

Abstract
Introduction
Epigenetics is the study of reversible modifications to chromatin and their extensive and profound effects on gene regulation. To date, the role of epigenetics in personalized medicine has been under-explored. Therefore, this review aims to highlight the vast potential that epigenetics holds.
Areas covered
We first review the cell-specific nature of epigenetic states and how these can vary with developmental stage and in response to environmental factors. We then summarize epigenetic biomarkers of disease, with a focus on diagnostic tests, followed by a detailed description of current and pipeline drugs with epigenetic modes of action. Finally, we discuss epigenetic biomarkers of drug response.
Expert commentary
Epigenetic variation can yield information on cellular states and developmental histories in ways that genotype information cannot. Furthermore, in contrast to fixed genome sequence, epigenetic patterns are plastic, so correcting aberrant, disease-causing epigenetic marks holds considerable therapeutic promise. While just six epigenetic drugs are currently approved for use in the United States, a larger number is being developed. However, a drawback to current therapeutics is their non-specific effects. Development of locus-specific epigenetic modifiers, used in conjunction with epigenetic biomarkers of response, will enable truly precision interventions.
Keywords: Chromatin remodeling, DNA methylation, DNMT inhibitors, Epigenetic biomarkers, Epigenetic drugs, HDAC inhibitors, Histone modifications, Oncology, Neuroscience, Pharmacokinetics
1. Introduction
Personalized medicine is founded upon the concept that individual differences in therapeutic success are the norm among patients that require pharmacological treatment. This concept is not new. Hippocrates writing in the 5th century BCE is known to have commented, “give different ones [drugs] to different patients, for the sweet ones do not benefit everyone, nor do the astringent ones, nor are all the patients able to drink the same things.” (see [1]). Thus, the concept of variable response to drugs has been discussed for at least two and a half millennia. However, being able to predict who will respond to a given drug has proven an enduring challenge. With the advent of modern genomic technologies, which enable us to read each patient’s genetic make-up, the idea of personalized medicine is becoming a reality.
Pharmacogenetics, the core discipline of personalized medicine, has already delivered some profound and meaningful successes. The effectiveness of Cytochrome P450 (CYP450) genotypes in predicting an individual’s drug metabolizing phenotype is a notable example [2]. This has led to several of these biomarkers being approved for clinical use by regulatory bodies such as the US Federal Drug Administration (FDA) (www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm). Beyond drug metabolism, genetic variants at numerous other loci have shown robust associations indicative of clinical relevance, with commercial kits and services now available to deliver this information to health providers and consumers [3].
In the last decade, pharmacogenetics has harnessed the power of genome-wide association studies (GWAS). This has enabled the field to move beyond the study of candidate genes to scanning hundreds of thousands of genetic markers for each subject. Several promising new leads have been discovered. Arguably, however, the success of GWAS in pharmacogenomics has not mirrored that of complex disease studies. Primarily this may be an issue of statistical power, whereby the clinical trials necessary to measure drug response are costly and so sample sizes currently tend to be small. As studies grow in size and number and meta-analyses are conducted across samples, we can expect GWAS to yield additional insight over time [4]. However, GWAS will not yield all the answers for any given drug response phenotype. Beyond the limitation where GWAS focuses on common polymorphisms, even if all the relevant variants for response to a given drug were mapped, we would still be unable to explain all the phenotypic variation in drug response [5]. Drug response is complex and, like other complex traits, it likely arises from the interplay of multiple genetic and environmental factors over the life course [6]. DNA sequence is just one component of this complexity.
Most genotype associations in complex traits such as drug response are probabilistic indicators of phenotype, which typically say little of certainty about the state of the organism at the time of sampling. When treating an individual patient with a specific drug, substantial supporting information in addition to genotype information may be required before making a clinical decision. Even phenotypes that are strongly influenced by genetics, such as the CYP450 drug metabolism phenotypes, will be modified by the effects of concurrent medications or alcohol and tobacco use that may inhibit or interfere with CYP enzyme activity [7,8]. This further illustrates the need to consider information beyond genotype alone.
There are two broad complexities to living organisms that are not addressed by genotype information. These are 1) spatial and 2) temporal variation in biological function or phenotypic expression within the same organism. Consider that humans are composed of multiple cell types with a diverse array of functions (spatial, or cell-specific variation) and that we take on very different macroscopic forms in early versus later life (temporal, or developmental variation). Yet essentially the same genome is present in all nucleated cells at all time points. In this review, we will show how the processes that lead to cellular diversity and organismal development, i.e. epigenetics, can be harnessed to provide more nuanced DNA-based biomarkers and novel treatment strategies [9]. Indeed, epigenetics may also yield an environmental exposure record of the patient that we are just beginning to comprehend [10]. Epigenetic biomarkers are therefore fundamentally different to studies of gene expression, proteins or metabolites, which provide snapshots of functional state at a single time point. Epigenetics provides layers of regulatory and environmental exposure information on top of each individual’s unique genome [11]. Thus, it indicates what happened to you and you alone, and from this we may be able to determine your truly personal drug regimen design and success, disease susceptibility and cure.
2. Epigenetics Overview
The term “epigenetics” was first described by the British developmental biologist Conrad Waddington in the 1940s as “the branch of biology which studies the causal interactions between genes and their products, which bring the phenotype into being” [12,13]. Waddington’s definition therefore predates the discovery of DNA and so the term “epigenetics” has developed over time. Waddington was focused on organismal development, whereby cells starting with the fertilized egg follow trajectories of increasing specialization until terminal differentiation, which cannot be reversed. One of Waddington’s visual metaphors for this process, where the cell is conceptualized as a marble rolling down a rolling hillside with ravines and valleys, has an enduring intuitive appeal and is explained in Figure 1.
Figure 1.

Waddington represented the developmental process as a series of “decisions” made by differentiating cells that could be represented as forks in the valleys of the “developmental landscape”. Panels A and B represent the alternate fates of the cell, or ball by analogy. As the pluripotent stem cell of the egg (ball at the top), begins to specialize, the differentiation “decisions” made are irreversible. Its pattern of epigenetic regulation is established by the point of terminal differentiation at the bottom of the landscape. With epigenetic drugs and therapies, the aim is to artificially reverse maladaptive epigenetic states and essentially “push the ball back up the hill”. Figure from Noble (2015) [13] reproduced with permission.
Today, backed by knowledge of the genome and some core molecular processes, epigenetics can be defined as the study of mitotically stable changes in genetic regulation that do not involve changes to nucleotide sequence [14]. Mitotic stability, in this sense, means that the epigenetic state of the parent cell is written to the daughter cell after mitosis, thereby continuing the developmental trajectory of the parent. This regulation is enacted via epigenetic marks, which are reversible regulatory modifications to chromatin.
2.1 Epigenetic modifications to chromatin
The most intensively studied epigenetic mark is the methylation of DNA cytosine residues at the carbon 5 position (5mC). This mark is made via the DNA N-methyl transferase (DNMT) enzymes and is most often found in the sequence context CpG [15]. DNA methylation is one of the core epigenetic marks essential for regulating gene expression in normal cell development and differentiation [16]. While 5mC is the most well-characterized, other cytosine modifications have now been discovered, such as 5-hydroxymethycytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [17,18]. The functions of these exotic marks are still being elucidated, but 5hmC may play an important role in the central nervous system, where it is prevalent, and in the regulation of pluripotency in stem cells [19,20].
Another major class of epigenetic mark involves the post-translational modification of histones, the proteins that package DNA into nucleosomes [21]. Histones are the chief protein components of chromatin, whereby 146 bp of DNA is wound around each histone octamer [22]. There are five major classes of canonical histones, where each octamer is typically formed of two H2A-H2B dimers and a H3-H4 tetramer while H1 serves as a linker protein between nucleosomes. H3 and H4 have long tails that protrude from the nucleosome that can be covalently modified in several places, while other histones can also be modified to a lesser degree. The best characterized modifications include mono-, di- and tri-methylation, acetylation and phosphorylation, although a growing number continue to be reported [23]. Standard nomenclature abbreviates the histone, the modified residue and the type of modification, such that histone 3 lysine 27 acetylation is written as “H3K27Ac”. These modifications are written and erased by specific enzyme families, such as histone acetyltransferases (HATs) and histone deacetylases (HDACs) in the case of acetylation marks, or histone methyltransferases (HMTs) and demethylases (HDMs) in the case of methylation marks [23].
In addition to histone modifications, histone variants can have significant transcriptional regulatory roles. Histone variants replace canonical histones to alter nucleosome structure and ultimately DNA accessibility [24]. An example histone variant is H2A.Z, which replaces nucleosomal H2A to perform several complex regulatory roles in gene expression and development [25]. Finally, for the purposes of this article, we also mention polycomb epigenetic repressors and bromodomain-containing proteins. Polycomb proteins can remodel chromatin and typically function as epigenetic gene silencers [26], while bromodomain proteins are transducers of the acetylation signal on histones [27]. These chromatin-interacting proteins are relevant for epigenetic personalized medicine because they are targets for epigenetic drugs that we mention below in Section 3.2. Other putatively epigenetic regulatory mechanisms exist, most notably the non-coding RNAs (ncRNAs), which are beyond the scope of the current article. NcRNAs primarily function as post-transcriptional regulators of gene expression, but also play roles in regulating chromatin accessibility. They have been extensively reviewed elsewhere (http://www.cell.com/cell/collections/noncoding-rna).
2.2 Epigenetic effects on gene expression and regulation
The “textbook”, or classic, view of epigenetic regulation is focused on DNA methylation at gene promoters. In this view, hypomethylated CpGs are typically associated with active, expressed genes, while hypermethylated CpGs are typically associated with silenced genes. This effect arises because methylation of cytosine inhibits transcription factor binding [28]. Subsequent research has indicated that methylated cytosine, in addition to methylated histone H3K9, and deacetylated H3 combine to form a repressive epigenetic signature, while unmethylated DNA, methylated H3K4, and acetylated H3 combine to form an activating epigenetic signature [29], although not all histone modifications are coupled with DNA methylation [30]. An overview is provided in Figure 2. During development, epigenetic patterns change and differentiated cells develop a stable and unique epigenetic pattern that regulates tissue-specific gene transcription. While this view is broadly consistent with current findings, waves of new genomic data have yielded a more nuanced view.
Figure 2.
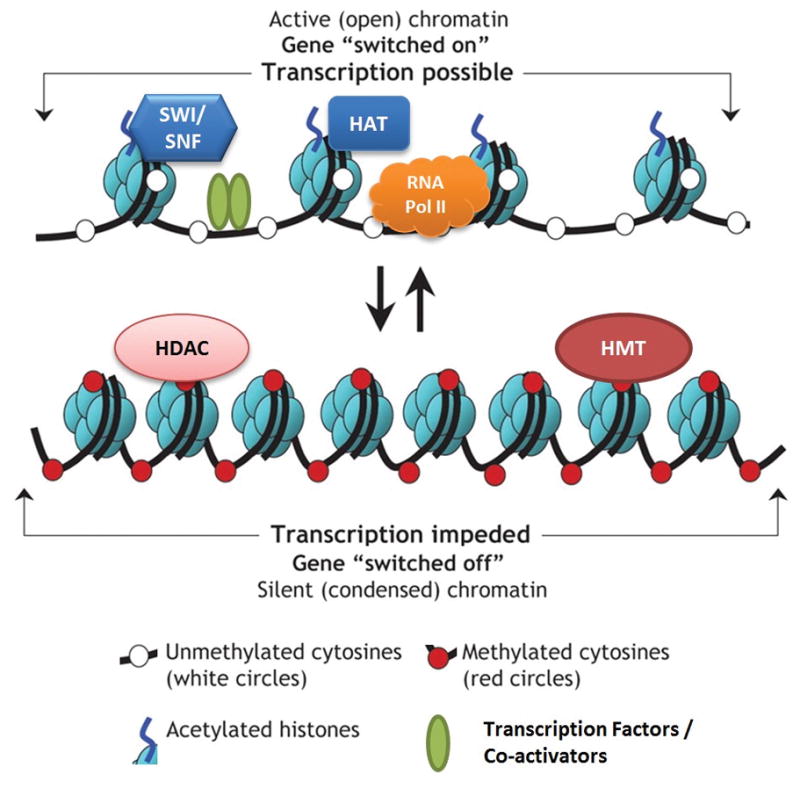
Epigenetic regulation of gene expression via chromatin remodeling. The diagram shows two generic chromatin activity states. At the top, active chromatin is open and accessible to transcription factors and polymerases, with nucleosomes spread apart, DNA typically in an unmethylated state and acetylation marks on histones. HAT is histone acetyltransferase, SWI/SNF is a nucleosome remodeling complex, RNA Pol II is RNA polymerase II. The lower panel shows the opposite inactive chromatin scenario, where the nucleosomes are tightly packed, the DNA is methylated and inaccessible to transcription factors, while histones have their acetylation marks removed. HDAC is histone deacetylase, HMT is histone methyltransferase. Figure is adapted from Luong, P. Basic Principles of Genetics, Connexions Web site. [http://cnx.org/content/m26565/1.1/] (2009) under a Creative Commons Attribution License ([http://creativecommons.org/licenses/by/3.0/CC-BY 3.0]).
Massive studies such as ENCODE [31] and Roadmap Epigenomics [32] have significantly advanced our understanding of genetic and epigenetic regulation. The ENCODE project aims to identify all functional elements in the genome, while RoadMap Epigenomics aims to elucidate epigenetic processes that contribute to human biology and disease. Both projects make extensive use of next-generation sequencing (NGS) to profile reference epigenomes and genome-wide protein-DNA binding patterns, including binding patterns for specific modified histones. The most recent culmination of these efforts was the publication of 111 reference epigenomes by RoadMap Epigenomics [32]. This study revealed epigenetic regulatory modules of coordinated activity, which are specific combinations of DNA methylation, histone modifications and other proteins that shape chromatin structure, which in turn determine transcriptional activity. These multi-layer data were used to classify genomic regions according to functional state [33]. The working models produced by RoadMap Epigenomics include a core 15 chromatin state model [32] and an expanded 18 chromatin state model (http://egg2.wustl.edu/roadmap/web_portal/chr_state_learning.html), the latter including twelve active and six inactive states. Active states include transcribed regions, active transcription start sites and their flanking regions, active enhancers and zinc finger protein binding sites. Inactive states include heterochromatin and repressed polycomb regions. This model, although complex, has already proven powerful for understanding regulation of gene expression.
2.3 Individual differences in epigenetic states and developmental plasticity
Epigenetic modifications to chromatin are affected by exposure to environmental factors, and any changes so induced are inherited mitotically in somatic cells [11]. Studies in human twins have shown that, while their epigenomes are very similar in early life, they diverge as the twins become older as a result of differing environmental exposures across the life course, in addition to stochastic effects [34]. Epigenetic changes in response to environmental factors may have evolved to provide plasticity in adaptation to environmental cues [11]. Through the phenomena of de novo epigenetic writing and mitotic stability, the effects of environmental factors can become embedded in the genome and persist to produce long-term phenotypic changes [35]. Example environmental factors with demonstrated developmental consequences include diet, toxins and stress. There is increasing recognition of the importance of this phenomenon for epigenetic translational research, because it provides concrete biological pathways that are involved in the persistence of environmental effects [36].
Epigenetic states can also vary between individuals because of genetic differences. In the case of methylation, one of the simplest examples involves polymorphic CpG sites [37]. If a nucleotide substitution ablates a CpG in some individuals, those individuals cannot be methylated at that locus. There are several examples of disease-associated polymorphic CpGs, suggesting that this is a significant contributor to individual differences in disease risk [38,39]. In addition to polymorphic CpGs, DNA sequence variation may also affect the binding of chromatin-interacting proteins and thus influence epigenetic states [40]. Thus, individual differences in epigenetic states, whether arising via genotype or maladaptive responses to environmental factors, can lead to disease.
3. Epigenetic Applications in Personalized Medicine
Epigenetic disease associations provide not only mechanistic clues to disease etiology, but can also function as diagnostic biomarkers. The developmental stage- and tissue-specificity of epigenetic marks has led to considerable interest in developing biomarkers that capitalize on these unique properties [41]. Furthermore, the fact that epigenetic marks are reversible has led to significant interest in the development of drugs with epigenetic modes of action [42,43].
3.1 Epigenetic biomarkers of disease
The largest body of work in disease epigenetics to date is on cancer. Since the first links between DNA methylation and cancer were established in the early 1980s, a number of epigenetic findings have been described, implicating several aspects of the epigenetic machinery. Some excellent reviews of cancer epigenetics have been published recently [44,45], so here we limit ourselves to epigenetic marks in cancer showing evidence or potential as diagnostic or prognostic biomarkers.
Current epigenetic biomarker applications predominantly involve DNA methylation [46]. In the United States, nucleic acid based tests intended for general clinical use are regulated by the Federal Drug Administration (FDA) as medical devices. Currently there are no FDA-approved tests that rely exclusively on epigenetic biomarkers. However, one commercially available test with an epigenetic component has received full FDA approval. This is ColoGuard®, a screening test for colorectal cancer in adults over 50. The test uses DNA methylation levels at BMP3 and NDRG4, in combination of mutated KRAS and an immunochemical assay for hemoglobin (Table 1). This test was reported to have superior sensitivity but slightly lower specificity for colorectal cancer compared to the traditional screening method, fecal immunochemical testing (FIT) [47]. However, more recent results suggest FIT may be more effective and less costly than ColoGuard®, the latter necessitating either very high patient uptake or a 60% reduction in cost per test to become the preferred testing method [48]. This illustrates the economic barriers that diagnostic tests must overcome, beyond the demonstration of efficacy and reproducibility, in order to become widespread.
Table 1.
Commercially available epigenetic diagnostic tests in the United States.
| Product | Proprietor/Launch year | Disease | Specimen | Epigenetic Targets | Regulation |
|---|---|---|---|---|---|
| Cologuard | Exact sciences/2014 | Colorectal cancer | Stool | DNA methylation of NDRG4 and BMP3 (plus other genetic markers) | FDA |
| ConfirmMDx | MDxHealth/2012 | Prostate cancer | Tissue | DNA methylation of GSTP1, RASSF1 and APC. | LDT/CLIA |
| AssureMDx | MDxHealth/2016 | Bladder cancer | Urine | DNA methylation of TWIST, ONECUT2 and OTX1 (plus other genetic markers) | LDT/CLIA |
Two other epigenetic tests are currently available in the US, classified as Laboratory Developed Tests (LDTs) and regulated under the Clinical Laboratory Improvement Amendments (CLIA) program. This means that the test may only be conducted “in house” in the laboratory where it was developed, once the lab meets CLIA performance standards. The two tests are ConfirmMDx and AssureMDx, for prostate cancer and bladder cancer respectively. Hypermethylation of the glutathione S-transferase gene (GSTP1) promoter in prostrate cancer was first shown in the 1990s [49]. This marker, plus APC and RASSF1, are now components of the ConfirmMDx test (Table 1), which is used to address false-negative prostate biopsy concerns [50]. The AssureMDx test for bladder cancer involves the analysis of DNA methylation levels of three genes (TWIST1, ONECUT2 and OTX1) in combination with mutation analysis of three others [51].
In lung cancer, the DNA methylation of the SHOX2 gene was reported to be an accurate marker for identifying lung cancer based on analysis of bronchial aspirates [52]. In Europe, this biomarker is now commercially available as the Epi proLungVR BL Reflex Assay [53]. However, this test has not yet received regulatory approval for use in the USA.
In breast and ovarian cancer, hypermethylation of the BRCA1 promoter region has been observed repeatedly [54,55]. BRCA1 is also thought to epigenetically repress expression of the oncogenic microRNA miR-155 via a mechanism involving histone deacetylase 2 (HDAC2) [56]. A recent study by Anjum et al. (2014) identified a blood cell DNA methylation signature at BRCA1 that was able to predict breast cancer risk several years prior to diagnosis [57]. However, this biomarker is not yet available in a commercial kit or test.
The biomarker potential of circulating cell-free DNA (cfDNA) has been investigated in breast cancer and other cancers. Circulating cfDNA is extracted from plasma or serum and is derived from dying tumor cells that release their DNA into the bloodstream. Kloten et al (2013) used a panel of three genes (ITIH5, DKK3 and RASSF1A) that showed hypermethylation in serum cfDNA from breast cancer patients and found these could discriminate between patients and controls with a sensitivity of 67% and specificity of 69% [58]. Fackler et al. (2014) followed this with a panel of 10 genes and cancer-specific DNA was detected in sera with a sensitivity of 91% and specificity of 96% in the test samples [59]. The researchers of the latter study are reportedly working with the diagnostics company Cepheid to bring this test to market [60].
While epigenetic studies of cancer are arguably the most advanced relative to other areas, several diseases have shown promising findings, particularly with respect to DNA methylation. These include neurological disorders such as Alzheimer’s Disease [61,62] and Parkinson’s Disease [63], autoimmune disorders such as systemic lupus erythematosus [64], and psychiatric disorders such as schizophrenia [65,66] and autism [67]. Despite these advances, there are no currently available diagnostic kits for these diseases that employ epigenetic markers. Nevertheless, it is hoped that the clinical value of epigenomics already seen in oncology will be replicated in these areas [68].
3.2 Epigenetic drugs
The dynamic and reversible nature of epigenetic modifications is of particular relevance to drug development, as it implies that specific disease-associated epigenetic states may be reversible with pharmacological treatment [69]. This segment will summarize current and potential “epidrugs”, or drugs with epigenetic modes of action. Epidrugs are classified according to their respective target enzymes, and include the following: DNA N-methyl transferase inhibitors (DNMTi), histone acetyltransferase inhibitors (HATi/KATi), histone methyltransferase inhibitors (HMTi/KMTi), histone N-methyl lysine demethylase inhibitors (HDMi/KDMi), histone deacetylase inhibitors (HDACi/KDACi), and bromodomain inhibitors. Currently there are two classes of epigenetic drugs that have been approved by FDA for clinical use in the United States: DNMTi and HDACi (see Table 2).
Table 2.
Classification of US FDA-approved epigenetic drug classes according to mechanism of action.
| Mechanism of Action | Active Ingredient (Trade name®, Proprietor) | Date of Approval | Indication(s) |
|---|---|---|---|
| DNA N-Methyltransferase Inhibitor (DNMTi) | Azacitidine (Vidaza®, Celgene) | May 19, 2004 | Chronic Myelomonocytic Leukemia. Myelodysplastic Syndrome i. |
| Decitabine(Dacogen®, Otsuka) | May 2, 2006 | Chronic Myelomonocytic Leukemia. Myelodysplastic Syndromes ii. | |
|
| |||
| Histone Deacetylase inhibitors (HDACi) | Vorinostat(Zolinza®, Merck) | October 6, 2006 | Cutaneous manifestations in patients with cutaneous T-Cell Lymphoma (CTCL) who have progressive, persistent or recurrent disease on or following two systemic therapies. |
| Romidepsin(Istodax®, Celgene) | November 5,2009 | Cutaneous T-cell Lymphoma (CTCL) iii, Peripheral T-cell Lymphoma (PTCL) iii | |
| Belinostat(Beleodaq®, Spectrum Pharmaceuticals) | July 3, 2014 | Relapsed or Refractory Peripheral T-cell Lymphoma (PTCL) | |
| Panobinostat(Farydak®, Novartis) | February 23, 2015 | Multiple Myeloma after receiving at least 2 prior regimens, including bortezomib and an immunomodulatory agent iv | |
Subtypes: refractory anemia or refractory anemia with ringed sideroblasts (if accompanied by neutropenia or thrombocytopenia or requiring transfusions), refractory anemia with excess blasts, refractory anemia with excess blasts in transformation.
Including 90 previously treated and untreated, de novo and secondary MDS of all French-American-British subtypes 91 (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, 92 refractory anemia with excess blasts in transformation, and 93 intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups).
In patients who have received at least one prior systemic therapy.
In combination with bortezomib and dexamethasone.
The first approved epidrug in the US was azacitidine (Vidaza, Azadine), a DNMTi indicated to treat chronic myelomonocytic leukemia and myelodysplastic syndrome. Azacitidine was approved in 2004 and quickly followed by decitabine (Dacogen) with same indication two years later. Both drugs cause broad hypomethylation that leads to cellular dysregulation that most seriously affects rapidly dividing cells. It is important to note that these drugs are not highly locus-specific and these agents can cause hypomethylation at many genomic sites. Even though current drugs are designed to favorably induce genes that have been silenced in cancer [70], they may also activate the expression of prometastatic genes as well as oncogenes [71]. There remains a need to develop more selective DNMTi to improve the efficacy and reduce side effects for this class of drug.
The potential application of DNMTi to other diseases is also under investigation and examples include multiple sclerosis [72], HIV [73], pain [74] and memory [75]. For example, DNMT activity was observed in HIV-1 infection of CD4(+) T-cells in vitro and induced hypermethylation of distinct cellular promoters [73]. Studies from Rajasethupathy et al. suggested that DNA methylation is necessary for serotonin-dependent long-term facilitation in memory formation [76]. For a curative therapy of AIDS patients, a combination of antiretroviral drugs and epidrugs has been suggested for the reactivation of latent HIV-1 genomes. These epidrugs include DNMTi, HDACi, histone methyltransferase inhibitors (HMTi) and histone demethylase inhibitors [73].
The HDAC inhibitors suberoylanilide hydroxamic acid (SAHA, vorinostat, in 2006) and romidepsin (depsipeptide, in 2009) have proven to be successful in cancer therapeutics [77,78]. These agents cause the accumulation of acetylated histones and prevent progression of tumor cells. Vorinostat was the first HDACi to be approved by the FDA, indicated for cutaneous manifestations in patients with cutaneous T-cell lymphoma (CTCL). Panobinostat is the latest HDACi approved by the FDA in 2015 and is indicated for the treatment of multiple myeloma in combination with bortezomib and dexamethasone. Outside the US, HDACi approvals vary. In Europe, for example, only panobinostat has been approved for general clinical use (to treat multiple myeloma), while belinostat received orphan designation for peripheral T-cell lymphoma (PTCL). In China, an additional HDACi known as chidamide (Epidaza®), was approved for treatment of PTCL by the Chinese FDA in 2015. Although most HDACi are approved for cancer type indications, studies have suggested potential roles in schizophrenia [79] and Type2 diabetes [80]. However, similar to the DNMTi drugs, current HDACi have broad effects across the genome and lack locus-specificity. These drugs can have serious side effects [43] and use of currently approved HDACi in cancer is often indicated only after other treatments have failed, or as combination therapies (Table 2).
Besides these two approved epidrug classes, HMTi and bromodomain inhibitors are other emerging epidrug classes under development. Pinometostat is a small molecule inhibitor of the histone methyltransferase DOT1L for the treatment of MLL-r leukemia [81]. Tazemetostat is an orally administered, first-in-class small molecule HMTi that targets the EZH2 transcriptional repressor to treat multiple types of hematological malignancies and genetically defined solid tumors [82]. GSK3326595, an inhibitor of the transcriptional regulator protein arginine methyltransferase 5 (PRMT5), is also in phase 1 clinical trial. Bromodomain proteins are readers that recognize acetylated lysine and transduce the gene activation signal [27]. OTX-015 and CPI-0610 are bromodomain protein inhibitors both in phase I trials for cancers. These drugs target a specific family of bromodomain proteins, known as Bromodomain Extra-Terminal motif (BET) proteins [83]. Another BET inhibitor, Apabetalone (RVX-208), is in Phase III clinical trials for cardiovascular events in Type 2 diabetes subjects with coronary artery disease. These example epidrugs, and several more are advancing through the clinical trial pipeline, are summarized in Table 3. In this table, we focus only on epidrugs in active or planned clinical trials registered in the US (clinicaltrials.gov) and show the latest phase trial for each drug, plus any trials for indications outside oncology. We restrict our listing of early phase cancer indications because these are too numerous to list concisely.
Table 3.
Classification of epigenetic drug classes in active clinical trials registered in the US (clinicaltrials.gov) according to mechanism of action.
| Mechanism of action | Active ingredient (Proprietor) | Indication | Clinical Trial Phase | Trial Ref ID | |
|---|---|---|---|---|---|
| BET Bromodomain Inhibitors | Apabetalone/RVX-208 (Resverlogix) | High-risk type 2 diabetes mellitus with coronary artery disease | Phase III | NCT02586155 | |
|
| |||||
| CPI-0610 (Constellation Pharmaceuticals) | Malignant Peripheral Nerve Sheath Tumor | Phase II | NCT02986919 | ||
|
| |||||
| INCB054329 (Incyte) | Advanced malignancies | Phase I/II | NCT02431260 | ||
|
| |||||
| GSK525762 (GlaxoSmithKline) | Carcinoma and hematological malignancies | Phase I | NCT01587703, NCT01943851 | ||
| GSK2820151 (GlaxoSmithKline) | Advanced or recurrent solid tumors | Phase I | NCT02630251 | ||
| ZEN-3694 (Zenith Epigenetics) | Metastatic castration-resistant prostate cancer (mCRPC) i | Phase I | NCT02711956i, NCT02705469 | ||
| OTX015/MK-8628 (Merck) | Selected advanced solid tumors | Phase I | NCT02698176 | ||
| TEN-010/RO6870810 (Hoffmann-La Roche) | Advanced Solid Tumors; Acute Myeloid Leukemia | Phase I | NCT01987362, NCT02308761 | ||
| FT-1101 (Forma therapeutics) | Relapsed/Refractory Acute Leukemia | Phase I | NCT02543879 | ||
| BMS-986158 (Bristol-Myers Squibb) | Advanced Solid Tumors | Phase I | NCT02419417 | ||
| Mivebresib/ABBV-075 (AbbVie) | Advanced cancers | Phase I | NCT02391480 | ||
|
| |||||
| DNA N-Methyltransferase Inhibitor (DNMTi) | Guadecitabine (Astex) | Acute Myeloid Leukemia | Phase III | NCT02920008 | |
|
| |||||
| TdCyd (NCI) | Advanced Solid Tumors | Phase I | NCT02423057 | ||
|
| |||||
| Histone Deacetylase inhibitors (HDACi) | Non-Selective | Entinostat (Syndax) | Advanced Hormone Receptor positive (HR+) Breast Cancer ii | Phase III | NCT02115282 |
|
| |||||
| Givinostat/ITF2357 (Italfarmaco) | Chronic Myeloproliferative Neoplasms | Phase II | NCT01761968 | ||
| Resminostat (4SC AG) | Advanced Stage Mycosis Fungoides or Sézary Syndrome | Phase II | NCT02953301 | ||
| Quisinostat/JNJ-26481585 (Janssen) | Ovarian cancer iii | Phase II | NCT02948075 | ||
| Pracinostat/SB939 (NCIC Clinical Trials Group) | Acute Myeloid Leukemia iv, Myelofibrosis v | Phase II | NCT01912274iv NCT02267278v | ||
|
| |||||
| Tefinostat (Chroma) | Hepatocellular Carcinoma | Phase I/II | NCT02759601 | ||
|
| |||||
| AR-42 (Celgene) | Relapsed multiple myeloma vi | Phase I | NCT02569320 | ||
| CUDC-907 (Curis) | Multiple Myeloma | Phase I | NCT01742988 | ||
|
| |||||
| HDAC 1&4 Selective | Mocetinostat (Mirati) | Non-Small Cell Lung Cancer vii | Phase II | NCT02954991 | |
|
| |||||
| HDAC 6 Selective | ACY 241 (Acetylon) | Non-Small Cell Lung Cancer vii | Phase I | NCT02635061 | |
| KA2507 (Karus) | Solid tumor | Phase I | NCT03008018 | ||
|
| |||||
| Histone Lysine De-methylases (KDM/HDM) | LSD1 inhibitors | GSK2879552 (GlaxoSmithKline) | Myelodysplastic Syndrome | Phase II | NCT02929498 |
|
| |||||
| Tranylcypromine (Martin Luther Universität) | Relapsed/Refractory Acute Myeloid Leukemia viii | Phase I/II | NCT02261779 | ||
| INCB059872 (Incyte) | Advanced Malignancies | Phase I/II | NCT02712905 | ||
|
| |||||
| IMG-7289 (Imago BioSciences) | Acute Myeloid Leukemia | Phase I | NCT02842827 | ||
|
| |||||
| Histone Lysine Methyl Transferase (KMT/HMT) | DOT1L inhibitor | Pinometostat/EPZ-5676 (Epizyme) | Relapsed/Refractory Leukemias ix | Phase I | NCT01684150 |
|
| |||||
| EZH1/2 inhibitor | DS-3201b (Daiichi Sankyo) | Lymphomas | Phase I | NCT02732275 | |
|
| |||||
| EZH2 inhibitor | Tazemetostat/EPZ-6438 (Epizyme) | Advanced Solid Tumors or B-cell lymphomas | Phase I/II | NCT01897571 | |
| MAK683 (Novartis) | Diffuse Large B-cell Lymphoma | Phase I/II | NCT02900651 | ||
|
| |||||
| GSK2816126 (GlaxoSmithKline) | Lymphomas, Multiple Myeloma, Solid Tumors | Phase I | NCT02082977 | ||
|
| |||||
| PRMT5 inhibitor | GSK3326595 (GlaxoSmithKline) | Solid Tumors and Non-Hodgkin’s Lymphoma | Phase I | NCT02783300 | |
In combination with Enzalutamide.
In combination with Aromasin (Exemestene).
In combination with Paclitaxel and Carboplatin chemotherapy.
In combination with Azacitidine.
In combination with Ruxolitinib.
In combination with Pomalidomide.
In Combination with Nivolumab.
In combination with all-trans-retinoic acid (ATRA) chemotherapy.
Only patients with rearrangements involving the MLL gene.
LSD1: Lysine (K)-specific demethylase 1A, DOT1L: Disruptor of telomeric silencing 1-like, EZH1 or EZH2: Enhancer Of Zeste Homolog 1 or 2 Polycomb Respressive Complex 2 Subunit, PRMT5: Protein Arginine Methyl Transferase 5.
Finally, several HATi are in preclinical studies at time of writing. Aberrant function of HATs, also called lysine acetyltransferases (KATs), is correlated with cancer and other diseases [84]. HATi are great candidates with potential therapeutic utility, but current HATi only have moderate potency and specificity and none are in clinical trial at time of writing. Nevertheless, some HATi have shown efficacy in preclinical studies. Compound C646 is a pyrazolone-containing small molecule inhibitor of the p300/CBP HAT subfamily [85]. It has been shown to cause growth arrest in melanoma cell lines and inhibit cancer cell growth in prostate and lung cancer cell lines. PU139 is a pyridoisothiazolone that inhibits several HAT subfamilies and was shown to block neuroblastoma xenograft growth in mice [86]. These agents and others in development are indicative that HATi are still in infancy relative to other epigenetic drugs, but they show enormous promise and need further investment to reach their potential as therapeutic compounds.
3.3 Epigenetic biomarkers of drug response
As a natural extension of pharmacogenetics, it is possible to use epigenetic biomarkers to predict drug response. While none have yet achieved regulatory approval for clinical use, a small number of examples are established in the literature. Among the best known is DNA methylation of the MGMT promoter. This gene encodes a DNA repair enzyme (O6-alkylguanine DNA alkyltransferase). Methylation in the promoter region of MGMT is associated with better response to alkylating neoplastic agents like temozolomide, as first shown in glioblastoma by Esteller et al. (2000) [87] and later by Hegi et al. (2005) [88]. The mechanism of effect is as follows. Temozolamide alkylates or methylates DNA at the N-7 or O-6 positions of guanine residues and the resulting DNA damage triggers tumor cell death. Hypomethylation of MGMT leads to expression of O6-alkylguanine DNA alkyltransferase, which can repair the DNA damage, whereas hypermethylation leads to silencing of the gene and thus greater susceptibility to the drug. In addition to glioma, a role for MGMT in predicting response to metastatic colorectal cancer (mCRC) treatment has also been suggested [89].
Other published epigenetic biomarker examples include GSTP1 and BRCA1. Methylation of the promoter of GSTP1 is correlated with survival in breast cancer patients and may be predictive of treatment efficacy with doxorubicin [90] or DNA methyltransferase (DNMT) inhibitors [91]. The BRCA1 gene plays a role in DNA damage response and hypermethylation of the BRCA1 promoter region may be predictive of enhanced sensitivity to platinum-derived drugs in cancer cell lines and xenografted tumors; it also may be predictive of increased time to relapse and survival in ovarian cancer patients under cisplatin treatment [92].
The impact of epigenetics in drug response has been investigated beyond oncology. For example, methylation of the P2 promoter of the IGF1 gene affects transcriptional response to growth hormone (GH) [93]. GH is mainly used to treat children with short stature due to growth hormone deficiency. Ouni et al. [93] measured P2-driven and total IGF1 transcripts before and 12 h after the GH injection and found an increase in P2-driven transcripts with a very strong inverse correlation with CG-137 methylation. This correlation accounted for ~ 25% of the variability in the response to GH.
3.4 Epigenetic modification of drug absorption, distribution, metabolism and excretion (ADME) genes
ADME genes encode transporters, plasma proteins, and drug metabolizing enzymes that are responsible for absorption, distribution, metabolism and excretion of xenobiotics. Genetic variation at ADME genes has proven extremely successful in predicting individual differences in pharmacokinetics, particularly in the case of drug metabolizing phenotypes associated with the CYP450s, as mentioned above. However, there remain large individual differences in drug metabolism unexplained by genetic variation that have led to the suggestion that epigenetics may substantially influence these phenotypes [94]. Unfortunately, research to date has not yet directly addressed this question, but individual variation in epigenetic states of ADME genes has been correlated with a range of outcomes. For example, Parkinson’s disease has been associated with hypomethylation of the CYP2E1 gene promoter in the brain [95]. Methylation levels at CYP1B1 [96] and CYP1A1 [97] have been associated with prostate cancer, and CYP2W1 with colon cancer [98]. Methylation levels at the drug transporter genes OCT1 [99] and OCT2 [100], responsible for the renal excretion of drugs, have been associated with renal carcinoma. These findings demonstrate the existence of inter-patient variability in ADME gene epigenetic states, some of which have functional effects on gene expression. However, the extent of normal epigenetic variation at these loci in the population and the extent to which it will affect pharmacokinetic phenotypes remains to be determined.
3.5 Conclusion
Epigenomic medicine is already here, with numerous epigenetic disease associations reported, six epidrugs and a handful of epigenetic biomarker tests available the US, plus a small number of other products available worldwide. The largest number of findings and applications to date is in the field of oncology. However, the field of epigenetics is only a few decades old and epigenomic medicine is a very recent arrival, so we are still in early days. The perceived benefits that epigenomics will bring to healthcare are emphatically illustrated by the large number of epidrugs currently in development and the large sums of research dollars spent on large-scale discovery efforts such as RoadMap Epigenomics. To drive the field forward, epigenomic medicine needs to expand beyond cancer. Also, while significant efforts are being devoted to bringing new epidrugs to market, more efforts must be devoted to developing new epigenetic biomarkers, of which there are few.
4. Expert Commentary
Several factors are currently driving innovation in epigenomic medicine. First is the general level of interest in the field, which is high. Second is the ongoing characterization of reference epigenomes to enrich and accelerate research efforts. Third is the availability of powerful methods such as next-generation sequencing (NGS) to characterize epigenomes. Discussion of technical methods is largely outside the scope of this article and reviews have been published elsewhere [101,102]. However, with NGS approaches already in use to characterize genome-wide DNA methylation and protein-DNA binding patterns, we would argue that technology is not a bottleneck for the advancement of epigenomic medicine.
Considering epigenomic biomarker research, among the most significant difficulties are data complexity and the clean interpretation of findings [103]. Unlike studies of genotype, epigenomics has a direction of causality problem. While epigenetic biomarkers may be predictive of disease state or drug response, epigenetic changes are also inducible by pharmacological treatments [11,104]. As a result, there is the risk that epigenetic differences between cases and controls in an epigenome-wide association study could be the result of drug treatment in cases, rather than causal variation. Furthermore, evidence from genome-wide studies suggests that not all epigenetic changes are functional or cause identifiable changes to gene expression [105]. Targeting specific populations, such as drug-naïve patients, may go some way to solving issues related to the direction of causality, but it is certain that experimental model systems will be needed to adequately disentangle causality and establish functionality of epigenetic changes.
Another complexity is that epigenetic modifications are cell-specific. While this is in many ways an advantage, and can give precise insight into the workings of the cell of origin, it also leads to some challenges in sample collection, particularly with respect to clinical studies. Blood DNA is the most readily-accessible source from humans, but the extent to which blood DNA methylation is reflective of methylation changes in other tissues is debated and it seems there may not be a hard and fast rule with respect to which changes are reflected in blood as compared to which are not. Aging epigenetic signatures, also known as the “epigenetic clock”, appear to transcend tissue barriers [106], but the extent to which a blood DNA methylation mark is informative about a disease of, for example, the lung or heart remains an open question. Circulating cfDNA is an exception, since it is sourced from the diseased tissue of interest and is merely liberated into the bloodstream.
While these considerations apply to the discovery of novel epigenetic biomarkers, a separate set of considerations apply to novel epigenetic drugs. Paramount among priorities for future epidrug development is improving target specificity. This can be viewed in two ways. First, as mentioned above, current drugs lack genomic locus specificity and affect DNA methylation or histone modifications somewhat indiscriminately. To truly enable precision correction of aberrant changes, some sort of nucleic acid targeting adjunct is likely to be required. While antisense RNA (MG98) has already been used to modulate DNMT activity with some success [107], it is difficult to speculate how this could be used to target epigenetic modifications at specific target loci. On the other hand, it may be possible to capitalize on the locus targeting abilities of CRISPR/Cas9 systems to deliver epigenetic modifying agents to specific loci. Indeed, epigenome editing has already been demonstrated using this broad approach [108]. A second consideration involves the specificity of epidrugs to specific members of families of chromatin modifying enzymes. For example, there are numerous human DNMTs and HDAC enzymes with somewhat different functions and substrate specificities but currently available DNMTi and HDACi are non-selective and inhibit many isozymes. However, some drugs currently in clinical trials appear to be more selective, e.g. mocetinostat that inhibits only HDAC 1 and 4 (see Table 3). Thus, the problem of specificity does not appear to be insurmountable. To conclude, we mention two areas, one technological and one clinical, that we consider to be of significant interest going forward.
The advent of chromatin conformation capture (3C) sequencing technology marked the beginning of a new era in precision medicine and our understanding of epigenomic regulation. Its evolution into Hi-C technology allows insight into the well-organized 3D structure of the human genome within the nucleus [109]. Numerous studies demonstrated highly conserved topologically associated domains (TADs) - spatially close units of chromatin bringing together enhancers, promoters of genes, and other regulatory elements. These TADs have well-defined boundaries marked by strongly interacting chromatin regions (chromatin loops) [110]. TADs harbor multiple active RNA polymerases anchored to a nuclear substructure, with genes within TADs showing co-expression patterns [111]. TADs are increasingly recognized as regulatory units orchestrating expression of thousands of genes, thus implying a new “druggable nucleosome” paradigm. Disruption of TAD boundaries due to genetic variants leads to fusion of TADs and/or formation of smaller TADs [112,113]. This is a frequent event in cancer, leading to coordinated expression of oncogenes [114,115]. With the dropping costs of sequencing using personalized TAD abnormalities for diagnostic, prognostic and, potentially, treatment purposes will soon complement traditional gene expression and epigenetic tests.
In the clinical arena, aging is an area where epigenomic medicine may make an impact. Older adults are at increased risk for adverse drug events and this may be partly because aging is associated with changes in physiology that can affect drug pharmacokinetics and pharmacodynamics [116]. The clinical challenge is to identify those patients who are more likely to experience an adverse drug event or altered drug response among the older adult population when weighing the risk versus the benefit of a drug therapy. Chronologic age alone is insufficient as an indicator that dosage adjustment or avoidance of a particular therapeutic agent is warranted. Pharmacogenetic information alone is also insufficient, as altered drug response and risk of adverse drug events changes across the lifespan while genotype remains constant [117,118]. Epigenetic alterations may be a better indicator than chronological age for personalizing drug therapy for the older population. For example, it has been proposed that epigenetic regulation of cytochrome P450 (CYP) enzymes responsible for drug metabolism through DNA methylation may result in altered drug exposure in geriatric patients [119]. More research is needed to elucidate the relationships between epigenetics and drug exposure and response during senescence, but is a promising alternative to chronologic age for adjusting pharmacotherapy in older adults.
Five-year view
We expect to see the introduction of several new epidrugs in the next five years, given that many are currently in later stage clinical trials. Perhaps the most significant change in this area will be the introduction of epidrugs with indications outside cancer. For example, Apabetalone (RVX-208) from Resverlogix is in Phase III trials for high risk Type 2 diabetes mellitus patients with coronary artery disease (Table 3). Resverlogix also reports the planning of Phase II trials for Alzheimer’s disease (www.resverlogix.com/programs/rvx-208-clinical-development/). Successful approval for either of these indications would be a first step for epidrugs outside oncology. We also expect to see new classes of epidrug enter clinical trials. As mentioned above, several HATi are in preclinical development and show therapeutic promise. There is growing recognition of the importance of histone acetylation in many diseases, including cancer, so we expect the first HATi to enter clinical trials in three to five years.
Given the high level of current research interest, we expect a number of epigenetic biomarker tests to become commercially available on a five-year horizon. There are numerous clinical trials currently ongoing to evaluate epigenetic biomarkers. In addition to the developments cited in Sections 3.1 and 3.3 above, clinical trials of DNA methylation biomarkers for disease diagnosis, prediction of treatment efficacy and even quantifying toxin exposure (see for example clinical trial NCT01815385) are ongoing or planned. One area that should see significant growth is the use of epigenetic markers on cfDNA. As mentioned in Section 3.1, cfDNA is sourced from dead cells that have released their contents into the bloodstream. Diseased tissue DNA contributes significantly to circulating cfDNA, so cfDNA is a more direct assay of the diseased tissue methylation levels than surrogate markers from readily accessible cell types such as lymphocytes. Double stranded DNA from tumor exosomes also has this property [120]. This is important given the cell-specific nature of methylation patterns. With cfDNA biomarker tests under development (Section 3.1), we expect more research in this area and perhaps clinical introduction of cfDNA methylation tests within five years.
As researchers continue to seek out new epigenetic biomarkers, we will see more large-scale epigenome-wide studies of complex diseases. These studies will primarily focus on DNA methylation but we expect the current, prevailing use of high density microarrays to largely give way to NGS as costs continue to fall. Whole genome bisulfite shotgun sequencing (WGBS) is poised to become the dominant method of choice, because it is the only way to assay DNA methylation at single base resolution. Correlation between neighboring methylated sites is low beyond very short distances [40], so this level of resolution is necessary for a comprehensive genome-wide DNA methylation analysis. While cost is the primary factor limiting large-scale implementation of WGBS, improved analysis methods will be needed to extract maximum information from these complex data. Research into whole genome sequence analysis methods will intensify over the next five years with efforts such as the US Precision Medicine Initiative, a >$200 million program to develop individualized therapy on a large scale. Analytical developments made in sequence variant analysis will likely bleed over to advance NGS methods for epigenetic studies.
Although epigenetic biomarker research will continue to focus on DNA methylation, it is of note that the first epigenome-wide association study focusing on histone modifications was recently published. In this study, Sun et al. (2016) compared genome-wide histone acetylation levels in autism spectrum disorder cases to control subjects [121]. Following this initial demonstration, it is certain that epigenome-wide association studies of histone modifications will follow shortly for other disorders. In addition to histones, inroads may be made into large-scale epidemiological studies of 5-hydroxymethylcytosine or other methylation variants.
In sum, the next five years should see intensifying research efforts in personalized epigenetic medicine, the introduction of several new epidrugs, initiation of clinical trials for new drug classes, and the clinical introduction of novel DNA methylation biomarker tests. Overall it should prove an exciting time for this nascent area.
Key issues.
Epigenetic data has distinct information content to genotype information because epigenetic marks, including DNA methylation and histone modifications, are developmentally dynamic and tissue-specific.
Epigenetic marks have profound effects on genetic regulation.
Aberrant epigenetic regulation may cause disease. Aberrant epigenetic marks may arise from genetic differences or may arise over the life course via maladaptive response to environmental factors.
There are numerous specific examples of epigenetic marks associated with disease and some are validated as diagnostic biomarkers for cancer.
Epigenetic marks are reversible, therefore much effort has been expended in developing drugs with epigenetic modes of action. All current epigenetic drugs are indicated for cancer but many more are in development.
A small number of epigenetic biomarkers of drug response have been reported but none are yet approved for general clinical use.
Next-generation sequencing will enable the discovery of new biomarkers, with the expectation this area will continue to focus on DNA methylation in the near future
A major challenge in epigenetic drug development is targeting specific genetic loci. Solutions may involve adaptations of the CRISPR/dCas9 system of epigenome editing.
Future directions may include the “druggable nucleosome” concept and life stage-specific biomarkers of drug response.
Acknowledgments
Funding
JL McClay was partially supported by grant R21MH099419 from the US National Institutes of Health. MG Dozmorov was supported in part by the Virginia Commonwealth University Presidential Research Quest award. R Huang was supported by an American Cancer Society Institutional Research Grant. Sponsors had no role in the preparation of this manuscript.
Footnotes
Declaration of Interest
JL McClay received honoraria from the American College of Clinical Pharmacy to present on the topic of pharmacoepigenomics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Sykiotis GP, Kalliolias GD, Papavassiliou AG. Pharmacogenetic Principles in the Hippocratic Writings. J Clin Pharmacol. 2005;45:1218–1220. doi: 10.1177/0091270005281091. [DOI] [PubMed] [Google Scholar]
- 2.Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004;25:193–200. doi: 10.1016/j.tips.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 3.McCarthy JJ, McLeod HL, Ginsburg GS. Genomic medicine: a decade of successes, challenges, and opportunities. Sci Transl Med. 2013;5:189sr4. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- 4.Ritchie MD. The success of pharmacogenomics in moving genetic association studies from bench to bedside: study design and implementation of precision medicine in the post-GWAS era. Hum Genet. 2012;131:1615–1626. doi: 10.1007/s00439-012-1221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardon LR, Harris T. Precision medicine, genomics and drug discovery. Hum Mol Genet. 2016;25:R166–R172. doi: 10.1093/hmg/ddw246. [DOI] [PubMed] [Google Scholar]
- 7.Tracy TS, Chaudhry AS, Prasad B, et al. Interindividual Variability in Cytochrome P450-Mediated Drug Metabolism. Drug Metab Dispos Biol Fate Chem. 2016;44:343–351. doi: 10.1124/dmd.115.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Z-X, Chen X-W, Zhou Z-W, et al. Impact of physiological, pathological and environmental factors on the expression and activity of human cytochrome P450 2D6 and implications in precision medicine. Drug Metab Rev. 2015;47:470–519. doi: 10.3109/03602532.2015.1101131. [DOI] [PubMed] [Google Scholar]
- 9.Bock C. Epigenetic biomarker development. Epigenomics. 2009;1:99–110. doi: 10.2217/epi.09.6. [DOI] [PubMed] [Google Scholar]
- 10.Ladd-Acosta C. Epigenetic Signatures as Biomarkers of Exposure. Curr Environ Health Rep. 2015;2:117–125. doi: 10.1007/s40572-015-0051-2. [DOI] [PubMed] [Google Scholar]
- 11.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 12.Goldberg AD, Allis CD, Bernstein E. Epigenetics: A Landscape Takes Shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Noble D. Conrad Waddington and the origin of epigenetics. J Exp Biol. 2015;218:816–818. doi: 10.1242/jeb.120071. [DOI] [PubMed] [Google Scholar]
- 14.Riggs A, Russo V, Martiensen R. Epigenetic Mechanisms of Gene Regulation. Plainview, NY: Cold Spring Harbor Laboratory Press; 1996. [Google Scholar]
- 15.Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 17.Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bachman M, Uribe-Lewis S, Yang X, et al. 5-Formylcytosine can be a stable DNA modification in mammals. Nat Chem Biol. 2015;11:555–557. doi: 10.1038/nchembio.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastor WA, Pape UJ, Huang Y, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 22.Luger K, Mäder AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 23.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014;28:672–682. doi: 10.1101/gad.238873.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marques M, Laflamme L, Gervais AL, et al. Reconciling the positive and negative roles of histone H2A.Z in gene transcription. Epigenetics. 2010;5:267–272. doi: 10.4161/epi.5.4.11520. [DOI] [PubMed] [Google Scholar]
- 26.Entrevan M, Schuettengruber B, Cavalli G. Regulation of Genome Architecture and Function by Polycomb Proteins. Trends Cell Biol. 2016;26:511–525. doi: 10.1016/j.tcb.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 28.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 29.Ivanov M, Barragan I, Ingelman-Sundberg M. Epigenetic mechanisms of importance for drug treatment. Trends Pharmacol Sci. 2014;35:384–396. doi: 10.1016/j.tips.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Hansen K, Helin K. Epigenetic inheritance through self-recruitment of the polycomb repressive complex 2. Epigenetics. 2009;4:133–138. doi: 10.4161/epi.4.3.8483. [DOI] [PubMed] [Google Scholar]
- 31.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32**.Roadmap Epigenomics Consortium. Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. A tour de force revealing baseline epigenetic patterns and regulation in human tissues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ernst J, Kheradpour P, Mikkelsen TS, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 36.Rutter M. Why is the topic of the biological embedding of experiences important for translation? Dev. Psychopathol. 2016;28:1245–1258. doi: 10.1017/S0954579416000821. [DOI] [PubMed] [Google Scholar]
- 37.Zhi D, Aslibekyan S, Irvin MR, et al. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics. 2013;8:802–806. doi: 10.4161/epi.25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou D, Li Z, Yu D, et al. Polymorphisms involving gain or loss of CpG sites are significantly enriched in trait-associated SNPs. Oncotarget. 2015;6:39995–40004. doi: 10.18632/oncotarget.5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cazaly E, Charlesworth J, Dickinson JL, et al. Genetic Determinants of Epigenetic Patterns: Providing Insight into Disease. Mol Med. 2015;21:400–409. doi: 10.2119/molmed.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClay JL, Shabalin AA, Dozmorov MG, et al. High density methylation QTL analysis in human blood via next-generation sequencing of the methylated genomic DNA fraction. Genome Biol. 2015;16:291. doi: 10.1186/s13059-015-0842-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.García-Giménez JL. Epigenetic Biomarkers and Diagnostics. Academic Press; 2015. [Google Scholar]
- 42.Szyf M. Epigenetics, DNA methylation, and chromatin modifying drugs. Annu Rev Pharmacol Toxicol. 2009;49:243–263. doi: 10.1146/annurev-pharmtox-061008-103102. [DOI] [PubMed] [Google Scholar]
- 43.Hunter P. The second coming of epigenetic drugs: a more strategic and broader research framework could boost the development of new drugs to modify epigenetic factors and gene expression. EMBO Rep. 2015;16:276–279. doi: 10.15252/embr.201540121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feinberg AP, Koldobskiy MA, Göndör A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–299. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amacher DE. A 2015 survey of established or potential epigenetic biomarkers for the accurate detection of human cancers. Biomarkers. 2016;21:387–403. doi: 10.3109/1354750X.2016.1153724. [DOI] [PubMed] [Google Scholar]
- 47.Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget stool DNA testing for colorectal-cancer screening. N Engl J Med. 2014;370:1287–1297. doi: 10.1056/NEJMoa1311194. [DOI] [PubMed] [Google Scholar]
- 48.Ladabaum U, Mannalithara A. Comparative Effectiveness and Cost Effectiveness of a Multitarget Stool DNA Test to Screen for Colorectal Neoplasia. Gastroenterology. 2016;151:427–439. e6. doi: 10.1053/j.gastro.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 49.Lee WH, Morton RA, Epstein JI, et al. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A. 1994;91:11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Partin AW, Van Neste L, Klein EA, et al. Clinical validation of an epigenetic assay to predict negative histopathological results in repeat prostate biopsies. J Urol. 2014;192:1081–1087. doi: 10.1016/j.juro.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Kessel KEM, Neste LV, Lurkin I, et al. Evaluation of an Epigenetic Profile for the Detection of Bladder Cancer in Patients with Hematuria. J Urol. 2016;195:601–607. doi: 10.1016/j.juro.2015.08.085. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt B, Liebenberg V, Dietrich D, et al. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer based on bronchial aspirates. BMC Cancer. 2010;10:600. doi: 10.1186/1471-2407-10-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dietrich D, Kneip C, Raji O, et al. Performance evaluation of the DNA methylation biomarker SHOX2 for the aid in diagnosis of lung cancer based on the analysis of bronchial aspirates. Int J Oncol. 2012;40:825–832. doi: 10.3892/ijo.2011.1264. [DOI] [PubMed] [Google Scholar]
- 54.Stefansson OA, Esteller M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am J Pathol. 2013;183:1052–1063. doi: 10.1016/j.ajpath.2013.04.033. [DOI] [PubMed] [Google Scholar]
- 55.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang S, Wang R-H, Akagi K, et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat Med. 2011;17:1275–1282. doi: 10.1038/nm.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anjum S, Fourkala E-O, Zikan M, et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2014;6:47. doi: 10.1186/gm567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kloten V, Becker B, Winner K, et al. Promoter hypermethylation of the tumor-suppressor genes ITIH5, DKK3, and RASSF1A as novel biomarkers for blood-based breast cancer screening. Breast Cancer Res BCR. 2013;15:R4. doi: 10.1186/bcr3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.Fackler MJ, Lopez Bujanda Z, Umbricht C, et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014;74:2160–2170. doi: 10.1158/0008-5472.CAN-13-3392. An important demonstration of circulating cell-free DNA methylation levels as diagnostic biomarkers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Butkus B. Hopkins Lab, Cepheid Developing Methylated DNA Panel for Breast Cancer Dx, Monitoring. Genome Web [Internet] 2015 Available from: https://www.genomeweb.com/molecular-diagnostics/hopkins-lab-cepheid-developing-methylated-dna-panel-breast-cancer-dx.
- 61.Lunnon K, Smith R, Hannon E, et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat Neurosci. 2014;17:1164–1170. doi: 10.1038/nn.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jowaed A, Schmitt I, Kaut O, et al. Methylation Regulates Alpha-Synuclein Expression and Is Decreased in Parkinson’s Disease Patients’ Brains. J Neurosci. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Absher DM, Li X, Waite LL, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. 2013;9:e1003678. doi: 10.1371/journal.pgen.1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pidsley R, Viana J, Hannon E, et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 2014;15:483. doi: 10.1186/s13059-014-0483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aberg KA, McClay JL, Nerella S, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014;71:255–264. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ladd-Acosta C, Hansen KD, Briem E, et al. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. 2014;19:862–871. doi: 10.1038/mp.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–692. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 69.DeWoskin VA, Million RP. The epigenetics pipeline. Nat Rev Drug Discov. 2013;12:661–662. doi: 10.1038/nrd4091. [DOI] [PubMed] [Google Scholar]
- 70.Liang G, Gonzales FA, Jones PA, et al. Analysis of gene induction in human fibroblasts and bladder cancer cells exposed to the methylation inhibitor 5-aza-2′-deoxycytidine. Cancer Res. 2002;62:961–966. [PubMed] [Google Scholar]
- 71.Cheishvili D, Boureau L, Szyf M. DNA demethylation and invasive cancer: implications for therapeutics. Br J Pharmacol. 2015;172:2705–2715. doi: 10.1111/bph.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peedicayil J. Epigenetic Drugs for Multiple Sclerosis. Curr Neuropharmacol. 2016;14:3–9. doi: 10.2174/1570159X13666150211001600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Abdel-Hameed EA, Ji H, Shata MT. HIV-Induced Epigenetic Alterations in Host Cells. Adv Exp Med Biol. 2016;879:27–38. doi: 10.1007/978-3-319-24738-0_2. [DOI] [PubMed] [Google Scholar]
- 74.Sun Y, Sahbaie P, Liang D, et al. DNA Methylation Modulates Nociceptive Sensitization after Incision. PloS One. 2015;10:e0142046. doi: 10.1371/journal.pone.0142046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh P, Konar A, Kumar A, et al. Hippocampal chromatin-modifying enzymes are pivotal for scopolamine-induced synaptic plasticity gene expression changes and memory impairment. J Neurochem. 2015;134:642–651. doi: 10.1111/jnc.13171. [DOI] [PubMed] [Google Scholar]
- 76.Rajasethupathy P, Antonov I, Sheridan R, et al. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell. 2012;149:693–707. doi: 10.1016/j.cell.2012.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lane AA, Chabner BA. Histone Deacetylase Inhibitors in Cancer Therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- 78.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 79.Kurita M, Holloway T, García-Bea A, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma S, Taliyan R. Histone deacetylase inhibitors: Future therapeutics for insulin resistance and type 2 diabetes. Pharmacol Res. 2016;113:320–326. doi: 10.1016/j.phrs.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 81*.Daigle SR, Olhava EJ, Therkelsen CA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. An interesting paper demonstrating the application of an epidrug to a genetically distinct cancer subset. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kurmasheva RT, Sammons M, Favours E, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2016 doi: 10.1002/pbc.26218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chung C, Coste H, White JH, et al. Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 84.Khan SN, Khan AU. Role of histone acetylation in cell physiology and diseases: An update. Clin Chim Acta Int J Clin Chem. 2010;411:1401–1411. doi: 10.1016/j.cca.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 85.Bowers EM, Yan G, Mukherjee C, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem Biol. 2010;17:471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gajer JM, Furdas SD, Gründer A, et al. Histone acetyltransferase inhibitors block neuroblastoma cell growth in vivo. Oncogenesis. 2015;4:e137. doi: 10.1038/oncsis.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87*.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-Repair Gene MGMT and the Clinical Response of Gliomas to Alkylating Agents. N Engl J Med. 2000;343:1350–1354. doi: 10.1056/NEJM200011093431901. A landmark study that was one of the first to show association between epigenetic factors and therapeutic drug response. [DOI] [PubMed] [Google Scholar]
- 88.Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 89.Fornaro L, Vivaldi C, Caparello C, et al. Pharmacoepigenetics in gastrointestinal tumors: MGMT methylation and beyond. Front Biosci Elite Ed. 2016;8:170–180. doi: 10.2741/E758. [DOI] [PubMed] [Google Scholar]
- 90.Chiam K, Centenera MM, Butler LM, et al. GSTP1 DNA Methylation and Expression Status Is Indicative of 5-aza-2′-Deoxycytidine Efficacy in Human Prostate Cancer Cells. In: Agoulnik I, editor. PLoS ONE. Vol. 6. 2011. p. e25634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dejeux E, Rønneberg J, Solvang H, et al. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stefansson OA, Villanueva A, Vidal A, et al. BRCA1 epigenetic inactivation predicts sensitivity to platinum-based chemotherapy in breast and ovarian cancer. Epigenetics. 2012;7:1225–1229. doi: 10.4161/epi.22561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ouni M, Belot MP, Castell AL, et al. The P2 promoter of the IGF1 gene is a major epigenetic locus for GH responsiveness. Pharmacogenomics J. 2016;16:102–106. doi: 10.1038/tpj.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ivanov M, Kacevska M, Ingelman-Sundberg M. Epigenomics and interindividual differences in drug response. Clin Pharmacol Ther. 2012;92:727–736. doi: 10.1038/clpt.2012.152. [DOI] [PubMed] [Google Scholar]
- 95.Kaut O, Schmitt I, Wüllner U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics. 2012;13:87–91. doi: 10.1007/s10048-011-0308-3. [DOI] [PubMed] [Google Scholar]
- 96.Tokizane T, Shiina H, Igawa M, et al. Cytochrome P450 1B1 is overexpressed and regulated by hypomethylation in prostate cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11:5793–5801. doi: 10.1158/1078-0432.CCR-04-2545. [DOI] [PubMed] [Google Scholar]
- 97.Okino ST, Pookot D, Li L-C, et al. Epigenetic inactivation of the dioxin-responsive cytochrome P4501A1 gene in human prostate cancer. Cancer Res. 2006;66:7420–7428. doi: 10.1158/0008-5472.CAN-06-0504. [DOI] [PubMed] [Google Scholar]
- 98.Gomez A, Karlgren M, Edler D, et al. Expression of CYP2W1 in colon tumors: regulation by gene methylation. Pharmacogenomics. 2007;8:1315–1325. doi: 10.2217/14622416.8.10.1315. [DOI] [PubMed] [Google Scholar]
- 99.Schaeffeler E, Hellerbrand C, Nies AT, et al. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 2011;3:82. doi: 10.1186/gm298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Y, Zheng X, Yu Q, et al. Epigenetic activation of the drug transporter OCT2 sensitizes renal cell carcinoma to oxaliplatin. Sci Transl Med. 2016;8:348ra97. doi: 10.1126/scitranslmed.aaf3124. [DOI] [PubMed] [Google Scholar]
- 101.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 102.Krueger F, Kreck B, Franke A, et al. DNA methylome analysis using short bisulfite sequencing data. Nat Methods. 2012;9:145–151. doi: 10.1038/nmeth.1828. [DOI] [PubMed] [Google Scholar]
- 103.Ledford H. Epigenetics: The genome unwrapped. Nature. 2015;528:S12–S13. doi: 10.1038/528S12a. [DOI] [PubMed] [Google Scholar]
- 104.Wang Y, Krishnan HR, Ghezzi A, et al. Drug-induced epigenetic changes produce drug tolerance. PLoS Biol. 2007;5:e265. doi: 10.1371/journal.pbio.0050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105*.Stricker SH, Köferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2016 doi: 10.1038/nrg.2016.138. advance online publication. An outstanding and up-to-date review of complex epigenomic regulation. [DOI] [PubMed] [Google Scholar]
- 106.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reid GK, Besterman JM, MacLeod AR. Selective inhibition of DNA methyltransferase enzymes as a novel strategy for cancer treatment. Curr Opin Mol Ther. 2002;4:130–137. [PubMed] [Google Scholar]
- 108.Thakore PI, Black JB, Hilton IB, et al. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods. 2016;13:127–137. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109*.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. A landmark paper in understanding the 3D genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Symmons O, Uslu VV, Tsujimura T, et al. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014;24:390–400. doi: 10.1101/gr.163519.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fudenberg G, Imakaev M, Lu C, et al. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016;15:2038–2049. doi: 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sanborn AL, Rao SSP, Huang S-C, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112:E6456–6465. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rickman DS, Soong TD, Moss B, et al. Oncogene-mediated alterations in chromatin conformation. Proc Natl Acad Sci U S A. 2012;109:9083–9088. doi: 10.1073/pnas.1112570109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Valton A-L, Dekker J. TAD disruption as oncogenic driver. Curr Opin Genet Dev. 2016;36:34–40. doi: 10.1016/j.gde.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Slattum P, Peron E, Ogbonna K. The Pharmacology of Aging. In: Fill HM, Rockwood K, Woodhouse K, Young JB, editors. Brocklehursts Textb Geriatr Med Gerontol. 8. Philadephia PA: Saunders/Elsevier; 2016. p. Chapter 26. [Google Scholar]
- 117.Pal S, Tyler JK. Epigenetics and aging. Sci Adv. 2016;2:e1600584. doi: 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brunet A, Berger SL. Epigenetics of aging and aging-related disease. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S17–20. doi: 10.1093/gerona/glu042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Seripa D, Panza F, Daragjati J, et al. Measuring pharmacogenetics in special groups: geriatrics. Expert Opin Drug Metab Toxicol. 2015;11:1073–1088. doi: 10.1517/17425255.2015.1041919. [DOI] [PubMed] [Google Scholar]
- 120.Thakur BK, Zhang H, Becker A, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766–769. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121*.Sun W, Poschmann J, Cruz-Herrera Del Rosario R, et al. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell. 2016;167:1385–1397. e11. doi: 10.1016/j.cell.2016.10.031. The first study, to our knowledge, to systematically compare epigenome-wide histone modification profiles in disease cases versus controls. [DOI] [PubMed] [Google Scholar]