

Abstract
Sepsis, a life-threatening organ dysfunction caused by infection, is a major public health concern with limited therapeutic options. We provide evidence to support a role for anaplastic lymphoma kinase (ALK), a tumor-associated receptor tyrosine kinase, in the regulation of innate immunity during lethal sepsis. The genetic disruption of ALK expression diminishes the stimulator of interferon genes (STING)–mediated host immune response to cyclic dinucleotides in monocytes and macrophages. Mechanistically, ALK directly interacts with epidermal growth factor receptor (EGFR) to trigger serine-threonine protein kinase AKT phosphorylation and activate interferon regulatory factor 3 (IRF3) and nuclear factor κB (NF-κB) signaling pathways, enabling STING-dependent rigorous inflammatory responses. Moreover, pharmacological or genetic inhibition of the ALK-STING pathway confers protection against lethal endotoxemia and sepsis in mice. The ALK pathway is up-regulated in patients with sepsis. These findings uncover a key role for ALK in modulating the inflammatory signaling pathway and shed light on the development of ALK-targeting therapeutics for lethal systemic inflammatory disorders.
INTRODUCTION
Sepsis is among the most common causes of death in hospitals and one of the most elusive syndromes in medicine (1). Although the word “sepsis” was first introduced by Hippocrates, clinical criteria for the definition of sepsis and septic shock remain challenging (2). Sepsis is now defined as life-threatening organ dysfunction due to a dysregulated host response to infection (3). Pathogenesis of the sepsis syndrome relies critically on the activation of innate immunity by a large family of pattern recognition receptors (PRRs) in response to microbial pathogens, including especially Gram-negative bacilli (Escherichia coli and Pseudomonas aeruginosa) (4). Mechanistically, immune chemicals (cytokines, chemokines, and growth factors) released by various innate immune cells trigger both pro- and anti-inflammatory immune responses, which can lead to organ dysfunction or failure, and even death (5). In these contexts, pharmacological inhibition of key inflammatory regulators that control the overwhelming immune response could be useful for therapy.
The stimulator of interferon genes (STING) is a transmembrane adaptor protein critically involved in the innate immune response to cyclic dinucleotides (CDNs) that are produced by bacteria or metabolized from double-stranded DNA (dsDNA) by cyclic guanosine monophosphate–adenosine monophosphate (cGAMP) synthase (cGAS) (6–12). Impairment of the STING pathway has been associated with the pathogenesis of several human diseases, including infections, inflammatory and autoimmune diseases, and cancers (13–15). Structurally, STING forms a complex with the TANK-binding kinase 1 (TBK1) to enable its phosphorylation, which results in activation of both interferon regulatory factor 3 (IRF3) and nuclear factor κB (NF-κB) signaling pathways (16–18), leading to the consequent production of type I interferons (IFNs) and other proinflammatory cytokines. Despite substantial investigation of the signaling pathways leading to STING activation, other key regulators of the STING signaling pathway remain to be elucidated.
Here, we screened a library of 464 kinase inhibitors for STING-modulating capacities and found that some inhibitors specific for anaplastic lymphoma kinase (ALK) displayed the strongest suppression of the STING-mediated type I IFN immune response in macrophages and monocytes. We provide evidence to support a critical role of ALK in the regulation of STING activation in monocytes and macrophages, which contribute to dysregulation of the innate immune response and pathogenesis of experimental and clinical sepsis. The second-generation ALK inhibitor LDK378, a U.S. Food and Drug Administration (FDA)–approved oral anticancer drug, exhibited promising anti-inflammatory activity in animal models of lethal sepsis, and the ALK pathway was up-regulated in patients with sepsis. Our data indicate a paradigm for how host innate immunity is regulated through the ALK signaling pathway in innate immune cells, and suggest that ALK inhibitors could be potential therapeutic agents for lethal systemic inflammatory diseases.
RESULTS
Bioactive compounds screen identifies STING modulators
To ensure a timely response to bacteria-derived CDN, an effective innate recognition system consisting of STING and other unknown transmembrane regulators has evolved in mammals (19). The 3′3′-cGAMP is a type of CDN and serves as a canonical STING ligand to induce the production of type I IFNs (IFNα and IFNβ) (20, 21). To identify other potential endogenous regulators of the STING signaling pathway in innate immune cells, we screened a library of 464 compounds that selectively target 174 signaling molecules in immortalized bone marrow–derived macrophages (iBMDMs) from B6 mice. Each compound was selected on the basis of its ability to principally interact with a single target, leading to minimal off-target activity. We found that 3′3′-cGAMP–induced IFNβ release in iBMDMs was changed by several target-selective inhibitors (Fig. 1A). The top five compounds that promoted the 3′3′-cGAMP–induced IFNβ release included lidocaine (a selective inverse peripheral histamine H1 receptor agonist), stavudine [a nucleoside analog reverse transcriptase inhibitor (NARTI) active against HIV], thiazovivin [a novel Rho-associated coiled-coil containing protein kinase (ROCK) inhibitor], AM1241 (a selective cannabinoid CB2 receptor agonist), and AS-252424 [a novel and potent phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit γ (PI3Kγ) inhibitor] (Fig. 1B). In contrast, the top five compounds that blocked 3′3′-cGAMP–induced IFNβ release in iBMDMs included AZD3463 (a novel orally bioavailable ALK inhibitor), KPT-330 (an orally bioavailable selective exportin-1 inhibitor), KU-60019 [a potent and specific ataxia telangiectasia mutated (ATM) inhibitor], PRT062607 [a novel and highly selective spleen-associated tyrosine kinase (Syk) inhibitor], and LDK378 (an inhibitor against ALK) (Fig. 1B). These top five bioactive compounds were further tested in primary peritoneal macrophages (pPMs) from B6 mice and human primary peripheral blood mononuclear cells (pPBMCs), which confirmed their inhibitory properties in mouse iBMDMs (Fig. 1C), mouse pPMs (Fig. 1D), and human pPBMCs (Fig. 1E). Although STING plays divergent and stimulus-dependent roles in innate immunity (13–15), a recent study revealed that activation of STING by bacteria accelerated the inflammatory response, organ dysfunction, and death in a mouse model of septic shock (22). Thus, the STING pathway seems to be a viable target for pharmacologic intervention during bacterial sepsis. After further analysis of the 174 molecular targets, we found that ALK was the top-ranked signaling molecule that promoted 3′3′-cGAMP–induced STING activation, based on IFNβ release from iBMDMs (Fig. 1F). Together, these findings suggest that ALK is a possible key modulator of STING activation during bacterial infections.
Fig. 1. Identification of bioactive compounds modulating STING activation.
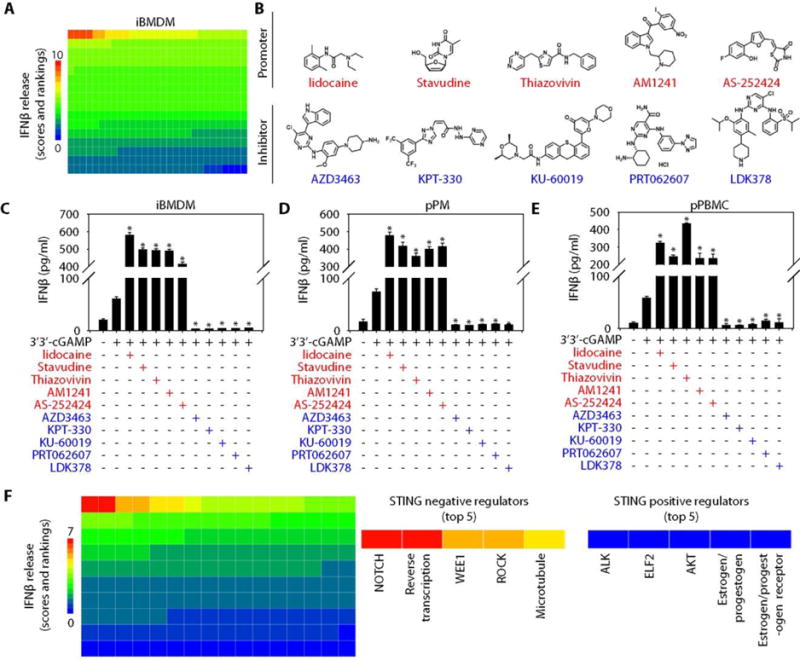
(A) Heatmap of STING activity changes based on IFNβ release from iBMDMs after 3′3′-cGAMP (10 μg/ml, 16 hours) stimulation in the absence or presence of 464 bioactive compounds (10 μM). (B) Structure of the compound identified to inhibit (blue) or promote (red) STING activity. (C to E) IFNβ release assayed using enzyme-linked immunosorbent assay (ELISA) from iBMDMs (C), pPMs (D), and pPBMCs (E) treated with 3′3′-cGAMP (10 μg/ml) in the absence or presence of indicated bioactive compounds (10 μM) for 16 hours [n = 3; data are means ± SD; *P < 0.05 versus 3′3′-cGAMP group, analysis of variance (ANOVA) least significant difference (LSD) test]. (F) Heatmap of STING activity changes as judged by IFNβ release from iBMDMs after 3′3′-cGAMP (10 μg/ml, 16 hours) stimulation in the absence or presence of 174 signaling modulating compounds. The top five negative (inhibitory) and positive (agonistic) regulators are noted.
Pharmacologic inhibition of ALK blocks STING activation
As secretory cells, monocytes and macrophages are vital to the regulation of immune responses and the development of inflammation (23). However, little information is available concerning the expression and activity of ALK in innate immune cells. We observed that ALK was abundantly expressed in primary or immortalized monocytes and macrophages (iBMDMs, pPMs, and pPBMCs; RAW264.7, J774A.1, and THP1 cells) from mice or humans (fig. S1A). Functionally, all three ALK inhibitors (AZD3463, LDK378, and AP26113) from a target-selective inhibitory library diminished 3′3′-cGAMP–induced IFNβ release in iBMDMs (fig. S1B). With respect to the tumor-killing activity of ALK inhibitors (24), we addressed whether AZD3463, LDK378, and AP26113 inhibit STING activation in macrophages through triggering cell death. AZD3463 exhibited cytotoxicity against iBMDMs, pPMs, and RAW264.7 and THP1 cells (fig. S1C). In contrast, LDK378 and AP26113 did not affect cell viability in these cells (fig. S1C), suggesting that the suppressive effect of LDK378 and AP26113 on STING activation in innate immune cells was not dependent on their cytotoxic capacities. In addition to 3′3′-cGAMP, a number of natural or enzymatically synthesized STING ligands [2′3′-cGAMP, 2′2′-cGAMP, cyclic dimeric adenosine monophosphate (c-di-AMP), cyclic dimeric guanosine monophosphate (c-di-GMP), cyclic dimeric inosine monophosphate (c-di-IMP), and 5,6-dimethylxanthenone-4-acetic acid (DMXAA)] with different structures also induce type I IFNs (8, 20, 25, 26). Both LDK378 and AP26113 inhibited IFNβ release induced by these different STING ligands in iBMDMs (Fig. 2, A and B), RAW264.7 cells (Fig. 2B and fig. S2A), J774A.1 cells (Fig. 2B and fig. S2B), THP1 cells (Fig. 2B and fig. S2C), or pPMs (Fig. 2B and fig. S2D). Notably, only DMXAA (also known as vadimezan or ASA404) targets the STING pathway in a mouse-specific manner (fig. S2C) (27, 28). Consistent with their inhibition of IFNβ protein release, pharmacologic inhibition of ALK by LDK378 and AP26113 also resulted in the attenuation of STING ligand–induced IFNβ mRNA expression in iBMDMs (Fig. 2, C and D), RAW264.7 cells (Fig. 2D and fig. S2E), J774A.1 cells (Fig. 2D and fig. S2F), THP1 cells (Fig. 2D and fig. S2G), or pPMs (Fig. 2D and fig. S2H). Thus, ALK seems to play an important role in the regulation of STING pathway activation in response to a wide array of STING ligands.
Fig. 2. Pharmacologic inhibition of ALK impairs STING activation.
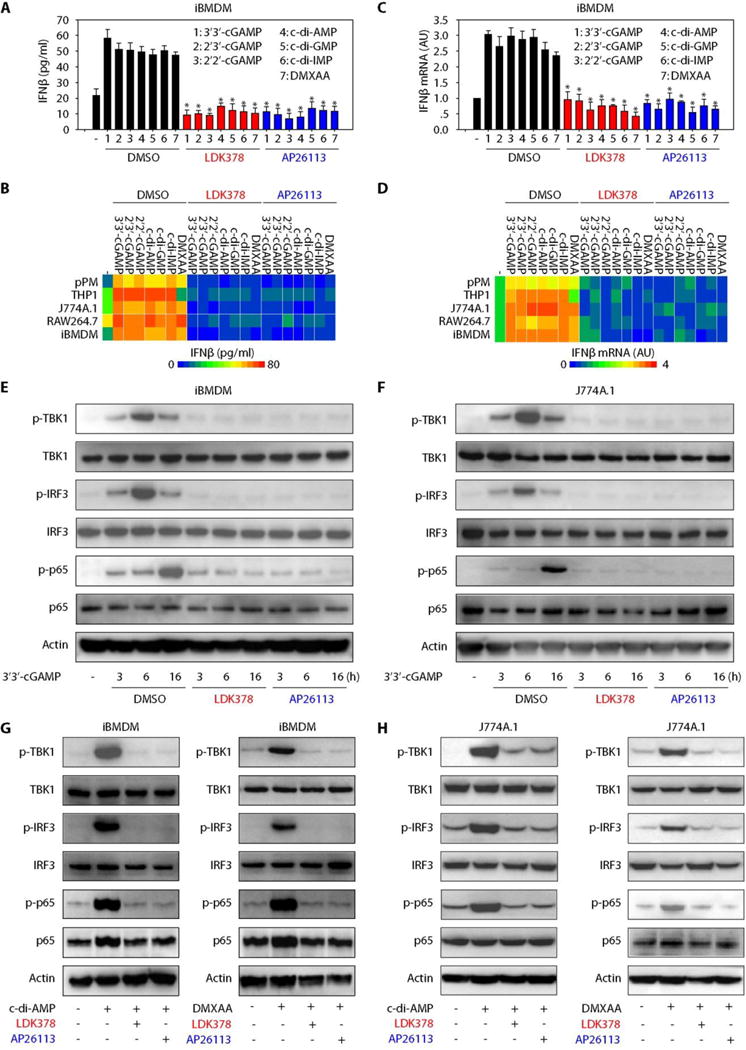
(A) iBMDMs were stimulated with indicated STING ligands (10 μg/ml) in the absence or presence of LDK378 (10 μM), AP26113 (10 μM), or control vehicle [dimethyl sulfoxide (DMSO)] for 16 hours, and the release of IFNβ was assayed using ELISA (n = 3; data are means ± SD; *P < 0.05 versus DMSO group, ANOVA LSD test). (B) Heatmap of IFNβ release changes in macrophages or monocytes after STING ligand (10 μg/ml) stimulation in combination with LDK378 (10 μM), AP26113 (10 μM), or vehicle (DMSO) for 16 hours. (C) iBMDMs were stimulated with indicated STING ligands (10 μg/ml) in the absence or presence of LDK378 (10 μM), AP26113 (10 μM), or vehicle (DMSO) for 16 hours, and IFNβ mRNA was assayed with quantitative polymerase chain reaction (n = 3; data are means ± SD; *P < 0.05 versus DMSO group, ANOVA LSD test). (D) Heatmap of IFNβ mRNA changes in macrophages or monocytes after STING ligand (10 μg/ml) stimulation in combination with LDK378 (10 μM), AP26113 (10 μM), or vehicle (DMSO) for 16 hours. AU, arbitrary units. (E and F) Western blot analysis of indicated protein expression in iBMDMs (E) or J774A.1 cells (F) after 3′3′-cGAMP (10 μg/ml) stimulation in combination with LDK378 (10 μM), AP26113 (10 μM), or vehicle (DMSO) for 3 to 16 hours. (G and H) Western blot analysis of indicated protein expression in iBMDMs (G) or J774A.1 cells (H) after c-di-AMP (10 μg/ml) or DMXAA (10 μg/ml) stimulation in combination with LDK378 (10 μM), AP26113 (10 μM), or vehicle (DMSO) for 16 hours.
A common event in STING activation by different ligands is the phosphorylation of TBK1 (p-TBK1) (16). We therefore examined the effect of ALK inhibition on the expression and phosphorylation of TBK1. Both LDK378 and AP26113 time-dependently reduced 3′3′-cGAMP–induced p-TBK1, but not total TBK1, in iBMDMs (Fig. 2E) and J774A.1 (Fig. 2F), RAW264.7 (fig. S3A), and THP1 (fig. S3B) cells. Similar to 3′3′-cGAMP, LDK378 and AP26113 also inhibited c-di-AMP– or DMXAA-induced p-TBK1, but not total TBK1, in iBMDMs (Fig. 2G) and J774A.1 (Fig. 2H), RAW264.7 (fig. S3C), and THP1 (fig. S3D) cells. Direct downstream targets of p-TBK1 include phosphorylation of IRF3 (p-IRF3) and NF-κB (p-p65) (16–18). Both LDK378 and AP26113 also inhibited 3′3′-cGAMP–, c-di-AMP–, and DMXAA-induced p-IRF3 and p-p65 in iBMDMs (Fig. 2, E and G) and J774A.1 (Fig. 2, F and H), RAW264.7 (fig. S3, A and C), and THP1 (fig. S3, B and D) cells. These results strongly suggest that pharmacologic inhibition of ALK blocks STING pathway activation through interfering with TBK1-mediated signaling transduction in monocytes and macrophages.
Genetic inhibition of ALK limits STING activation
Because pharmacological inhibitors often have undesirable off-target effects, we thus determined whether ALK gene knockdown has a similar impact on STING activation. We generated stable ALK knockdown macrophages using two different specific short hairpin RNA (shRNAs) and achieved 85% ~ 95% ALK knockdown after antibiotic selection in iBMDMs (Fig. 3A), RAW264.7 (fig. S4A), J774A.1 (fig. S4A), and THP1 (fig. S4A) cells, as confirmed using western blot analysis.
Fig. 3. Genetic silencing of ALK limits STING activation.
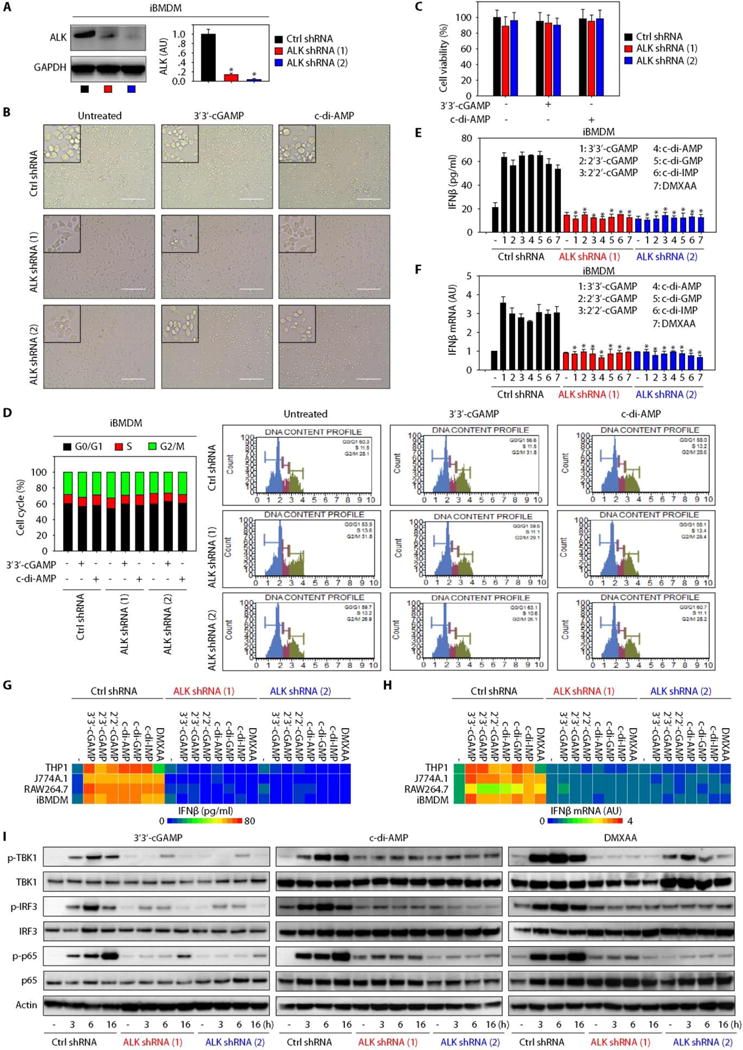
(A) Western blot analysis of ALK expression in ALK stable knockdown iBMDMs (n = 3; data are means ± SD; *P < 0.05 versus control shRNA group, t test). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B to D) Indicated iBMDMs were stimulated with 3′3′-cGAMP (10 μg/ml) or c-di-AMP (10 μg/ml) for 16 hours, and cell morphology (B), viability (C), and cell cycle phase (D) were assayed (scale bars, 200 μm). (E and F) Indicated iBMDMs were stimulated with indicated STING ligands (10 μg/ml) for 16 hours, and IFNβ protein release (E) and IFNβ mRNA (F) were assayed [n = 3; data are means ± SD; *P < 0.05 versus control (Ctrl) shRNA group, ANOVA LSD test]. (G and H) Heatmap of IFNβ protein release (G) and IFNβ mRNA expression (H) changes in indicated ALK-WT (wild-type) and ALK-knockdown macrophages or monocytes after STING ligand (10 μg/ml) stimulation for 16 hours. (I) Western blot analysis of indicated protein expression in ALK-WT and ALK-knockdown iBMDMs after stimulation with 3′3′-cGAMP (10 μg/ml), c-di-AMP (10 μg/ml), or DMXAA (10 μg/ml) for 16 hours.
As aforementioned, different ALK inhibitors exhibit divergent impacts on macrophage cell viabilities. To assess this behavior, we carefully analyzed cell morphology, cell viability, and cell proliferation with or without STING ligand (3′3′-cGAMP and c-di-AMP) treatment in ALK knockdown iBMDMs. Like control shRNA cells, these ALK knockdown iBMDM cell lines were not associated with a change in cell morphology (Fig. 3B), cell viability (Fig. 3C), and cell cycle (Fig. 3D) after treatment with 3′3′-cGAMP and c-di-AMP, suggesting that ALK depletion may not lead to macrophage death upon STING activation. Similar to pharmacological ALK inhibition, genetic inhibition of ALK by shRNA also attenuated STING ligand (3′3′-cGAMP, 2′3′-cGAMP, 2′2′-cGAMP, c-di-AMP, c-di-GMP, c-di-IMP, and DMXAA)–induced IFNβ expression and release in iBMDMs (Fig. 3, E to H) and RAW264.7 (Fig. 3, G and H, and fig. S4, B and C), J774A.1 (Fig. 3, G and H, and fig. S4, D and E), and THP1 (Fig. 3, G and H, and fig. S4, F and G) cells, indicating that ALK expression is required for STING activation. To corroborate these findings, we further examined protein phosphorylation of key STING signaling molecules in ALK knockdown and control cells. Consistent with pharmacologic inhibition of ALK by LDK378 and AP26113, genetic suppression of ALK expression also reduced 3′3′-cGAMP–, c-di-AMP–, and DMXAA-induced p-TBK1, p-IRF3, and p-p65 in iBMDMs (Fig. 3I) and RAW264.7 (fig. S4H), J774A.1 (fig. S4I), and THP1 (fig. S4J) cells. These data strongly support ALK as an important regulator of activation of the STING signaling pathway.
ALK-EGFR-AKT pathway promotes STING activation
It has been suggested that the phosphorylation of ALK (p-ALK) at Tyr1078 is required for its activation in tumor cells (30). To determine whether ALK is similarly phosphorylated by STING ligands in innate immune cells, we analyzed the expression of p-ALK and ALK in iBMDMs and RAW264.7 and THP1 cells after STING activation. Expression of p-ALK (but not total ALK) was enhanced in these cells by 3′3′-cGAMP, c-di-AMP, and DMXAA (Fig. 4A), suggesting that ALK is possibly activated by a wide array of STING ligands.
Fig. 4. ALK/EGFR binding triggers AKT-dependent STING activation.
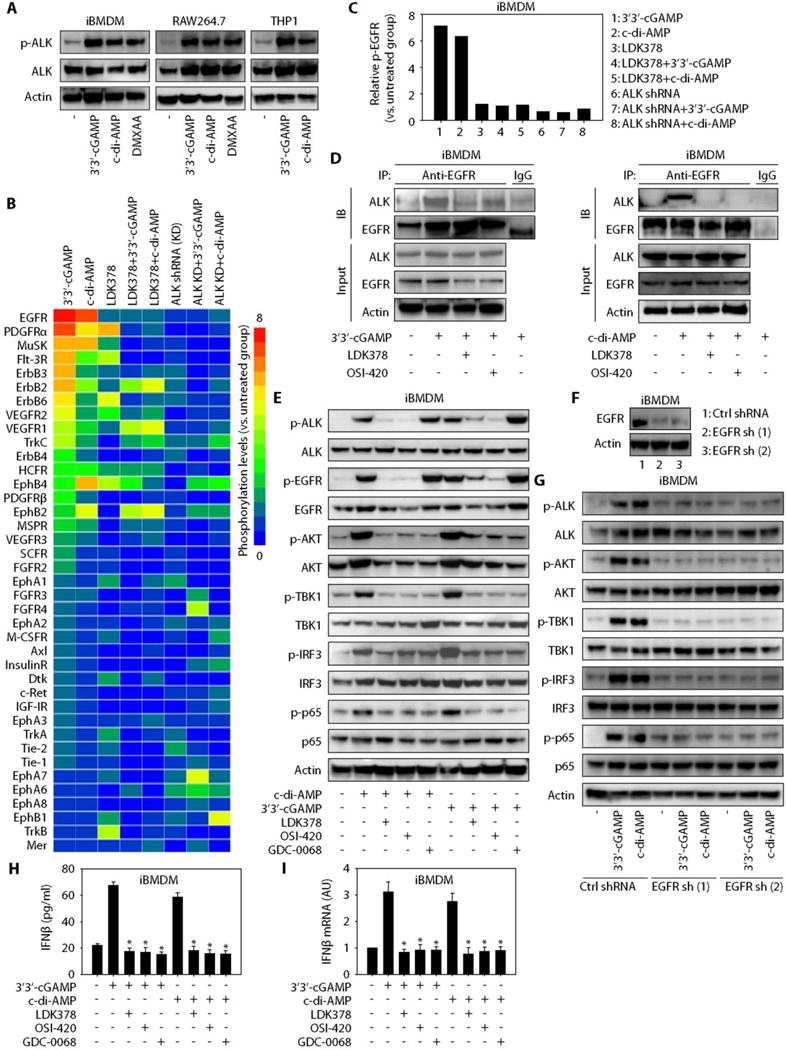
(A) Western blot analysis of indicated protein expression in iBMDMs and RAW264.7 and THP1 cells after stimulation with 3′3′-cGAMP (10 μg/ml), c-di-AMP (10 μg/ml), or DMXAA (10 μg/ml) for 16 hours. (B) Heatmap of RTKs phosphorylation changes in iBMDMs after 3′3′-cGAMP (10 μg/ml) or c-di-AMP (10 μg/ml) stimulation for 16 hours with or without pharmacologic (LDK378, 10 μM) or genetic inhibition of ALK. (C) Relative EGFR phosphorylation assayed in parallel to (B). (D) Immunoprecipitation (IP) analysis of the interaction between ALK and EGFR in iBMDMs after 3′3′-cGAMP (10 μg/ml) or c-di-AMP (10 μg/ml) stimulation for 16 hours with or without LDK378 (10 μM) or OSI-420 (10 μM). IB, immunoblotting. (E) Western blot analysis of indicated protein expression in iBMDMs after treatment with 3′3′-cGAMP (10 μg/ml) or c-di-AMP (10 μg/ml) for 16 hours with or without LDK378 (10 μM), OSI-420 (10 μM), or GDC-0068 (10 μM). (F) Western blot analysis of EGFR expression in EGFR stable knockdown iBMDMs. (G) Western blot analysis of indicated protein expression in EGFR-WT and EGFR-knockdown iBMDMs after stimulation with 3′3′-cGAMP (10 μg/ml) or c-di-AMP (10 μg/ml) for 16 hours. (H and I) iBMDMs were treated with 3′3′-cGAMP (10 μg/ml) or c-di-AMP (10 μg/ml) for 16 hours with or without LDK378 (10 μM), OSI-420 (10 μM), or GDC-0068 (10 μM), and IFNβ protein release (H) and IFNβ mRNA expression (I) were assayed (n = 3; data are means ± SD; *P < 0.05 versus 3′3′-cGAMP or c-di-AMP group, ANOVA LSD test).
To elucidate the possible role of ALK in the regulation of the STING signaling pathway, we tested whether ALK promotes the phosphorylation of core components of the STING pathway (STING, TBK1, and cGAS) through protein-protein interaction using coimmunoprecipitation techniques. We did not observe a direct interaction between ALK and these cytosolic signaling molecules in iBMDMs (fig. S5A) and RAW264.7 (fig. S5B) and THP1 (fig. S5C) cells after treatment with 3′3′-cGAMP and c-di-AMP. Immunoprecipitation analysis also did not reveal evidence of ALK binding to other recently identified cytosolic STING-interacting partners such as TIR [Toll/interleukin-1 (IL-1) receptor] domain–containing adapter-inducing IFNβ (TRIF) (30) or the ribosomal protein S6 kinase (fig. S5, A to C) (31). These findings suggest that ALK-mediated STING activation may not be dependent on direct binding to known cytosolic regulators of the STING pathway in macrophages and monocytes.
It is still possible that ALK, a member of the receptor tyrosine kinases (RTKs), may interact with other RTKs in the cell surface to mediate signal transduction into the cytoplasm under STING activation. To test this possibility, we used a proteome profiler antibody array to survey the phosphorylation of 49 RTKs in iBMDMs after stimulation with 3′3′-cGAMP or c-di-AMP. The phosphorylation of certain RTKs was changed by exposure to 3′3′-cGAMP and c-di-AMP (Fig. 4B and fig. S6). Among them, the phosphorylation of epidermal growth factor receptor (EGFR) was up-regulated by 3′3′-cGAMP and c-di-AMP (Fig. 4C and fig. S6), whereas LDK378 or knockdown of ALK impaired the STING ligand–induced up-regulated phosphorylation of various RTKs, including EGFR, in iBMDMs (Fig. 4C and fig. S6). These data suggest that RTK phosphorylation might be widely implicated in the STING pathway.
Previous observation demonstrated that the interplay between ALK and EGFR coordinately regulates AKT phosphorylation to promote tumor growth (32, 33). We therefore tested whether the ALK-EGFR-AKT pathway is also a critical driver of STING activation in innate immune cells. First, the interaction between ALK and EGFR was found to be increased in iBMDMs (Fig. 4D) and THP1 cells (fig. S7) after treatment with 3′3′-cGAMP or c-di-AMP but was reduced by inhibitors specific for both ALK (LDK378) and EGFR (OSI-420) (Fig. 4D and fig. S7). Second, these ALK (LDK378) and EGFR (OSI-420) inhibitors also attenuated the 3′3′-cGAMP– or c-di-AMP–induced phosphorylation of ALK, EGFR, and AKT in iBMDMs (Fig. 4E) and RAW264.7 (fig. S8A) and THP1 (fig. S8B) cells. In contrast, the pan-AKT inhibitor GDC-0068 only blocked the phosphorylation of AKT, but not ALK or EGFR, in activated iBMDMs (Fig. 4E) and RAW264.7 (fig. S8A) and THP1 (fig. S8B) cells. Third, all inhibitors (LDK378, OSI-420, and GDC-0068) similarly diminished the STING ligand–induced phosphorylation of TBK1, IRF3, and p65 in iBMDMs (Fig. 4E) and RAW264.7 (fig. S8A) and THP1 (fig. S8B) cells. Fourth, the ability of EGFR and AKT to promote the phosphorylation of TBK1, IRF3, and p65 was similarly impaired by stable genetic knockdown of EGFR via two different shRNAs in iBMDMs (Fig. 4, F and G) and RAW264.7 cells (fig. S9). Finally, pharmacologic inhibition of the ALK-EGFR-AKT pathway attenuated STING ligand (3′3′-cGAMP and c-di-AMP)–induced IFNβ release and mRNA expression in iBMDMs (Fig. 4, H and I), RAW264.7 cells (fig. S10, A and B), THP1 cells (fig. S10, C and D), and pPMs (fig. S10, E and F). Together, these data indicate that the interplay between ALK and EGFR contributes to the AKT-dependent STING activation in macrophages and monocytes.
ALK and STING regulates immune chemical release
The production and release of various immune chemicals such as cytokines, chemokines, and growth factors, as well as damage-associated molecular patterns, are generally considered a final effector during an immune response. Several clinical studies have suggested that immune chemical profiles, especially cytokines, are markers of disease severity, prognosis, and potential future therapeutic targets in patients with sepsis (34–36). Thus, to correlate symptoms with different phases of sepsis, it is important to survey the profile of multiple cytokines (37).
Like bacteria-derived CDN, lipopolysaccharide (LPS), the major component of the outer membrane of Gram-negative bacteria, is also a critical pathogen-associated molecular pattern (PAMP) involved in the pathogenesis of sepsis (38). Using a proteome profiler antibody array of 111 immune chemicals, we observed that stimulation with 3′3′-cGAMP, c-di-AMP, or LPS led to the release of different proteins from iBMDMs (Fig. 5A and fig. S11A). For example, all three stimuli induced the release of TNFα (tumor necrosis factor–α), IL-6, and serpin E1/PAI-1 (plasminogen activator inhibitor-1) (Fig. 5B), whereas only 3′3′-cGAMP and c-di-AMP markedly increased E-selectin release (Fig. 5B). In contrast, both 3′3′-cGAMP and LPS up-regulated the release of P-selectin and resistin (Fig. 5B), and only c-di-AMP or LPS enhanced the release of GDF-15 and CXCL10 (Fig. 5B). Collectively, these findings support the notion that different PAMPs might cause the release of different immune chemicals during innate immunity activation.
Fig. 5. ALK and STING have overlapping and distinct immune functions in immune chemical release.
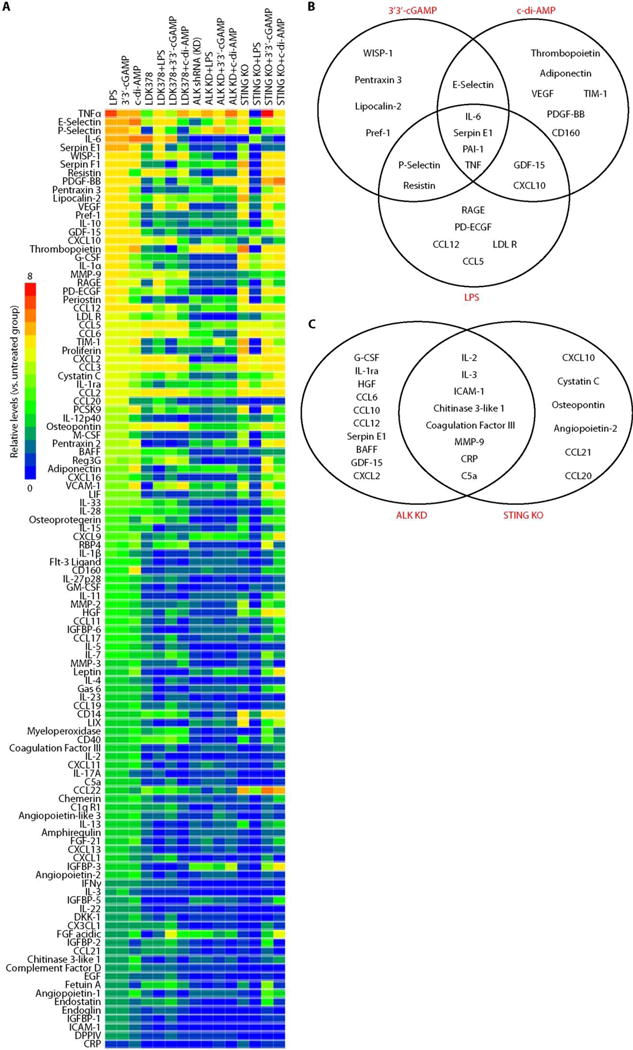
(A) Heatmap of immune chemical profile in wild type (WT), ALK-knockdown (KD), or STING-knockout (KO) iBMDMs after stimulation with LPS (1 μg/ml), 3′3′-cGAMP (10 μg/ml), or c-di-AMP (10 μg/ml) for 16 hours with or without LDK378 (10 μM). (B) Changes in immune chemical release in WT iBMDMs after LPS, 3′3′-cGAMP, and c-di-AMP treatment. (C) Changes in immune chemical release between ALK-KD and STING-KO iBMDMs in response to 3′3′-cGAMP, c-di-AMP, or LPS.
Given that signal transduction elements interact through complex biochemically related networks, we next sought to identify the overlapping and distinct immune effects of ALK and STING on immune chemical release in response to 3′3′-cGAMP, c-di-AMP, or LPS (Fig. 5A and fig. S11A). The knockdown of ALK and STING impaired the release of CRP (complement-reactive protein), IL-2, IL-3, C5a (complement component 5a), ICAM1 (intercellular adhesion molecule 1), coagulation factor III, chitinase 3–like 1, and MMP9 (matrix metalloproteinase 9) (Fig. 5C). Notably, most of these ALT (alanine aminotransferase)– and STING-related common immune chemicals play important roles in mediating sepsis-associated disseminated intravascular coagulation and thromboembolic disease. In addition, the expression profiling assays also revealed that ALK and STING occupied distinct roles in the regulation of most chemokines released in activated iBMDMs (Fig. 5C), suggesting that other unknown signaling molecules or feedback loops may also contribute to the regulation of different chemokines. LPS also has the ability to activate TBK1 to coordinate the activation of the IRF3 and NF-κB pathway in immune cells (39, 40). LDK378 also inhibited the LPS-induced phosphorylation of TBK1, IRF3, and p65 (fig. S11B) as well as IFNβ release (fig. S11C) in iBMDMs. These findings indicate that ALK contributes to LPS-induced activation of the TBK1–IRF3–NF-κB pathway in macrophages.
Inhibition of the ALK-STING pathway protects mice against septic death
One ultimate aim is to evaluate the therapeutic potential of ALK-STING pathway–targeting agents in sepsis and septic shock in vivo. Over the years, multiple animal models of sepsis have been developed, of which the cecal ligation and puncture (CLP) model is the most relevant to clinical sepsis (41, 42). To determine the effect of the ALK inhibitor LDK378 on polymicrobial sepsis, B6 mice were subjected to CLP with 22-gauge syringe needles (Fig. 6A). Repetitive administration of LDK378 [20 mg/kg, intraperitoneally (ip)] 2, 24, 48, and 72 hours after the onset of CLP conferred protection against lethality (Fig. 6B), which was associated with reduced injury in the lung, small intestine, and other tissues (Fig. 6C and fig. S12). For instance, septic lungs showed alveolar septal wall thickening, increase in leukocyte infiltrates, and alveolar congestion and edema, whereas septic intestines exhibited signs of injury characterized by the loss of goblet cells and loss of villi (Fig. 6C). These CLP-induced pathological changes were attenuated by LDK378 administration in septic mice (Fig. 6C). Biochemical measurement of tissue enzymes also revealed protective effects of LDK378 against dysfunction of the heart [creatine kinase (CK)], pancreas [amylase (AMYL)], kidney [blood urea nitrogen (BUN)], and liver (ALT) (Fig. 6D). Moreover, LDK378 administration reduced CLP-induced TNFα, IFNβ, MCP1 (monocyte chemoattractant protein-1), and IL-7 mRNA expression (Fig. 6E) and systemic release and accumulation in the serum (Fig. 6, F and G, and fig. S13). In an animal model of lethal endotoxemia (LPS, 10 mg/kg, ip) (Fig. 7A), LDK378 promoted similar protection against endotoxemic lethality (Fig. 7B), endotoxemia-induced tissue injury (Fig. 7C and fig. S14), organ dysfunction (Fig. 7D), and proinflammatory cytokine expression (Fig. 7E) and release (Fig. 7, F and G, and fig. S15). Thus, LDK378 protects mice against polymicrobial sepsis and lethal endotoxemia.
Fig. 6. Inhibition of the ALK-STING pathway protects mice against CLP-induced polymicrobial sepsis.
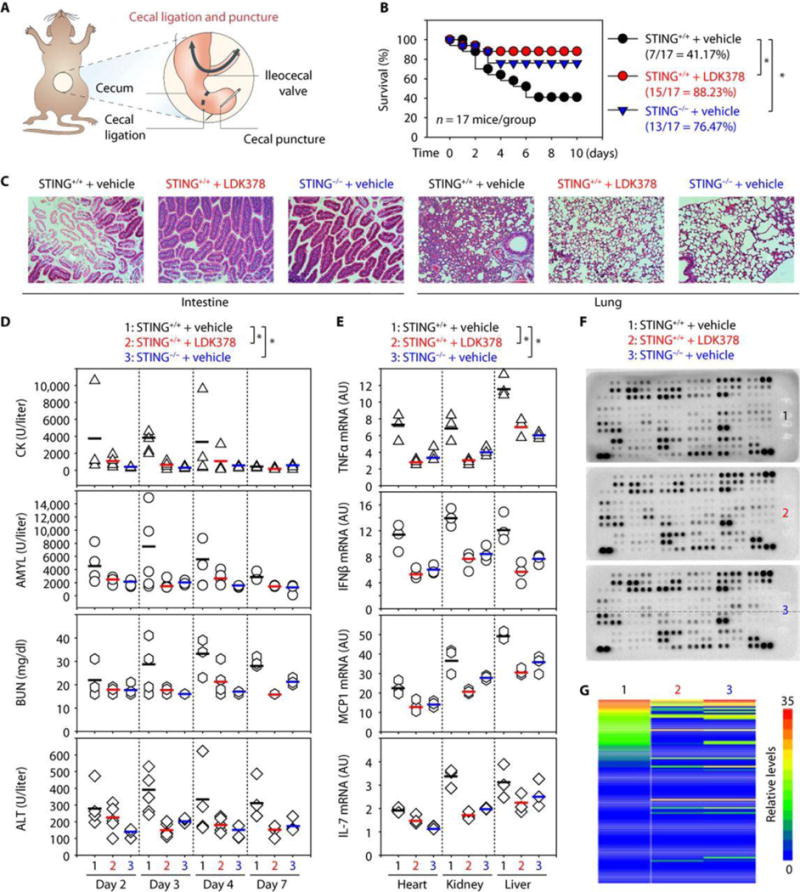
(A) Schematic depiction of the CLP model. (B) Administration of LDK378 or depletion of STING in mice prevented CLP (22-gauge needle)–induced animal death (n = 17 mice per group; *P < 0.05, Kaplan-Meier survival analysis). (C to G) In parallel, tissue hematoxylin and eosin staining (day 3; scale bars, 200 μm) (C), serum enzyme activity (days 2 to 7) (D), cytokine mRNA (day 3) (E), serum antibody array (day 3) (F), and heatmap of immune chemical profile (day 3) (G) were assayed (n = 3 to 5 mice per group; each bar represents the mean of the data; *P< 0.05, ANOVA LSD test). The top five down-regulated circulating immune chemical mediators in LDK378 and STING−/− groups compared with control group included IL-10, serpin E1, serpin F1, TIM-1, and CXCL2. High-resolution images related to (C), (F), and (G) are shown in figs. S12 and S13.
Fig. 7. Inhibition of the ALK-STING pathway protects mice against LPS-induced endotoxemia.
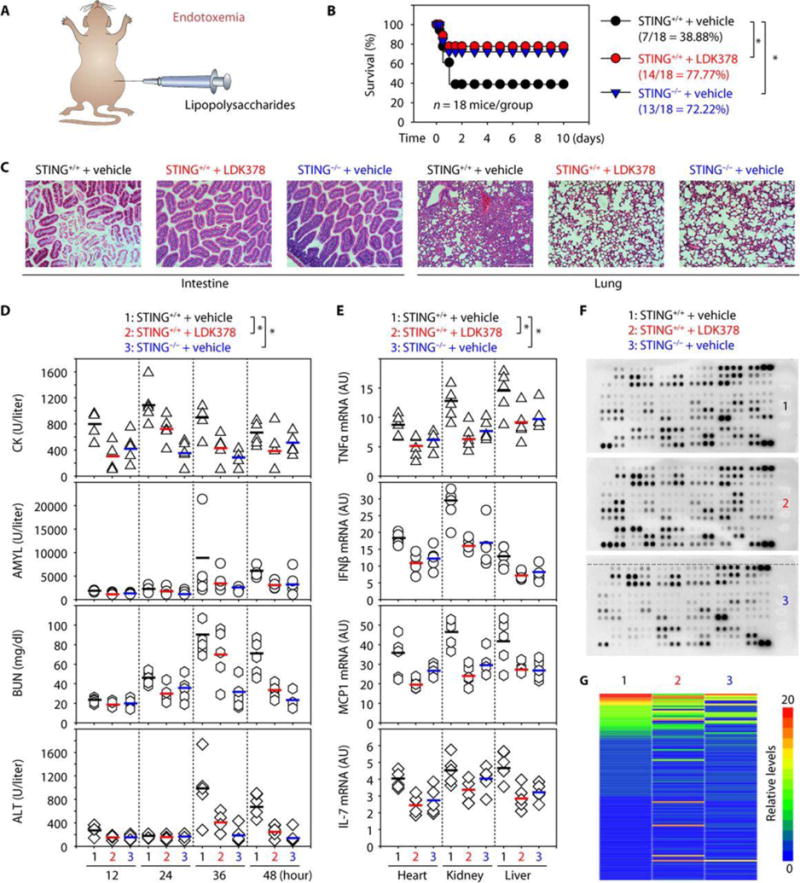
(A) Schematic depicting the endotoxemia model. (B) Administration of LDK378 or depletion of STING in mice prevented LPS (10 mg/kg)–induced animal death (n = 18 mice per group; *P < 0.05, Kaplan-Meier survival analysis). (C to G) In parallel, tissue hematoxylin and eosin staining (24 hours; scale bars, 200 μm) (C), serum enzyme activity (12 to 48 hours) (D), cytokine mRNA (24 hours) (E), serum antibody array (24 hours) (F), and heatmap of immune chemical profile (24 hours) (G) were assayed (n = 3 to 5 mice per group; each bar represents the mean of the data; *P < 0.05, ANOVA LSD test). The top five down-regulated circulating immune chemical mediators in LDK378 and STING−/− groups compared with control group included EGF, CD14, CXCL1, endoglin, and CCL22. High-resolution images related to (C), (F), and (G) are shown in figs. S14 and S15.
We next sought to test whether STING−/− mice are more resistant to polymicrobial sepsis and lethal endotoxemia. Like pharmacologic inhibition of ALK by LDK378, genetic STING depletion reduced animal death in both polymicrobial sepsis (Fig. 6B) and lethal endotoxemia models (Fig. 7B), which was associated with attenuated tissue injury such as tissue destruction, necrosis, and leukocyte infiltration (Figs. 6C and 7C), organ dysfunction (Figs. 6D and 7D), and proinflammatory cytokine expression (Figs. 6E and 7E) and release (Figs. 6, F and G, and 7, F and G). Collectively, these in vivo data agree with the in vitro data obtained in macrophages and monocytes and suggest that activation of the ALK-STING pathway mediates the pathophysiology of sepsis and septic shock.
It is possible that the ALK-STING pathway may still be needed to instill effective innate immunity against pathogens in response to mild or sublethal infections. During severe or lethal infections, the dysregulated overactivation of the ALK-STING pathway may tilt the balance toward promoting overzealous inflammatory responses that may contribute to the pathogenesis of sepsis. To determine whether the ALK-STING pathway’s contribution to mouse sepsis depends on the severity of the disease model, B6 mice were subjected to CLP with syringe needles with gauges ranging from 17 to 27. Increasing the needle thickness decreased the percent survival from 86.66% (using a 27-gauge needle, “low-grade sepsis”) to 41.17% (using a 22-gauge needle, “middle-grade sepsis”) to 0% (using a 17-gauge needle, “high-grade sepsis”). Treatment with LDK378 prolonged animal survival in low-, middle-, and high-grade sepsis models (fig. S16, A to C). In contrast, STING-deficient mice had prolonged survival only in high- and middle-grade sepsis models (fig. S16, B and C), suggesting that the STING signaling pathway might still be needed for the host to instill an appropriate innate immunity against pathogens in response to mild and sublethal infections. Notably, LDK378 conferred further protection to STING-deficient mice in response to high-grade sepsis (fig. S16C), indicating that ALK and STING have both overlapping and distinct functions in septic death. Genetic or pharmacologic inhibition of ALK or STING also led to changes in the release of overlapping and distinct immune chemicals in response to CDN, LPS, or CLP in vitro or in vivo (Figs. 5, 6, F and G, and 7, F and G).
ALK-STING pathway is changed in human sepsis
Although the murine endotoxemia and CLP models mimic many features of human sepsis, the translation of findings and inferences from these animal sepsis models to human sepsis remains a challenge (41, 42). Thus, we next determined whether the ALK-STING pathway is similarly altered in the PBMCs of patients with sepsis. Compared with a healthy control group, the mRNA expression of ALK, EGFR, STING, TBK1, and IRF3 in PBMCs was increased in the sepsis group (Fig. 8, A and B). Moreover, the expression of total and phosphorylated ALK, EGFR, STING, TBK1, and IRF3 proteins was also increased in the sepsis group (Fig. 8C), indicating an overall activation of ALK-STING signaling pathways during human sepsis. These findings further support a potential pathogenic role of the ALK-STING pathway in human sepsis.
Fig. 8. Gene and protein changes in ALK-dependent STING pathways in human sepsis.

(A) Box plots comparing measures of ALK, EGFR, STING, TBK1, and IRF3 mRNA in PBMC samples of sepsis patients (n=16) and healthy controls (n=10). The mRNA are presented as median value (black line), interquartile range (box), and minimum and maximum of all data (black line). *, P<0.05 versus control group, t test. (B) Table depicting clinical characteristics of sepsis patients and healthy control individuals. (C) Western blot analysis of indicated protein expression in PBMC samples of sepsis patients and healthy controls.
DISCUSSION
The innate immune system constitutes the first line of defense against pathogen invasion. However, insufficient, excessive, or poorly controlled activation of PRRs can cause an imbalance in the inflammation-immune network, leading to sepsis, septic shock, and ultimately death. The elucidation of this complex network can shed light on critical pathways and key molecules driving sepsis progression. We provide evidence to support ALK as a regulator of innate immune STING activation that contributes to the pathogenesis of microbial sepsis (fig. S17). Genetic or pharmacological inhibition of the ALK-STING signaling pathway corrected excessive host response to infection and rendered mice more resistant to sepsis and septic shock, supporting the therapeutic potential of ALK inhibitors in the treatment of human sepsis.
ALK, a tyrosine kinase receptor belonging to the insulin receptor superfamily, was originally discovered as a fusion protein with nucleophosmin in anaplastic, large-cell non-Hodgkin’s lymphoma in 1994 (43). Various alterations in the ALK gene or protein have been implicated in human cancer tumorigenesis, especially in non–small-cell lung cancer (NSCLC) (44). In cancer cells, ALK initiates several signal transduction pathways [Janus kinase (JAK)–signal transducer and activator of transcription 3 (STAT3), EGFR-AKT, and RAS–mitogen-activated protein kinase (MAPK)] involved in cell proliferation and transformation (44). In normal healthy tissues, the expression of ALK is relatively low, with the exception of the nervous system (45, 46). Despite this, global knockout of ALK in mice does not cause serious behavioral phenotypes, which hinders our understanding of ALK’s physiological role in mammals (47, 48). Here, we demonstrated that ALK is abundantly expressed in innate immune cells (monocytes and macrophages) and instigates proinflammatory responses to PAMPs, including DNAs, during lethal infection. This may partly explain why ALK-positive cancer patients have an increased risk of developing infections (49).
Clinically, patients with sepsis frequently have elevated circulating DNA from invading pathogens or damaged host cells, which is often associated with poor outcomes (50–52). Although the underlying causes remain elusive, it has been suggested that the inability to efficiently eliminate DNA or abnormal DNA-sensing pathways contributes to dysregulated systemic inflammation in sepsis. Almost two decades ago, STING was suggested as a key adaptor protein for most DNA-sensing signaling pathways (6, 10). Our present study has established the critical involvement of another transmembrane tyrosine kinase, ALK, in the STING-dependent innate recognition of microbial DNA during sepsis. In septic patients, we found that the expression of ALK and STING is increased in circulating PBMCs. Thus, pharmacologically blocking the ALK-STING signaling pathway may therapeutically modulate the DNA-induced excessive inflammation response in sepsis.
Bacterial CDN has long been shown to gain access to the inside of innate immune cells and directly bind cytosolic STING to initiate IRF3- and NF-κB–dependent immune responses (8, 20). Host self-DNA passively released by injured cells can also enter and accumulate in the cytoplasm of innate immune cells to bind and activate STING (53). Here, we provided evidence for an alternative transmembrane receptor–dependent pathway, by which extracellular DNA activates STING through transmembrane ALK/EGFR-dependent mechanisms (fig. S17). Our finding supports the notion that cell surface receptors can mediate extracellular DNA activity in inflammation and immune response (54, 55). We propose that various types of STING ligands trigger ALK/EGFR interaction and activation (phosphorylation), leading to subsequent AKT phosphorylation and consequent activation of the cytosolic STING pathway. We did not observe direct interaction between ALK and STING or its downstream signaling components (TBK1, cGAS, TRIF, and S6), suggesting that ALK is likely not a direct adaptor for cytosolic STING. Given the possible interaction between EGFR, AKT, TBK1 and IRF3 (56, 57), it will be important to determine whether the ALK/EGFR complex could recruit TBK1, IRF3, or AKT to trigger their phosphorylation and activation, thereby activating the STING and TRIF pathways. Previous studies have established the involvement of AKT in the positive regulation of the STING signaling pathway and IFNβ production (58, 59), although a recent study suggested a negative regulatory role of AKT in the regulation of STING signaling pathways during viral infection (60). It is not yet known whether AKT divergently regulates STING activation during viral and bacterial infections. Nevertheless, it has been suggested that STING occupies different roles in innate immune responses to viral versus bacterial infections (13–15), supporting the notion that innate immune cells can distinguish between viral and bacterial pathogens to mount a divergent immune responses (61). Regardless, phosphorylation of STING has been considered an essential and conserved mechanism of innate immune activation to both viral and bacterial infections (16, 62, 63), which now appears to depend on ALK activation.
During the last few decades, numerous phase 3 clinical trials of single-target therapies have failed to show efficacy in the clinical management of human sepsis. For complex systemic inflammatory syndromes, it is difficult to translate successful animal studies into clinical applications, partly because of the pitfalls in the selection of nonfeasible therapeutic targets or nonrealistic clinical outcome measures such as survival rates. However, the investigation of pathogenic cytokines in animal models of diseases has led to the development of successful cytokine-targeting therapeutic strategies for autoimmune diseases like rheumatoid arthritis, such as the chimeric anti-TNF monoclonal antibody infliximab and the soluble TNF receptor–Fc fusion protein etanercept (64, 65). Here, we demonstrated that LDK378, an ALK inhibitor also known as ceritinib, FDA-approved for the treatment of metastatic ALK-positive NSCLC (66), conferred significant protection against both lethal endotoxemia and sepsis in mice. Because LDK378 also inhibited LPS-induced phosphorylation of TBK1, IRF3, and p65 of NF-κB, and conferred further protection against both low- and high-grade sepsis, it is possible that LDK378 confers protection against lethal sepsis potentially via inhibiting both TRIF and STING pathways. With its well-defined pharmacokinetics, safety profile, and tolerability in cancer patients (66), it is possible and important to explore its therapeutic potential for the treatment of human sepsis.
In summary, we have revealed a role for ALK in regulating the STING-mediated innate immune response and provided a potential ALK-targeting strategy for the clinical management of sepsis. Activation of the STING pathway by ALK may be a crucial step for its immunological activity and may contribute to the pathogenesis of sepsis and septic shock. Inhibition of ALK and STING led to divergent changes of immune chemical expression profiles, suggesting that nonoverlapping functions of ALK and STING exist in innate immune responses. ALK regulated both bacterial CDN- and LPS-induced macrophage activation in vitro, and ALK inhibition increased septic animal survival in STING-deficient mice in vivo. The underlying molecular mechanism responsible for the immune functional differences between ALK and STING awaits further investigation.
Material and Methods
Study design
The objective of our study was to identify potential drugs targeting the STING pathway for the treatment of sepsis. Through a kinase inhibitor library screen, we identified ALK inhibitors as the top inhibitors for STING activation in monocytes and macrophages. We next defined the molecular mechanism by which ALK promotes STING activation through EGFR and AKT. We also evaluated the efficacy of ALK-targeting strategies for the management of polymicrobial sepsis and lethal endotoxemia in mice. Finally, we determined whether the ALK-STING pathway is similarly altered in the PBMCs of patients with sepsis. In all experiments, animals were randomized to different treatment groups without blinding. Sample size in experiments was specified in each figure legend. We did not exclude samples or animals. For every figure, statistical tests are justified as appropriate. All data meet the assumptions of the tests. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally used in the field. All reagent sources are listed in table S1.
Animal models of endotoxemia and sepsis
C57BL/6J wild-type mice (#000664) and STING−/− mice [strain name: B6(cg)-Tmem173tm1.2Cambgt/J, #017537] were purchased from the Jackson Laboratory. All mice were maintained in the University of Pittsburgh animal care facility. Mice were housed with their littermates in groups of four animals per cage and kept on a regular 12-hour light and dark cycle (7:00 to 19:00 light period). Food and water were available ad libitum. Experiments were carried out under pathogen-free conditions, and the health status of mouse lines was routinely checked by veterinary staff. Experiments were carried out with randomly chosen littermates of the same sex and matched age and body weight. We conducted all animal care and experimentation in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (www.aaalac.org) and with approval from the University of Pittsburgh Institutional Animal Care and Use Committees.
CLP model. Sepsis was induced in male or female C57BL/6J mice (8 to 10 weeks old, 22 to 26 g) using a surgical procedure as previously described (67). Briefly, anesthesia was induced with ketamine (80 to 100 mg/kg, ip) and xylazine (10 to 12.5 mg/kg, ip). A small midline abdominal incision was made, and the cecum was exteriorized and ligated with 4-0 silk immediately distal to the ileocecal valve without causing intestinal obstruction. The cecum was then punctured twice with a 17- to 27-gauge needle. The abdomen was closed in two layers, and mice were injected subcutaneously with 1 ml of Ringer’s solution including analgesia (buprenorphine, 0.05 mg/kg). LDK378 (20 mg/kg) was dissolved in vehicle [10% dimethyl sulfoxide, 20% cremophor/ethanol (3:1), and 70% phosphate-buffered saline (PBS)] and repeatedly administered orally to mice at 2, 24, 48, and 72 hours after CLP.
Endotoxemia model. LPS (E. coli 0111:B4, 10 mg/kg, #L4391, Sigma) was dissolved in PBS. Male or female C57BL/6J mice (8 to 10 weeks old, 22 to 26 g) received a single oral dose of LDK378 (20 mg/kg) or vehicle and then an infusion of LPS (10 mg/kg, ip) 60 min later. Mice were retreated with LDK378 or vehicle 6, 12, and 24 hours after LPS infusion.
Patient samples
PBMCs from patients with sepsis (n = 16) and healthy controls (n = 16) were collected from Daping Hospital and Xiangya Hospital. Collection of samples was approved by the Institutional Review Board. Sepsis was identified according to The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) (3).
Cell culture
iBMDMs (a gift from K. Fitzgerald at University of Massachusetts Medical School), murine macrophage-like RAW264.7 [American Type Culture Collection (ATCC), #TIB-71], murine J774A.1 (ATCC, #TIB-67), and human monocytic THP1 cell lines (ATCC, #TIB-202) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 1× penicillin-streptomycin. Human PBMCs were purchased from Lifeline Cell Technology (#1207). Macrophages/monocytes were maintained in a 5% CO2 incubator at 37°C. All cells were mycoplasma-free and authenticated with short tandem repeat DNA profiling analysis.
Mouse pPMs were isolated from C57BL/6J mice as previously described (68). In brief, 8-week-old female or male C57BL/6J mice were injected with 3.0% thioglycollate medium (2 ml per mouse) into the peritoneum. Three days after injection, mice were sacrificed and injected with 3 ml of 0.05% EDTA-PBS into the peritoneum to harvest peritoneal macrophages. Collected cells were centrifuged at 1000 rpm for 5 min at 4°C, and the cell pellet was washed with PBS and centrifuged again. The cell pellet was then suspended in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and 1× penicillin-streptomycin and cultured in a culture dish.
Immunoblotting and immunoprecipitation
For immunoblotting, cells were lysed in cell lysis buffer (#9803, Cell Signaling Technology) with protease inhibitor cocktail (Promega), phosphatase inhibitor cocktail (Sigma), and 1 mM Na3VO4 (69). Cleared lysates were resolved by SDS-PAGE (polyacrylamide gel electrophoresis; #3450124, Bio-Rad) and then transferred onto polyvinylidene difluoride membranes (#1704273, Bio-Rad). The membranes were blocked with tris-buffered saline Tween 20 (TBST) containing 5% skim milk for 1 hour at room temperature and then incubated with the indicated primary antibodies (1:1000 to 1:5000) overnight at 4°C. After being washed with TBST, the membranes were incubated with a horseradish peroxidase (HRP)–linked anti-mouse immunoglobulin G (IgG) secondary antibody (#7076, Cell Signaling Technology) or HRP-linked anti-rabbit IgG secondary antibody (#7074, Cell Signaling Technology) for 1 hour at room temperature. The membranes were washed three times in TBST and then visualized and analyzed with a ChemiDoc Touch Imaging System (#1708370, Bio-Rad). The intensities of bands were analyzed with Image Lab software.
For immunoprecipitation, cells were lysed at 4°C in ice-cold modified radioimmunoprecipitation lysis buffer (#9806, Cell Signaling Technology), and cell lysates were cleared by centrifugation (12,000g, 10 min). Concentrations of proteins in the supernatant were determined by bicinchoninic acid assay. Before immunoprecipitation, samples containing equal amounts of proteins were precleared with protein A/G agarose/Sepharose beads (#20423, Thermo Fisher Scientific Inc.) (4°C, 3 hours) and subsequently incubated with various irrelevant IgG or anti-ALK (#ab190934, Abcam) or anti-EGFR antibodies (#2232S, Cell Signaling Technology) in the presence of protein A/G agarose/Sepharose beads overnight at 4°C with gentle shaking. After incubation, agarose/Sepharose beads were washed extensively with PBS, and proteins were eluted by boiling in 2× SDS sample buffer before SDS-PAGE electrophoresis.
The antibodies to p-STING (#85735; 1:1000), STING (#13647; 1:1000), p-TBK1/NAK (#S172; 1:1000) (#5483; 1:1000), TBK1/NAK (#3504; 1:1000), IRF3 (#4962; 1:1000), p-IRF3 (S396; #4947S; 1:1000), p-p65 (S536; #3033P; 1:1000), p-EGFR (Y1068; #3777T; 1:1000), EGFR (#2232; 1:1000), p-AKT (S473; #4060; 1:1000), and S6 (#2317; 1:1000) were obtained from Cell Signaling Technology. The antibody to p-ALK (Y1057; #ab192809; 1:1000) was obtained from Abcam. The antibody to ALK (#sc-398791; 1:100) was obtained from Santa Cruz Biotechnology. The antibody to TRIF (#A01872; 1:500) was obtained from Boster.
Statistical analysis
Data are expressed as means ± SD. Unpaired Student’s t tests were used to compare the means of two groups. One-way ANOVA was used for comparison among the different groups. When ANOVA was significant, post hoc testing of differences between groups was performed using the LSD test. The Kaplan-Meier method was used to compare differences in mortality rates between groups. A P value of <0.05 was considered statistically significant. Individual subject-level data are shown in table S2.
Supplementary Material
Materials and methods
Fig. S1. ALK inhibitors block STING activation.
Fig. S2. Pharmacologic inhibition of ALK blocks STING ligand-induced IFNβ release and expression.
Fig. S3. Pharmacologic inhibition of ALK blocks STING activation.
Fig. S4. Genetic inhibition of ALK limits STING activation.
Fig. S5. ALK does not bind known STING regulators.
Fig. S6. Inhibition of ALK limits receptor tyrosine kinase phosphorylation in STING activation.
Fig. S7. ALK binds EGFR during STING activation.
Fig. S8. ALK-EGFR-AKT pathway mediates STING activation.
Fig. S9. Knockdown of EGFR inhibits STING activation.
Fig. S10. ALK-EGFR-AKT pathway mediates STING ligand-induced IFNβ release and expression.
Fig. S11. ALK mediates LPS-induced macrophage activation.
Fig. S12. Histological analysis of tissue injury in CLP-treated mice.
Fig. S13. Heat map of circulating immune chemicals profile in the indicated mice.
Fig. S14. Histological analysis of tissue injury in LPS-treated mice.
Fig. S15. Heat map of circulating immune chemicals profile in the indicated mice.
Fig. S16. Effects of targeting the ALK-STING pathway on CLP-induced septic death.
Fig. S17. Schematic depicting the pathologic role of ALK-dependent STING pathways in lethal sepsis.
Table S1. Reagent sources
One-sentence summary.
FDA-approved ALK-inhibiting drugs protect mice against sepsis through attenuating STING signaling activation in innate immune cells.
Acknowledgments
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript.
Funding: This work was supported by grants from the US National Institutes of Health (R01GM115366, R01CA160417, R01AT005076, R01GM063075, and R01GM44100), the National Key Technology R&D Program in China (2012BAI11B01), the Natural Science Foundation of Guangdong Province (2016A030308011), the National Natural Science Foundation of China (31671435, 81400132, and 81772508), and Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2017). This project partly utilized University of Pittsburgh Cancer Institute shared resources supported by award P30CA047904.
Footnotes
Author contributions: Experiments were conceived and designed by D.T. and J.J. Experiments were performed by L.Z., R.K., S.Z., X.W., L.C., J.J. and D.T. Data was analyzed by L.Z., H.W., T.R.B., J.J., and D.T. The paper was written by D.T. H.W. and T.R.B. edited and commented on the manuscript.
Competing interests: No potential conflicts of interest were disclosed.
Data and materials availability: All data and materials are available from the authors upon reasonable request.
References and notes
- 1.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. doi: 10.1056/NEJMra1208623. [DOI] [PubMed] [Google Scholar]
- 2.Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions: time for change. Lancet. 2013;381:774–775. doi: 10.1016/S0140-6736(12)61815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. 2013;13:551–565. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 5.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- 14.Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 15.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 18.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 19.Danilchanka O, Mekalanos JJ. Cyclic dinucleotides and the innate immune response. Cell. 2013;154:962–970. doi: 10.1016/j.cell.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Shi H, Wu J, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies BW, Bogard RW, Young TS, Mekalanos JJ. Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell. 2012;149:358–370. doi: 10.1016/j.cell.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heipertz EL, Harper J, Walker WE. STING and TRIF Contribute to Mouse Sepsis, Depending on Severity of the Disease Model. Shock. 2016 doi: 10.1097/SHK.0000000000000771. [DOI] [PubMed] [Google Scholar]
- 23.Johnston RB., Jr Current concepts: immunology. Monocytes and macrophages. N Engl J Med. 1988;318:747–752. doi: 10.1056/NEJM198803243181205. [DOI] [PubMed] [Google Scholar]
- 24.Pall G. The next-generation ALK inhibitors. Curr Opin Oncol. 2015;27:118–124. doi: 10.1097/CCO.0000000000000165. [DOI] [PubMed] [Google Scholar]
- 25.Jin L, Hill KK, Filak H, Mogan J, Knowles H, Zhang B, Perraud AL, Cambier JC, Lenz LL. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol. 2011;187:2595–2601. doi: 10.4049/jimmunol.1100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem. 2012;287:39776–39788. doi: 10.1074/jbc.M112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim S, Li L, Maliga Z, Yin Q, Wu H, Mitchison TJ. Anticancer flavonoids are mouse-selective STING agonists. ACS Chem Biol. 2013;8:1396–1401. doi: 10.1021/cb400264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, Vogel SN, Vance RE, Fitzgerald KA. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol. 2013;190:5216–5225. doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roskoski R., Jr Anaplastic lymphoma kinase (ALK): structure, oncogenic activation, and pharmacological inhibition. Pharmacol Res. 2013;68:68–94. doi: 10.1016/j.phrs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Majumdar T, Kessler P, Ozhegov E, Zhang Y, Chattopadhyay S, Barik S, Sen GC. STING Requires the Adaptor TRIF to Trigger Innate Immune Responses to Microbial Infection. Cell Host Microbe. 2016;20:329–341. doi: 10.1016/j.chom.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F, Alain T, Szretter KJ, Stephenson K, Pol JG, Atherton MJ, Hoang HD, Fonseca BD, Zakaria C, Chen L, Rangwala Z, Hesch A, Chan ES, Tuinman C, Suthar MS, Jiang Z, Ashkar AA, Thomas G, Kozma SC, Gale M, Jr, Fitzgerald KA, Diamond MS, Mossman K, Sonenberg N, Wan Y, Lichty BD. S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat Immunol. 2016;17:514–522. doi: 10.1038/ni.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi N, Lucena-Araujo AR, Nakayama S, de Figueiredo-Pontes LL, Gonzalez DA, Yasuda H, Kobayashi S, Costa DB. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer. 2014;83:37–43. doi: 10.1016/j.lungcan.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, Nakamura T, Matsumoto K, Soda M, Mano H, Uenaka T, Yano S. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res. 2012;18:3592–3602. doi: 10.1158/1078-0432.CCR-11-2972. [DOI] [PubMed] [Google Scholar]
- 34.Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, Bozza MT, Castro-Faria-Neto HC, Bozza PT. Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care. 2007;11:R49. doi: 10.1186/cc5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Presterl E, Staudinger T, Pettermann M, Lassnigg A, Burgmann H, Winkler S, Frass M, Graninger W. Cytokine profile and correlation to the APACHE III and MPM II scores in patients with sepsis. Am J Respir Crit Care Med. 1997;156:825–832. doi: 10.1164/ajrccm.156.3.9607131. [DOI] [PubMed] [Google Scholar]
- 36.Surbatovic M, Popovic N, Vojvodic D, Milosevic I, Acimovic G, Stojicic M, Veljovic M, Jevdjic J, Djordjevic D, Radakovic S. Cytokine profile in severe Gram-positive and Gram-negative abdominal sepsis. Sci Rep. 2015;5:11355. doi: 10.1038/srep11355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ulloa L, Tracey KJ. The “cytokine profile”: a code for sepsis. Trends Mol Med. 2005;11:56–63. doi: 10.1016/j.molmed.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 38.Munford RS. Endotoxemia-menace, marker, or mistake? J Leukoc Biol. 2016;100:687–698. doi: 10.1189/jlb.3RU0316-151R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gatot JS, Gioia R, Chau TL, Patrascu F, Warnier M, Close P, Chapelle JP, Muraille E, Brown K, Siebenlist U, Piette J, Dejardin E, Chariot A. Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKepsilon-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J Biol Chem. 2007;282:31131–31146. doi: 10.1074/jbc.M701690200. [DOI] [PubMed] [Google Scholar]
- 40.Bakshi S, Taylor J, Strickson S, McCartney T, Cohen P. Identification of TBK1 complexes required for the phosphorylation of IRF3 and the production of interferon beta. Biochem J. 2017;474:1163–1174. doi: 10.1042/BCJ20160992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–865. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 42.Dejager L, Pinheiro I, Dejonckheere E, Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19:198–208. doi: 10.1016/j.tim.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 43.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’ lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 44.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 45.Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- 46.Vernersson E, Khoo NK, Henriksson ML, Roos G, Palmer RH, Hallberg B. Characterization of the expression of the ALK receptor tyrosine kinase in mice. Gene Expr Patterns. 2006;6:448–461. doi: 10.1016/j.modgep.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Bilsland JG, Wheeldon A, Mead A, Znamenskiy P, Almond S, Waters KA, Thakur M, Beaumont V, Bonnert TP, Heavens R, Whiting P, McAllister G, Munoz-Sanjuan I. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology. 2008;33:685–700. doi: 10.1038/sj.npp.1301446. [DOI] [PubMed] [Google Scholar]
- 48.Weiss JB, Xue C, Benice T, Xue L, Morris SW, Raber J. Anaplastic lymphoma kinase and leukocyte tyrosine kinase: functions and genetic interactions in learning, memory and adult neurogenesis. Pharmacol Biochem Behav. 2012;100:566–574. doi: 10.1016/j.pbb.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 49.Savan R, McFarland AP, Reynolds DA, Feigenbaum L, Ramakrishnan K, Karwan M, Shirota H, Klinman DM, Dunleavy K, Pittaluga S, Anderson SK, Donnelly RP, Wilson WH, Young HA. A novel role for IL-22R1 as a driver of inflammation. Blood. 2011;117:575–584. doi: 10.1182/blood-2010-05-285908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rhodes A, Cecconi M. Cell-free DNA and outcome in sepsis. Crit Care. 2012;16:170. doi: 10.1186/cc11508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saukkonen K, Lakkisto P, Pettila V, Varpula M, Karlsson S, Ruokonen E, Pulkki K. Cell-free plasma DNA as a predictor of outcome in severe sepsis and septic shock. Clin Chem. 2008;54:1000–1007. doi: 10.1373/clinchem.2007.101030. [DOI] [PubMed] [Google Scholar]
- 52.Kung CT, Hsiao SY, Tsai TC, Su CM, Chang WN, Huang CR, Wang HC, Lin WC, Chang HW, Lin YJ, Cheng BC, Su BY, Tsai NW, Lu CH. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J Transl Med. 2012;10:130. doi: 10.1186/1479-5876-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol. 2014;31:121–126. doi: 10.1016/j.coi.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 54.Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, Mian A, Jiang J, Schrum J, Bossaller L, Pelka K, Garbi N, Brewah Y, Tian J, Chang C, Chowdhury PS, Sims GP, Kolbeck R, Coyle AJ, Humbles AA, Xiao TS, Latz E. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210:2447–2463. doi: 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hefeneider SH, Bennett RM, Pham TQ, Cornell K, McCoy SL, Heinrich MC. Identification of a cell-surface DNA receptor and its association with systemic lupus erythematosus. J Invest Dermatol. 1990;94:79S–84S. doi: 10.1111/1523-1747.ep12875170. [DOI] [PubMed] [Google Scholar]
- 56.Chakraborty S, Li L, Puliyappadamba VT, Guo G, Hatanpaa KJ, Mickey B, Souza RF, Vo P, Herz J, Chen MR, Boothman DA, Pandita TK, Wang DH, Sen GC, Habib AA. Constitutive and ligand-induced EGFR signalling triggers distinct and mutually exclusive downstream signalling networks. Nat Commun. 2014;5:5811. doi: 10.1038/ncomms6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, Tan SL, White MA. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaur S, Sassano A, Dolniak B, Joshi S, Majchrzak-Kita B, Baker DP, Hay N, Fish EN, Platanias LC. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc Natl Acad Sci U S A. 2008;105:4808–4813. doi: 10.1073/pnas.0710907105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaur S, Sassano A, Joseph AM, Majchrzak-Kita B, Eklund EA, Verma A, Brachmann SM, Fish EN, Platanias LC. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J Immunol. 2008;181:7316–7323. doi: 10.4049/jimmunol.181.10.7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seo GJ, Yang A, Tan B, Kim S, Liang Q, Choi Y, Yuan W, Feng P, Park HS, Jung JU. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep. 2015;13:440–449. doi: 10.1016/j.celrep.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124:767–782. doi: 10.1016/j.cell.2006.01.034. [DOI] [PubMed] [Google Scholar]
- 62.Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. doi: 10.1126/science.aaa2630. [DOI] [PubMed] [Google Scholar]
- 63.Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weber GF, Chousterman BG, He S, Fenn AM, Nairz M, Anzai A, Brenner T, Uhle F, Iwamoto Y, Robbins CS, Noiret L, Maier SL, Zonnchen T, Rahbari NN, Scholch S, Ameln A Klotzsche-von, Chavakis T, Weitz J, Hofer S, Weigand MA, Nahrendorf M, Weissleder R, Swirski FK. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science. 2015;347:1260–1265. doi: 10.1126/science.aaa4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang H, Ward MF, Sama AE. Targeting HMGB1 in the treatment of sepsis. Expert Opin Ther Targets. 2014;18:257–268. doi: 10.1517/14728222.2014.863876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li S, Qi X, Huang Y, Liu D, Zhou F, Zhou C. Ceritinib (LDK378): a potent alternative to crizotinib for ALK-rearranged non-small-cell lung cancer. Clin Lung Cancer. 2015;16:86–91. doi: 10.1016/j.cllc.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 67.Rittirsch D, Huber-Lang MS, Flierl MA, Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc. 2009;4:31–36. doi: 10.1038/nprot.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andujar I, Rios JL, Giner RM, Miguel Cerda J, Recio Mdel C. Beneficial effect of shikonin on experimental colitis induced by dextran sulfate sodium in BALB/c mice. Evid Based Complement Alternat Med. 2012;2012:271606. doi: 10.1155/2012/271606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deng W, Wang Y, Liu Z, Cheng H, Xue Y. HemI: A toolkit for illustrating heatmaps. PLOS ONE. 2014;9:e111988. doi: 10.1371/journal.pone.0111988. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods
Fig. S1. ALK inhibitors block STING activation.
Fig. S2. Pharmacologic inhibition of ALK blocks STING ligand-induced IFNβ release and expression.
Fig. S3. Pharmacologic inhibition of ALK blocks STING activation.
Fig. S4. Genetic inhibition of ALK limits STING activation.
Fig. S5. ALK does not bind known STING regulators.
Fig. S6. Inhibition of ALK limits receptor tyrosine kinase phosphorylation in STING activation.
Fig. S7. ALK binds EGFR during STING activation.
Fig. S8. ALK-EGFR-AKT pathway mediates STING activation.
Fig. S9. Knockdown of EGFR inhibits STING activation.
Fig. S10. ALK-EGFR-AKT pathway mediates STING ligand-induced IFNβ release and expression.
Fig. S11. ALK mediates LPS-induced macrophage activation.
Fig. S12. Histological analysis of tissue injury in CLP-treated mice.
Fig. S13. Heat map of circulating immune chemicals profile in the indicated mice.
Fig. S14. Histological analysis of tissue injury in LPS-treated mice.
Fig. S15. Heat map of circulating immune chemicals profile in the indicated mice.
Fig. S16. Effects of targeting the ALK-STING pathway on CLP-induced septic death.
Fig. S17. Schematic depicting the pathologic role of ALK-dependent STING pathways in lethal sepsis.
Table S1. Reagent sources