

Abstract
The Response Evaluation Criteria in Solid Tumors (RECIST) were developed and published in 2000, based on the original World Health Organization (WHO) guidelines first published in 1981. In 2009, revisions were made (RECIST 1.1) incorporating major changes, including a reduction in the number of lesions to be assessed, a new measurement method to classify lymph nodes as pathologic or normal, the clarification of the requirement to confirm a complete response (CR) or partial response (PR) and new methodologies for more appropriate measurement of disease progression. The purpose of this paper is to summarize the questions posed and the clarifications provided as an update to the 2009 publication.
Keywords: RECIST, clarifications, tumor response
Introduction
World Health Organization (WHO) response guidelines were first published in 1981 [1,2]. The RECIST criteria were based on those criteria, and were themselves updated in a 2009 in the European Journal of Cancer (RECIST 1.1) [3]. The revised guidelines incorporated major changes to the original RECIST criteria [2], including a reduction in the number of lesions to be assessed, a new measurement method to classify lymph nodes as pathologic or normal, the clarification of the requirement to confirm a complete response (CR) or partial response (PR) and new recommendations for the assessment of disease progression. Supplementary information provided included imaging guidelines, which better defined image acquisition and interpretation.
The RECIST criteria have gained widespread adoption and are widely used in oncology clinical trials. The RECIST 1.1 paper has been cited 3881 times as of December 2015 Web of Science. Endpoints categorized by the RECIST criteria have been used as either primary or supportive data for regulatory approval of new therapeutics by both the FDA and EMA [4]. RECIST provides a standardized set of rules for response assessment using tumour shrinkage, based upon imaging modalities that are globally available and interpretable by most clinicians. This standardisation, and the rules and criteria established, provide a framework for reproducible analysis and reporting of changes in tumour size. The reproducibility of these criteria and the correlations with historical trial results serve an important purpose in drug discovery.
Despite the widespread acceptance of RECIST, the RECIST Working Group continues its work. RECIST and RECIST 1.1 were developed and tested using data from clinical trials testing cytotoxic drugs. In the last decade, there have been substantial changes in the mechanism of action of cancer therapeutics (targeted agents, immunotherapies), as well as advances in imaging and clinical trial design and endpoints. Although the majority of clinical trials continue to use RECIST as an adjunct to the gold standard endpoints of survival and quality of life, the RECIST Working Group continues to build data warehouses (e.g. data from clinical trials involving targeted agents and immunotherapies, FDG PET/CT data) to review the criteria, update them periodically as required and validate any changes in a standardized, methodical manner in response to both therapeutic and imaging technology advances. Critically, the global oncology community must be able to implement and adopt any changes proposed to RECIST in a timely and cost effective manner.
Since the original publication, users have submitted questions on the use and interpretation of the guidelines through the RECIST website (http://www.eortc.org/recist/). These questions are reviewed by the Steering Committee and replies provided based on the 2009 publication. If not addressed in the publication, the Working Group is consulted to prepare appropriate replies. Relevant and/or frequently asked questions (FAQs) are posted on a regular basis on the RECIST website.
Here we summarize the questions and clarifications posed as an update to the 2009 publication.
Commonly asked questions regarding RECIST 1.1
-
What is the frequency of tumour evaluation?
The schedule and frequency of tumour response re-evaluation is protocol-specific and is based on the therapeutic, the disease, the anticipated time to response and progression as well as practical considerations such as cost and patient convenience.
However, in the context of phase II studies where the beneficial effect of therapy is not known, follow-up every 6–8 weeks (timed to coincide with the end of a cycle) is reasonable. Shorter or longer time intervals than these could be justified in specific regimens or circumstances. The protocol should specify which areas (chest, abdomen) and organs are to be evaluated at baseline (usually those most likely to be involved with metastatic disease for the tumour type under study) and how often evaluations should be repeated [2]. The method of assessment should be stated and, ideally, repeated using the same imaging technique, equipment and assessor each time. Additional assessments should be performed if there is suspicion of new site of metastasis, or of disease progression based upon clinical symptoms. The presence of a new lesion(s) should be documented on an imaging study. All potential sites of metastases should be evaluated at each time point rather than following only sites of disease identified at baseline.
-
Should lesions smaller than 5 mm be reported as the actual size or reported as a default value of 5 mm?
All lesions, both nodal and non-nodal, must be evaluated, accurately measured (if measurable/target) and recorded at all timepoints. If a lesion is no longer seen, it should be recorded as zero (or absent if non-measurable /non-target). If a lesion is smaller than 5 mm and the radiologist believes the lesion can be accurately measured then the actual size should be recorded.
It is recognized that lesions become small and ill-defined on a CT scan such that the radiologist cannot accurately measure them. Some radiologists use the term “too small to measure” to describe this phenomena. When this occurs, the radiologist may decide that the lesion is present but he/she does not feel comfortable providing the oncologist with a precise measurement. If this occurs, the radiologist may assign the lesion a value of 5 mm by default. This default value is nominally derived from the 5 mm CT slice thickness (but should not be changed with varying CT slice thickness). The measurement of this type of lesion is potentially non-reproducible, and providing this default value will prevent a false assessment of response or progressive disease due to measurement error.
-
If there are three or more measurable lesions in one organ, and we select two of them as target lesions, how should the third lesion be considered?
The third lesion should be considered a non-target lesion and should be recorded and followed as part of the non-target disease.
-
Is a single target lesion measurable if a patient has multiple non-target non-measurable disease?
Yes, a single target lesion is considered measurable disease provided the lesion meets the definition of measurability as described in RECIST 1.1.
-
How should the limitation of two target lesions per organ be applied to lymph nodes? Can individual chains/regions be considered one organ or are lymph nodes (all locations included) a single organ?
Lymph nodes are considered one organ. Only two lymph nodes should be measured per patient as target lesions. Other involved lymph nodes should be assessed and followed as non-target lesions. The analysis of data from the RECIST warehouse was performed in this manner.
-
Should double the slice thickness/interval be applied to lymph nodes as well when a CT slice thickness of 10 mm is used?
It is strongly recommended that CT slice thickness of 5 mm be used. Although RECIST 1.1 (2009) recommends the following: “As is described in Appendix II, when CT scans have slice thickness greater than 5 mm, the minimum size for a measurable lesion should be twice the slice thickness”, in 2016 contemporary CT scanners globally should be able to acquire images with a slice thickness of 5 mm or less. This is recommended by the Quantitative Imaging Biomarkers Alliance (QIBA) guidelines for standardizing image acquisition for CT (http://rsna.org/QIBA_.aspx) [5]. There are many disadvantages and no real advantages in obtaining 10 mm slices. Not only are lesions more difficult to measure, but new lesion conspicuity is significantly less at 10 mm [6, 7].
-
When lymph nodes coalesce forming a conglomerate mass, which axis should be measured to assess the response: short or long axis?
The short axis of lymph nodes should always be measured. As nodal lesions coalesce, a plane between them may be maintained that would aid in obtaining maximal short axis diameter measurements of each individual lesion. If the nodal lesions have truly coalesced such that they are no longer separable, the vector of the longest diameter in this instance should be used to determine the perpendicular vector for the maximal short axis diameter of the coalesced lesion (Fig. 1). Non-nodal lesions that coalesce should similarly be assessed by the longest diameter.
-
Clarification of the definition of Stable Disease (SD).
The definition of stable disease is clarified as follows: “Neither sufficient shrinkage (compared to baseline) to qualify for partial or complete response (CR or PR) nor sufficient increase (taking as reference the smallest sum of diameters at baseline or while on study, whichever is smallest) to qualify for progressive disease (PD)”.
It is important to recognize that the classification of a response (either CR or PR) occurs in comparison to the sum of diameters at baseline, while progression is based on a comparison to the smallest of the sum of diameters at baseline or the smallest sum of diameters during the trial (nadir). Most protocols require the criteria for SD for a specified period (for example at least 4 weeks) before SD can be concluded. Thus, if imaging is conducted at 2 weeks and 4 weeks on-study, and at 2 weeks the criteria for CR, PR or PD are not met, but at 4 weeks meets the criteria for PD, the best overall response is PD, not SD as the subject was not on-study long enough to qualify for SD.
-
If an abnormal lymph node (or non-nodal disease) ‘disappears’ but then ‘reappears’ should this considered to be PD?
In general, significant lesions that completely regress and then reappear are indicative of PD. However, it is important to consider the patient's entire tumor burden in order to make certain that the patient is not falsely classified as having PD based upon a single measurement or lesion, especially when those lesions are small or there is a change in optimal imaging assessment. This holds true for all types of metastases (Fig 2)
If the response was previously considered to be CR, with resolution of all sites of disease, then the reappearance of any lesion, or the development of a new lesion considered to be malignant, would generally be considered PD (see comments below regarding lymph nodes).
In the case of PR or SD, if a previously resolved lesion reappears, then PD should not be assigned purely based on the reappearance of the lesion; rather, other criteria for PD must be met as well, such as the appearance of new lesions or a sum measurement of target lesions that has increased more than 20%.
It is important to remember that lymph nodes must meet the criteria for malignancy (defined pragmatically as ≥ 10mm) in order to be considered to have reappeared. A node which is less than 10 mm is considered benign and is not PD. In a subject with CR, a new node that meets the size criteria for a pathologic node is considered a new site of disease and is therefore consistent with PD. In subjects with PR or SD:- A previously abnormal target node that became normal and subsequently enlarged in size meeting the criteria for a pathologic and measurable node (a short axis of ≥ 15 mm) should be added to the Sum of the Diameters (SOD) to determine if the criteria for PD are met based on target lesions.
- A previously abnormal non-target node that became normal and subsequently recurred must meet the criteria for PD based on NT lesions to call progression.
- A normal node at baseline that subsequently becomes pathologic is considered a new lesion and results in PD.
In the circumstances illustrated above, where a single pathologic node is driving the progression event, continuation of treatment/follow-up and confirmation by a subsequent exam should be contemplated. If it becomes clear that the “new node” has not resolved, or has significantly increased in size, and truly represents PD, the date of PD would be the date the new node was first documented.
-
How should patients be classified who have had surgery/radiotherapy during trials for which they are being followed by RECIST?
For most clinical trials using RECIST, surgery or radiotherapy after trial inclusion and prior to disease progression is a protocol deviation, and if a target lesion has been surgically removed or treated with radiotherapy, then the patient's response is not evaluable. If surgery or radiotherapy is part of the clinical trial, then the protocol must define in advance how response and progression will be handled and ‘censored’ in the analyses. If the treatment has resulted in inoperable lesions being suitable for resection, the researcher may wish to capture that separately as an indicator of ‘activity’.
-
The section on FDG-PET mentions that correlation with CT is warranted for new lesions, and that PD should be declared if the hot spot on PET corresponds to a progressing lesion on CT. How should this correlation be made if the hot spot on PET corresponds to a target lesion that has increased in size but the sum of the measurements does not show an increase that is sufficient for PD (other target lesions have not enlarged or have actually decreased). Is this PD?
The RECIST 1.1 guidelines state “a. Negative FDG-PET at baseline, with a positive FDG-PET at follow-up is a sign of PD based on a new lesion. b. No FDG-PET at baseline and a positive FDG-PET at follow- up: If the positive FDG-PET at follow-up corresponds to a new site of disease confirmed by CT, this is PD. If the positive FDG-PET at follow-up is not confirmed as a new site of disease on CT, additional follow-up CT scans are needed to determine if there is truly progression occurring at that site (if so, the date of PD will be the date of the initial abnormal FDG-PET scan). If the positive FDG-PET at follow-up corresponds to a pre-existing site of disease on CT that is not progressing on the basis of the anatomic images, this is not PD”.
Therefore, the scenario is not PD. Currently PET scanning is considered a complementary modality primarily to assess for new tumor lesions. Therefore, if a target lesion is FDG avid and even if the standardized uptake value (SUV) on PET has increased, the patient would only be considered to be PD if the CT metrics for progression were met, including a greater than 20% increase in the sum measurement of lesions, non-target unequivocal PD or new lesions on CT (regardless of their presence or SUV on PET).
-
If there is a hot spot on FDG-PET (baseline PET is not available) that is not associated with a new CT (or MRI) lesion, the article states that PD should not be declared. What should the response be, not evaluable (NE)?
FDG-PET is a complementary modality and should be used in adjunct to the CT or MRI. Therefore, in this example, the patient's response should be based solely upon the measurements and findings on CT and/or MRI. Since there is no PET at baseline and no corresponding new lesion on CT or MRI then the patient can only be assessed on the existing lesions and their change from baseline to follow-up on CT or MRI. Providing the scan was evaluable and all lesions visible/measurable, the response would therefore be CR, PR or SD as no new lesions were seen on CT/MRI.
-
Can the CT information from PET-CT be used as the basis of CT assessments? Is the technical quality of such images sufficient for quantification as required in RECIST?
At present, the low dose or the attenuation correction CT portion of a combined PET-CT is not of optimal diagnostic CT quality for use with RECIST measurements. However, if the site has documented that a CT with appropriate radiation dose for diagnostic quality and IV and oral contrast was used (if not medically contraindicated) the CT portion of the PET-CT can be used for RECIST measurements.
-
Can we use coronal or sagittal imaging to measure lesions in CT if the largest diameter is in a plane other than axial?
It is recommended that the axial imaging plane be used in all cases on CT scans for consistency and for ease of measurement especially since reconstructions or advanced workstations are not always available globally. It is recognized that the other planes may represent the true long axis of the tumor but depending on the CT acquisition parameters across timepoints this may be difficult to consistently and reproducibly measure.
Fig 1.
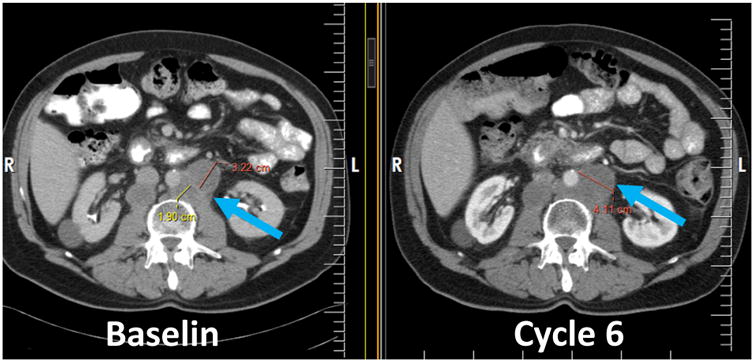
Note optimal manner for measuring the short axis of coalescing lymph nodes (blue arrow). At baseline, there are two distinct nodes, therefore the short axis is measured for each (red and yellow lines) and at cycle 6 the single short axis of the coalesced node is now measured as a single line (red).
Fig. 2.
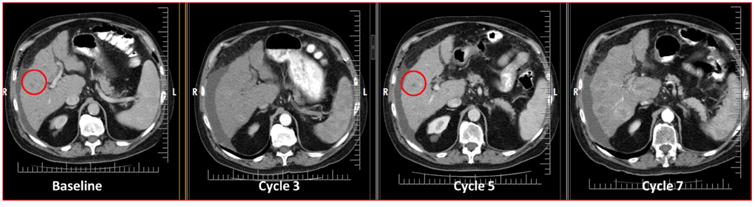
Patient with colorectal cancer. Liver metastases at baseline (red circle) appear to resolved at cycle 3 with “reappearance” at cycle 5 (red circle). However, notice that the imaging techniques are different and the cycle 3 is of poor image quality to visualize these metastases. Therefore, these lesions have not truly reappeared and the patient should not be considered to have progressive disease at cycle 5. There is true progression at cycle 7.
Conclusions
The RECIST Working Group is currently testing the Criteria for their applicability with modern therapeutics, imaging techniques and trial endpoints. The current RECIST 1.1 criteria remain widely used. A number of users have asked questions regarding the interpretation of the 2009 Criteria and we have attempted to summarize those questions, and the answers, in this update. For further updates on the criteria, please visit the RECIST website at http://www.eortc.org/recist/
Highlights.
Clarification on how to select target lesions and what to do with measurable lesions which are not selected for target disease response assessment
Clarification on the definition of stable disease
Clarification on the role of FDG-PET/PET-CT in the context of RECIST 1.1
Acknowledgments
Canadian Cancer Trials Group participation was supported by the Canadian Cancer Society Research Institute (grant #021039). This publication was supported by the EORTC Cancer Research Fund and by the NCI grant number 5U10-CA11488-45.
E de Vries: research grants from Roche/Genentech, Amgen, Novartis, Pieris and Servier to the institute, data monitoring committee Biomarin, advisory board Synthon.
R Ford: Dr. Ford is not a stockholder in any pharmaceutical company and holds no options, grants or patents. He is currently or was previously a consultant for the following companies (either directly through Clinical Trials Imaging Consulting, LLC., or indirectly through other pharmaceutical service companies for which he consults) including: ACR Image Metrix, AbbVie, Amgen, Aptiv, Aragon, Bioclinica, Biomedical Systems, Bristol Myers Squibb, Celldex, Celgene, Celsion, Covance, DNAtrix, Eisai, Exelixis, Genentech/Roche, ICON Medical Imaging, Image Endpoints, Janssen, Kyowa, Merck, Mirati Therapeutics, Novartis, Novocure, Oncothyreon, Ono, Optimer, OrbiMed Advisors, Pfizer, Radiant Sage, Quintiles, Red Hill Pharma, Sun Advanced Pharma Research Company, Tokai Pharmaceuticals, Vascular Biogenics and Virtualscopics.
S Hodi: Consultant for Merck, Novartis, Genentech, Synta; research support to institution from Bristol-Myers Squibb
N Lin: Research funding for clinical trials from (no direct salary support): Genentech, GSK, Novartis, Synta, Array biopharma, Kadmon, Puma
J.D. Wolchok: research grants from Bristol-Myers Squibb. Advisory board member for Brisol-Myers Squibb, Merck, Medimmune, Genentech.
Footnotes
Conflict of interest statement: Author and all other co-authors: none relevant
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47:207–14. doi: 10.1002/1097-0142(19810101)47:1<207::aid-cncr2820470134>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 3.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Janku F, Berry DA, Gong J, Parsons HA, Stewart DJ, Kurzrock R. Outcomes of phase II clinical trials with single-agent therapies in advanced/metastatic non–small cell lung cancer published between 2000 and 2009. Clin Cancer Res. 2012;18:6356–63. doi: 10.1158/1078-0432.CCR-12-0178. [DOI] [PubMed] [Google Scholar]
- 5.http://www.rsna.org/uploadedFiles/RSNA/Content/Science_and_Education/QIBA/QIBA_CT%20Vol_TumorVolumeChangeProfile_v2.2_PubliclyReviewedVersion_08AUG2012.pdf
- 6.Zhao B, Tan Y, Bell DJ, Marley SE, Guo P, Mann H, Scott ML, Schwartz LH, Ghiorghiu DC. Exploring intra- and inter-reader variability in uni-dimensional, bi-dimensional, and volumetric measurements of solid tumors on CT scans reconstructed at different slice intervals. Eur J Radiol. 2013;82:959–68. doi: 10.1016/j.ejrad.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sullivan DC, Schwartz LH, Zhao B. The imaging viewpoint: How imaging affects determination of progression-free survival. Clin Cancer Res. 2013;19:2621–8. doi: 10.1158/1078-0432.CCR-12-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]