

Abstract
Myelodysplastic syndromes (MDS) are clonal disorders of hematopoietic stem and progenitor cells and represent the most common cause of acquired marrow failure. Hallmarked by ineffective hematopoiesis, dysplastic marrow, and risk of transformation to acute leukemia, MDS remains a poorly treated disease. Although identification of hematopoietic aberrations in human MDS has contributed significantly to our understanding of MDS pathogenesis, evidence now identify the bone marrow microenvironment (BMME) as another key contributor to disease initiation and progression. With improved understanding of the BMME, we are beginning to refine the role of the hematopoietic niche in MDS. Despite genetic diversity in MDS, interaction between MDS and the BMME appears to be a common disease feature, and therefore represents an appealing therapeutic target. Further understanding of the interdependent relationship between MDS and its niche is needed to delineate the mechanisms underlying hematopoietic failure and how the microenvironment can be clinically targeted. This review will provide an overview of data from human MDS and murine models supporting a role for BMME dysfunction at several steps of disease pathogenesis. While no models or human studies so far have combined all these findings, we will review current data identifying BMME involvement in each step of MDS pathogenesis, organized to reflect the chronology of BMME contribution as the normal hematopoietic system becomes myelodysplastic and MDS progresses to marrow failure and transformation. Although microenvironmental heterogeneity and dysfunction certainly add complexity to this syndrome, data are already demonstrating that targeting microenvironmental signals may represent novel therapeutic strategies for MDS treatment.
Keywords: myelodysplastic syndrome, bone marrow microenvironment, mesenchymal stem cell, osteoblast, endothelial cell
Introduction
The myelodysplastic syndromes (MDS) are malignant disorders of hematopoietic stem and progenitor cells (HSPCs) hallmarked by bone marrow failure due to defective hematopoiesis and production of dysplastic cells. As the most commonly diagnosed myeloid neoplasm in the U.S. [1, 2], with a 3-year survival rate of 35–45% [3, 4], MDS leads to significant morbidity and mortality due to complications of multi-lineage cytopenias and a high risk of transformation to acute myelogenous leukemia (AML). Because MDS is most prevalent in the aged population such that 86% of patients are ≥60 years of age at diagnosis [5], most patients are ineligible for bone marrow transplantation due to older age-related co-morbidities. Consequently, the current standard of care relies largely on symptomatic management of cytopenias along with few agents with limited response rates and durability [1]. Notably, there have been no new FDA-approved drugs for MDS in the past decade [2]. Therefore, there is a critical need for new MDS therapies.
Although recent identification of numerous chromosomal, genetic, and epigenetic aberrations in patients with MDS has contributed significantly to our understanding of MDS pathogenesis [1, 6–8], there remains a paucity of MDS therapies due in part to a lack of knowledge on how to restore hematopoietic function. While MDS has been well-described to arise due to heterogeneous hematopoietic cell-intrinsic defects that drive clonal expansion, ineffective hematopoiesis, dysplasia, and leukemic progression, accumulating evidence now identify the bone marrow microenvironment (BMME) as a key mediator of MDS pathophysiology and, therefore, a potential therapeutic target.
The bone marrow is a complex tissue containing self-renewing HSPCs which generate progeny that progressively differentiate into mature blood and immune cells. Regulation of hematopoiesis and maintenance of the hematopoietic stem cell pool is mediated at least in part by constituents of the BMME including mesenchymal stromal cells [9–12], osteolineage cells [13, 14], and endothelial cells [15, 16] among many other diverse cell types [9–17]. These BMME cells produce key maintenance factors and receptors which support HSPC quiescence, proliferation, and migration [17–20]. Studies of genetic murine models have significantly advanced our ability to identify BMME cells along with the key maintenance genes they express to support HSPC activities under normal homeostatic conditions. For example, selective deletion of stem cell factor (SCF) or the chemokine C-X-C motif ligand 12 (CXCL12) in endothelial cells and leptin receptor (Lepr)+ mesenchymal stromal cells leads to HSPC depletion [17, 21, 22]. Furthermore, osteoblastic cell ablation [23, 24] or Cxcl12 deletion [21] in this population results in loss of lineage-restricted hematopoietic progenitors followed by loss of hematopoietic stem cells. Aside from maintaining HSC numbers, BMME cells are also essential for retaining HSPCs in the bone marrow as Cxcl12 deletion in mesenchymal-osteolineage cells leads to HSPC mobilization out of the marrow [21, 22].
Although numerous other cell types and maintenance factors participate in HSPC regulation (reviewed here[25]), these studies cumulatively demonstrate that specific BMME cells including mesenchymal stromal cells, osteoblastic lineage cells, and endothelial cells critically impact hematopoietic function under normal physiologic conditions. Therefore, dysfunction of such populations may also contribute to the pathophysiology of hematologic pathologies including MDS. Particularly, emerging evidence point to BMME abnormalities as central participants in the step-wise progression of MDS pathogenesis whereby, 1) BMME abnormalities contribute to the development and expansion of MDS clones, 2) MDS cells further modify the BMME via aberrant production of secreted factors such as cytokines, and 3) a dysfunctional BMME further promotes clonal expansion and disease progression (Figure 1). Further understanding of the multi-directional relationships between MDS and the diverse cells within the hematopoietic niche is needed to delineate the mechanisms underlying hematopoietic failure and how the microenvironment can be targeted for clinical benefit. In this review, we will discuss recent evidence identifying the BMME as a contributor to MDS pathogenesis in terms of disease initiation and progression. Our discussion first focuses on data from in vitro studies of human MDS and in vivo studies of murine MDS models supporting a role for dysfunction of mesenchymal stromal cells and osteolineage cells in MDS. We will also discuss data that point to vascular and endothelial abnormalities in MDS as another contributor to disease pathophysiology. For an overview of the hematopoietic niche in a broader range of myeloid malignancies, please refer to these excellent reviews [26, 27].
Figure 1. Role of the bone marrow microenvironment in MDS pathogenesis.
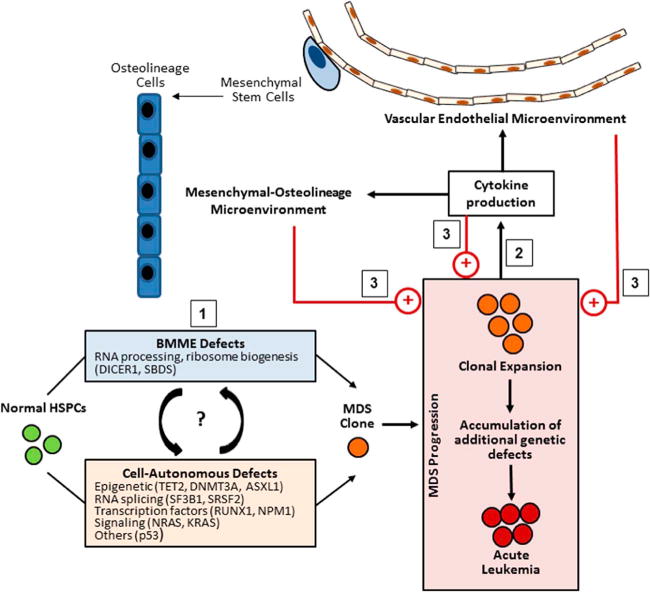
A proposed model of bone marrow microenvironment (BMME) involvement in MDS initiation and progression: 1) BMME defects may initiate or cooperate with intrinsic hematopoietic defects to lead to the development of MDS clonal cells. As MDS cells expand, they accumulate additional genetic defects that may lead to eventual progression to acute leukemia. 2) During this process, MDS cells secrete cytokines which modify the mesenchymal-osteolineage and vascular endothelial BMME. 3) The modified BMME along with autocrine signaling of secreted cytokines both promote further disease progression.
In vitro evidence for stromal abnormalities in MDS
Given the regulatory role of the HSPC niche, alterations in the microenvironment may contribute to hematopoietic failure in MDS. Early evidence of BMME abnormalities in MDS comes from in vitro studies of patient-derived bone marrow mesenchymal stromal cells. Mesenchymal stromal cell function can be assessed in vitro based on morphology, differentiation capacity, proliferative capacity, and ability to support co-cultured HSPCs. In terms of morphology, investigators have observed MDS-derived mesenchymal stromal cells to be disorganized in appearance compared to the fibroblastic-like morphology of normal donor-derived mesenchymal stromal cells [28–30]. However, several other groups reported no changes in the morphology of MDS-derived mesenchymal stromal compared to normal controls [31–36].
Assessments of osteogenic, adipogenic, and chondrogenic differentiation capacity are also conflicting. An early study of the bone biopsies from MDS patients revealed an adynamic bone phenotype with decreased bone matrix formation and mineralization, suggesting that hematopoietic abnormalities in MDS impair bone remodeling [37]. Subsequent reports identified no differences in the ability of MDS-derived mesenchymal stromal cells to generate osteolineage cells in vitro [31–33, 38–40]. However, Geyh et al. reasoned that marked variability in MDS along with the small sample size of prior studies are limiting factors in data interpretation [28]. To overcome this, they evaluated samples from 106 patient samples spanning a wide range of MDS subtypes and identified reduced osteogenic differentiation potential in mesenchymal stromal cells across all MDS subtypes evaluated, demonstrated by decreased alizarin red and von Kossa positive mineralization and decreased alkaline phosphatase activity upon stimulation by osteogenic media [28]. This was further corroborated by decreased mRNA expression of osteogenesis-associated genes such as Runx2, osterix, alkaline phosphatase, and osteocalcin [28, 41]. Additionally, reduced serum osteocalcin in MDS patients provides further support for decreased osteoblastic activity due to defective osteoblastic generation or function [28]. Another group also reported diminished osteogenic differentiation along with decreased oil red O staining upon culture in adipogenic media, suggesting that adipogenic differentiation capacity may also be impaired in MDS [29]. While others also observed decreased adipogenic potential in MDS-derived mesenchymal stromal cells [38, 41], several reports, including the study by Geyh et al. evaluating a large MDS cohort, identified no changes [28, 32, 33, 39, 40]. Finally, only a few published studies have examined chondrogenic differentiation and have revealed no differences in MDS-derived compared to normal subject-derived mesenchymal stromal cells [28, 39, 40].
Overall, osteogenic differentiation, compared to adipogenic and chondrogenic differentiation, has been the most widely assessed in MDS-derived mesenchymal stromal cells, yielding variable results ranging from no change to impaired differentiation capacity. These data indicate that although alterations of osteogenic differentiation are present in MDS, the level of impairment is heterogeneous and may be difficult to detect in small cohorts, especially since patient heterogeneity is an acknowledged feature of MDS.
Despite conflicting data on morphology and differentiation capacity, the majority of reports describe altered growth and survival patterns in MDS-mesenchymal stromal cells. This is most prominently characterized by a decreased proliferative rate, increased population doubling time, and impaired ability to reach confluence in culture compared to healthy subject-derived counterparts [29, 35, 38, 40–44]. Impaired growth may be due to a greater propensity for cellular senescence evidenced by increased β-galactosidase+ cells [28, 29, 44] and reduced number of maximum passages [28, 38]. However, one study reported decreased proliferative capacity in MDS-derived mesenchymal stromal cells in the absence of premature senescence [41]. The investigators attributed defective proliferation to increased non-canonical WNT signaling, implicated in inhibition of mesenchymal stem/progenitor cell proliferation, based on supportive transcriptional data [41]. This was accompanied by decreased gene expression of canonical WNT signaling pathway components and downstream transcriptional targets, suggesting that aberrancies in WNT signaling may explain the diminished proliferative capacity [30, 41]. Furthermore, increased cell death may also contribute to growth impairment as MDS-derived stromal cultures have decreased live cells [40] and increased TUNEL-positive apoptotic cells [34]. Consistent with a diminished proliferative capacity, multiple groups report that MDS-derived mesenchymal stromal cells have reduced clonogenic potential evidenced by the production of fewer fibroblastic colony-forming units (CFU-F) in vitro [28–30, 40, 42]. While two studies found no differences in the growth properties of MDS-derived mesenchymal stromal cells, they did identify other functional alterations such as an impaired capacity to support HSPCs in vitro [32, 45].
Evidence of functional deficits in proliferation and differentiation led to the question of whether MDS-mesenchymal stromal cells are also defective in HSPC support. To assess hematopoietic support in vitro, healthy donor-derived or umbilical cord blood CD34+ HSPCs are plated over a pre-established feeder layer of mesenchymal stromal cells which provide essential molecular and cellular signals to sustain HSPCs. Following a period of co-culture, HSPCs are assayed for their ability to generate hematopoietic colony-forming units (CFU). The length of co-culture dictates the type of HSPC function that can be assessed. For example, colonies formed after a shorter culture period (days versus weeks) are derived from lineage-restricted progenitor cells. In contrast, colonies formed after several weeks of culture, when mature progenitors have been exhausted, are derived from primitive HSPCs, termed long-term culture-initiating cells (LTC-IC) [46]. Cobblestone area forming cells (CAFC) can also be observed at this time point [47]. Many groups have used this in vitro paradigm to investigate the ability of MDS-mesenchymal stromal cells to maintain growth of normal HSPCs. Several studies have detected fewer LTC-ICs and CAFCs after long-term co-culture of normal HPSCs with MDS-derived stroma [28, 29, 32, 38, 48], indicating a diminished ability to sustain normal primitive HSPCs. Healthy CD34+ HSPCs co-cultured with MDS-stroma also exhibited a decreased capacity to form a variety of myeloid and erythroid CFUs [29, 41, 49]. These data indicate that MDS-derived mesenchymal stromal cells have deficient capacity to support hematopoietic cells ranging from primitive HSPCs to more mature lineage-restricted progenitors. Thus, defective microenvironmental support may contribute to hematopoietic failure and multi-lineage peripheral blood cytopenias in MDS. Of note, pre-treatment of MDS-derived stroma with lenalidomide decreased CAFCs but increased formation of erythroid and myeloid colonies [29], suggesting that microenvironment modulation may be an avenue to improve hematopoietic support in MDS.
Although data cumulatively demonstrate that MDS-derived mesenchymal stromal cells are functionally impaired in proliferation, differentiation, and HSPC support, discrepancies exist in the literature. Several factors contribute to this, including inconsistencies in the methodology of mesenchymal stromal cell isolation and potential for biologic artifacts arising during long-term ex vivo expansion. While studying primary patient samples provides the important advantage of directly studying human disease, it is associated with limitations that make data interpretation difficult. First, there is variability in mesenchymal stromal cell identification and isolation due to the lack of a specific definition for such a population despite attempts at standardization. In 2006 the International Society for Cellular Therapy (ISCT) recommended three minimal criteria for defining human multipotent mesenchymal stromal cells: 1) plastic adherence, 2) expression of universally accepted mesenchymal cell surface antigens (CD105, CD73, and CD90) and lack of expression of hematopoietic-specific surface antigens (CD45, CD34, CD14 or CD11b, CD79α or CD19, and HLA-DR), and 3) capacity for osteogenic, adipogenic, and chondrogenic differentiation in vitro [50]. This definition enriches for mesenchymal stem/progenitor cells, but the population identified is still heterogeneous. While most studies define mesenchymal stromal cells as the population of marrow-derived, plastic-adherent cells, there is inconsistency regarding depletion of hematopoietic cells prior to culture and the assessment of cell surface antigens before or after culture to assess cell purity. Secondly, mesenchymal stromal cells in the marrow are scarce and require ex vivo expansion prior to analysis, a process which disrupts physiologic interactions with MDS hematopoietic cells and other cell populations in the BMME. Mesenchymal stromal cells are one component of the complex BMME and evaluating their function in isolation does not address changes that may be occurring in other populations such as their osteolineage-derivatives and neighboring endothelial cells. Finally, another key consideration is that MDS is known to be a genetically and clinically heterogeneous disease. Consequently, such heterogeneity may extend into the degree of niche involvement wherein discrepant findings of different investigators may reflect the variability in the level of microenvironmental dysfunction in MDS. Such variability make it more difficult to rigorously detect differences in the microenvironment when evaluating a small cohort of patient samples and this may be further complicated by inconsistent mesenchymal stromal cell identification.
A question that remains is whether the observed stromal changes are primary or secondary defects in MDS. Although cytogenetic changes have been identified in patient-derived mesenchymal stromal cells [33, 35, 39, 51–54] (discussed in the next section, “Bone marrow microenvironment defects and MDS initiation”), their etiology and functional relevance are still unclear. BMME dysfunction may increase the susceptibility of hematopoietic cells to acquire and accumulate intrinsic defects that drive MDS. Alternatively, current evidence also suggests that microenvironmental defects arise in response to hematopoietic derangements in MDS. These two theories are not necessarily mutually exclusive and may contribute to a feed-forward cycle of disease progression. An implication of the latter theory is that BMME dysfunction may depend on continuous exposure to pathologic stimuli from malignant MDS cells. This correlates with MDS patients who are able to regain normal hematopoietic function following bone marrow transplantation, indicating that BMME dysfunction is reversible upon normalization of the hematopoietic compartment. The potential interdependency of MDS and BMME pathologies raises the possibility that findings from in vitro studies in which MDS-derived mesenchymal stromal cells were cultured in isolation may not sufficiently reflect functional alterations present in vivo. Despite the limitations of in vitro studies, they have provided critical insight into the extent and consequences of microenvironmental derangements in MDS, identifying the microenvironment as a potential mediator of ineffective hematopoiesis and thus a therapeutic target.
Bone marrow microenvironment defects and MDS initiation
In addition to supporting the life-long production of blood and immune cells, the BMME may also be important in preventing neoplastic disease in the hematopoietic system. Landmark studies in murine models have revealed that defects in non-hematopoietic BMME populations are sufficient to drive myeloid neoplasms, providing some of the most compelling evidence for a BMME role in the pathophysiology of hematologic malignancies. Raaijmakers et al. demonstrated that Cre recombinase-mediated deletion of Dicer1 in osterix-expressing osteoprogenitors cells (Osx-GFP-Cre+ Dicerfl/fl) resulted in osteoblastic dysfunction and development of MDS [55]. Mice exhibited multi-lineage cytopenias and granulocytic and megakaryocytic dysplasia with a subset progressing to secondary leukemia, features strongly correlating with human MDS [55]. Transplantation studies revealed that hematopoietic cells from Osx-GFP-Cre+ Dicerfl/fl mice failed to propagate disease in wild-type recipient mice while Osx-GFP-Cre+ Dicerfl/fl mice transplanted with wild-type marrow developed MDS, further supporting niche-initiated disease as a central feature of MDS in this model [55]. Although Raaijmakers et al. are the only investigators to report MDS induced by extrinsic defects in the BMME, several other groups have shown that microenvironmental alterations can initiate both acute myelogenous leukemia (AML) [56] and myeloproliferative disorders [57–60].
While these murine studies show that primary microenvironment defects can induce MDS and other myeloid malignancies, whether this occurs in human disease is not established. In human MDS, the widely-accepted etiology of MDS pathogenesis is attributed to cell-intrinsic genetic and epigenetic lesions of the hematopoietic compartment. However, the loss of Dicer1 in osteoprogenitors was associated with downregulation of the Shwachman-Bodian-Diamond syndrome (Sbds) gene, whose deficiency in humans leads to a clinical syndrome of skeletal defects and a high propensity to develop MDS and secondary leukemia [55].
The SBDS protein is involved in ribosome biogenesis and its loss results in a ribosomapathy [61–63] thought to explain the hematopoietic dysfunction long observed in patients with SDS. While knocking down or deleting Sbds in mouse hematopoietic progenitors impairs myeloid differentiation, decreased Sbds expression did not generate other pathologic features of MDS [64, 65]. Consequently, it has been suggested that BMME dysfunction may also contribute to the SDS hematopoietic phenotype as normal human CD34+ cells demonstrated reduced myeloid potential following co-culture with marrow stroma from SDS patients compared to normal stroma [66]. In contrast, CD34+ cells from SDS patients co-cultured with normal stroma exhibited increased myeloid potential compared to co-culture with SDS-derived stroma [66]. Therefore, in addition to cell-autonomous effects, Sbds deficiency in the BMME may also play a role in the pathogenesis of MDS and secondary leukemia in SDS. Indeed, decreased Dicer1 and Sbds expression has been identified in mesenchymal stromal cells from MDS patients [28, 67]. Therefore, it is reasonable to consider that certain MDS-inducing gene mutations identified in the mouse BMME may have clinical correlation in a subset of MDS patients. Taken together, there is increasing evidence that dysfunction of the microenvironment may initiate or cooperate with hematopoietic defects to permit MDS development. Specifically, it may be reasonable to postulate that the healthy BMME restrains the hematopoietic compartment to prevent the selection and expansion of malignant clones.
In human MDS, the presence of genetic aberrations in BMME cells such as mesenchymal stromal cells remains controversial. While some groups identified chromosomal abnormalities in MDS-derived stromal cells [33, 35, 39, 51–54], others have not [32, 36]. Cytogenetic aberrations identified in mesenchymal stromal cells from MDS patients are different from those of the hematopoietic counterparts, indicating that they are not derived from the malignant MDS clone [31, 35, 40, 51, 52, 54]. However, one cannot exclude the possibility of cytogenetic abnormalities arising in vitro as patient-derived mesenchymal stromal cells were passaged multiple times prior to karyotypic analysis and most studies did not compare the cytogenetic profiles before and after in vitro expansion. Indeed, longitudinal cytogenetic analyses of MDS-derived mesenchymal stromal cells post-propagation identified chromosomal aberrations at later passages that were not detected at earlier passages [54]. Chromosomal alterations have also been observed in normal donor-derived stromal cultures [40, 54, 68], indicating that genetic abnormalities can arise in long-term cultures of mesenchymal stromal cells. Moreover, given that mesenchymal stromal cells may be functionally impaired in aging and MDS, they may be more susceptible to accumulating genetic defects in culture [28]. Currently, the presence, timeline, and pathologic significance of mesenchymal cytogenetic aberrations remain unclear and their elucidation requires further evaluation of highly purified patient samples. Of note, one study of combined MDS and AML patients found that abnormal mesenchymal stromal cell karyotypes were associated with decreased survival and that unfavorable hematopoietic cytogenetics correlated with stromal aberrations, indicating that stromal genetic abnormalities may be of prognostic value [51].
Microenvironmental support is required for propagation of human MDS in xenotransplantation models
In vivo investigation of MDS is crucial to better understand the bi-directional interactions between malignant MDS cells and their bone marrow microenvironment. Importantly, in vivo models offer the advantage of more closely recapitulating the physiologic microenvironment of human MDS and allow for longitudinal evaluation of hematopoietic and BMME function. However, development of human MDS xenotransplantation models in mice has proven to be challenging. Unlike certain subsets of human acute myelogenous leukemia (AML) cells which engraft in immunodeficient mice [69, 70], human MDS cells either fail to engraft or engraft at low, transient and inconsistent levels [71–76]. Benito et al. found that engrafted human cells in NOD/SCID mice transplanted with patient-derived MDS samples were derived from residual normal cells rather than the malignant MDS clone [75]. Using NOD/SCID-β2m−/− mice as recipients, Thanopoulou et al. observed engraftment for 9 of 11 human MDS samples in which 4 cases generated cells expressing the same clonal cytogenetic markers as the original patient sample [77]. However, for 6 of the 9 engrafting MDS samples (including 2/4 samples regenerating clonal MDS cells), human cells were detected only transiently at 3 weeks post-transplant and progressively declined in number by 19 weeks [77].
The inability of human MDS cells to engraft and reconstitute immunodeficient mice suggests that they may require extrinsic support from the microenvironment to potentiate disease. Indeed, MDS cell engraftment was enhanced by co-transplantation with mesenchymal stromal cells [73, 76, 78–80]. By co-injecting patient-derived MDS cells with a mixture of HS5 and HS27a cells (two human stromal cell lines), Kerbauy et al. observed variable but improved human cell engraftment in NOD/SCID-β2m−/− mice compared to Thanopoulou et al., which they attributed to direct intramedullary injections (versus intravenous injections) and the co-administration of stromal cells [78]. In a follow-up report, the same group found that stromal support from HS27a and not HS5 cells were responsible for enhancing engraftment of CD34+ human MDS cells in NSG mice [79]. Furthermore, CD146 (or MCAM, melanoma cell adhesion molecule) plays an important role in facilitating clonal MDS cell propagation as it is highly expressed by HS27a cells compared to HS5 cells [79]. Co-transplantation of human MDS cells with HS5 cells overexpressing CD146 increased engraftment to levels observed when the same sample was co-transplanted with HS27a cells [79]. Notably, CD146 expression marks a population of human perivascular mesenchymal stromal cells enriched for CFU-F activity and able generate hematopoiesis-supporting bony ossicles when transplanted heterotropically in mice [81, 82]. Therefore, these data show that osteolineage-generating mesenchymal stromal cells play a critical role in facilitating or promoting MDS propagation.
Muguruma et al. observed engraftment of CD34+ human MDS cells with co-injection of human-derived mesenchymal stromal cells into the tibia of NOD/SCID-IL2Rγ−/− mice [80]. Engrafted human CD34+ cells were noted to be clustered along the endosteal surface and surrounding irregular fibronectin networks in the central marrow, indicating infiltration and disruption of the murine hematopoietic microenvironment [80]. There were fewer mouse hematopoietic cells in the injected tibia compared to the non-injected contralateral tibia, a phenomenon not noted when healthy human cells were injected, suggesting that MDS cells suppress murine hematopoiesis [80]. The concurrent presence of microenvironmental and hematopoietic derangements suggests that engrafted MDS cells disrupt the endogenous stromal architecture to impair normal hematopoietic support and contribute to murine hematopoietic inhibition.
Available data provide strong evidence that MDS cells require support from microenvironment components to propagate disease in xenograft models. While co-transplantation with normal mesenchymal stromal cells (either cell line-derived or primary donor-derived) improved engraftment of MDS clones, Medyouf et al. found that MDS patient-derived mesenchymal stromal cells confer superior support for MDS cells [76]. Engraftment of CD34+ MDS cells was significantly augmented when identical patient samples were co-transplanted with MDS-derived mesenchymal stromal cells compared to those derived from age-matched normal donors [76]. They further demonstrated that MDS-derived mesenchymal stromal cells are molecularly distinct from their healthy counterparts, exhibiting aberrant expression of genes associated with extracellular matrix remodeling, cytokine signaling, and secreted factors—features which can be directly induced in normal mesenchymal stromal cells by MDS cells [76]. The presence of an abnormal molecular profile in MDS-derived mesenchymal stromal cells was further corroborated in a recent transcriptional analysis of prospectively purified patient-derived cells, revealing upregulation of inflammation-associated genes implicated in hematopoietic inhibition [83]. Thus, MDS cell may initiate alterations in their microenvironment by “reprogramming” healthy mesenchymal stromal cells to take on pathologic features that preferentially support MDS cells, possibly at the expense of normal hematopoiesis.
Mesenchymal-osteolineage dysfunction in murine models of MDS
Elucidation of cytogenetic and genetic aberrations in patients with MDS has enabled the development of murine models of MDS via knock-in or knock-out of implicated genes. Genetic mouse models recapitulate hallmark features of human MDS—including multi-lineage peripheral blood cytopenias, dysmyelopoiesis, and variable propensities for transformation to acute leukemia [84]—and therefore represent useful in vivo models to investigate the reciprocal interactions between MDS cells and their BMME. Moreover, recent studies of the hematopoietic niche in mice have identified specific populations of mesenchymal stromal cells, osteoblastic lineage cells, and endothelial cells that are critical for HSPC support [9–16]. Therefore, studying genetically engineered mice as preclinical models of MDS provides the important advantage of assessing specific populations of immunophenotypically-defined cells in the MDS BMME. In particular, the Levesque and Passegue groups have characterized microenvironmental populations within the lineage−/CD45−/CD31− non-hematopoietic and non-endothelial compartment, identifying osteoblastic lineage cells (OBCs, CD51+/Sca1−) and precursor multipotent stromal cells (MSCs, CD51+/Sca1+) which exhibit gene expression profiles and functional properties consistent with osteolineage and mesenchymal progenitor cells, respectively [85, 86].
NUP98-HOXD13 (NHD13) transgenic mice are an established murine model of MDS which recapitulate human disease due to Vav-driven, hematopoietic cell-specific expression of the NHD13 fusion gene, originally identified in a patient with MDS (Table 1) [87, 88]. NHD13 mice exhibit pathologic features of human MDS evidenced by multi-lineage blood cytopenias, dyspoiesis of erythroid, megakaryocytic and granulocytic cells and progression to acute leukemia [84, 88]. In studies of the NHD13 model, MSCs and OBCs were increased in the BMME of 18–23 week old NHD13 mice [89]. However, NHD13 mice exhibited no changes in skeletal structure nor osteoblastic bone formation and osteoclastic bone resorption, indicating that the expanded populations do not generate functional bone-forming cells [89]. Recently, Weidner et al. reported decreased trabecular bone volume in younger NHD13 mice at 2 month of age when cytopenias are not yet prominent in this model [90]. This suggests that mesenchymal-osteolineage dysfunction in NHD13 mice alters skeletal structure in a time-dependent manner, resulting in early disruption of bone microarchitecture that precedes overt multi-lineage cytopenias by approximately 4 months of age [88]. Such osteoblastic disruption is consistent with work by Frisch et al. demonstrating bone and osteoblastic cell loss in a murine model of myeloid leukemia in which osteoblastic inhibition is already present even at early stages of disease when leukemic burden is low [91]. Frisch et al. found that osteoblastic inhibition in murine myeloid leukemia was associated with leukemic cell production of CCL3 (or MIP-1α), a chemokine previously reported as a mediator of osteoblastic dysfunction in multiple myeloma and chronic myelogenous leukemia (CML) [85, 92, 93]. Of note, CCL3 was also increased in NHD13 mice and in a cohort of patients with MDS [89, 94], suggesting that factors produced by MDS cells may drive mesenchymal-osteolineage abnormalities.
Table 1.
Murine models of MDS exhibiting bone marrow microenvironment involvement
| Model | Genetic Alteration | Model Features | Ref | ||
|---|---|---|---|---|---|
| Gene | Location | MDS | Microenvironment | ||
| Osx-GFP- Cre+ Dicerfl/fl mice | Dicer1 deletion | Microenvironment (osteoprogenitors targeted by Osterix) |
|
|
[55] |
| Osx-GFP- Cre+ Sbdsfl/fl mice | Sbds deletion | Microenvironment (osteoprogenitors targeted by Osterix) |
|
|
[55, 96] |
| NUP98- HOXD13 (NHD13) transgenic mice | NHD13 fusion transgene expression | Hematopoietic cells (targeted by Vav regulatory elements) |
|
|
[88–90] |
| S100A9 transgenic mice | S100A9 over- expression | Hematopoietic cells (targeted by H2K promoter) |
|
|
[134–142] |
A link between bone alterations and MDS also exists in humans as osteoblast numbers and serum osteocalcin are lower in patients with MDS compared to age-match healthy individuals [95] and MDS is more prevalent in elderly individuals with osteoporosis compared to those without osteoporosis [90]. Moreover, skeletal defects due to inactivating SBDS mutations in Shwachman-Diamond syndrome (SDS) are associated MDS and AML development early in life, while osteoprogenitor-specific knockout of Sbds in mice induced myelodysplasia [55]. Therefore, abnormalities in the mesenchymal-osteolineage BMME may contribute to structural bone defects to promote progression of the MDS phenotype.
In NHD13 mice, NHD13 transgene expression is driven by Vav regulatory elements to be specific to hematopoietic cells and excluded from stromal populations [88, 89], further supporting the presence of MDS-dependent BMME changes in this model. This is in line with work by Medyouf et al. demonstrating MDS-induced “re-programming” of healthy stromal cells to take on pathologic features of MDS-derived stromal cells [76]. Moreover, Schepers et al. showed that neoplastic cells in a murine model CML induced MSCs to overproduce OBCs with a pro-inflammatory and pro-fibrotic phenotype, effects mediated in part by CCL3 [85]. OBCs in CML mice were also functionally defective in their ability to support normal hematopoietic stem cells but not neoplastic cells [85]. Cumulatively, these studies indicate that neoplastic cells in myeloid malignancies including MDS and CML induce functional alterations in the mesenchymal-osteolineage cells, impairing normal hematopoiesis while supporting disease progression.
Other murine models with well-characterized bone marrow microenvironment dysfunction include Osx-GFP-Cre+ Dicerfl/fl and Osx-GFP-Cre+ Sbdsfl/fl mice (Table 1) [55, 96]. As described earlier (please refer to the section, “Bone marrow microenvironment defects and MDS initiation”), selective Dicer1 deletion in Osx+ osteoprogenitors demonstrated for the first time that primary BMME defects can be sufficient to drive MDS. MDS pathogenesis was mediated by decreased Sbds expression in Dicer1-deficient osteoprogenitors as Osx-GFP-Cre+ Sbdsfl/fl mice recapitulated skeletal defects and MDS features of Osx-GFP-Cre+ Dicerfl/fl mice [55]. Subsequent studies found that production of the pro-inflammatory molecules S100A8 and S100A9 by Sbds-deficient Osx+ cells drives genotoxic stress in HSPCs, characterized by mitochondrial dysfunction, oxidative stress, and DNA damage [96]. Therefore, a dysfunctional mesenchymal-osteolineage niche can be a source of pro-inflammatory signals which promote MDS pathogenesis (please refer to the section “Immune Dysregulation in MDS”). In human MDS, S100A8/9 overexpression by mesenchymal-lineage cells in a subset of patients correlates with shorter time to leukemic progression [96].
Therefore, murine MDS models represent powerful tools with which to identify BMME defects that play a role in human MDS pathogenesis and progression. Of particular interest will be to determine whether microenvironment abnormalities lead to development of MDS clones and the mechanism of such processes. Currently, driver mutations in non-hematopoietic BMME cells have not yet been established in human MDS; rather, MDS is widely-accepted to arise from cell-autonomous genetic abnormalities. Given this, the majority of existing murine models have been genetically engineered to express hematopoietic defects identified in human disease due to cytogenetic aberrations (NHD13 translocation [88]; CD74-Nid67 [97] and Nmp1 [98] loss in del5q) and mutations or aberrant expression of epigenetic regulators (Asxl1 [99] and Dnmt3a [100]) and transcription factors (Runx1 [101], Npm1 [98], and Evi1 [102]). Examining the BMME in such models may provide crucial insight into how MDS initiates BMME dysfunction to promote disease progression. Of critical importance will be to identify: 1) the MDS-derived signals mediating BMME dysfunction, 2) the subsequent BMME signals impairing hematopoietic function and promoting disease progression, and 3) how such interactions may be clinically modulated to improve hematopoietic function in patients with MDS.
Vascular and endothelial alterations in MDS and other hematologic malignancies
VEGF-mediated angiogenesis is well-established as a critical mediator of solid tumor progression by promoting tumor growth, dissemination, and metastasis. More recently, a role for angiogenesis in the pathophysiology of hematologic malignancies was brought into light based on increased vascularity observed in bone marrow and lymph node biopsies from patients with MDS [103–107], AML [105, 107–109], myeloproliferative disorders [110, 111], and a number of lymphoid neoplasms [103, 112–116]. In line with increased vascularity, circulatory levels of VEGF and other pro-angiogenic factors including bFGF and HGF are elevated both in patients with MDS and other myeloid and lymphoid malignancies as well as in murine models of MDS [89, 103]. While the mechanism by which angiogenesis contributes to progression of MDS and other hematologic malignancies are not completely understood, increased vascularity across a broad range of hematologic malignancies suggests that one function of neoangiogenesis may be to deliver oxygen and nutrients required for expansion of malignant cells, similar to solid tumors. Consistent with this, vascular alterations and levels of angiogenic factors have been found to correlate with disease progression and prognosis [105, 117]. However, one group found that plasma level of VEGF has more prognostic significance in AML compared to MDS [118]. While increased microvessel density (MVD) has been described for both MDS and AML, studies report both decreased and increased MVD in MDS relative to AML [104, 107]. One study of de novo AML patients and paired specimens from MDS patients before and after transformation to AML observed lower MVD upon progression to AML compared to de novo AML [105]. Evaluation of different MDS subtypes revealed lower MVD in refractory anemia (RA), RA with ring sideroblasts (RARS), and RA with excess blasts (RAEB) compared to RAEB in transformation (RAEB-t) and chronic myelomonocytic leukemia (CMML) [104]. Furthermore, another group reported that RA and RARS exhibited larger caliber vessels compared to RAEB [107]. Therefore, distinct vascular features may be present in different MDS subtypes and upon transformation to acute leukemia, suggesting that alterations in angiogenesis may be involved in disease progression.
In solid tumors, neoplastic cells are a major source of VEGF wherein upregulation occurs downstream of proto-oncogene activation, resulting in tumor-induced neoangiogenesis. In hematologic malignancies, however, increased VEGF may originate from a number of cellular sources given the complexity of the bone marrow and its microenvironment. Evaluation of primary patient samples identified increased VEGF mRNA and protein in bulk marrow, while immunohistochemical analyses revealed that VEGF is expressed by myeloid precursors in MDS and myeloblasts in AML, identifying malignant cells as one source of VEGF [117, 119–122]. One study observed strong VEGF expression in megakaryocytes of MDS patients [104]. Transcriptional analyses have identified upregulation of VEGF mRNA expression in human MDS-derived mesenchymal stromal cells [76, 83]. Overall, aberrant VEGF production may originate from both hematopoietic and microenvironmental cells in MDS. The co-expression of VEGFR1 and VEGFR2 with VEGF in MDS myeloid precursors and AML myeloblasts indicates that VEGF participates both in paracrine signaling to mediate changes in the vascular microenvironment and in autocrine signaling pathways that can directly stimulate malignant cells [119, 120, 122]. Indeed, in vitro assays demonstrated that VEGF stimulation increased colony-forming unit-leukemia (CFU-L) generation by primary patient-derived CMML and RAEB-t samples, while treatment with a VEGF neutralizing antibody inhibited CFU-L formation [120].
As discussed above, a major function of VEGF paracrine signaling is to recruit and promote proliferation of endothelial cells for new blood vessel formation [123, 124]. Thus, VEGF upregulation in MDS and numerous other hematologic malignancies is associated with increased vascularity, which may augment delivery of oxygen and nutrients for proliferating malignant clones. Another consequence of increased vascularity is increased endothelial cells observed in the marrow of a murine MDS model and in the circulation of patients with MDS [89, 106, 125]. VEGF stimulation of human umbilical vein endothelial cells has been reported to induce secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) [122, 126]. This suggests that paracrine activities of VEGF may include both expansion of endothelial cells and further stimulating them to generate growth factors for malignant cells. As other cell populations in the bone marrow microenvironment such as osteolineage cells also express VEGF receptors [127], it is possible that VEGF-mediated activity of other cell types may also contribute malignant clone proliferation. Therefore, the pro-survival and growth effects of VEGF may occur via both autocrine and paracrine signaling pathways to promote the proliferation and self-renewal of malignant clones, contributing to the progression of MDS and other hematologic malignancies.
Along with its trophic effects, VEGF is also known to induce vascular permeability which is normally important in inflammatory responses following infection or tissue damage. Thus, elevated VEGF in the MDS bone marrow microenvironment may lead to greater vascular leakage in addition to increased vascularity. This may have functional consequences on HSPC maintenance as subsets of marrow blood vessels possess distinct permeability properties that differentially regulate hematopoiesis [128]. Greater permeability, attributed to marrow sinusoids compared to arteriolar vessels, promotes HSPC activation, leading to increased cell cycling, migration, and differentiation, but at the expense of HSPC survival and function [128]. This effect is thought to be mediated by exposure to blood plasma leading to increased reactive oxygen species (ROS) in HSPCs as both in vitro exposure of HSPCs to blood plasma and in vivo modeling of impaired vascular integrity resulted in increased apoptosis and myeloid skewing of hematopoietic output following transplantation [128]. Since increased vascular permeability can impair HSPC maintenance and induce MDS features of increased apoptosis and myeloid skewing in the non-malignant setting, it is possible that VEGF-mediated vessel leakage may contribute to such features in MDS as well.
Endothelial cell abnormalities may also contribute to alterations in the vascular microenvironment. In a study of CD45−/CD146+/CD34+ circulating endothelial cells isolated from MDS patients with del5q and trisomy 8, 39–84% of the endothelial cells demonstrated the same chromosomal abnormalities as hematopoietic cells on FISH analysis [106]. The finding that endothelial cells harbor the same genetic aberrations as the neoplastic clone is not specific to MDS and has been noted in other hematologic malignancies [129–131]. In B-cell lymphomas, immunohistochemical and FISH analyses revealed that 15–85% of vascular endothelial cells demonstrated the same cytogenetic lesions as lymphoma cells [129]. In CML, patient-derived endothelial cells were found to express BCR-ABL gene fusion, suggesting that the neoplastic clone and a portion of the endothelial population may arise from a common hemangioblastic progenitor cell [130]. A subsequent study demonstrated that CML patient-derived, BCR-ABL expressing progenitor cells generated both malignant hematopoietic cells and phenotypic endothelial cells, both harboring BCR-ABL [131]. Overall, these data suggest that endothelial cell and vascular expansion may be in part mediated by the neoplastic clone. Since endothelial cells with and without neoplasia-specific genetic aberrations co-exist in vascular structures [129], it is possible that abnormal endothelial cells may recruit or coordinate neoangiogenesis by normal endothelial cells. Furthermore, as endothelial cells are critical for normal HSPC support [17], abnormalities in this population may contribute to features of MDS.
In the non-malignant setting, induction of arteriole formation via Notch signaling activation led to increased perivascular stromal cells and mesenchymal stromal cells [132]. This suggests that increased vascularity may be associated with or coordinate expansion of perivascular niches which house populations of mesenchymal stromal cells that are important for HSPC support. Therefore, in the setting of MDS, it is possible that increased vasculature along with endothelial cell abnormalities may contribute to or provide a permissible niche for the expansion of dysfunctional mesenchymal stromal cells. On the other hand, as MDS-derived mesenchymal stromal cells demonstrate elevated VEGF protein secretion compared to age-matched healthy counterparts [76], microenvironmental cells may be another important mediator of endothelial and vascular increases in MDS. While expansion of non-functional mesenchymal-osteolineage cells are associated with increased endothelial cells and vascular structures in a mouse model of MDS [89], the molecular and cellular interactions coordinating such alterations in MDS requires further investigation.
Immune dysregulation in MDS
Immune dysregulation has been proposed as a key component of MDS pathophysiology [133]. The presence of aberrant inflammatory signaling in MDS is supported by numerous studies describing altered levels of inflammatory cytokines and signaling factors in MDS bone marrow [96, 134–138]. Moreover, upregulation of cell surface receptors for pro-inflammatory molecules also contribute to perturbed immune responses. For example, the IL-1 receptor accessory protein (IL1RAP) is overexpressed in HSPCs from patients with AML and high-risk MDS and correlates with poor survival [139]. Overexpression of toll-like receptors (TLR), key regulators of the innate immune response, and increased TLR-mediated signaling in HSPCs of MDS patients have been implicated in development of hematopoietic dysfunction [140, 141]. Therefore, a pro-inflammatory milieu in the MDS microenvironment in conjunction with increased susceptibility of HSPCs to respond to inflammatory stimuli likely both coordinate aberrant immune signaling that impair normal hematopoiesis and promote expansion of malignant cells.
Recently, danger-associated molecular pattern (DAMP) proteins such as S100A8 and S100A9 are emerging as central regulators of inflammation-mediated hematopoietic dysfunction in MDS. S100A9 drives expansion of CD33+ myeloid-derived suppressor cells, reported to contribute to ineffective hematopoiesis via secretion of suppressive cytokines such as TGF-β [142]. S100A9 overexpression in transgenic mice led to increased CD33+ myeloid-derived suppressor cells and induced an MDS-like phenotype characterized by multi-lineage cytopenias and dysplasia [142]. In addition, elevated S100A9 in MDS was identified as a critical regulator of inflammasome-mediated pyroptosis [134]. In contrast to apoptosis, pyroptosis is a pro-inflammatory and caspase-1-dependent form of programmed cell death that can be initiated under various pathologic conditions by DAMP-activation of TLRs [143]. An early step in pyroptosis involves formation of cytosolic inflammasome complexes containing NLRs (nucleotide-binding domain and leucine-rich repeat pattern recognition receptor), leading to downstream activation of caspase-1 to trigger pyroptosis [143]. Strikingly, human MDS HSPCs exhibit inflammasome activation and pyroptosis despite differences in genetic aberrations [134]. S100A9 is sufficient to induce pyroptosis in normal hematopoietic cells, whereas S100A9 neutralization decreases pyroptosis and rescues hematopoietic function of MDS hematopoietic cells, indicating that S100A9-mediated pyroptosis may be a central feature of MDS contributing to hematopoietic failure [134]. In addition to driving pyroptosis, caspase-1 activation was found to increase production of IL-1β [134], previously reported to be produced by MDS HSPCs in correlation with the level of marrow cell death observed in MDS patient specimens [144].
While overexpression by MDS cells has been described as one source of S100A9 in MDS [134], microenvironmental cell-derived S100A9 was recently identified as a driver of HSPC genotoxic stress in MDS [96]. Gene expression of S100A9 and its heterodimer partner S100A8 was upregulated in CD271+ mesenchymal stromal cells from patients with low-risk MDS and Shwachman-Diamond syndrome (SDS), a disorder characterized by skeletal abnormalities and increased propensity to develop MDS and AML [96]. In a mouse model of SDS, generated via Osterix-Cre-mediated Sbds deletion in osteoprogenitor cells, Sbds-deficient Osterix+ cells also overexpressed S100A9 and S100A8, leading to increased marrow plasma levels of both proteins [96]. S100A8/9 heterodimer activation of their canonical receptor, TLR4, on mouse HSPCs induced reactive oxygen species generation and subsequent DNA damage [96]. Such DNA damage may contribute to the genomic instability in MDS thought to underlie the accumulation of additional genetic defects and clonal evolution to acute leukemia [145]. This reveals an additional mechanism of MDS pathophysiology as S100A8/9 treatment of normal human CD34+ cord blood cells induced DNA damage and apoptosis and impaired hematopoietic function [96]. Notably, S100A8/9 overexpression in mesenchymal stromal cells correlates with shorter time to leukemic transformation in patients with MDS [96].
Taken together, the DAMP proteins S100A9 and S100A8 are specific pro-inflammatory molecules within the MDS bone marrow microenvironment that can drive key pathologic features of MDS including impaired hematopoiesis, accumulation of genetic damage, and progression to leukemia. Although MDS cells are a source of such pro-inflammatory signals, emerging data now point to abnormal MDS microenvironmental cells another key contributor to immune perturbations in MDS [83].
Cytokines in the MDS microenvironment
Another component of the MDS microenvironment is dysregulation of homeostatic signaling pathways. The balance between stimulatory and inhibitory cytokines in the BMME plays a critical role in regulating hematopoiesis whereby alterations of the signaling milieu may contribute to MDS-initiated BMME abnormalities and BMME-mediated disease progression. Abnormal expression of numerous cytokines have been reported in MDS and the effects of aberrant CCL3, S100A8/9, IL-1β, and TGF-β have been discussed in other sections and are summarized in Table 2.
Table 2.
Aberrant signaling molecules in the MDS microenvironment
| Signaling Molecule | Involvement in MDS |
|---|---|
| CCL3 | Mesenchymal-osteolineage dysfunction [85, 89, 91–93] |
| TGF-β | Hematopoietic suppression [146] |
| Secreted by myeloid cells downstream of S100A9-CD33 signaling [142] | |
| S100A9 and S100A8 | Production by mesenchymal stromal cells in human MDS correlates with leukemic progression [96] |
| Induces expansion of CD33+ myeloid-derived suppressor cells and induces secretion of suppressive cytokines TGF-b and IL-10 [142] | |
| Induces caspase-1-mediated pyroptotic cell death and IL-1β production [134] | |
| Induces HSPC genotoxic stress [96] | |
| IL-1β | Increased cell death in MDS marrow [144] |
| Increased production downstream of S100A9-mediated caspase-1 activation [134] | |
| VEGF | Increases marrow vascularity [103–107] to potentially increase O2 and nutrient delivery to malignant cells |
| Direct stimulation of malignant cell proliferation [120] | |
| Overexpressed by myeloid precursors in MDS [120], megakaryocytes [104], and mesenchymal stromal cells [76, 83] |
Briefly, the chemokine CCL3 has been reported as a mediator of osteoblastic dysfunction in myeloid malignancies and multiple myeloma [85, 91–93]. In the NHD13 murine model of MDS, mesenchymal-osteolineage dysfunction is associated with increased serum levels of CCL3, suggesting that it may also disrupt the BMME in MDS [89]. CCL3 has also been reported to be overexpression by patients with MDS [94], indicating that it may be a beneficial clinical target in a subset of human MDS.
Upregulation of the pro-inflammatory DAMP protein S100A9 has recently been reported to mediate numerous features of MDS including induction of capase-1-dependent pyroptotic cell death and production of IL-1β [134]. Furthermore, activation of TLR4 by S100A9 and its heterodimer partner, S100A8, induces genotoxic stress in HSPCs [96], identifying a mechanism for the progressive genomic instability in MDS underlying clonal evolution to acute leukemia. In addition, S100A9 may contribute to hematopoietic failure by stimulating CD33+ myeloid to produce suppressive cytokines such as TGF-β [142].
Studies of TGF-β signaling in MDS illustrate how clinical targeting of aberrant signal transduction pathways may be a beneficial therapeutic strategy in MDS. Over-activation of the TGF-β signaling pathway has been identified as mediator of ineffective hematopoiesis in MDS [146]. While TGF-β production may be upregulated downstream of other dysregulated signaling pathways in MDS, such as S100A9-activation of CD33+ myeloid cells [142], Zhou et al. found that decreased expression of the TGF-β receptor I (TBRI) kinase inhibitor SMAD7 leads to myelosuppression in MDS patient-derived HSPCs [146]. Notably, administration of Galunisertib, a TBRI inhibitor, improved the hematopoietic function in preclinical studies of human MDS marrow and a murine model of TGF-β-mediated marrow failure, demonstrating that clinical targeting of aberrant signaling pathways may be a beneficial therapeutic strategy in MDS [146]. Indeed, a phase 2 clinical trial demonstrated hematologic improvement in 26% of MDS patients treated with Galunisertib [147].
Myeloid skewing
Aside from the hallmark pathologic features of blood cytopenias and myeloid dysplasia, another aspect of hematopoietic dysfunction in MDS is myeloid skewing of hematopoietic output described in murine MDS models and in human MDS xenograft models [76, 89]. Myeloid skewing is characterized by an increased proportion of myeloid cells relative to lymphoid cells. In NHD13 mice, the proportion of CD11b+ myeloid cells is increased within peripheral blood CD45+ leukocytes compared to WT littermates [89]. When WT marrow (CD45.1) was transplanted into NHD13 or WT recipient mice (CD45.2), hematopoietic output by CD45.1+ WT cells exhibited increased myeloid production in NHD13 recipients compared to WT recipients, suggesting that the abnormal MDS BMME can induce normal hematopoietic cells to develop an MDS phenotype of myeloid skewing [89]. Osteolineage dysfunction may be one component of the MDS BMME contributing to myeloid skewing. Osteoblasts have been well-characterized to play an essential role in B lymphopoiesis. In vitro, osteoblasts support all stages of B cell development, and in vivo, genetic osteoblastic defects or ablation in mouse models reduce B cell precursors in the marrow and mature B cells in the peripheral blood [24, 148]. Studies of human MDS have described B cell lineage defects, evidenced by lower expression of B cell lineage-related genes in CD34+ HSPCs and reduced B cell progenitors in unfractionated marrow from low-risk MDS patients [149]. Therefore, it may be reasonable to postulate that osteolineage abnormalities lead to B cell lineage defects, resulting in a loss of lymphoid populations to contribute to myeloid bias in MDS.
Regulation of progression to acute leukemia
Osteoblasts have been described to play a critical role in regulating leukemic progression in murine models myeloid malignancies [95, 150, 151]. When NHD13 marrow was transplanted into NHD13 mice or WT littermates, WT recipients had lower rate of death and/or leukemic progression, suggesting that normalization of the mesenchymal-osteolineage compartment can improve disease outcome [89]. Osteoblast ablation via ganciclovir treatment of double transgenic Col2.3kb-Δtk SCL-tTA/BCR-ABL mice accelerated CML development and decreased survival compared to control mice with intact osteoblasts [150]. Furthermore, myeloid leukemia cells exhibited increased proliferation when transplanted into mice with Col2.3kb-Cre-mediated osteoblast depletion, leading to higher leukemic burden and shorter survival—effects which were reversed via pharmacological restoration of osteoblast numbers [95]. Taken together, these reports suggest that osteoblastic cells restrain disease progression in MDS and other myeloid malignancies. Therefore, the osteolineage hematopoietic niche represents a viable target of clinical intervention to improve disease outcome. Indeed, osteoblast-specific activation of parathyroid hormone (PTH) receptor-mediated signaling augments survival in mouse models of CML and reduces engraftment of human CML cells in mouse xenotransplantation models, possibly via TGF-β1-mediated suppression of CML proliferation [151]. Consistent with this, TGF-β1 expression has been reported to be downregulated in CML-modified OBCs [85]. However, activation of PTH receptor signaling in osteoblastic cells accelerated MLL-AF9-mediated AML in mouse transplantation models [151]. Therefore, strong evidence supports an osteoblastic role in regulating disease progression in myeloid malignancies; however, further investigation is needed to elucidate their specific cellular and molecular interactions with different types of myeloid neoplasms to promote or restrain disease.
Concluding Remarks
Overall, MDS remains a poorly treated disease and the genetic heterogeneity of hematopoietic abnormalities represents a significant hurdle to specific targeting of cell-autonomous mutations. In spite of genetic diversity in MDS, interaction between MDS and the BMME appears to be a common disease feature and therefore represents an appealing therapeutic target, especially given the well-established role of the microenvironment in maintaining normal hematopoiesis. Therefore, clinical intervention via the microenvironment may be a novel therapeutic strategy complementary to specific targeting of malignant cells.
With improved understanding of the bone marrow microenvironment, we are beginning to refine the role of the hematopoietic niche in MDS. Current data support the emerging concept of interdependency between MDS and diverse cell populations in the BMME including but not limited to mesenchymal stromal cells, osteoblastic lineage cells, and endothelial cells. However, many technical and conceptual challenges must be resolved to better understand the BMME contribution to MDS initiation and progression. Specifically, we need to refine definitions and techniques to isolate purified stromal cell populations in both the human and murine BMME. This will help elucidate the localization and functional interactions of such populations with neoplastic cells and normal HSPC subsets in MDS and other hematologic malignancies. Specific questions to address include how may the BMME restrain neoplastic transformation under normal conditions? How then does BMME dysfunction arise in MDS and what is the temporal relationship relative to intrinsic hematopoietic abnormalities in MDS? Finally, what are the critical cellular or molecular signals mediating interactions between different microenvironmental cell populations and their interactions with malignant MDS cells? Addressing these questions will be critical to developing therapeutic strategies for BMME modulation to improve hematopoietic function and survival in individuals with MDS.
Highlights.
Microenvironmental defects can initiate myeloid neoplasms including MDS
MDS induce functional alterations in microenvironmental cells
Niche abnormalities impair normal hematopoiesis and facilitate MDS progression
Aberrant inflammatory signaling contribute to pathologic features of MDS
Vascular and endothelial abnormalities may contribute to MDS progression
Acknowledgments
A.J.L. is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (NIH) under award number F30DK113727. This work is also supported by funds from the University of Rochester CTSA award number UL1 TR002001 from the National Center for Advancing Translational Sciences, the National Institutes of Allergy and Infectious Diseases (U01 AI107276 to L.M.C), the National Cancer Institute (R01 CA166280 to L.M.C.), and the National Institute on Aging (R01 AG046293 to L.M.C).The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bejar R, Steensma DP. Recent developments in myelodysplastic syndromes. Blood. 2014;124:2793–2803. doi: 10.1182/blood-2014-04-522136. [DOI] [PubMed] [Google Scholar]
- 2.DeZern AE. Nine years without a new FDA-approved therapy for MDS: how can we break through the impasse? Hematology/the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2015;2015:308–316. doi: 10.1182/asheducation-2015.1.308. [DOI] [PubMed] [Google Scholar]
- 3.Ma X, Does M, Raza A, Mayne ST. Myelodysplastic syndromes: incidence and survival in the United States. Cancer. 2007;109:1536–1542. doi: 10.1002/cncr.22570. [DOI] [PubMed] [Google Scholar]
- 4.Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008;112:45–52. doi: 10.1182/blood-2008-01-134858. [DOI] [PubMed] [Google Scholar]
- 5.Ma X. Epidemiology of myelodysplastic syndromes. The American journal of medicine. 2012;125:S2–5. doi: 10.1016/j.amjmed.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epling-Burnette PK, List AF. Advancements in the molecular pathogenesis of myelodysplastic syndrome. Current opinion in hematology. 2009;16:70–76. doi: 10.1097/MOH.0b013e3283257ac7. [DOI] [PubMed] [Google Scholar]
- 7.Tefferi A, Vardiman JW. Myelodysplastic syndromes. The New England journal of medicine. 2009;361:1872–1885. doi: 10.1056/NEJMra0902908. [DOI] [PubMed] [Google Scholar]
- 8.Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nature reviews Cancer. 2017;17:5–19. doi: 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinho S, Lacombe J, Hanoun M, et al. PDGFRalpha and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. The Journal of experimental medicine. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morikawa S, Mabuchi Y, Kubota Y, et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. The Journal of experimental medicine. 2009;206:2483–2496. doi: 10.1084/jem.20091046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell stem cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–841. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 15.Kunisaki Y, Bruns I, Scheiermann C, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooper AT, Butler JM, Nolan DJ, et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell stem cell. 2009;4:263–274. doi: 10.1016/j.stem.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–161. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell stem cell. 2007;1:685–697. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenbaum A, Hsu YM, Day RB, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Visnjic D, Kalajzic Z, Rowe DW, Katavic V, Lorenzo J, Aguila HL. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103:3258–3264. doi: 10.1182/blood-2003-11-4011. [DOI] [PubMed] [Google Scholar]
- 24.Zhu J, Garrett R, Jung Y, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109:3706–3712. doi: 10.1182/blood-2006-08-041384. [DOI] [PubMed] [Google Scholar]
- 25.Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015;126:2443–2451. doi: 10.1182/blood-2015-07-533588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korn C, Mendez-Ferrer S. Myeloid malignancies and the microenvironment. Blood. 2017;129:811–822. doi: 10.1182/blood-2016-09-670224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medyouf H. The microenvironment in human myeloid malignancies: emerging concepts and therapeutic implications. Blood. 2017;129:1617–1626. doi: 10.1182/blood-2016-11-696070. [DOI] [PubMed] [Google Scholar]
- 28.Geyh S, Oz S, Cadeddu RP, et al. Insufficient stromal support in MDS results from molecular and functional deficits of mesenchymal stromal cells. Leukemia. 2013;27:1841–1851. doi: 10.1038/leu.2013.193. [DOI] [PubMed] [Google Scholar]
- 29.Ferrer RA, Wobus M, List C, et al. Mesenchymal stromal cells from patients with myelodyplastic syndrome display distinct functional alterations that are modulated by lenalidomide. Haematologica. 2013;98:1677–1685. doi: 10.3324/haematol.2013.083972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falconi G, Fabiani E, Fianchi L, et al. Impairment of PI3K/AKT and WNT/beta-catenin pathways in bone marrow mesenchymal stem cells isolated from patients with myelodysplastic syndromes. Experimental hematology. 2016;44:75–83. e71–74. doi: 10.1016/j.exphem.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Soenen-Cornu V, Tourino C, Bonnet ML, et al. Mesenchymal cells generated from patients with myelodysplastic syndromes are devoid of chromosomal clonal markers and support short- and long-term hematopoiesis in vitro. Oncogene. 2005;24:2441–2448. doi: 10.1038/sj.onc.1208405. [DOI] [PubMed] [Google Scholar]
- 32.Zhao ZG, Xu W, Yu HP, et al. Functional characteristics of mesenchymal stem cells derived from bone marrow of patients with myelodysplastic syndromes. Cancer letters. 2012;317:136–143. doi: 10.1016/j.canlet.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 33.Rathnayake AJ, Goonasekera HW, Dissanayake VH. Phenotypic and Cytogenetic Characterization of Mesenchymal Stromal Cells in De Novo Myelodysplastic Syndromes. Analytical cellular pathology (Amsterdam) 2016;2016:8012716. doi: 10.1155/2016/8012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flores-Figueroa E, Gutierrez-Espindola G, Montesinos JJ, Arana-Trejo RM, Mayani H. In vitro characterization of hematopoietic microenvironment cells from patients with myelodysplastic syndrome. Leukemia research. 2002;26:677–686. doi: 10.1016/s0145-2126(01)00193-x. [DOI] [PubMed] [Google Scholar]
- 35.Flores-Figueroa E, Arana-Trejo RM, Gutierrez-Espindola G, Perez-Cabrera A, Mayani H. Mesenchymal stem cells in myelodysplastic syndromes: phenotypic and cytogenetic characterization. Leukemia research. 2005;29:215–224. doi: 10.1016/j.leukres.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 36.Han Q, Sun Z, Liu L, et al. Impairment in immuno-modulatory function of Flk1(+)CD31(−)CD34(−) MSCs from MDS-RA patients. Leukemia research. 2007;31:1469–1478. doi: 10.1016/j.leukres.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 37.Mellibovsky L, Diez A, Serrano S, et al. Bone remodeling alterations in myelodysplastic syndrome. Bone. 1996;19:401–405. doi: 10.1016/s8756-3282(96)00210-4. [DOI] [PubMed] [Google Scholar]
- 38.Varga G, Kiss J, Varkonyi J, et al. Inappropriate Notch activity and limited mesenchymal stem cell plasticity in the bone marrow of patients with myelodysplastic syndromes. Pathology oncology research : POR. 2007;13:311–319. doi: 10.1007/BF02940310. [DOI] [PubMed] [Google Scholar]
- 39.Flores-Figueroa E, Montesinos JJ, Flores-Guzman P, et al. Functional analysis of myelodysplastic syndromes-derived mesenchymal stem cells. Leukemia research. 2008;32:1407–1416. doi: 10.1016/j.leukres.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Klaus M, Stavroulaki E, Kastrinaki MC, et al. Reserves, functional, immunoregulatory, and cytogenetic properties of bone marrow mesenchymal stem cells in patients with myelodysplastic syndromes. Stem cells and development. 2010;19:1043–1054. doi: 10.1089/scd.2009.0286. [DOI] [PubMed] [Google Scholar]
- 41.Pavlaki K, Pontikoglou CG, Demetriadou A, et al. Impaired proliferative potential of bone marrow mesenchymal stromal cells in patients with myelodysplastic syndromes is associated with abnormal WNT signaling pathway. Stem cells and development. 2014;23:1568–1581. doi: 10.1089/scd.2013.0283. [DOI] [PubMed] [Google Scholar]
- 42.Aanei CM, Flandrin P, Eloae FZ, et al. Intrinsic growth deficiencies of mesenchymal stromal cells in myelodysplastic syndromes. Stem cells and development. 2012;21:1604–1615. doi: 10.1089/scd.2011.0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tauro S, Hepburn MD, Bowen DT, Pippard MJ. Assessment of stromal function, and its potential contribution to deregulation of hematopoiesis in the myelodysplastic syndromes. Haematologica. 2001;86:1038–1045. [PubMed] [Google Scholar]
- 44.Liu Q, Zhu H, Dong J, Li H, Zhang H. Defective proliferative potential of MSCs from pediatric myelodysplastic syndrome patients is associated with cell senescence. International journal of clinical and experimental pathology. 2015;8:13059–13066. [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang YZ, Zhao DD, Han XP, Jin HJ, Da WM, Yu L. In vitro study of biological characteristics of mesenchymal stem cells in patients with low-risk myelodysplastic syndrome. Zhongguo shi yan xue ye xue za zhi. 2008;16:813–818. [PubMed] [Google Scholar]
- 46.Sutherland HJ, Eaves CJ, Eaves AC, Dragowska W, Lansdorp PM. Characterization and partial purification of human marrow cells capable of initiating long-term hematopoiesis in vitro. Blood. 1989;74:1563–1570. [PubMed] [Google Scholar]
- 47.Ploemacher RE, van der Sluijs JP, van Beurden CA, Baert MR, Chan PL. Use of limiting-dilution type long-term marrow cultures in frequency analysis of marrow-repopulating and spleen colony-forming hematopoietic stem cells in the mouse. Blood. 1991;78:2527–2533. [PubMed] [Google Scholar]
- 48.Tennant GB, Walsh V, Truran LN, Edwards P, Mills KI, Burnett AK. Abnormalities of adherent layers grown from bone marrow of patients with myelodysplasia. British journal of haematology. 2000;111:853–862. [PubMed] [Google Scholar]
- 49.Aizawa S, Nakano M, Iwase O, et al. Bone marrow stroma from refractory anemia of myelodysplastic syndrome is defective in its ability to support normal CD34-positive cell proliferation and differentiation in vitro. Leukemia research. 1999;23:239–246. doi: 10.1016/s0145-2126(98)00163-5. [DOI] [PubMed] [Google Scholar]
- 50.Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 51.Blau O, Baldus CD, Hofmann WK, et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood. 2011;118:5583–5592. doi: 10.1182/blood-2011-03-343467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blau O, Hofmann WK, Baldus CD, et al. Chromosomal aberrations in bone marrow mesenchymal stroma cells from patients with myelodysplastic syndrome and acute myeloblastic leukemia. Experimental hematology. 2007;35:221–229. doi: 10.1016/j.exphem.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 53.Lopez-Villar O, Garcia JL, Sanchez-Guijo FM, et al. Both expanded and uncultured mesenchymal stem cells from MDS patients are genomically abnormal, showing a specific genetic profile for the 5q-syndrome. Leukemia. 2009;23:664–672. doi: 10.1038/leu.2008.361. [DOI] [PubMed] [Google Scholar]
- 54.Kouvidi E, Stratigi A, Batsali A, et al. Cytogenetic evaluation of mesenchymal stem/stromal cells from patients with myelodysplastic syndromes at different time-points during ex vivo expansion. Leukemia research. 2016;43:24–32. doi: 10.1016/j.leukres.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Raaijmakers MH, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129:1081–1095. doi: 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim YW, Koo BK, Jeong HW, et al. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood. 2008;112:4628–4638. doi: 10.1182/blood-2008-03-148999. [DOI] [PubMed] [Google Scholar]
- 60.Dong L, Yu WM, Zheng H, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539:304–308. doi: 10.1038/nature20131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nature genetics. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 62.Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. 2011;118:4305–4312. doi: 10.1182/blood-2011-06-353938. [DOI] [PubMed] [Google Scholar]
- 63.Burwick N, Coats SA, Nakamura T, Shimamura A. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood. 2012;120:5143–5152. doi: 10.1182/blood-2012-04-420166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rawls AS, Gregory AD, Woloszynek JR, Liu F, Link DC. Lentiviral-mediated RNAi inhibition of Sbds in murine hematopoietic progenitors impairs their hematopoietic potential. Blood. 2007;110:2414–2422. doi: 10.1182/blood-2006-03-007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zambetti NA, Bindels EM, Van Strien PM, et al. Deficiency of the ribosome biogenesis gene Sbds in hematopoietic stem and progenitor cells causes neutropenia in mice by attenuating lineage progression in myelocytes. Haematologica. 2015;100:1285–1293. doi: 10.3324/haematol.2015.131573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dror Y, Freedman MH. Shwachman-Diamond syndrome: An inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94:3048–3054. [PubMed] [Google Scholar]
- 67.Santamaria C, Muntion S, Roson B, et al. Impaired expression of DICER, DROSHA, SBDS and some microRNAs in mesenchymal stromal cells from myelodysplastic syndrome patients. Haematologica. 2012;97:1218–1224. doi: 10.3324/haematol.2011.054437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tarte K, Gaillard J, Lataillade JJ, et al. Clinical-grade production of human mesenchymal stromal cells: occurrence of aneuploidy without transformation. Blood. 2010;115:1549–1553. doi: 10.1182/blood-2009-05-219907. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez PV, Perry RL, Sarry JE, et al. A robust xenotransplantation model for acute myeloid leukemia. Leukemia. 2009;23:2109–2117. doi: 10.1038/leu.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agliano A, Martin-Padura I, Mancuso P, et al. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. International journal of cancer. 2008;123:2222–2227. doi: 10.1002/ijc.23772. [DOI] [PubMed] [Google Scholar]
- 71.Nilsson L, Astrand-Grundstrom I, Arvidsson I, et al. Isolation and characterization of hematopoietic progenitor/stem cells in 5q-deleted myelodysplastic syndromes: evidence for involvement at the hematopoietic stem cell level. Blood. 2000;96:2012–2021. [PubMed] [Google Scholar]
- 72.Nilsson L, Astrand-Grundstrom I, Anderson K, et al. Involvement and functional impairment of the CD34(+)CD38(−)Thy-1(+) hematopoietic stem cell pool in myelodysplastic syndromes with trisomy 8. Blood. 2002;100:259–267. doi: 10.1182/blood-2001-12-0188. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Deeg HJ. Murine xenogeneic models of myelodysplastic syndrome: an essential role for stroma cells. Experimental hematology. 2014;42:4–10. doi: 10.1016/j.exphem.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martin MG, Welch JS, Uy GL, et al. Limited engraftment of low-risk myelodysplastic syndrome cells in NOD/SCID gamma-C chain knockout mice. Leukemia. 2010;24:1662–1664. doi: 10.1038/leu.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Benito AI, Bryant E, Loken MR, et al. NOD/SCID mice transplanted with marrow from patients with myelodysplastic syndrome (MDS) show long-term propagation of normal but not clonal human precursors. Leukemia research. 2003;27:425–436. doi: 10.1016/s0145-2126(02)00221-7. [DOI] [PubMed] [Google Scholar]
- 76.Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell stem cell. 2014;14:824–837. doi: 10.1016/j.stem.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 77.Thanopoulou E, Cashman J, Kakagianne T, Eaves A, Zoumbos N, Eaves C. Engraftment of NOD/SCID-beta2 microglobulin null mice with multilineage neoplastic cells from patients with myelodysplastic syndrome. Blood. 2004;103:4285–4293. doi: 10.1182/blood-2003-09-3192. [DOI] [PubMed] [Google Scholar]
- 78.Kerbauy DM, Lesnikov V, Torok-Storb B, Bryant E, Deeg HJ. Engraftment of distinct clonal MDS-derived hematopoietic precursors in NOD/SCID-beta2-microglobulin-deficient mice after intramedullary transplantation of hematopoietic and stromal cells. Blood. 2004;104:2202–2203. doi: 10.1182/blood-2004-04-1518. [DOI] [PubMed] [Google Scholar]
- 79.Li X, Marcondes AM, Ragoczy T, Telling A, Deeg HJ. Effect of intravenous coadministration of human stroma cell lines on engraftment of long-term repopulating clonal myelodysplastic syndrome cells in immunodeficient mice. Blood cancer journal. 2013;3:e113. doi: 10.1038/bcj.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muguruma Y, Matsushita H, Yahata T, et al. Establishment of a xenograft model of human myelodysplastic syndromes. Haematologica. 2011;96:543–551. doi: 10.3324/haematol.2010.027557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sacchetti B, Funari A, Michienzi S, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 82.Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell stem cell. 2015;16:239–253. doi: 10.1016/j.stem.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 83.Chen S, Zambetti NA, Bindels EM, et al. Massive parallel RNA sequencing of highly purified mesenchymal elements in low-risk MDS reveals tissue-context-dependent activation of inflammatory programs. Leukemia. 2016;30:1938–1942. doi: 10.1038/leu.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou T, Kinney MC, Scott LM, Zinkel SS, Rebel VI. Revisiting the case for genetically engineered mouse models in human myelodysplastic syndrome research. Blood. 2015;126:1057–1068. doi: 10.1182/blood-2015-01-624239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell stem cell. 2013;13:285–299. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Winkler IG, Sims NA, Pettit AR, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–4828. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- 87.Raza-Egilmez SZ, Jani-Sait SN, Grossi M, Higgins MJ, Shows TB, Aplan PD. NUP98-HOXD13 gene fusion in therapy-related acute myelogenous leukemia. Cancer research. 1998;58:4269–4273. [PubMed] [Google Scholar]
- 88.Lin YW, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 transgenic mice develop a highly penetrant, severe myelodysplastic syndrome that progresses to acute leukemia. Blood. 2005;106:287–295. doi: 10.1182/blood-2004-12-4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Balderman SR, Li AJ, Hoffman CM, et al. Targeting of the bone marrow microenvironment improves outcome in a murine model of myelodysplastic syndrome. Blood. 2016;127:616–625. doi: 10.1182/blood-2015-06-653113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Weidner H, Rauner M, Trautmann F, et al. Myelodysplastic syndromes and bone loss in mice and men. Leukemia. 2017 doi: 10.1038/leu.2017.7. [DOI] [PubMed] [Google Scholar]
- 91.Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM. Functional inhibition of osteoblastic cells in an in vivo mouse model of myeloid leukemia. Blood. 2012;119:540–550. doi: 10.1182/blood-2011-04-348151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vallet S, Pozzi S, Patel K, et al. A novel role for CCL3 (MIP-1alpha) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia. 2011;25:1174–1181. doi: 10.1038/leu.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang B, Ho YW, Huang Q, et al. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer cell. 2012;21:577–592. doi: 10.1016/j.ccr.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mavrogianni D, Tsaftaridis P, Terpos E, et al. Macrophage Inflammatory Protein-1 alpha (MIP-1alpha) is over-expressed in a cohort of patients with myelodysplastic syndromes. European journal of haematology. 2005;75:85–86. doi: 10.1111/j.1600-0609.2005.00444.x. [DOI] [PubMed] [Google Scholar]
- 95.Krevvata M, Silva BC, Manavalan JS, et al. Inhibition of leukemia cell engraftment and disease progression in mice by osteoblasts. Blood. 2014;124:2834–2846. doi: 10.1182/blood-2013-07-517219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zambetti NA, Ping Z, Chen S, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell stem cell. 2016 doi: 10.1016/j.stem.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 97.Barlow JL, Drynan LF, Hewett DR, et al. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nature medicine. 2010;16:59–66. doi: 10.1038/nm.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sportoletti P, Grisendi S, Majid SM, et al. Npm1 is a haploinsufficient suppressor of myeloid and lymphoid malignancies in the mouse. Blood. 2008;111:3859–3862. doi: 10.1182/blood-2007-06-098251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Inoue D, Kitaura J, Togami K, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. The Journal of clinical investigation. 2013;123:4627–4640. doi: 10.1172/JCI70739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mayle A, Yang L, Rodriguez B, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood. 2015;125:629–638. doi: 10.1182/blood-2014-08-594648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Watanabe-Okochi N, Kitaura J, Ono R, et al. AML1 mutations induced MDS and MDS/AML in a mouse BMT model. Blood. 2008;111:4297–4308. doi: 10.1182/blood-2007-01-068346. [DOI] [PubMed] [Google Scholar]
- 102.Buonamici S, Li D, Chi Y, et al. EVI1 induces myelodysplastic syndrome in mice. The Journal of clinical investigation. 2004;114:713–719. doi: 10.1172/JCI21716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Aguayo A, Kantarjian H, Manshouri T, et al. Angiogenesis in acute and chronic leukemias and myelodysplastic syndromes. Blood. 2000;96:2240–2245. [PubMed] [Google Scholar]
- 104.Pruneri G, Bertolini F, Soligo D, et al. Angiogenesis in myelodysplastic syndromes. British journal of cancer. 1999;81:1398–1401. doi: 10.1038/sj.bjc.6693515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keith T, Araki Y, Ohyagi M, et al. Regulation of angiogenesis in the bone marrow of myelodysplastic syndromes transforming to overt leukaemia. British journal of haematology. 2007;137:206–215. doi: 10.1111/j.1365-2141.2007.06539.x. [DOI] [PubMed] [Google Scholar]
- 106.Della Porta MG, Malcovati L, Rigolin GM, et al. Immunophenotypic, cytogenetic and functional characterization of circulating endothelial cells in myelodysplastic syndromes. Leukemia. 2008;22:530–537. doi: 10.1038/sj.leu.2405069. [DOI] [PubMed] [Google Scholar]
- 107.Korkolopoulou P, Apostolidou E, Pavlopoulos PM, et al. Prognostic evaluation of the microvascular network in myelodysplastic syndromes. Leukemia. 2001;15:1369–1376. doi: 10.1038/sj.leu.2402220. [DOI] [PubMed] [Google Scholar]
- 108.Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000;95:309–313. [PubMed] [Google Scholar]
- 109.Padro T, Ruiz S, Bieker R, et al. Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95:2637–2644. [PubMed] [Google Scholar]
- 110.Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. The American journal of pathology. 2000;157:15–19. doi: 10.1016/S0002-9440(10)64511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mesa RA, Hanson CA, Rajkumar SV, Schroeder G, Tefferi A. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood. 2000;96:3374–3380. [PubMed] [Google Scholar]
- 112.Ribatti D, Vacca A, Nico B, Fanelli M, Roncali L, Dammacco F. Angiogenesis spectrum in the stroma of B-cell non-Hodgkin’s lymphomas. An immunohistochemical and ultrastructural study. European journal of haematology. 1996;56:45–53. doi: 10.1111/j.1600-0609.1996.tb00293.x. [DOI] [PubMed] [Google Scholar]
- 113.Vacca A, Ribatti D, Roncali L, Dammacco F. Angiogenesis in B cell lymphoproliferative diseases. Biological and clinical studies. Leukemia & lymphoma. 1995;20:27–38. doi: 10.3109/10428199509054750. [DOI] [PubMed] [Google Scholar]
- 114.Molica S, Vacca A, Ribatti D, et al. Prognostic value of enhanced bone marrow angiogenesis in early B-cell chronic lymphocytic leukemia. Blood. 2002;100:3344–3351. doi: 10.1182/blood-2002-01-0084. [DOI] [PubMed] [Google Scholar]
- 115.Ridell B, Norrby K. Intratumoral microvascular density in malignant lymphomas of B-cell origin. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica. 2001;109:66–72. doi: 10.1111/j.1600-0463.2001.tb00015.x. [DOI] [PubMed] [Google Scholar]
- 116.Perez-Atayde AR, Sallan SE, Tedrow U, Connors S, Allred E, Folkman J. Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. The American journal of pathology. 1997;150:815–821. [PMC free article] [PubMed] [Google Scholar]
- 117.Aguayo A, Estey E, Kantarjian H, et al. Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood. 1999;94:3717–3721. [PubMed] [Google Scholar]
- 118.Aguayo A, Kantarjian HM, Estey EH, et al. Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer. 2002;95:1923–1930. doi: 10.1002/cncr.10900. [DOI] [PubMed] [Google Scholar]
- 119.Verstovsek S, Estey E, Manshouri T, et al. Clinical relevance of vascular endothelial growth factor receptors 1 and 2 in acute myeloid leukaemia and myelodysplastic syndrome. British journal of haematology. 2002;118:151–156. doi: 10.1046/j.1365-2141.2002.03551.x. [DOI] [PubMed] [Google Scholar]
- 120.Bellamy WT, Richter L, Sirjani D, et al. Vascular endothelial cell growth factor is an autocrine promoter of abnormal localized immature myeloid precursors and leukemia progenitor formation in myelodysplastic syndromes. Blood. 2001;97:1427–1434. doi: 10.1182/blood.v97.5.1427. [DOI] [PubMed] [Google Scholar]
- 121.Ghannadan M, Wimazal F, Simonitsch I, et al. Immunohistochemical detection of VEGF in the bone marrow of patients with acute myeloid leukemia. Correlation between VEGF expression and the FAB category. American journal of clinical pathology. 2003;119:663–671. doi: 10.1309/331Q-X7AX-KWFJ-FKXM. [DOI] [PubMed] [Google Scholar]
- 122.Fiedler W, Graeven U, Ergun S, et al. Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood. 1997;89:1870–1875. [PubMed] [Google Scholar]
- 123.Podar K, Anderson KC. The pathophysiologic role of VEGF in hematologic malignancies: therapeutic implications. Blood. 2005;105:1383–1395. doi: 10.1182/blood-2004-07-2909. [DOI] [PubMed] [Google Scholar]
- 124.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature medicine. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 125.Cortelezzi A, Fracchiolla NS, Mazzeo LM, et al. Endothelial precursors and mature endothelial cells are increased in the peripheral blood of myelodysplastic syndromes. Leukemia & lymphoma. 2005;46:1345–1351. doi: 10.1080/10428190500144235. [DOI] [PubMed] [Google Scholar]
- 126.Bellamy WT, Richter L, Frutiger Y, Grogan TM. Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer research. 1999;59:728–733. [PubMed] [Google Scholar]
- 127.Duan X, Murata Y, Liu Y, Nicolae C, Olsen BR, Berendsen AD. Vegfa regulates perichondrial vascularity and osteoblast differentiation in bone development. Development (Cambridge, England) 2015;142:1984–1991. doi: 10.1242/dev.117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Itkin T, Gur-Cohen S, Spencer JA, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016 doi: 10.1038/nature17624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Streubel B, Chott A, Huber D, et al. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. The New England journal of medicine. 2004;351:250–259. doi: 10.1056/NEJMoa033153. [DOI] [PubMed] [Google Scholar]
- 130.Gunsilius E, Duba HC, Petzer AL, et al. Evidence from a leukaemia model for maintenance of vascular endothelium by bone-marrow-derived endothelial cells. Lancet (London, England) 2000;355:1688–1691. doi: 10.1016/S0140-6736(00)02241-8. [DOI] [PubMed] [Google Scholar]
- 131.Fang B, Zheng C, Liao L, et al. Identification of human chronic myelogenous leukemia progenitor cells with hemangioblastic characteristics. Blood. 2005;105:2733–2740. doi: 10.1182/blood-2004-07-2514. [DOI] [PubMed] [Google Scholar]
- 132.Kusumbe AP, Ramasamy SK, Itkin T, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. 2016;532:380–384. doi: 10.1038/nature17638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ganan-Gomez I, Wei Y, Starczynowski DT, et al. Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes. Leukemia. 2015;29:1458–1469. doi: 10.1038/leu.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Basiorka AA, McGraw KL, Eksioglu EA, et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood. 2016;128:2960–2975. doi: 10.1182/blood-2016-07-730556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kitagawa M, Saito I, Kuwata T, et al. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia. 1997;11:2049–2054. doi: 10.1038/sj.leu.2400844. [DOI] [PubMed] [Google Scholar]
- 136.Gersuk GM, Beckham C, Loken MR, et al. A role for tumour necrosis factor-alpha, Fas and Fas-Ligand in marrow failure associated with myelodysplastic syndrome. British journal of haematology. 1998;103:176–188. doi: 10.1046/j.1365-2141.1998.00933.x. [DOI] [PubMed] [Google Scholar]
- 137.Deeg HJ, Beckham C, Loken MR, et al. Negative regulators of hemopoiesis and stroma function in patients with myelodysplastic syndrome. Leukemia & lymphoma. 2000;37:405–414. doi: 10.3109/10428190009089441. [DOI] [PubMed] [Google Scholar]
- 138.Allampallam K, Shetty V, Hussaini S, et al. Measurement of mRNA expression for a variety of cytokines and its receptors in bone marrows of patients with myelodysplastic syndromes. Anticancer research. 1999;19:5323–5328. [PubMed] [Google Scholar]
- 139.Barreyro L, Will B, Bartholdy B, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120:1290–1298. doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Maratheftis CI, Andreakos E, Moutsopoulos HM, Voulgarelis M. Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:1154–1160. doi: 10.1158/1078-0432.CCR-06-2108. [DOI] [PubMed] [Google Scholar]
- 141.Starczynowski DT, Kuchenbauer F, Argiropoulos B, et al. Identification of miR-145 and miR-146a as mediators of the 5q- syndrome phenotype. Nature medicine. 2010;16:49–58. doi: 10.1038/nm.2054. [DOI] [PubMed] [Google Scholar]
- 142.Chen X, Eksioglu EA, Zhou J, et al. Induction of myelodysplasia by myeloid-derived suppressor cells. The Journal of clinical investigation. 2013;123:4595–4611. doi: 10.1172/JCI67580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nature reviews Microbiology. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mundle SD, Venugopal P, Cartlidge JD, et al. Indication of an involvement of interleukin-1 beta converting enzyme-like protease in intramedullary apoptotic cell death in the bone marrow of patients with myelodysplastic syndromes. Blood. 1996;88:2640–2647. [PubMed] [Google Scholar]
- 145.Catenacci DV, Schiller GJ. Myelodysplasic syndromes: a comprehensive review. Blood reviews. 2005;19:301–319. doi: 10.1016/j.blre.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 146.Zhou L, McMahon C, Bhagat T, et al. Reduced SMAD7 leads to overactivation of TGF-beta signaling in MDS that can be reversed by a specific inhibitor of TGF-beta receptor I kinase. Cancer research. 2011;71:955–963. doi: 10.1158/0008-5472.CAN-10-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Valcarcel D, Verma A, Platzbecker U, et al. Phase 2 Study of Monotherapy Galunisertib (LY2157299 Monohydrate) in Very Low-, Low-, and Intermediate-Risk Patients with Myelodysplastic Syndromes. Blood. 2015;126:1669–1669. [Google Scholar]
- 148.Wu JY, Purton LE, Rodda SJ, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16976–16981. doi: 10.1073/pnas.0802898105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sternberg A, Killick S, Littlewood T, et al. Evidence for reduced B-cell progenitors in early (low-risk) myelodysplastic syndrome. Blood. 2005;106:2982–2991. doi: 10.1182/blood-2005-04-1543. [DOI] [PubMed] [Google Scholar]
- 150.Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, Bhatia R. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood. 2015;125:2678–2688. doi: 10.1182/blood-2014-06-582924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Krause DS, Fulzele K, Catic A, et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nature medicine. 2013;19:1513–1517. doi: 10.1038/nm.3364. [DOI] [PMC free article] [PubMed] [Google Scholar]