

Abstract
Background and Aims Allopolyploids exhibit both different levels and different patterns of genetic variation than are typical of diploids. However, scant attention has been given to the partitioning of allelic information and diversity in allopolyploids, particularly that among homeologous monoploid components of the hologenome. Sphagnum × falcatulum is a double allopolyploid peat moss that spans a considerable portion of the Holantarctic. With monoploid genomes from three ancestral species, this organism exhibits a complex evolutionary history involving serial inter-subgeneric allopolyploidizations.
Methods Studying populations from three disjunct regions [South Island (New Zealand); Tierra de Fuego archipelago (Chile, Argentina); Tasmania (Australia)], allelic information for five highly stable microsatellite markers that differed among the three (ancestral) monoploid genomes was examined. Using Shannon information and diversity measures, the holoploid information, as well as the information within and among the three component monoploid genomes, was partitioned into separate components for individuals within and among populations and regions, and those information components were then converted into corresponding diversity measures.
Key Results The majority (76 %) of alleles detected across these five markers are most likely to have been captured by hybridization, but the information within each of the three monoploid genomes varied, suggesting a history of recurrent allopolyploidization between ancestral species containing different levels of genetic diversity. Information within individuals, equivalent to the information among monoploid genomes (for this dataset), was relatively stable, and represented 83 % of the grand total information across the Holantarctic, with both inter-regional and inter-population diversification each accounting for about 5 % of the total information.
Conclusions Sphagnum ×falcatulum probably inherited the great majority of its genetic diversity at these markers by reticulation, rather than by subsequent evolutionary radiation. However, some post-hybridization genetic diversification has become fixed in at least one regional population. Methodology allowing statistical analysis of any ploidy level is presented.
Keywords: Allelic information, allelic diversity, allopolyploid, bryophytes, hologenome, populations, reticulation, Shannon partitioning, Sphagnum, Sphagnum × falcatulum
INTRODUCTION
Allopolyploids exhibit both different levels and different patterns of genetic variation than are typical of diploids, with accentuated differences in higher order allopolyploids such as Triticum × aestivum L. (bread wheat, Marcussen et al., 2014), Elymus × repens (L.) Gould (quackgrass or couchgrass, Mason-Gamer 2008), and the peat mosses Sphagnum × falcatulum Besch. (Karlin et al., 2009; Karlin, 2014) and S. × planifolium Müll. Hal. (Karlin et al., 2014), all of which have nuclear genomes from three ancestral species. The holoploid genome (entire chromosome complement) of an allopolyploid gametophyte consists of two or more homeologous monoploid genomes (Greilhuber et al., 2005), with potentially variable genetic diversity among the ancestral genomes. Gain or loss of homeologous chromosomes arising from meiotic or mitotic aberrations may lead to changes in the structure of a holoploid genome (Chester et al., 2012, 2015), with the balance between disomic and polysomic inheritance across homeologous chromosomes playing a key role in this regard. Thus, the holoploid genome of an allopolyploid may vary considerably across individuals and populations of the derivative organism (Chester et al., 2012, 2015). In marked contrast to the gametophytes of diploid vascular species and haploid bryophyte species, allopolyploid gametophytes harbour considerable internal genetic diversity, and that difference is particularly significant for bryophytes, whose gametophytes are the dominant phase of the life cycle. However, scant attention has been given to the partitioning of allelic information and diversity in allopolyploids, particularly that among monoploid components of the hologenome.
The origin of any allopolyploid lineage is associated with an extreme founder effect. In the case of monoicous bryophytes (with hermaphroditic gametophytes), it is possible for a single meiotically unreduced spore from a hybrid sporophyte to give rise to both the first gametophytic generation of a novel allopolyploid lineage and the first sporophytic generation, via intra-gametophytic selfing (Såstad, 2005). In all such cases, the genetic diversity included within the founding gametophyte, as well as that within individuals of the first generation of sporophytes, would represent the allelic divergence between the component homeologous monoploid genomes. Barring meiotic aberrations or mutation itself, all spores produced by such first-generation sporophytes would be genetically identical to the founding gametophyte, as well as to each other. Thus, the founding genotype of such allopolyploids would initially be maintained by both vegetative and sexual reproduction, with spore production allowing for a rapid and long-distance distribution of that lineage. Subsequent genetic novelty may arise via new mutation, recombination (including inter-genomic transfers) and change in hologenomic structure caused by gain or loss of homeologous chromosomes, so additional genetic diversity may augment that acquired by the initial reticulation (hybridization) event (Feldman et al., 2012). Sexual reproduction between different allopolyploid lineages, resulting from recurrent allopolyploidy, could augment genetic diversity among individuals (Soltis et al., 2010; Symonds et al., 2010).
Genetic markers with a unique allele from each homeologous genome in an individual are useful in the study of allopolyploid genomes, particularly for the assessment of allelic diversity within and among the component monoploid genomes. In addition, allopolyploid bryophyte gametophytes are good model organisms for such studies, because the assignment of alleles to monoploid genomes is facilitated by the absence of homologous chromosome pairs.
Objectives
Here, we use an existing microsatellite (simple sequence repeat, SSR) data set to explore the application of recently articulated Shannon information and diversity measures (cf. Jost, 2006, 2007; Sherwin et al., 2006; Sherwin, 2010; Smouse et al., 2015) to genetically allo-allo-triploid gametophytes in S. × falcatulum, as those manifest across levels of geographical organization, ranging from that within single individuals, representing the monoploid ancestral species (of the haplo-triploid), to that among extant individuals, spread across three Holantarctic regional populations, namely NZ: South Island, New Zealand; TdF: Tierra Del Fuego (Chile and Argentina); and Tas: Australian state of Tasmania (Macquarie Island and Tasmania). For comparison, we also examined the patterns of genetic information and diversity within the two immediate progenitor taxa.
We compared the relative contributions of allelic diversity captured by ancestral hybridization events with those emerging subsequently, addressing four specific questions: (1) How much of the total allelic information and diversity in S. × falcatulum was ‘captured’ from the progenitors? (2) How does allelic information and diversity vary among the three monoploid genomes? (3) To what extent has change in hologenomic structure occurred within S. × falcatulum? (4) How does the partitioning of allelic information and diversity vary across the Holantarctic geographical range of S. × falcatulum?
MATERIALS AND METHODS
The species
The genus Sphagnum (peat mosses) occurs on all continents except Antarctica and contains some 250–400 species (Shaw et al., 2016). Based on multigene phylogenetic analyses, Shaw et al. (2010) recognized five subgenera within the genus. The double allopolyploid S. × falcatulum (Fig. 1) is gametophytically allo-allo-triploid, with homeologous monoploid genomes (R, S and T), each captured from a different ancestral species; the evolutionary history involves both inter-subgeneric and serial inter-ploidal hybridization (Karlin et al., 2009, 2011, 2013; Karlin, 2014). The immediate progenitors are the gametophytically haploid S. cuspidatum Ehrh. ex Hoffm (subgen. Cuspidata), which contributed monoploid genome T, and the gametophytically allodiploid S. × irritans Warnst., an inter-subgeneric hybrid that contributed homeologous genomes R (associated with an unknown subgen. Cuspidata species) and S (associated with an unknown subgen. Subsecunda species) (Fig. 2; Karlin et al., 2009, 2011, 2013; Karlin, 2014; Karlin and Robinson, 2016). The genetic diversity within S. × falcatulum thus represents the initial tripling of evolutionary potential, via double reticulation, followed by subsequent innovation (mutations, changes in hologenomic structure, genetic recombination). With its history of inter-subgeneric hybridization, divergence among S. × falcatulum’s monoploid genomes is thought to be among the highest in Sphagnum allopolyploids (Karlin, 2014; Karlin et al., 2014). Based on nuclear nucleotide sequences, divergence between inter-subgenomic sequences in S. × falcatulum (S vs. R; S vs. T) was about twice as high as that between intra-subgeneric sequences (R vs. T) (Karlin, 2014).
Fig. 1.

The peat moss Sphagnum × falcatulum on South Island, New Zealand, with both allo-allo-triploid (3n) gametophytes and young, putatively allo-allo-hexaploid (6n) sporophytes (small brown spheres on plants near the centre of the picture) being present. Photo taken by E. F. Karlin.
Fig. 2.
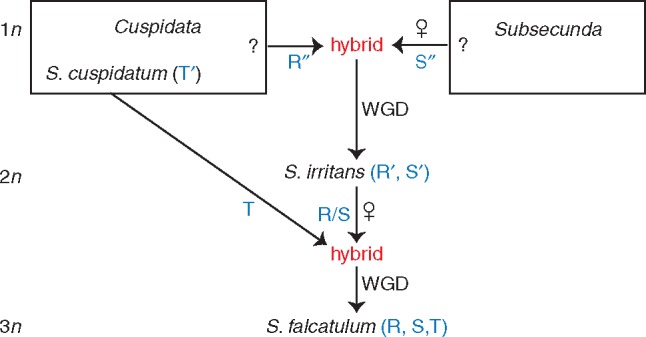
Putative evolutionary pathways for the allopolyploids S. × falcatulum and S. × irritans. The maternal parent of each hybridization event is indicated by ‘♀’. The gametophytic ploidy level is indicated on the left side of the diagram. Text in red is in the sporophytic phase. WGD, whole genome duplication. Adapted from Karlin et al. (2014).
Although a relatively young species (estimated origin = 23·5–116 kyr BP), putatively unisexual (dioecous) S. × falcatulum is the most widespread peat moss in the Holantarctic (Fig. 2; Karlin et al., 2013). Genetically documented populations occur in New Zealand (both North and South Islands), the Tierra del Fuego archipelago (Chile and Argentina), and the Australian states of Victoria and Tasmania (Karlin et al., 2013; Karlin and Robinson, 2016). Its precise geographical extent is unclear, by virtue of strong morphological overlap and sympatry with its immediate progenitors (Karlin et al., 2009, 2011; Karlin and Robinson, 2016). Separation of the three taxa at the macroscopic level is sometimes difficult, and they form a cryptic species complex in the Holantarctic (Karlin et al., 2013; Karlin and Robinson, 2016). Karlin et al. (2013) concluded that S. × falcatulum probably evolved in New Zealand, with subsequent colonization of Macquarie Island, Tierra del Fuego and possibly Tasmania, each via a single propagule or (at most) a small number of closely related individuals. Sexual reproduction is known to occur in S. × falcatulum, so genetic recombination plays at least some role in its evolution and population structure, but sexual assortment is absent from at least some isolated populations, both locally and regionally. Although common on South Island, sexual reproduction appears to be rare in Tasmania and lacking in both Tierra del Fuego and Macquarie Island (Karlin et al., 2013). Occasional sexuality aside, vegetative reproduction is ubiquitous in Sphagnum, so populations are typically composed of one to several gametophytic clones.
The allodiploid maternal ancestor (S. × irritans) of S. × falcatulum has been genetically documented only from two New Zealand islands: South Island (where it is sympatric with S. × falcatulum) and Chatham Island (Karlin et al., 2009; Karlin and Robinson, 2016). Sphagnum × irritans had been treated as a heterotypic synonym of S. × falcatulum (Andrews, 1949; Fife, 1996), but Karlin and Robinson (2016) found the type to be associated with allo-diploid plants instead. Thus, S. × irritans has been removed from the synonymy of allo-allo-triploid S. × falcatulum and is now recognized as a proper species. The paternal ancestor, S. cuspidatum (monoploid), was considered to be a Holarctic species with disjunct populations in Australia and New Zealand by Warnstorf (1911). It was subsequently excluded from Australasia by Andrews (1949), Sainsbury (1955), Scott et al. (1976), Catcheside (1980), Beever et al. (1992), Fife (1996) and Seppelt (2012). Recent genetic analyses, however, have documented its presence at two sites in Queensland, Australia, and S. cuspidatum is now reported for all continents except Antarctica (Karlin et al., 2011).
Taxon sampling
The SSR genotypes deployed for this study have been described by Karlin et al. (2009, 2011, 2013) and Karlin and Robinson (2016), and involved 138 individuals of S. × falcatulum gametophytes collected from South Island (New Zealand), Tierra del Fuego archipelago (Chile, Argentina) and the Australian state of Tasmania (Supplementary Data, Table S1). The most complete coverage was for South Island (NZ), with 97 individuals collected from 11 sites (= populations). The Tierra del Fuego archipelago (TdF) was represented by 24 individuals collected from 16 sites across eight islands and one mainland location. Eight of these sites were each represented by one individual and eight sites were represented by two individuals each. The state of Tasmania (Tas) was represented by one site on Macquarie Island (with 15 individuals) and two sites on Tasmania (with one individual each). To allow for Shannon analysis among and within regions, TdF and Tas were each divided into ‘super populations’ (i.e. sub-regions). TdF had three ‘super populations’: (1) Isla Grande de la Tierra del Fuego and one mainland site (eight individuals from six sites); (2) sites occurring south of Isla Grande de la Tierra del Fuego and north of 55·6°S (seven individuals from five sites across three islands); and (3) sites south of 55·6°S (nine individuals from five sites across four islands). There were two ‘super populations’ for Tas: Macquarie Island (15 individuals from one site) and Tasmania (two individuals from two sites). The SSR assay battery also included data for 17 individuals of S. × irritans (from four South Island sites) and 11 individuals of S. cuspidatum (from two Queensland, Australia, sites).
Microsatellite genotypes
DNA was extracted from one gametophyte stem selected from each specimen. DNA extractions were accomplished with the protocols described by Shaw et al. (2003). All samples were assayed across 14 SSR markers. Protocols for microsatellite (SSR) amplification and scoring are presented in Karlin et al. (2008). Fragments of different sizes (nucleotide pairs) were coded as alleles.
Here, we have used just five of these markers. They were selected because each had alleles (within any single individual) that could be unambiguously assigned to their respective monoploid genomes. Thus, these ‘ancestrally indicative’ markers allowed for a study of the allelic diversity among and within the three monoploid genomes present in S. × falcatulum. The markers were 1, 12, 18, 19 and 22 (numbered as in Shaw et al., 2008). Each of the other nine SSR markers, although easily assayed and reliable, had some, even many, alleles that could not be assigned to ancestral genomes.
The allelic data for the individuals are listed in Supplementary Data (Table S2). All five markers exhibited stable homeologous alleles, most of which occurred both within S. × falcatulum and within at least one of the three ancestral linages. Each individual would thus be expected to contain one allele from each of the three homeologous ‘loci’, so the five markers collectively present five ‘trios’ of homeologous alleles within each individual: (1R, 1S, 1T), (12R, 12S, 12T), (18R, 18S, 18T), (19R, 19S, 19T) and (22R, 22S, 22T). These five trios permit comparison of allelic diversity captured by the initial hybridization events (Fig. 3) with that emerging post-hybridization within S. × falcatulum itself.
Fig. 3.

Map showing the location of the three study regions: South Island, New Zealand (NZ), Tierra del Fuego (TdF; Chile, Argentina) and the State of Tasmania, Australia (Tas), which includes Macquaire Island (MI) and Tasmania (Tas).
Allelic information and diversity
To characterize the patterns of holoploid (triploid) allelic diversity within S. × falcatulum, we computed estimates of (allelic) Shannon information. Briefly, consider a single SSR marker (say the marker A), represented in the collective (S. × falcatulum) sample (of N) individuals by a series of alleles (say A1, … , AK). The kth allele (k = 1, … , K) is present in that collective sample with a relative frequency (in the N trios) of Pk = (Xk/3N). The total (within-species) Shannon information (HWS) for marker A is defined as
| (1) |
which is bounded below by ‘0’ and above by ln (3N). The lower bound is achieved when all 3N of the alleles are of the same type (monomorphic marker); the upper bound is achieved when all 3N alleles are different and appear once each. To convert from Shannon (allelic) information to allelic diversity, we exponentiate. The total holoploid within-species diversity (σWS) for this A-locus is thus defined as
| (2) |
obviously bounded below by 1 (= a single, monomorphic allele) and above by 3N (= all alleles different and equally frequent). Both information and diversity increase with the number of different alleles, and for any particular number of alleles, both information and diversity increase with ‘evenness’ of their frequencies. Computation of allelic diversity for each homeologous monoploid genome was the same, but with the upper bound set at N and the relative frequency of each allele being Pk = (Xk/N).
Our targets for allo-triploid Sphagnum gametophytes are the diversity components themselves, but Shannon information values are conveniently additive (see below), so we can partition information among and within regions (HAR and HWR), among and within populations (HAP and HWP) for single regions, as well as among and within allotriploid individuals (HAI and HWI) for single populations, for each of the five loci separately. We then average the assayed information values for the five marker loci. For a single region (say NZ), we compute the information and diversity within that stratum, as in eqn (1), but with 3N now defined as the theoretical maximum number of alleles within that region (HWR-NZ). Using a weighted average within-region information value for the three regions (HWR), we then compute the among-regions information component (HAR = HWS – HWR). We extract average within-population (HWP) and average within-(triploid)-individual (HWI) information values in similar fashion, and calculate the among-population (HAP = HWR – HWP) and among-individual (HAI = HWP – HWI) values. We compute the information partition, marker by marker, and then average. The information value is then exponentiated to obtain the diversity values, all measured in ‘effective number of ‘equi-frequent’ units. The total layout takes the following form:
Information analysis is similar to traditional analysis of variance (cf. Smouse and Ward, 1978; Smouse et al., 2015). Information is additive:
| (3) |
| (4) |
but the diversity components are multiplicative (cf. Tuomisto, 2010):
| (5) |
| (6) |
Allelic diversity within groups (γWG, σWS, ρWR, αWP, μWI) is interpreted as effective numbers of alleles per marker within a group. Allelic diversity among groups (γAS, δAR, βAP, λAI) is interpreted as effective numbers of non-overlapping allele sets, or ‘compositional units’, among strata per marker (Tuomisto, 2010). Information and diversity among groups measure the divergence (differentiation, distinctness) among the groups (strata) in question (Whittaker, 1977; Sherwin et al., 2006; Jost, 2010; Sherwin, 2010; Tuomisto, 2010; Smouse et al., 2015). For this dataset, allelic diversity within individuals (μWI) is equivalent to the divergence among monoploid genomes. Thus, μWI can be interpreted as being both the effective numbers of alleles (within an individual) and the effective number of non-overlapping allelic sets, or monoploid genomes (within an individual).
We used GenAlEx 6.5 (http://biology.anu.edu.au/GenAlEx/; Peakall and Smouse, 2006, 2012) to construct the analyses reported here. For each individual, data were arranged in three consecutive rows (one per ancestral monotype), with each of the five trios represented by a single column. This slight shift in GenAlEx data format allowed haplo-triplo data to be analysed as needed here. Shannon analysis was carried out using the ‘Partition’ option, with each individual defined as a ‘population’ of three sub-samples, with each sub-sample represented by one row of data. This approach was elaborated here to deal with additional regions across the Holantarctic, and could easily be extended to other ploidy levels. An example data sheet and a results sheet for triploid analysis are provided in Supplementary Data (Tables S3 and S4).
Three of the five markers typically had three different alleles per marker per individual in S. × falcatulum, with one allele from each monoploid genome; two markers had only a pair of alleles per individual (because of allelic overlap between R and T). These patterns appear to represent those directly inherited from the ancestor and thus allow for the calculation of the ‘founding’ (or original) HWI and μWI for S. × falcatulum. Averaging across the five markers yields a mean ‘founding’ information and diversity of [HWI = 0·914 ↔ μWI = 2·49]. For S. × irritans, which has (1) no allelic overlap between its two monoploid genomes (R′ and S′) at these markers, and (2) each marker typically has two alleles per individual, mean ‘founding’ information and diversity are [HWI = 0·693 ↔ μWI = 2·00]. For both allopolyploid species, a change in hologenomic structure manifests as a change in the within-individual components (HWI ↔ μWI). A tally of the HWI associated with each individual sampled was made to examine the extent of variation in HWI.
RESULTS
Monoploid genomes
A total of 26 different alleles were detected across the three monoploid genomes assayed with the five markers, with two monomorphic alleles (12R ≡ 12T) and (18R ≡ 18T) being shared by the (co-subgeneric) ancestral monoploid genomes R and T (Table 1). Nineteen of the alleles (76 %) co-occurred in the immediate progenitors, and we will refer to such alleles as ‘ancestral’. For each monoploid genome, one ancestral allele had a frequency ≥0·60 at each homeologous locus, and with a single exception (18S), these ‘most frequent’ alleles were also predominant in the immediate progenitors (Table 1). The six (of 19) ancestral alleles having a lower frequency in S. × falcatulum were also lower frequency polymorphs in the immediate ancestors. Moreover, only two of the seven putative ‘de novo’ (new) alleles detected in S. × falcatulum (i.e. not yet found in the progenitors) had frequencies ≥0·05 in the respective monoploid genomes. The ‘de novo’ alleles in S. × falcatulum were assigned to a homeologous genome, based on similarity in size to the ancestral alleles associated with that homeologous genome or if the ancestral alleles for the other two homeologues were also present in an individual. The mean total combined frequency of de novo alleles across the five markers was <0·01 in genomes T and R and ∼0·06 in S. The mean number of alleles per marker with frequency ≥0·05 and the mean number of alleles per marker shared with the two immediate progenitors were all lowest in T, highest in S and intermediate in R (Table 2). The numbers of monoploid haplotypes (across 15 homeologous loci) detected for R, S and T were seven, 13 and three, respectively, with the most common haplotypes having frequencies of 0·69, 0·46 and 0·98 for R, S and T, respectively. We infer that the ancestral alleles were captured by hybridization and that the de novo alleles have probably developed within S. × falcatulum, subsequent to formation. Most of the genetic information and diversity contained within these five markers for S. × falcatulum were inherited from the progenitors.
Table 1.
Frequency of the most common allele (F-MCA) per monoploid genome, the combined frequencies of all ‘ancestral alleles’ (CF-Ancestral) in each monoploid genome, and the total number of ‘ancestral alleles’ present in each monoploid genome/marker (given in square brackets) across three regional populations of Sphagnum × falcatulum, based on five ancestrally indicative markers
| Ancestral marker | Monoploid genome | F-MCA | CF-Ancestral |
|---|---|---|---|
| Marker - 1 | R | 0·89 (0·88) | 1·00 [2] |
| S | 1·00 (1·0) | 1·00 [1] | |
| T | 0·98 (1·00) | 0·98 [1] | |
| Marker - 12 | R | 1·00 (1·0)* | 1·00 [1] |
| S | 0·90 (0·71) | 1·00 [2] | |
| T | 1·00 (1·0)* | 1·00 [1] | |
| Marker - 18 | R | 1·00 (1·0)* | 1·00 [1] |
| S | 0·64 (0·29) | 0·83 [3] | |
| T | 1·00 (1·0)* | 1·00 [1] | |
| Marker - 19 | R | 0·96 (0·82) | 0·99 [2] |
| S | 1·00 (1·0) | 1·00 [1] | |
| T | 1·00 (1·0) | 1·00 [1] | |
| Marker - 22 | R | 0·99 (1·0) | 0·99 [1] |
| S | 0·84 (0·59) | 0·88 [3] | |
| T | 1·00 (1·0) | 1·00 [1] | |
| Mean ± s.e. | R | 0·97 ± 0·02 | 1·00 ± 0·00 |
| S | 0·87 ± 0·07 | 0·94 ± 0·04 | |
| T | 1·00 ± 0·00 | 1·00 ± 0·00 |
The frequency in the immediate ancestors for the MCA in each monoploid genome is given in parentheses. An asterisk (*) indicates that the same allele occurred in the R and T monoploid genomes.
Table 2.
Allelic sample composition for the holoploid and three monoploid genomes (R, S, T) across three regional populations of S. × falcatulum, as well as those from the ancestral species S. × irritans (R′, S′) and S. cuspidatum (T): number of individuals (N), number of alleles (Na), number of polymorphic alleles (Na ≥ 5 %), number of alleles inherited from ancestor (Ni), number of unique (private) alleles (Nu) and effective number of alleles (Ne), based on five ancestrally indicative markers
| Genome | N | Na | Na ≥ 5 % | Ni | Nu | Ne |
|---|---|---|---|---|---|---|
| Sphagnum × falcatulum | ||||||
| R | 133·2 | 1·8 | 1·2 | 1·4 | 0·4 | 1·12 |
| S | 137·0 | 2·4 | 1·8 | 1·8 | 0·6 | 1·47 |
| T | 138·0 | 1·4 | 1·0 | 1·0 | 0·4 | 1·02 |
| R,S | 270·2 | 4·2 | 2·8 | 3·2 | 1·0 | 2·58 |
| R,S,T | 408·2 | 5·2 | 3·0 | 3·8 | 1·2 | 2·97 |
| Sphagnum × irritans | ||||||
| R′ | 16·8 | 1·4 | 1·4 | – | 0·0 | 1·18 |
| S′ | 17·0 | 2·2 | 2·2 | – | 0·4 | 1·71 |
| R′,S′ | 33·8 | 3·6 | 3·4 | – | 0·4 | 2·84 |
| Sphagnum cuspidatum | ||||||
| T′ | 11·0 | 1·0 | 1·0 | – | 0·0 | 1·00 |
Allelic information and diversity.
Considerable divergence was detected among all three monoploid genomes based on the five markers. Allelic information and diversity between S (subgen. Subsecunda) and each of the two subgen. Cuspidata monoploid genomes R and T were [HAS = 0·693 ↔ θAS = 2·00]. Information and allelic diversity between R and T were [HAS= 0·415 ↔ θAS = 1·51]. A detailed partition of allelic information and diversity for each monoploid genome is presented in Table 3. Total allelic information and diversity (HWS ↔ σWS) across the three regions differed among the three monoploid genomes, with HWS(S) > HWS(R) > HWS(T). This ranking held for all other partitions, except for that within individuals, where [HWI = 0·000 ↔μWI = 1·00] for each of the three monoploid genomes (Table 3). Total allelic diversity ranged from σWS(S) = 1·47 to σWS(T) = 1·02. For each monoploid genome, allelic diversity among individuals within populations (λAI) was slightly higher than that detected among populations (βAP) and that among regions (δAR); HAI represented between 40 and 58 % of the respective HWS values.
Table 3.
Shannon diversity analysis of five ancestrally indicative markers for Sphagnum × falcatulum, both as separate monoploid (R, S, T) genomes, and combined as a holoploid (triploid) genome, across three regions (New Zealand, Tasmania, Tierra del Fuego); the R genome was based on four markers (see text for explanation)
| Information and diversity | Monoploid (R) | Monoploid (S) | Monoploid (T) | Holoploid (R, S, T) |
|---|---|---|---|---|
| Within species (S. × falcatulum) | ||||
| Information (HWS) | 0·131 | 0·385 | 0·024 | 1·089 |
| Diversity (σWS) | 1·14 | 1·47 | 1·02 | 2·97 |
| Among regions (NZ, Tas, TdF) within species | ||||
| Information (HAR) | 0·014 | 0·121 | 0·002 | 0·054 |
| Diversity (δAR) | 1·01 | 1·13 | 1·00 | 1·06 |
| Within regions | ||||
| Information (HWR) | 0·117 | 0·264 | 0·022 | 1·035 |
| Diversity (ρWR) | 1·12 | 1·30 | 1·02 | 2·82 |
| Among populations (within regions) | ||||
| Information (HAP) | 0·047 | 0·111 | 0·009 | 0·054 |
| Diversity (βAP) | 1·05 | 1·12 | 1·01 | 1·06 |
| Within populations | ||||
| Information (HWP) | 0·071 | 0·153 | 0·014 | 0·981 |
| Diversity (αWP) | 1·07 | 1·17 | 1·01 | 2·67 |
| Among individuals (within populations) | ||||
| Information (HAI) | 0·071 | 0·153 | 0·014 | 0·079 |
| Diversity (λAI) | 1·07 | 1·17 | 1·01 | 1·08 |
| Within individuals | ||||
| Information (HWI) | 0·000 | 0·000 | 0·000 | 0·901 |
| Diversity (μWI) | 1·00 | 1·00 | 1·00 | 2·46 |
All Shannon analyses were based on the five markers, with one exception. Because the TdF population lacked the (22R) allele, just four markers were used to determine the partitioning of information and diversity within and among regions for R. However, by treating all individuals as one population, a second value for just HWS(R) was calculated using all five markers. Based on five markers, [HWS(R)= 0·115 and ρWS(R) = 1·12], which were comparable to, but slightly lower than, the HWS(R) and ρWS(R) based on four markers (Table 3). With either approach, HWS(R) and ρWS(R) were intermediate between the corresponding values for T and S.
Holoploid genomes
Among-region allelic information and diversity.
A detailed partition of the holopoloid allelic information and diversity is presented in Table 3. Based on Shannon analysis of the holoploid genome (as triploid data) for the five markers, the grand total information for S. × falcatulum across the three regions was HWS(R,S,T) = 1·089, translating into an (effective number) diversity equivalent of σWS(R,S,T) ∼ 2·97 alleles per trio. By far the majority of allelic diversity was present within each individual, with μWI = 2·46 alleles per trio (marker). As this diversity also represents divergence among the monoploid genomes, it shows that the effective number of monoploid genomes present is 2·46. Allelic information within individuals HWI(R,S,T) = 0·901 represented ∼83 % of HWS(R,S,T). This is in stark contrast to the monoploid analysis, where for each monoploid genome, μWI = 1·00 and HWI represented 0·0 % of HWS. The next highest diversity among partitions was λAI(R,S,T) = 1·08, with HAI(R,S,T) representing ∼7 % of HWS(R,S,T). βAP(R,S,T) and δAR(R,S,T) were comparable, with HAR(R,S,T) and HAP(R,S,T) each representing ∼5 % of HWS(R,S,T). Thus, ∼95 % of HWS(R,S,T) was resident within regions and ∼ 90 % was resident within populations. For S. × falcatulum, ancestry is (almost) everything!
Within region allelic information and diversity.
New Zealand. A detailed partition of the holoploid allelic information and diversity for NZ is presented in Table 4 and Fig. 4. Shannon analysis of the holoploid genome within NZ yielded a pattern of diversity among partitions similar to the pattern detected for the analysis across the three regions, albeit with slightly higher allelic information and diversity values at all levels (Tables 3 and 4). Total information for New Zealand was HWR(NZ) = 1·097, translating into a diversity equivalent of 3·00 alleles per trio (marker). HWI(NZ) represented 83 % of HWR(NZ), HAP(NZ) represented 7 % and HAI(NZ) accounted for 10 %. Thus, some 93 % HWR(NZ) was resident within populations, and most of that was the ancestral diversity resident within any single individual.
Table 4.
Shannon information and diversity analyses of the holoploid (R,S,T) genomes for each of the regional collections: Tierra del Fuego (TdF), Tasmania (Tas), New Zealand (NZ), along with the average analyses, based on five ancestrally indicative markers
| Information and diversity | Separate within-region averages |
Overall average | ||
|---|---|---|---|---|
| TdF | Tas | NZ | ||
| Within regions | ||||
| Information (HWR) | 0·833 | 0·914 | 1·097 | 1·035 |
| Diversity (ρWR) | 2·30 | 2·49 | 3·00 | 2·82 |
| Among populations (within regions) | ||||
| Information (HAP) | 0·000 | 0·000 | 0·076 | 0·054 |
| Diversity (βAP) | 1·00 | 1·00 | 1·08 | 1·06 |
| Within populations | ||||
| Information (HWP) | 0·833 | 0·914 | 1·021 | 0·981 |
| Diversity (αWP) | 2·30 | 2·49 | 2·78 | 2·67 |
| Among individuals (within populations) | ||||
| Information (HAI) | 0·000 | 0·000 | 0·111 | 0·079 |
| Diversity (λAI) | 1·00 | 1·00 | 1·12 | 1·08 |
| Within individuals | ||||
| Information (HWI) | 0·833 | 0·914 | 0·911 | 0·901 |
| Diversity (μWI) | 2·30 | 2·49 | 2·49 | 2·46 |
Fig. 4.
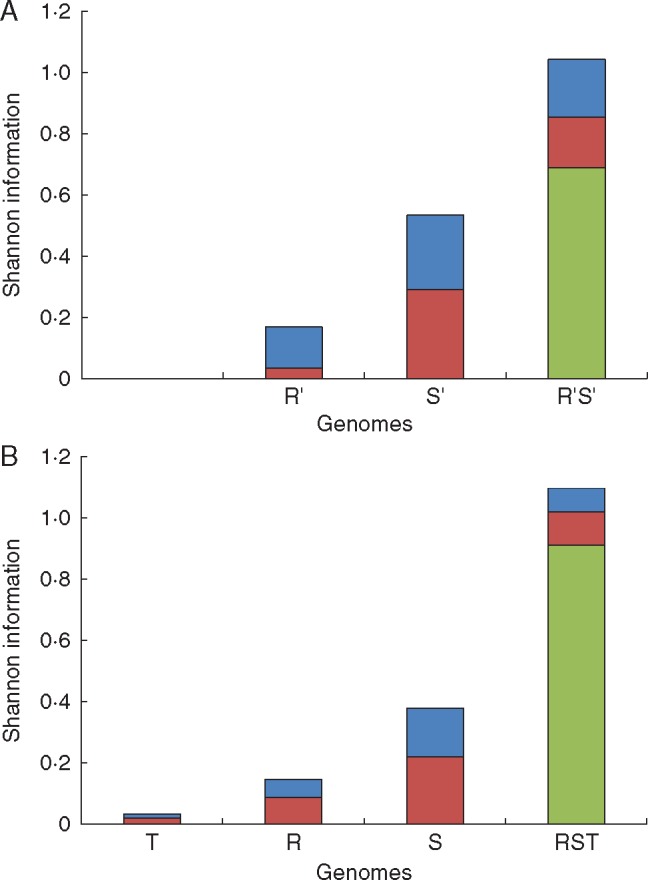
Partitioning of Shannon Information: (A) the two monoploid genomes (R′, S′) and the holoploid genome of S. × irritans (across four sites); and (B) the three monoploid genomes (R, S, T) and the holoploid genome of Sphagnum × falcatulum (across 11 sites) on South Island, New Zealand. The total height of each column represents the total information (HWR) for the respective genome (or holoploid genome) on South Island. Based on five ‘ancestrally indicative’ microsatellite markers: green for information within individuals (HWI); red for information among individuals within populations (HAI); blue for information among populations (HAP).
Tasmania and Tierra del Fuego. Shannon analysis of the holoploid genome for both these regions showed a starkly different partitioning of allelic information and diversity among levels than that detected for NZ (Table 4). For both regions, 100 % of holoploid HWR was associated with HWI; no information was present in any of the other components. Consequently, allelic information and diversity in these two regions was much lower than that detected for NZ, with ρWR(Tas) = 2·49 and ρWR(TdF) = 2·30. HWI differed among the three regions, being highest in Tas and lowest in TdF.
Changes in hologenomic structure.
The large majority (109/138) of the S. × falcatulum individuals in this study, including all of the Tasmanian (15) and 92 (out of 97) NZ individuals, had [HWI(R,S,T) = 0·914 ↔ μWI(R,S,T)I = 2·49 alleles] per average trio. As this value represents the holoploid ‘founding’ HWI, there appears to have been no change in allotriploid hologenomic structure in these individuals, at least for these markers. The remaining 29 individuals sampled in this study, representing all of the TdF individuals and the remaining five NZ individuals, had [HWI(R,S,T) = 0·833 ↔ μWI′(R,S,T) = 2·30], indicative of a change in hologenomic structure. All 24 of the TdF individuals lacked the (22R) allele, and four of the five South Island individuals, all ramets of one clonal lineage, lacked the (22S) allele instead. The fifth South Island individual (EK498) lacked (12S). These changes for markers (22 and 12) reflect post-hybridization evolution in the hologenomic structure of S. × falcatulum over evolutionary time and geographical space. It is also possible that the change in hologenomic structure occurred in S. × irritans and was directly inherited by S.× falcatulum via one of its multiple origins.
Allelic information and diversity in the immediate ancestors
Allelic information and diversity in the two immediate progenitors of S. × falcatulum, the South Island population of S. × irritans (R′ and S′) and the Queensland population of S. cuspidatum (T′) revealed three major points. (a) Total information and diversity within each of the homeologues (R′, S′, T′) were comparable to those detected within S. × falcatulum (R, S, T) (Table 2). We find [HWS(T′) ↔ σWS(T′)] < [HWS(R′) ↔ σWS(R′)] < [HWS(S′) ↔ σWS(S′)], so the pattern we see today in S. × falcatulum was probably established via the initial hybridization event(s). (b) There were no R′ alleles for these five markers in S. × irritans or T′ alleles in S. cuspidatum that were not also present in S. × falcatulum, but there were two S′ alleles in S. × irritans that have not been detected within S. × falcatulum. (c) Divergence between each monoploid genome in S. × falcatulum and its ancestral progenitor was slight (Table 5). Collectively, the data suggest that the majority of the allelic information and diversity associated with the markers in the two immediate progenitor species was captured by S. × falcatulum, almost certainly via repeated hybridization. In addition, [HWI = 0·689 ↔ μWI = 1·99] in S. × irritans, representing 66 % of HWS. All (but one) individuals of S. × irritans had the ‘founding’ HWI = 2·00 at these markers. The one exception lacked the allele for (1R) and had [HWI = 0·555 ↔ μWI = 1·74].
| Component | Information | Diversity | Effective number units |
|---|---|---|---|
| Within genus | HWG | γWG = exp{HWG} | Alleles per genus |
| Among species | HAS | θAS = exp{HAS} | Species per genus |
| Within species | HWS | σWS = exp{HWS} | Alleles per species |
| Among regions | HAR | δAR = esp{HAR} | Regions per species |
| Within regions | HWR | ρWR =exp{HWR} | Alleles per region |
| Among populations | HAP | βAP = exp{HAP} | Populations per region |
| Within populations | HWP | αWP =exp{HWP} | Alleles per population |
| Among individuals | HAI | λΑΙ = exp{HAI} | Individuals per population |
| Within individuals | HWI | μWI = exp{HWI} | Alleles per individual |
Table 5.
Within-species information (HWS) and diversity (σWS) for Sphagnum × falcatulum (R,S,T) monoploid genomes, and of its ancestral (allo-diploid) S. × irritans (R′,S′) and (haploid) S. cuspidatum (T′) progenitors, in addition to the among-species components (HAS and θAS), based on five ancestrally indicative markers
| Information and diversity | Ancestral species | Derivative species | Ancestral vs. derivative |
|---|---|---|---|
| S. × irritans → S. × falcatulum | |||
| HWS (R or R′) = | 0·169 | 0·131 | 0·005 = HAS (R′ vs. R) |
| σWS (R or R′) = | 1·20 | 1·14 | 1·01 = θAS (R′ vs. R) |
| S. × irritans → S. × falcatulum | |||
| HWS (S or S′) = | 0·534 | 0·385 | 0·048 = HAS (S′ vs. S) |
| σWS (S or S′) = | 1·70 | 1·47 | 1·05 = θAS (S′ vs. S) |
| S. cuspidatum → S. × falcatulum | |||
| HWS (T′ or T) = | 0·000 | 0·024 | 0·053 = HAS (T′ vs. T) |
| σWS (T′ or T) = | 1·00 | 1·02 | 1·05 = θAS (T′ vs. T) |
A comparison of the partition of information in the New Zealand regional collections of S. × falcatulum and S. × irritans is shown in Fig. 4. Total holoploid information HWR(R,S,T) in S. × falcatulum was slightly higher than HWI(R′S′) in S. × irritans. However, HWI(R,S,T) in S. × falcatulum was much greater than HWI(R′,S′) in S. × irritans, while holoploid HAP and HAI contributed a much larger portion of the total information in S. × irritans than in S. × falcatulum.
DISCUSSION
Allelic information and diversity capture from progenitors
There is considerable diversity (divergence) among the monoploid genomes contributing to S. × falcatulum, as captured subsequently in its current information and diversity. For these five markers, divergence among monoploid genomes (represented by HWI and μWI here) was by far the major source of allelic diversity in S. × falcatulum. Large differences in allelic information and diversity within the (R,S,T) homeologues are emblematic of the fact that multiple origins and ploidy-level changes have played a large role in the evolution of S. × falcatulum. Recurrent hybridization has evidently increased allelic diversity within both R and S homeologues, but it has not increased the allelic novelty within T. These patterns suggest recurrent hybridization between a genetically depauperate haploid T′ progenitor (S. cuspidatum) and an allodiploid R′ and S′ progenitor (S. × irritans), containing genetically diverse monoploid genomes. The pattern of allelic information and diversity detected in the two latter species supports this hypothesis.
In turn, the large difference in allelic diversity between the R genomes and the S genomes in S. × irritans strongly suggests recurrent hybridization between S. × irritan’s (monoploid) ancestors, with the ancestor contributing genome S′ harbouring greater internal genetic diversity than the ancestor contributing R′. Our results show that the frequency of hybridization between divergent progenitors, plus the diversity within each of the participants, can be expected to influence both the monoploid and the holoploid allelic diversity within allopolyploid derivative species, with diverse ancestors leaving separable signatures. In the case of S. × falcatulum, the addition of monoploid genome T increased holoploid μWI, but decreased holoploid βAP and λΑΙ.
We conclude that the large majority of genetic information and diversity contained within these five markers in S. × falcatulum were ‘captured’ directly from the ancestral progenitors, via serial and recurrent hybridization. Clearly, a trio of monoploid ancestors introduced novelty into S. × falcatulum, with monoploid S contributing the most diversity and T the least. A small number of subsequent mutations and changes in hologenomic structure have also contributed to genetic diversification within S. × falcatulum. Given its relatively recent origin (Karlin et al., 2013), the current genetic information and diversity within each of the three homeologous genomes in S. × falcatulum largely reflects the intense filtering of alleles associated with two serial rounds of recurrent reticulation, the first round giving rise to S. × irritans and the second yielding S. × falcatulum. However, our data show only slight subsequent divergence between S. × falcatulum and its two immediate progenitors.
Post-reticulate innovation
De novo alleles.
As the data sets for each progenitor species were small, separating ancestral from de novo alleles was inevitably a bit tentative. We note, however, that the numbers of de novo alleles per locus (within S. × falcatulum) were comparable among the three monoploid genomes (Table 2), suggesting the appearance of (post-hybridization) allelic novelty, rather than representing alleles captured via hybridization. It is possible, of course, that at least some of these putative de novo alleles, not currently detectable in immediate progenitors, were introduced via hybridization. While more elaborate sampling of the progenitors may eventually provide additional evidence for or against this interpretation, the data in hand argue for post-establishment generation of at least some genetic novelty.
Change in hologenomic structure.
The hologenomic structure in S. × falcatulum based on these five markers has been relatively stable, both within and across regions, and there appears to have been little change in the balance among monoploid genomes, in spite of multiple origins. But some hologenomic change has occurred, either in S. × irritans (subsequently inherited by S. × falcatulum) or post-origin, within S. × falcatulum. The clonal nature of gametophytic Sphagnum allows such changes to replicate with fidelity and to persist for extensive periods of time (Karlin et al., 2012). If a single individual exhibits loss of a particular allele, that might represent no more than a non-amplifying (null) allele in that individual. But when several individuals from a single clonal lineage (or an entire population) lack the allele, absence probably represents a change in holoploid genomic structure. The strongest evidence of hologenomic loss was associated with the TdF population. The evidence for hologenomic change in S.× irritans was ambiguous, as all individuals but one had the ‘founding’ HWI(R′,S′) = 0·693.
Regional vs. subregional diversification
Based on five ancestrally indicative markers, allelic divergence for S. × falcatulum was slight among these three highly disjunct Holantarctic regions, and also low among populations within those regions. Indeed, divergence among individuals within populations (HAI) exceeded divergence among regions (HAR). However, in spite of the low regional divergence, markedly different patterns of holoploid allelic information and diversity occurred among individuals within NZ [HAP(NZ) + HAI(NZ)] compared with those in Tas and TdF. The lack of information among individuals within the five sets of three homeologous trios for each of the Tas and TdF regions clearly indicates that their respective histories differ greatly from that associated with NZ. Given the stability of the alleles at these five markers, HAI(NZ) > 0 indicates that at least two genets are present in at least some NZ populations. Likewise, HAP(NZ) > 0 indicates that there are at least frequency differences among genets for different NZ populations. In stark contrast, our data show no evidence supporting the occurrence of more than one genet in either Tas or TdF. This possibility is supported by the frequent occurrence of sexual reproduction in NZ, and its virtual absence from TdF and from Macquarie Island (Karlin et al., 2013). The occurrence of a sporophyte on only one Tasmanian herbarium specimen suggests that there may be more than one genet present on that island, but we see no evidence of that in our limited Tas samples. Additional study is required to fully explore this question. Our data indicate that NZ was the probable source region and that TdF and Tas represent later colonizations by one or a few propagules each. Sphagnum × falcatulum’s capacity for long-distance dispersal, its unisexuality and its clonal nature allow for the development of populations, even at the regional level, within which there is no divergence among individuals, based on these five markers. Although a young species, S. × falcatulum has managed to span much of the Holantarctic, more or less clonally.
Appropriate statistical analysis for allopolyploids
With two or more homeologous loci per marker, allopolyploids have a greater genetic potential than that found in diploids (or haploids in the case of bryophytes). Based on the five markers used in this study, the majority of this genetic potential in both S. × falcatulum and S. × irritans, and probably other allopolyploids having highly divergent monoploid genomes, occurs within individuals, and not among individuals. Thus, it becomes important to consider HWI in the context of (a) how much genetic novelty is present within the species, (b) where it is localized and (c) how it is bundled. Finally, changes in HWI may signal changes in hologenomic structure. We conclude that it is particularly valuable to elucidate HWI (and μWI) when studying allopolyploids.
Meirmans and Van Tienderen (2013) note that subgenomes (homeologous monoploid genomes) should be analysed as separate loci when strict disomic inheritance occurs in allopolyploids. That approach was utilized by the first author in two previous studies of Sphagnum allopolyploids, one on the allo-diploid S. × palustre (Karlin et al., 2012) and one on S. × falcatulum (Karlin et al., 2013). However, focusing solely on allelic information and diversity among alleles for the constituent homeologous loci misses the larger and perhaps more important story. Although accommodating ‘among-individual’ comparisons, the ‘separate loci’ approach ignores the fact that allelic diversity among homeologous loci (represented by HWI and μWI in S. × falcatulum and S. × irritans) may represent the bulk of allelic information and diversity resident within many allopolyploid species.
CONCLUDING THOUGHTS
As shown here, Shannon information and diversity measures provide a powerful and effective approach to the study of allelic diversity at many levels (Sherwin et al., 2006). Even when applied to a genetically complex species, Shannon analysis yields an interconnected and seamless link from the allelic diversity within individuals to that present within and among regions.
This study focused on five markers that facilitated comparison of allelic diversity captured from multiple origins with that emerging from post-hybridization within S. × falcatulum itself. Although such stable markers are often overlooked by studies focused on one species, we show that they can be quite informative about the evolutionary history of allopolyploids. SSR markers which are evolutionarily more labile, and thus unsuitable for this study, would be expected to provide a different perspective on the allelic information and diversity in S. × falcatulum, notably the recent history of these same populations and regions. Analyses of the nine other SSR markers that have been sampled from this same material are currently underway. It will prove challenging (but nevertheless informative) to compare the information and diversity of those more labile markers with those found for the highly stable markers used here, and we expect to ‘flesh out the story’.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Table S1: voucher data for Sphagnum × falcatulum, S. ×irritans and S. cuspidatum. Table S2: allelic data for Sphagnum × falcatulum, S. × irritans and S. cuspidatum at five ancestrally indicative markers. Table S3: data sheet showing format for triploid analysis of the Tasmanian hologenomic data in GenAlEx. Table S4: results of Shannon analysis of the Tasmanian triploid data using the ‘Partition’ option.
Supplementary Material
ACKNOWLEDGMENTS
DNA extraction and analysis was done at The Shaw Bryology lab at Duke University and we thank Jonathan Shaw for the use of the lab and Sandra Boles for help with the lab work. Allelic data for one individual of S. × irritans was provided by the lab of Sean Robinson (New York State University College at Oneonta). P.E.S. was supported byUSDA/NJAES-17111 and 17160. We appreciate the constructive comments provided by the anonymous reviewers and the Handling Editor (Anne Brysting).
LITERATURE CITED
- Andrews AL. 1949. Studies in the Warnstorf Sphagnum herbarium V. The Group Cuspidata in South America. Bryologist 52: 124–130. [Google Scholar]
- Beever J, Allison KW, Child J.. 1992. The moss flora of New Zealand, 2nd edn. Dunedin: University of Otago Press. [Google Scholar]
- Catcheside DG. 1980. Mosses of South Australia. Handbook of the flora and fauna of South Australia. South Australia: Government Printer. [Google Scholar]
- Chester M, Gallagher JP, Symonds VV, et al. 2012. Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proceedings of the Natural Academy of Sciences, USA 109: 1176–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester M, Riley RK, Soltis PS, Soltis DE.. 2015. Patterns of chromosomal variation in natural populations of the neoallotetraploid Tragopogon mirus (Asteraceae). Heredity 114: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman M, Levy A, Chalhoub B, Kashkush K.. 2012. Genomic plasticity in polyploid wheat In: Soltis PS, Soltis DE, eds. Polyploidy and genome evolution. Berlin: Springer-Verlag. doi:10.1007/978-3-642-31442-1_7 [Google Scholar]
- Fife AJ. 1996. A synopsis of New Zealand Sphagna, with a description of S. simplex sp. nov. New Zealand Journal of Botany 34: 309–328. [Google Scholar]
- Greilhuber J, Doležel J, Lysák MA, Bennett MD.. 2005. The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C-value’ to describe nuclear DNA contents. Annals of Botany 95: 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost L. 2006. Entropy and diversity. Oikos 113: 363–375. [Google Scholar]
- Jost L. 2007. Partitioning diversity into independent alpha and beta components. Ecology 88: 2427–2439. [DOI] [PubMed] [Google Scholar]
- Jost L. 2010. Independence of alpha and beta diversities. Ecology 91: 1969–1974. [DOI] [PubMed] [Google Scholar]
- Karlin EF. 2014. Subgenome analysis of two Southern Hemisphere allotriploid species in Sphagnum (Sphagnaceae). Journal of Bryology 36: 165–179. [Google Scholar]
- Karlin EF, Robinson SC.. 2016. Update on the Holantarctic Sphagnum × falcatulum s.l. (Sphagnaceae) complex: S. irritans is associated with the allo-diploid plants. Journal of Bryology (in press) http://dx.doi.org/10.1080/03736687.2016.1218674. [Google Scholar]
- Karlin EF, Boles SB, Shaw AJ.. 2008. Systematics of Sphagnum section Sphagnum in New Zealand: a microsatellite-based analysis. New Zealand Journal of Botany 46: 105–118. [Google Scholar]
- Karlin EF, Boles SB, Ricca M, Temsch E, Greilhuber J, Shaw AJ.. 2009. Three-genome mosses: complex double allopolyploid origins for triploid gametophytes in Sphagnum. Molecular Ecology 18: 1439–1454. [DOI] [PubMed] [Google Scholar]
- Karlin EF, Boles SB, Seppelt RD, Terracciano S, Shaw AJ.. 2011. The peat moss Sphagnum cuspidatum in Australia: microsatellites provide a global perspective. Systematic Botany 26: 22–32. [Google Scholar]
- Karlin EF, Hotchkiss SC, Boles SB, et al. 2012. High genetic diversity in a remote island population system: sans sex. New Phytologist 193: 1088–1097. [DOI] [PubMed] [Google Scholar]
- Karlin EF, Buck WR, Seppelt RD, Boles SB, Shaw AJ.. 2013. The double allopolyploid Sphagnum × falcatulum (Sphagnaceae) in Tierra del Fuego, a Holantarctic perspective. Journal of Bryology 36: 165–179. [Google Scholar]
- Karlin EF, Temsch EM, Bizuru E, et al. 2014. Invisible in plain sight: recurrent double allopolyploidy in the African Sphagnum ×planifolium (Sphagnaceae). The Bryologist 117: 187–201. [Google Scholar]
- Marcussen T, Sandve SR, Heier L, et al. 2014. Ancient hybridizations among the ancestral genomes of bread wheat. Science 345: doi:10.1126/science.1250092. [DOI] [PubMed] [Google Scholar]
- Mason-Gamer RJ. 2008. Allohexaploidy, introgression, and the complex phylogenetic history of Elymus repens (Poaceae). Molecular Phylogenetics and Evolution 47: 598–611. [DOI] [PubMed] [Google Scholar]
- Meirmans PG, Van Tienderen PH.. 2013. Statistical challenges for population genetics of polyploids: the effects of inheritance in tetraploids on genetic diversity and population divergence. Heredity 110: 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE.. 2006. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6: 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE.. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics 28, 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury G.K. 1955. A handbook of New Zealand mosses. Bulletin of the Royal Society of New Zealand 5: 1–490. [Google Scholar]
- Såstad SM. 2005. Patterns and mechanisms of polyploid speciation in bryophytes In: Bakker T, Chatrou Gravendeel B, Pelser P, eds. Plant species level systematics: new perspectives on pattern and process. Ruggell, Liechtenstein: Gantner Verlag, 317–333. [Google Scholar]
- Scott GAM, Stone IG, Rosser C.. 1976. The mosses of southern Australia. London: Academic Press. [Google Scholar]
- Seppelt RD. 2012. Australian Mosses Online 52. Sphagnaceae. Australian Biological Resources Study, Canberra. Version 22 June 2012. http://www.anbg.gov.au/abrs/Mosses_online/ 52_ Sphagnaceae.html. last accessed 10 August 2016.
- Shaw AJ, Cox CJ, Boles SB.. 2003. Polarity of peatmoss (Sphagnum) evolution: who says mosses have no roots? American Journal of Botany 90: 1777–1787. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, Cao T, Wang L-S, et al. 2008. Genetic variation in three Chinese peat mosses based on microsatellite markers, with primer information and analysis of ascertainment bias. The Bryologist 111: 271–281. [Google Scholar]
- Shaw AJ, Cox CJ, Buck WR, et al. 2010. Newly resolved relationships in an early land plant lineage: Bryophyta class Sphagnopsida (peat mosses). American Journal of Botany 97: 1511–1531. [DOI] [PubMed] [Google Scholar]
- Shaw AJ, Devos N, Liu Y, et al. 2016. Organellar phylogenomics of an emerging model system: Sphagnum (peat moss). Annals of Botany 118: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin WB. 2010. Entropy and information approaches to genetic diversity and its expression: genomic geography.Entropy 12: 1765–1798. [Google Scholar]
- Sherwin WB, Jobot F, Rush R, Rossetto M.. 2006. Measurement of biological information with applications from genes to landscapes. Molecular Ecology 15: 2857–2869. [DOI] [PubMed] [Google Scholar]
- Smouse PE, Ward RH.. 1978. A comparison of the genetic infrastructure of the Ye'cuana and the Yanomama: a likelihood analysis of genotypic variation among populations. Genetics 88: 611–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smouse PE, Whitehead MR, Peakall R.. 2015. An informational diversity analysis framework, illustrated with sexually deceptive orchids in early stages of speciation. Molecular Ecology Resources 15: 1375–1384. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Buggs RJA, Doyle JJ, Soltis PS.. 2010. What we still don’t know about polyploidy. Taxon 59: 1387–1403. [Google Scholar]
- Symonds VV, Soltis PS, Soltis DE.. 2010. Dynamics of polyploid formation in Tragopogon (Asteraceae): recurrent formation, gene flow, and population structure. Evolution 64: 1984–2003. [DOI] [PubMed] [Google Scholar]
- Tuomisto H. 2010. A diversity of beta diversities: straightening up a concept gone awry. I. Defining beta diversity as a function of alpha and gamma diversity. Ecography 33: 2–22. [Google Scholar]
- Warnstorf C. 1911. Sphagnales–Sphagnaceae (Sphagnologia universalis) In Engler A. (ed.) Das Pflanzenreich: regni vegetablilis conspectus, Vol 51 Leipzig: W. Engelmann. [Google Scholar]
- Whittaker RH. 1977. Evolution of species diversity in land communities. Evolutionary Biology 10: 1– 67. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.