screening of a library of peptidyl bis-boroxoles as low molecular weight receptors for complex oligosaccharides in water: Identification of a receptor for the tumour marker TF-antigen. Angew Chem, Int Ed. 2010;49:1492–1495. doi: 10.1002/anie.200906620. [DOI] [PubMed] [Google Scholar]
- 17.Wade CR, Broomsgrove AEJ, Aldridge S, Gabbaï FP. Fluoride ioncomplexation and sensing using organoboron compounds. Chem Rev. 2010;110:3958–3984. doi: 10.1021/cr900401a. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Li Z, Chen XX, Fossey JS, James TD, Jiang YB. Selective sensing of saccharides using simple boronic acids and their aggregates. Chem Soc Rev. 2013;42:8032–8048. doi: 10.1039/c3cs60148j. [DOI] [PubMed] [Google Scholar]
- 19.Guan Y, Zhang Y. Boronic acid-containing hydrogels: synthesis and their applications. Chem Soc Rev. 2013;42:8106–8121. doi: 10.1039/c3cs60152h. [DOI] [PubMed] [Google Scholar]
- 20.You L, Zha D, Anslyn EV. Recent Advances in supramolecular analytical chemistry using optical sensing. Chem Rev. 2015;115:7840–7892. doi: 10.1021/cr5005524. [DOI] [PubMed] [Google Scholar]
- 21.Bunnett JF. Aromatic substitution by the Srn1 mechanism. Acc Chem Res. 1978;11:413–420. [Google Scholar]
- 22.Uyeda C, Tan YC, Fu GC, Peters JC. A new family of nucleophiles forphotoinduced, copper-catalyzed cross-couplings via single-electron transfer:Reactions of thiols with aryl halides under mild conditions (0 °C) J Am Chem Soc. 2013;135:9548–9552. doi: 10.1021/ja404050f. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Liu W, Zeng H, Mu X, Cosa G, Mi Z, Li CJ. Photoinduced metal-catalyst-free aromatic Finkelstein reaction. J Am Chem Soc. 2015;137:8328–8331. doi: 10.1021/jacs.5b03220. [DOI] [PubMed] [Google Scholar]
- 24.Chen K, He P, Zhang S, Li P. Synthesis of aryl trimethylstannanes from aryl halides: an efficient photochemical method. Chem Commun. 2016;52:9125–9128. doi: 10.1039/c6cc01135g. [DOI] [PubMed] [Google Scholar]
- 25.Mella M, Coppo P, Guizzardi B, Fagnoni M, Freccero M, Albini A. photoinduced, ionic Meerwein arylation of olefins. J Org Chem. 2001;66:6344–6352. doi: 10.1021/jo010469s. [DOI] [PubMed] [Google Scholar]
- 26.Dichiarante V, Fagnoni M, Albini A. Metal-free synthesis of sterically crowded biphenyls by direct Ar–H substitution in alkyl benzenes. Angew Chem Int Ed. 2007;46:6495–6498. doi: 10.1002/anie.200701462. [DOI] [PubMed] [Google Scholar]
- 27.Grimshaw J, de Silva AP. Photochemistry and photocyclization of aryl halides. Chem Soc Rev. 1981;10:181–203. [Google Scholar]
- 28.Lu SC, Zhang XX, Shi ZJ, Ren YW, Li B, Zhang W. Intramolecularphotochemical cross-coupling reactions of 3-acyl-2-haloindoles and 2-chloropyrrole-3-carbaldehydes with substituted benzenes. Adv Synth Cat. 2009;351:2839–2844. [Google Scholar]
- 29.Mfuh AM, Doyle JD, Chhetri B, Arman HD, Larionov OV. Scalable, Metal-and additive-free, photoinduced borylation of haloarenes and quaternary arylammonium salts. J Am Chem Soc. 2016;138:2985–2988. doi: 10.1021/jacs.6b01376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen K, Zhang S, He P, Li P. Efficient metal-free photochemical borylation of aryl halides under batch and continuous-flow conditions. Chem Sci. 2016;7:3676–3680. doi: 10.1039/c5sc04521e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Cheung MS, Lin Z, Li P. Metal-free borylation of electron-rich aryl(pseudo)halides under continuous-flow photolytic conditions. Org Chem Front. 2016;3:875–879. [Google Scholar]
- 32.Mfuh AM, Nguyen VT, Chhetri B, Burch JE, Doyle JD, Nesterov VN, Arman HD, Larionov OV. Additive- and metal-free, predictably 1,2- and 1,3-regioselective, photoinduced dual C–H/C–X borylation of haloarenes. J Am Chem Soc. 2016;138:8408–8411. doi: 10.1021/jacs.6b05436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Schoneker D, Ulman KL, Sturm JJ, Thackery LM, Kauffman JF. Elemental impurities in pharmaceutical excipients. J Pharm Sci. 2015;104:4197–4206. doi: 10.1002/jps.24650. [DOI] [PubMed] [Google Scholar]
- 34.McDaniel FD, Datar SA, Nigam M, Ravi Prasad GV. Impurity measurementsin semiconductor materials using trace element accelerator mass spectrometry. Nucl Instrum Methods Phys Res, Sect B. 2002;190:826–830. [Google Scholar]
- 35.Ishiyama T, Murata M, Miyaura N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: A direct procedure for arylboronic esters. J Org Chem. 1995;60:7508–7510. [Google Scholar]
- 36.Murata M, Watanabe S, Masuda Y. Novel palladium(0)-catalyzed coupling reaction of dialkoxyborane with aryl halides: convenient synthetic route to arylboronates. J Org Chem. 1997;62:6458–6459. [Google Scholar]
- 37.Ishiyama T, Miyaura N. Chemistry of Group 13 element-transition metal linkage —the platinum- and palladium-catalyzed reactions of (alkoxo)diborons. J Organomet Chem. 2000;611:392–402. [Google Scholar]
- 38.Chow WK, Yuen OY, Choy PY, So CM, Lau CP, Wong WT, Kwong FY. A decade advancement of transition metal-catalyzed borylation of aryl halides and sulfonates. RSC Adv. 2013;3:12518–12539. [Google Scholar]
- 39.Cho JY, Tse MK, Holmes D, Maleczka RE, Smith MR. Remarkably selective iridium catalysts for the elaboration of aromatic C–H bonds. Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 40.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. C-HActivation for the construction of C-B bonds. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 41.Hartwig JF. Borylation and silylation of C–H bonds: A platform for diverse C–Hbond functionalizations. Acc Chem Res. 2012;45:864–873. doi: 10.1021/ar200206a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.