

Abstract
Hypertrophic cardiomyopathy (HCM) is primarily caused by mutations in β-cardiac myosin and myosin-binding protein-C (MyBP-C). Changes in the contractile parameters of myosin measured so far do not explain the clinical hypercontractility caused by such mutations. We propose that hypercontractility is due to an increase in the number of myosin heads (S1) that are accessible for force production. In support of this hypothesis, we demonstrate myosin tail (S2)-dependent functional regulation of actin-activated human β-cardiac myosin ATPase. In addition, we show that both S2 and MyBP-C bind to S1 and that phosphorylation of either S1 or MyBP-C weakens these interactions. Importantly, the S1-S2 interaction is also weakened by four myosin HCM-causing mutations but not by two other mutations. To explain these experimental results, we propose a working structural model involving multiple interactions, including those with myosin’s own S2 and MyBP-C, that hold myosin in a sequestered state.
Myosin is the force-producing element of cardiac muscle, and the overall contractile motion of a cardiac sarcomere is generated by many regulated protein-protein interactions among various proteins including myosin, actin, tropomyosin, troponin complexes, MyBP-C, and titin. The myosin thick filament alone is a complex structure containing myosin, MyBP-C, titin, and other proteins. Important low-resolution structures have been determined by a number of investigators using both skeletal1–6 and cardiac muscle7–9 isolated thick filaments. However, the complexity of this multiprotein assembly makes the locations of its various components difficult to determine. Constantly improving high-resolution cryo-EM techniques10–13 hold great promise for generating better-defined thick-filament structures in the future14.
HCM is a cardiac muscle disorder and a leading cause of sudden death in people under age 35 (refs. 15,16). Approximately half of all HCM patients have mutations in genes encoding cardiac sarcomeric proteins, predominantly human β-cardiac myosin (~40%) and MyBP-C (~40%)17–22. A leading hypothesis for the cause of clinical hyper-contractility in HCM is that these mutations increase one or more key parameters that determine the power output of the heart23,24. Because power output is the product of force and velocity, the key parameters are the velocity of actin movement along an ensemble of motors and the ensemble force produced (Fensemble), which can be estimated as:
in which Fintrinsic is the intrinsic force of a single myosin motor, Na is the number of myosin heads that are functionally accessible for interaction with actin in the sarcomere, and dr is the duty ratio or fraction of those accessible myosin heads strongly bound to actin at any point during contraction.
Most HCM-related mutations are found in S1 (or subfragment 1) of myosin. For two human β-cardiac S1 HCM mutations, D239N and H251N (early onset, pathologically severe), intrinsic force, velocity (as measured by in vitro motility assays), and ATPase activity are 30–90% higher than those in wild-type (WT) S1 (ref. 25) (Table 1), thus easily accounting for clinically observed hypercontractility. However, human β-cardiac S1 HCM mutations that typically result in adult-onset disease, such as R403Q26 and R453C27, show less severe effects in vitro, as compared with previously described results in a mouse α-cardiac myosin model23, with the largest observed change being an ~50% increase in Fintrinsic compared with that of WT27 (Table 1). Furthermore, these changes for any particular mutant occur in both directions, thus contributing to hypercontractility in some parameters and hypocontractility in others (Table 1).
Table 1.
Fundamental parameters of the actin–sS1 system with sS1 HCM mutants relative to wild-type sS1
| HCM mutation | Intrinsic force (Fintrinsic) | Velocity (v) | ATPase (kcat) |
|---|---|---|---|
| R403Q | 0.8 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.1 |
| R453C | 1.5 ± 0.1 | 0.8 ± 0.1 | 0.7 ± 0.1 |
| R719W | 0.8 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 |
| R723G | 0.8 ± 0.1 | 1.1 ± 0.1 | 1.0 ± 0.1 |
| G741R | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 |
| D239Na | 1.4 ± 0.1 | 1.9 ± 0.1 | 1.5 ± 0.1 |
| H251Na | 1.3 ± 0.1 | 1.4 ± 0.1 | 1.4 ± 0.1 |
One hotspot for HCM mutations in S1 is the converter domain28, which connects the catalytic domain of S1 to its light-chain-bound lever arm and is instrumental in converting the chemical energy of ATP hydrolysis to mechanical movement. Recent studies of three converter-domain mutations in human β-cardiac myosin that also typically result in adult-onset disease, R719W, R723G, and G741R, have indicated similar small changes in Fintrinsic, velocity, and ATPase relative to those of WT myosin, with most changes being 20% or less in either hypercontractile or hypocontractile direction and with G741R showing no significant changes in these three parameters29 (Table 1). Therefore, for all five of these typically adult-onset HCM mutant myosin proteins examined, the changes in these fundamental parameters were 50% or less, and some contributed to hypercontractility, whereas others contributed to hypocontractility, thus making it difficult to determine the net change in contractility.
One often-ignored parameter in the Fensemble equation is Na, the number of myosin heads functionally accessible for interaction with actin. This parameter may be key in fine-tuned regulation of the heart30–33, and, as previously hypothesized25,28,29,34–37, may be pivotal in determining the hypercontractility known to arise from HCM mutations in human β-cardiac myosin.
We have previously observed that a relatively flat surface of the myosin motor domain, which we named the myosin mesa, is almost completely conserved in all cardiac species from mouse α-cardiac myosin to human β-cardiac myosin36. The myosin mesa is yet another hotspot for myosin HCM mutations28,36; ~70% of the variants in the human population that map to this region produce disease28, and many of these mutations alter arginine residues36. We have proposed that the cluster of arginine residues on the mesa that are mutated in HCM are likely to act as a binding domain for another protein, thereby holding the myosin heads in an inactive form36. We noted two candidate binding partners near S1 in the sarcomere36,37: (i) the proximal 126 residues of the coiled-coil subfragment 2 (S2) tail region of myosin (proximal S2) and (ii) MyBP-C, which is in myosin thick filaments in a ratio of one MyBP-C to approximately six myosin molecules38. In comparison with cardiac myosin, most residues on the mesa are conserved in fast skeletal muscle, which has MyBP-C, whereas many mesa residues have been changed in smooth muscle myosin and nonmuscle myosin II, which lack MyBP-C36. This observation supports our hypothesis that MyBP-C might be a binding partner for the mesa region. Regarding possible proximal S2 binding to the mesa, outside of the myosin head domain, the one hotspot for HCM mutations is the proximal S2 region19,28, where mutations primarily occur in glutamate and aspartate residues, which would be complementary to the arginine residue charges on the mesa.
Structural evidence for interaction of S1 with the proximal S2 tail from the same molecule has been observed for isolated heavy meromyosin (HMM, a soluble fragment of myosin consisting of two S1 heads attached to S2)39–42, myosin molecules40–43, and intact thick filaments1–3,7,8. The first 3D structure of a folded state of myosin, named the interacting heads motif (IHM), came from a cryo-EM study of unphosphorylated smooth muscle myosin39 and describes a 2.0-nm-resolution map of an asymmetric interaction between the S1 domains. In this folded ‘off state’ of the motor domains, a binding surface of the ‘blocked head’ binds to the converter domain of the ‘free head’. In the folded-back IHM structure, the blocked head may also be in structural contact with the proximal S2 (refs. 1,2,25,40,43,44). The IHM has also been observed in other classes of muscle myosin, including striated muscle myosins41, and in images of intact thick filaments isolated from a variety of muscle types1–6. Thick-filament structures of 2.0- to 4.0-nm resolution have been obtained with cryo-EM for tarantula skeletal muscle1,2, Limulus telson muscle3, and vertebrate cardiac muscle7,8. The solved crystal structure of human β-cardiac proximal S2 reveals possible charge interactions between the S2 and the S1 (ref. 34), and these have been discussed as part of a general mechanism of shutting down S1 heads from interaction with actin40,41. All of this evidence is consistent with the concept that Na is highly regulated in the heart.
Here we report functional ATPase data and binding data that support our mesa hypothesis. Importantly, we show effects of various HCM mutations on the affinity of the S1-S2 interactions that are consistent with a homology model of a human β-cardiac folded-back myosin structure that we built using a tarantula structural template. We present arguments suggesting that many, if not most, myosin HCM mutations effectively weaken interactions in this sequestered complex, thereby liberating more heads for subsequent interaction with actin during systolic contraction and resulting in hypercontractility of the heart.
RESULTS
Model of a sequestered complex of human β-cardiac myosin
On the basis of the folded-back sequestered heads previously described by low-resolution EM, we built a homology model for a folded-back, closed structure of human β-cardiac myosin45 (Fig. 1), using the 3D-reconstructed structure of tarantula skeletal muscle thick filaments2. We emphasize that this homology model is not an actual structure of the folded-back state of human β-cardiac myosin, but it serves as a structural model to guide experiments. Inasmuch as similar folded-back structures have been described for other myosins1–7,9,14,39–41,43, including human cardiac myosin8, we found that our homology model provided a good working hypothesis for our studies.
Figure 1.
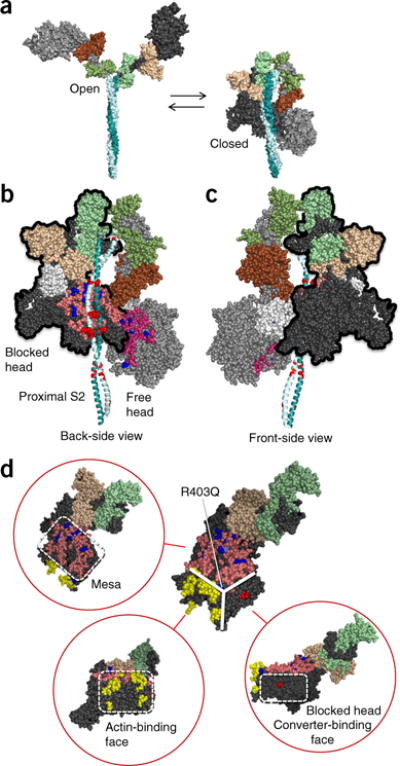
Structural model of sequestered heads of human β-cardiac myosin. The model is based on the 3D reconstruction of tarantula skeletal myosin thick filaments (PDB 3DTP)2. (a) A short version of myosin HMM, showing the first 123 residues of the coiled-coil S2 domain, is illustrated in its open and closed states, which are in equilibrium. The templates used to model the poststroke structure were obtained from the human β-cardiac myosin motor domain45 supplemented with the rigor structure from the squid myosin motor domain63 (Online Methods). (b) The back-side view (side facing the myosin bipolar thick filament) of the sequestered state, showing the potential interaction between different domains of the two heads. The heavy-chain residues of the S1 head on the left (blocked head, outlined in black) are colored pink (mesa residues), dark blue (arginine HCM mutations), light blue (a histidine HCM mutation), white (the converter domain), and dark gray (all remaining residues). The ELC is colored light brown, and the RLC is light green. The heavy-chain residues of the S1 head on the right (free head) are colored dark pink (mesa residues), dark blue (arginine HCM mutations), light blue (a histidine HCM mutation), and light gray (all remaining residues). The ELC is colored dark brown, and the RLC is colored dark green. The proximal S2 tail is shown in teal, with glutamate and aspartate residues mutated in HCM in red. (c) The front-side view of the sequestered state showing the blocked head (outlined in black) interacting with the converter domain (white) of the free head (left). (d) The catalytic domain of human β-cardiac myosin forms a pyramidal structure (thick white solid lines). The three surfaces are the mesa (pink residues with arginine HCM mutations shown in dark blue), the actin-binding interface (yellow, according to the residues involved, as discussed64–67), and the blocked-head surface (dark gray) that abuts the converter domain of the free head in the folded, sequestered-head state (marked by the red residue Asp382, whose mutation to tyrosine is an early-onset HCM mutation). These different interfaces are shown in different orientations of the myosin molecule in the red circles. Coordinates of the homology modeled folded-back human β-cardiac myosin are available for download (MS01, Supplementary Data Set 1).
The open and closed configurations of this structure are proposed to be in equilibrium (Fig. 1a). One head is called the ‘blocked head’ (Fig. 1b, left), because its actin-binding interface is not available for interaction with actin, and the second head is called the ‘free head’39 (Fig. 1b, right). When the structure is viewed from its back side (the side facing the shaft of the thick filament in the 3D reconstructions2), the proximal S2 is cradled by the mesa domains of the S1 heads (Fig. 1b). These regions contain a considerable number of pathogenic HCM mutations28. Several positively charged mesa residues (Fig. 1b, blue) are in proximity to a cluster of negatively charged residues on the proximal S2 that are mutated in HCM28 (Fig. 1b, red). This observation suggests a possible electrostatic interaction between the mesa and the proximal S2 that is important for sarcomere function. Notably, the S2 tail in this model is in closest contact with the mesa of the blocked head (Fig. 1b).
Rotating this model 180° about its vertical axis to show the front side indicates a surface of the blocked head (Fig. 1c, dark-gray residues) that abuts the converter domain of the free head (Fig. 1c, white residues). The converter is another hotspot for HCM mutations28. Interestingly, there are virtually no HCM mutations on the surfaces of the S1 catalytic domains seen from this front view28. The region of the blocked-head surface (dark gray, bottom right of the molecule) that binds to the free-head converter surface is distinct from the actin-binding region (yellow), which in turn is distinct from the mesa surface, as we have defined it (pink) (Fig. 1d). Interestingly, the R403Q mutation lies near the apex of the resulting pyramidal structure (Fig. 1d and Supplementary Video 1).
Actin-activated β-cardiac myosin ATPase regulation depends on the presence of proximal S2
Our mesa hypothesis36,37 states that interactions between S1 and proximal S2 and/or MyBP-C would sequester heads from being able to interact with actin. To set up a functional assay, we took advantage of observations that phosphorylation of Ser15 on the myosin regulatory light chain (RLC) has considerable effects on cardiac muscle force production, shortening velocity and peak power output, and is altered during cardiac disease states46–48.
To test our mesa hypothesis and explore the effects of RLC phosphorylation on the function of cardiac myosin, we created two constructs that resembled conventional heavy meromyosin (HMM) but with shortened S2 tails. The first was a GFP-tagged two-headed human β-cardiac myosin construct that lacked the S2 domain, except for the first two heptads (2-hep HMM, Fig. 2a), and therefore presumably could not form a sequestered state with proximal S2 interacting with the S1 heads, which would inhibit the myosin ATPase. It dimerizes via a leucine zipper moiety49 and was found to migrate as a dimer by native gel electrophoresis (data not shown). Moreover, it showed excellent activity in in vitro motility assays, propelling actin filaments at 1.5 μm s−1 at 23 °C, and showed an intrinsic force of 1.8 pN in laser-trap single- molecule assays. The stoichiometry of the complex was 1:1:1 for HC/ essential light chain (ELC)/RLC. The second HMM-like construct was a 25-hep HMM version of human β-cardiac myosin, which had its proximal S2 as part of the same molecule as the myosin heads (Fig. 2b).
Figure 2.
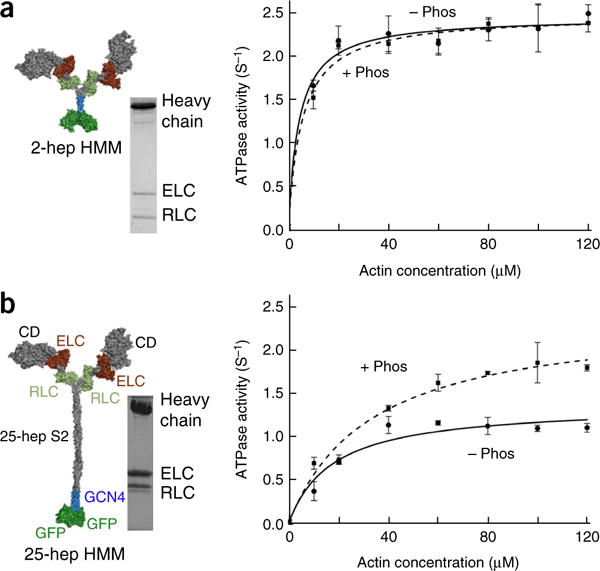
Actin-activated ATPase activities of phosphorylated- and nonphosphorylated-RLC forms of 2-hep and 25-hep HMM. (a) Actin-activated ATPase activity of 2-hep HMM, phosphorylated (+ Phos, dashed curve) and nonphosphorylated (− Phos, solid curve). An SDS–PAGE gel and a structural schematic of the architecture of the 2-hep HMM structure are shown on the left. This construct has 2-heptad repeats of the S2 region. Gray, heavy-chain residues of the S1 heads and S2; brown, ELCs; light green, RLCs; blue, GCN4; green, GFP. (b) Actin-activated ATPase activity of 25-hep HMM, phosphorylated (+ Phos, dashed curve) and nonphosphorylated (− Phos, solid curve). An SDS–PAGE gel of 25-hep HMM and a structural schematic of its architecture are shown on the left. The two S1 heads (1–841 residues of each, containing the catalytic domain (CD) and both the human cardiac ELC- and RLC-binding sites) are linked by 25-heptad repeats of the S2 region followed by a GCN4 leucine zipper to ensure dimerization. C-terminal to the GCN4 are two GFPs. Data points are mean and s.e.m. from n = 2 independent experiments. Source data are available online.
In agreement with all heads being active in the phosphorylated state, the phosphorylated forms of 25-hep and 2-hep HMM showed similar kcat values (2.4 ± 0.2 s−1 and 2.5 ± 0.1 s−1, respectively) (Fig. 2a,b). However, in the case of the 25-hep HMM, the nonphosphorylated form (1.4 ± 0.1 s−1) showed an ~40% decrease in kcat compared with its phosphorylated form (Fig. 2b), in agreement with S1 heads being sequestered into a nonfunctional state dependent on the presence of proximal S2. This conclusion was supported by the finding that no such decrease in kcat was observed with nonphosphorylated 2-hep HMM (2.5 ± 0.1 s−1), for which there was no proximal S2 with which the S1 heads could interact (Fig. 2a).
Human β-cardiac short S1 binds to the proximal part of human cardiac S2
We next sought biochemical evidence that proximal S2 binds to the myosin head in the absence of RLC phosphorylation. Our first experiments involved short S1 (sS1), a truncated form of S1 lacking the RLC and its heavy-chain-binding domain. We investigated interactions between sS1 and proximal S2 by using microscale thermophoresis (MST), a technique that follows the diffusion of a fluorescent probe along a temperature gradient and can be used to measure bimolecular interactions over a broad range of affinities, including a Kd in the double-digit micromolar range50–52.
We first validated that MST was able to measure the affinities of four known protein–protein interactions involving sS1, actin, proximal S2, and MyBP-C (Fig. 3a). We then used sS1 fluorescently tagged with a C-terminal GFP to measure binding to proximal S2 (Fig. 3b). A control with GFP alone showed no binding to S2. In contrast, we observed binding between sS1 and proximal S2 with a Kd of 4 ± 1 μM in 25 mM KCl and a Kd of 46 ± 7 μM (P < 0.05) in 100 mM KCl, thus indicating an electrostatic interaction between the sS1 and proximal S2. Similar results were observed with sS1 fluorescently tagged with Cy5-labeled cysteine residues (Supplementary Fig. 1, Kd of 1.5 ± 0.5 μM at 25 mM KCl and 34 ± 4 μM at 100 mM KCl). Notably, because the S1 and S2 domains are physically connected to each another in the sarcomere, the effective concentrations of S1 and S2 were estimated to be ~60 μM (estimation in Supplementary Note), and thus a Kd in the double-digit micromolar range may be physiologically relevant.
Figure 3.
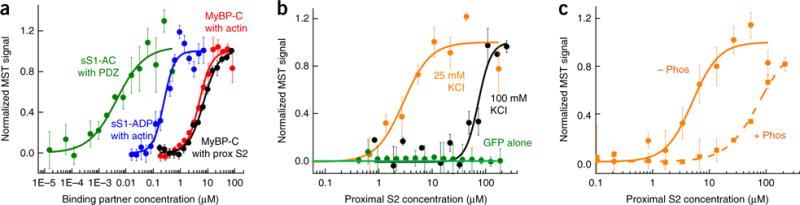
Binding of sS1 and 2-hep HMM to proximal S2 by using MST. (a) MST assays for four protein-protein interactions yielded Kd values within two-fold of those reported in earlier studies using other techniques: sS1 with a C-terminal eight-residue affinity tag (RGSIDTWV) (sS1-AC) binding to PDZ (~5 nM, green)68, sS1 binding to actin in the ADP state (~250 nM, blue)26,69, MyBP-C binding to actin (~5 μM, red)70, and MyBP-C binding to proximal S2 (~7 μM, black)53. (b) Binding of GFP-tagged sS1 to proximal S2 at 25 mM (orange) and 100 mM (black) KCl. GFP alone showed no binding (green circles). (c) Binding of GFP-labeled 2-hep HMM to proximal S2 at 25 mM KCl. Nonphosphorylated (− Phos) and phosphorylated (+ Phos) 2-hep HMM are compared. All data shown in graphs are mean and s.e.m. from n = 3 measurements from a single set of protein preparations; source data are available online. Data from multiple preparations are summarized in Supplementary Table 1.
Next, we used MST binding assays to test whether the binding of 2-hep HMM to S2 was weakened by phosphorylation of its RLC (Fig. 3c). Whereas the nonphosphorylated 2-hep HMM bound proximal S2 with a Kd of 8 ± 3 μM at 25 mM KCl, the phosphorylated 2-hep HMM bound with a much lower affinity (Kd = 65 ± 5 μM; P < 0.05 by two-sided unpaired Student’s t test) (Fig. 3c). At 100 mM KCl, the Kd for nonphosphorylated 2-hep HMM was 20 ± 3 μM, whereas the phosphorylated 2-hep HMM again bound with a much lower affinity (Kd >60 μM) (Supplementary Fig. 2).
HCM mutations decrease the affinity of sS1-S2 binding
If the myosin mesa interacts with proximal S2, thereby sequestering myosin heads, we hypothesized that the S1 mesa arginine and lysine residues and the proximal S2 glutamate and aspartate residues that give rise to HCM when mutated would shift the equilibrium of the S1-S2 interaction to a more open state, thereby increasing the number of functionally accessible heads and causing hypercontractility of the sarcomere, in agreement with clinical observations. To this end, we investigated the binding of sS1with HCM-causing mutations (R453C or R249Q) to proximal S2, because these mutations in the blocked head lie near the proposed position of proximal S2 binding in the sequestered-complex model (Fig. 4a and Supplementary Video 2). Both R453C sS1 and R249Q sS1 binding to proximal S2 was much weaker (Kd >90 μM for R453C and Kd >300 μM for R249Q) than WT sS1 binding (Kd of 46 ± 7 μM) (Fig. 4b). In a separate study25, the early-onset HCM mutation H251N, which is next to R453C and R249Q in the myosin mesa in our working model (Fig. 4a), has also been found to substantially weaken the binding of sS1 to S2 (green curve in Fig. 4b; data from Adhikari et al.25).
Figure 4.
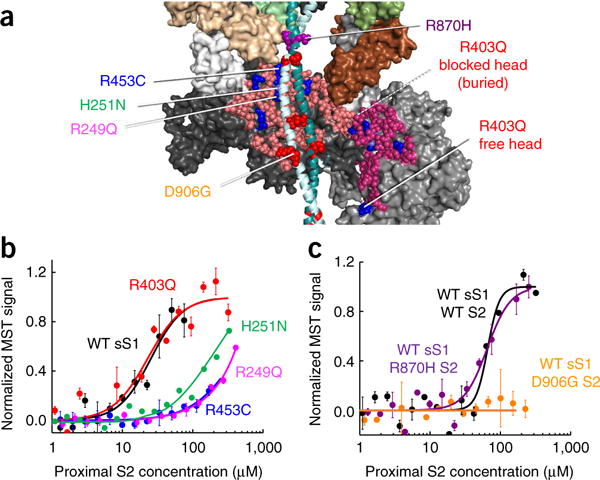
Effects of HCM mutations on the interaction of human β-cardiac sS1 with proximal S2. (a) Model of S1 of the blocked head interacting with proximal S2, marking the positions of four mesa HCM mutations and two proximal-S2 mutations used for binding experiments. Also shown is the free head interacting with the blocked head. Arg453 lies in the mesa domain and is predicted to interact with the proximal S2, whereas Arg403 is buried and away from the interacting site. Similarly, Asp906 on the proximal S2 is predicted to interact with the mesa, but Arg870 is not. (b) Binding of GFP-labeled WT (black), R403Q (red), R453C (blue) R249Q (magenta), and H251N (green curve; data reproduced from Adhikari et al.25) sS1 to proximal S2. (c) Binding of GFP-labeled WT sS1 to WT proximal S2 (black), and R870H (purple) and D906G (orange) mutant proximal S2. Graphs in b and c show mean and s.e.m. from n = 3 measurements from a single set of protein preparations; source data are available online. Data from multiple preparations are summarized in Supplementary Table 1.
Controls for the observed effects on sS1-S2 binding were the examination of HCM mutations that are not structurally adjacent to S2 in our model. For example, R403Q, which is on the far outer edge of the blocked-head mesa and is buried in the interaction between the two heads (Fig. 4a), was not predicted to affect the sS1-S2 interaction. The R403Q sS1 showed an affinity for S2 (Kd of 54 ± 4 μM) similar to that of WT sS1 (Kd of 46 ± 7 μM) (Fig. 4b). Similarly, D239N in sS1, on the other side of the molecule from the mesa, has no effect on the affinity of sS1 for proximal S2 compared with WT sS1 (ref. 25). However, these mutations may well affect other interactions holding the complex together.
To further test our hypothesis, we examined the aspartate and glutamate HCM mutations in the proximal S2 domain that are near the mesa to determine whether they would also weaken the binding of sS1 to S2. D906G S2, which overlies the blocked-head mesa, exhibited no binding to sS1 up to 250 μM proximal S2, in contrast to WT S2 (Kd of 46 ± 7 μM) (Fig. 4c). A control for the lack of binding of the D906G S2 was the proximal S2 HCM mutation R870H, which is outside the range of the mesa (Fig. 4a and Supplementary Video 1). R870H S2 and WT S2 binding to sS1 showed similar affinities (Kd of 59 ± 2 μM and 46 ± 7 μM, respectively) (Fig. 4c).
β-cardiac sS1 binds MyBP-C, and binding is weakened by MyBP-C phosphorylation
As discussed above, we hypothesized that the myosin mesa might serve as an interacting platform for MyBP-C36. Results showing binding of the C1–C2 fragment of MyBP-C to proximal S2 (ref. 53) and the proximity of proximal S2 to the myosin mesa (Figs. 1b and 4a) are consistent with this hypothesis.
We directly tested for binding of MyBP-C to sS1 by using MST. The human cardiac MyBP-C, homology modeled on the basis of known structures of some of the domains (Online Methods), is shown in Figure 5a. We found that full-length recombinant human nonphosphorylated MyBP-C bound sS1 (which does not contain the RLC that was previously shown to bind the C0 domain of MyBP-C54) with a Kd of 17 ± 2 μM (Fig. 5b). Phosphorylation of the MyBP-C with protein kinase A (PKA) significantly decreased the binding affinity (Kd of 37 ± 2 μM, P < 0.05, by two-sided unpaired Student’s t test) (Fig. 5b). Similar but somewhat weaker binding (Kd of 33 ± 2 μM, P < 0.05) was seen with nonphosphorylated C0–C2 (Fig. 5c). As for the full-length MyBP-C, binding was weaker after phosphorylation of C0–C2 with PKA (Kd of 63 ± 3 μM, P < 0.05) (Fig. 5c).
Figure 5.
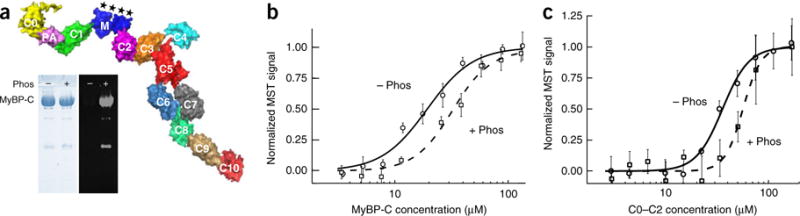
Binding of human β-cardiac sS1 to human cardiac MyBP-C. (a) Surface rendition of full-length human cardiac MyBP-C (details of the model in Online Methods). The C0–C10 domains were connected C-terminal to N-terminal in PyMOL to obtain the image shown. There are four serine phosphorylation sites (asterisks) on the M domain (blue) that regulate MyBP-C function71. The left panel of the gel shows a Coomassie gel of dephosphorylated (−) and phosphorylated (+) full-length human cardiac MyBP-C. The right panel shows ProQ-Diamond staining of the same gel. Lower-molecular-weight contaminants are also phosphorylated (probably proteolytic fragments of MyBP-C). (b) Binding of Cy5-labeled sS1 to dephosphorylated (− Phos, solid curve) and phosphorylated (+ Phos, dashed curve) full-length MyBP-C. (c) Binding of Cy5-labeled sS1 to dephosphorylated (− Phos, solid curve) and phosphorylated (+ Phos, dashed curve) C0–C2. Data points in graphs in b and c are mean and s.e.m. from n = 3 measurements from a single set of protein preparations; source data are available online. Data from multiple preparations are summarized in Supplementary Table 1.
All converter HCM mutations lie near the interface of the two folded-back S1 heads
In addition to the biochemical interactions demonstrated here and previously53,54, there is a predicted S1–S1 interaction that involves various associations, most prominently between a blocked-head surface adjacent to the mesa (Fig. 1d) and the converter domain of the free head (Fig. 6a,b). The converter domain is of great interest in HCM because it is the hottest spot for HCM mutations, for which all reported variants to date in humans have proven to be HCM-causing pathogenic mutations28. We therefore modeled the converter HCM mutations into the homology-modeled folded-back human β-cardiac myosin shown in Figure 1. Strikingly, all nine of the converter-domain mutations examined are positioned near the interface of the free-head converter and the blocked-head binding face (Fig. 6b–e). Furthermore, R719W on the converter (Fig. 6d,e, middle cyan residue) of the free head is potentially in direct contact with the severe early-onset HCM-mutant residue D382Y55,56 on the binding face of the blocked head (Fig. 6e, red residue). This possible proximity, however, must be confirmed in an actual high-resolution structure of the folded state of human β-cardiac HMM. Additionally, this view of Arg453 on the mesa and Glu875 on S2 shows their proximity in our working model.
Figure 6.
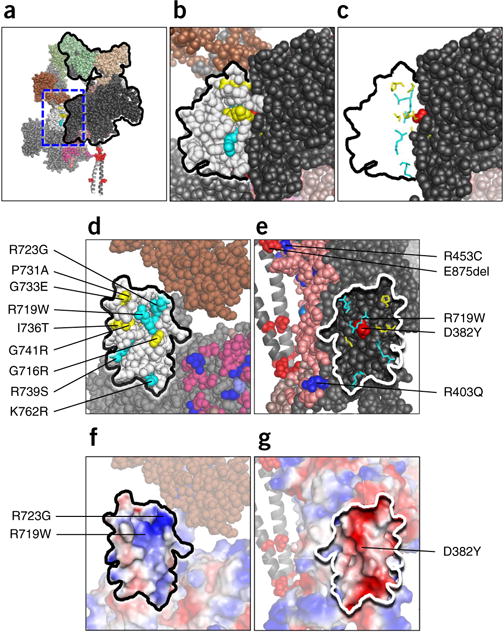
Structural features of the S1-S1 interaction in the homology-modeled human β-cardiac S1. (a) The front view of the sequestered complex shows the association of a surface adjacent to the mesa of the blocked head (black outline) with the converter domain of the free head (white). (b) The area in the dashed blue box in a is enlarged to focus on the interface between the blocked head (dark gray) and the converter (white residues, black outline) of the free head. Converter mutations are cyan for arginine and lysine residues and yellow for uncharged residues. (c) The nine HCM residues shown in stick format with the remainder of the converter-domain residues removed. The alignment along the binding face of the two S1 heads is shown. (d) The image in c rotated 90° counterclockwise about the vertical axis defining the binding interface. All nine HCM residues near the surface of the converter at the S1-S1 junction. (e) The image in c, rotated 90° clockwise about the vertical axis defining the binding interface. The positions of the nine HCM residues of the converter domain of the free head are mapped onto the black binding interface of the blocked head. (f) The same image as in d, but shown in vacuum-electrostatics mode in PyMOL. The converter-binding interface is generally positively charged. (g) The same image as in e, but shown in vacuum-electrostatics mode in PyMOL. The blocked-head binding interface is generally negatively charged.
Examination of the binding interface with the vacuum electrostatics mode in PyMOL suggested an electrostatic interaction in which the binding interface of the converter domain of the free head is generally positively charged (Fig. 6f), Arg723 and Arg719 are key residues, and the binding interface of the blocked-head domain is generally negatively charged (Fig. 6g). Beyond weakening a general charge–charge interaction between the blocked and free head, any one of these converter HCM mutations is likely to perturb the structure of the domain in which it resides, and such a structural change may weaken the interaction between the two domains. The same can be said for HCM mutations in other regions of the molecule that may be involved in the proposed interactions holding the complex in a closed, sequestered state.
DISCUSSION
Notably, the resolution of all available IHM models is only ~2 nm, and resolutions better than 3 Å are necessary to determine how side chains are oriented and how they may affect myosin function12,13. It is not possible to know how the high-resolution information of the myosin heads changes after they fold back into the IHM state. Furthermore, because ~40% of the residues of the S1 domains differ between human β-cardiac myosin and tarantula skeletal myosin, the actual human β-cardiac folded structure will be different in high-resolution detail. Thus, these models and our homology model of human β-cardiac myosin can serve only as a guide and should not be treated as actual high-resolution structures. High-resolution EM reconstruction images or X-ray crystallography structures with purified human β-cardiac HMM will therefore be necessary to begin to assess the more detailed aspects of the effects of HCM mutations. Inferences such as specific side chain interactions, must be delayed until after these true structures become available.
Similar caution should be taken regarding the relationship between the folded-back sequestered state and the very interesting super-relaxed state (SRX) of myosin molecules described in both skeletal and cardiac fiber studies31,32,57. SRX is defined as a subpopulation of myosin molecules in the sarcomere that release nucleotides from the myosin active site extremely slowly (half-life of ~100 s)31,32,57. Although the molecular nature of this state has not been identified, the folded-back sequestered state observed through EM has been ascribed to the SRX state5,58. This relationship seems highly likely, but the role of thick-filament proteins such as MyBP-C or titin in decreasing basal-level ATPase rates to the SRX level needs to be explored. There are data for purified dephosphorylated smooth muscle HMM indicating a very slow ATPase activity (<0.004 s−1)59, but phosphorylation/dephosphorylation is an on/off switch for smooth muscle, unlike striated muscle. To understand the role of SRX in HCM, it is therefore very important to biochemically demonstrate with purified β-cardiac myosin (or HMM) whether dephosphorylated myosin alone results in a decrease in basal ATPase relative to the levels in the SRX in fibers. If not, then which proteins must be added (MyBP-C, titin, and others) to myosin to decrease the ATPase activity to the low SRX values?
The binding of MyBP-C to sS1 (Fig. 5) places little constraint on where the binding occurs. However, C0 binds to the myosin RLC54, and C1–C2 binds to proximal S2 (ref. 53). One model consistent with these results is shown in Figure 7a. The model can also account for the observation that the R870H S2 HCM mutation decreases the binding affinity of this domain for the C1–C2 domain of MyBP-C by over an order of magnitude53 (Figs. 4a and 7a). The model shown in Figure 7b is consistent with our observation that the binding of C0–C2 to sS1 was two-fold weaker than that of full-length MyBP-C (Fig. 5). This result was consistent in duplicate experiments from different protein preparations and may suggest that the C4–C10 domains play a role in sequestered-complex formation. These models may be used to direct future experimental work to test their validity.
Figure 7.
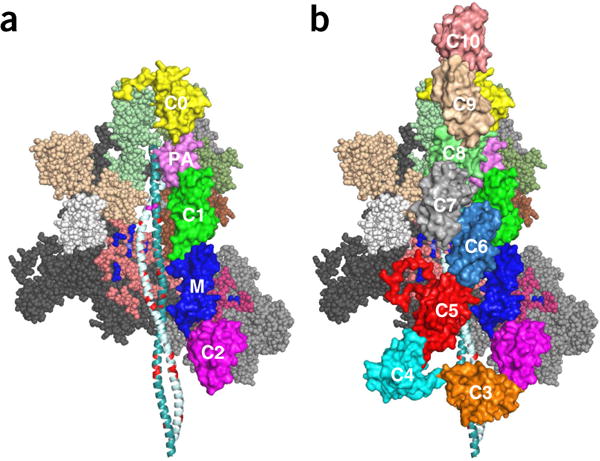
Hypothetical models of the interaction of C0 and C2 and full-length MyBP-C with folded-back, sequestered S1 heads. (a) The backside view of the sequestered state, showing the myosin mesa domains of the two heads cradling the C0–C2 domains of MyBP-C with potential interactions between the C0–C2 and the mesa of the free head (right), illustrating potential interactions between proximal S2 and C1–C2 (ref. 53). The C0 domain (yellow) is bound to the RLCs54, and the PA domain (pink) connects to the C1(green)–M (blue)–C2 (magenta) domains, which are bound to proximal S2. (B) Domains C3–C10 have been added to illustrate potential interactions among C5 and C7 with the mesa of the blocked head and possibly proximal S2. These structures, which were assembled by manual placement of the MyBP-C domains onto our folded-back homology model by using PyMOL, should serve as working models for future experiments. All domains of MyBP-C are marked. Coordinates of the homology modeled folded-back human β-cardiac myosin bound to C0-C2 and C0-C10 are available as MS01C0C2 (Supplementary Data Set 2) and MS01C0C10 (Supplementary Data Set 3), respectively.
We propose that the sequestered state is held together by a series of relatively weak interactions between the two S1 heads; S1 and proximal S2; MyBP-C and S1; and MyBP-C and S2. Additional interactions in the sarcomere may be between the myosin head of one molecule and that of its neighboring molecule in the helical array of the thick filament1,5,8; between the folded heads and the light meromyosin (LMM) backbone; and possibly between myosin and titin. Arg403 is in a position to potentially interact with MyBP-C, actin, the converter, or near the converter of the free head (Fig. 1d). The R403Q mutation has very little effect on actin interaction26, so we propose that it may be involved in either weakening the C0-C2 interaction with the mesa or the blocked-head interaction with the free head. Similarly, the D382Y mutation (Fig. 1d) was predicted to interrupt the S1–S1 interaction. Future experiments will be directed at testing these predictions.
The decreases in Kd that we observed for sS1 binding to proximal S2 when comparing R453C and R249Q human β-cardiac sS1 and WT sS1 were nearly an order of magnitude, a decrease that on its own would be predicted to release a substantial fraction of heads in the sequestered complex for interaction with actin (Supplementary Note). If a substantial portion of the myosin heads were in the sequestered state under normal conditions of heart function, such a large change might not be well tolerated. Our hypothesis, however, is that beyond the sS1-S2 interaction, the sequestered complex is held together by a number of other associations, as described above. Thus, an order-of-magnitude decrease or increase in binding affinity of any one of these interactions would lead to a smaller change in the fraction of heads released than would be expected if there were only one type of intra- or intermolecular interaction.
In summary, it is becoming increasingly clear that many myosin heads in the sarcomere are in a sequestered state, held in equilibrium with actively cycling heads (Fig. 8). This equilibrium is shifted toward more active heads by phosphorylation of the myosin RLC and the M domain of MyBP-C, thus providing fine-tuned control of cardiac function by adrenergic stimulation. In the cardiac sarcomere, we additionally hypothesize that the open-to-closed myosin equilibrium is shifted toward open active heads by a large number of myosin HCM mutations, thereby causing hypercontractility of the heart. Notably, none of the HCM mutations studied here strengthen the sS1–S2 interactions, but they weaken binding, in accordance with our hypothesis and structural model. A majority of MyBP-C HCM mutations are truncations, which may lead to loss of the protein and to haploinsufficiency60–62. The considerations described here may suggest that the MyBP-C HCM truncation mutations simply decrease the level of the tightly bound state involving MyBP-C (Fig. 8), thus causing a general shift of the sequestered state toward open active heads and resulting in hypercontractility.
Figure 8.
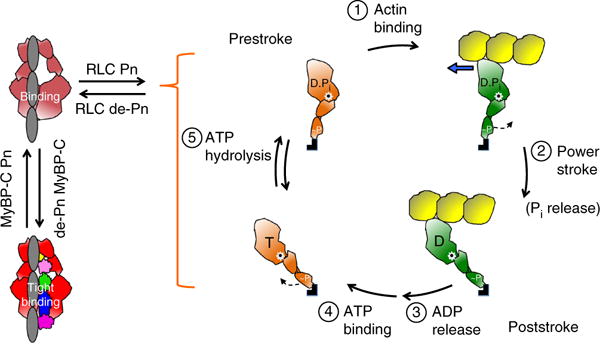
Schematic drawings of the actin-myosin chemomechanical cycle and hypothesized sequestered states of myosin heads. Pn, phosphorylation. (1) The prestroke S1 (orange) with bound ADP (D) and phosphate (Pi) binds to actin (yellow). (2) While bound to actin (green head), the lever arm swings to the right about a fulcrum point (black dot on white star) to the poststroke position, thus moving the actin filament to the left (blue arrow) with respect to the myosin thick filament. (3) ADP release frees the active site for binding of ATP (T). (4) ATP binding weakens the interaction of S1 with actin and frees the lever arm so that it can cock into the prestroke state. (5) ATP hydrolysis locks the head into the prestroke state. The heads in the cycle are phosphorylated (~P) on the RLC. Heads that are sequestered into a nonfunctional state are shown in two states on the left side of the figure: RLC nonphosphorylated and bound to the S2 tail (light red) and complexed with nonphosphorylated MyBP-C and more firmly locked into the sequestered state (bright red). Other than the interactions shown here, many other interactions, for example those involving the LMM and titin, are probably involved in the sequestered state of myosin, and a common theme for HCM mutations may be that they shift the equilibrium away from the sequestered folded-back state of the myosin heads to the open state, which is functionally accessible for interaction with actin, thus producing the hypercontractility observed clinically.
Note added in proof: A homology model of the folded-back human β-cardiac myosin based on a more recent tarantula template (PDB 3JBH, Alamo et al.5) as well as two alternative models of the MyBP-C-bound form of the folded back state are available for download on our website (http://spudlab.stanford.edu/homology-models/). Both homology models (based on PDB 3DTP or PDB 3JBH) are consistent with the conclusions and considerations discussed in this manuscript.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Protein constructs
All protein constructs, expression, purification, and labeling procedures are described in the Supplementary Note.
Development of human β-cardiac myosin and MyBP-C protein models
We developed our models on the basis of known human β-cardiac myosin S1 motor-domain structural data45 as previously described28. In brief, we retrieved the protein sequence of human β-cardiac myosin and the human cardiac light chains from the UniProt database72: myosin heavy-chain motor domain (MYH7) P12883, myosin essential light chain (MYL3) P08590, and myosin regulatory light chain (MYL2) P10916. We used a multitemplate homology modeling approach to build the structural coordinates of MYH7 (residues 1–840), MYL3 (residues 1–195), MYL2 (residues 1–166), and S2 (residues 841–1280). The templates used to model the post-stroke structure were obtained from the human β-cardiac myosin motor domain solved by Winkelmann et al.45 (PDB 4P7H, no nucleotide in the active site) and were supplemented with the rigor structure from the squid myosin motor domain63 (PDB 3I5G, no nucleotide in the active site) to model the converter domain, lever arm, and light chains. The S2 region is a long coiled-coil structure; thus, we used the template from the Myosinome database73. We obtained the 3D structural model of the human β-cardiac myosin folded-back state by integrating the known structural data from solved crystal structures. The folded-back homology model was built by using the tarantula myosin heavy-chain structure (PDB 3DTP) as a template, and the light chains that were modeled earlier28 were aligned to the light chains of the tarantula template structure to obtain the full model. The model was obtained for the head domain (1–961) and the two light chains. Modeling was done with the MODELLER package. Visualizations were performed with PyMOL version 1.7.4 (http://www.pymol.org/). Coordinates of the folded-back human β-cardiac myosin are available as MS01 (Supplementary Data Set 1).
For MyBP-C modeling, we used the protein sequence MYBPC3–Q14896 from the UniProt database72. The structures of C0, C1, C2, C3, C5, and M2 domains were obtained individually with structural homologs from the PDB files for C0 (PDB 2K1M), C1 (PDB 2AVG), C2 (PDB 1PD6), C3 (PDB 2MQ0), C5 (PDB 1GXE), and M2 (PDB 2LHU). The other domain structures (C4, C6, C7, C8, C9, C10, M1, and PA loop) were modeled independently with ab initio and template-based prediction methods74,75. All structures were energy minimized with SYBYL7.2 (Tripos) to remove potential short contacts and were validated with RAMPAGE76, which provides a detailed check of the stereochemistry of the protein structure on the basis of the Ramachandran map. The models of MyBP-C domains bound to the folded-back structure were assembled by manual placement of the MyBP-C domains onto our folded-back homology model by using PyMOL. Coordinates of the folded-back human β-cardiac myosin complexed with C0–C2 or C0–C10 are available as MS01C0C2 (Supplementary Data Set 2) and MS01C0C10 (Supplementary Data Set 3), respectively.
Microscale thermophoresis binding assays
Protein-protein interactions were characterized by MST77,78. For all interactions, the unlabeled protein partner was titrated against a fixed concentration of the fluorescently labeled partner (~50–150 nM). 16 such serially diluted titrations of the unlabeled protein partner were prepared to generate one full binding isotherm. All binding reactions were carried out in a buffer containing 10 mM imidazole, 2 mM MgCl2, 1 mM EGTA, 1 mM DTT, 100 mM KCl, and 0.05% Tween 20, pH 7.5, unless otherwise stated. All binding experiments involving human β-cardiac sS1 and 2 hep-HMM had 500 μM ADP in the binding buffer. The binding interactions for sS1 and 2 hep-HMM with proximal S2 were verified at two different KCl concentrations, 100 mM and 25 mM. All the other binding experiments were carried out at 100 mM KCl. Samples were loaded into NT.115 premium treated capillaries (Nanotemper Technologies) after the reactions were incubated in the dark at 23 °C for 30 min to 1 h. Then the samples were mounted in a Monolith NT.115 apparatus (Nanotemper Technologies) for binding measurements. The data for micro-scale thermophoresis analysis were recorded at 23 °C. All binding experiments were assessed on the basis of GFP, Cy5, or Alexa Fluor 647 fluorescence signal. The binding isotherms of sS1 with PDZ18 were followed by monitoring the sS1-Cy5 fluorescence. The sS1-actin interaction with ADP was followed by sS1-GFP fluorescence, whereas the MyBP-C interaction with either actin or proximal S2 was followed by Alexa Fluor-647 phalloidin and proximal S2-Cy5 fluorescence, respectively. The sS1–proximal S2 binding experiments in either 100 mM or 25 mM KCl were followed by either sS1-GFP or sS1-Cy5 fluorescence to validate the interaction with two different probes. The 2 hep-HMM–proximal S2 binding was followed by assessment of 2 hep-HMM-GFP fluorescence. All mutant sS1–proximal S2, sS1–mutant S2 and sS1–C0–C2 binding experiments were assessed on the basis of sS1-GFP fluorescence. For binding of sS1 to MyBP-C, sS1-Cy5 was used because GFP alone nonspecifically bound to MyBP-C. For the GFP fluorescence, a blue LED at 30% excitation power (blue filter, excitation 460–480 nm, emission 515–530 nm) and IR-laser power at 60% was used, whereas for the Cy5 and Alexa Fluor 647 fluorescence, the red LED at 30% excitation power (red filter, excitation 605–645 nm, emission 680–685 nm) and IR-laser power at 60% was used. Data analysis was performed with software NTAffinityAnalysis (NanoTemper Technologies), in which the binding isotherms were derived from the raw fluorescence data. The binding isotherms were then fitted with both the NanoTemper software and Origin 9.0 software (Origin Lab), with the Hill equation for cooperativity, to estimate the apparent Kd, with a linear regression method. The binding affinities and stoichiometries determined by the two software programs were similar. For each mutant-protein preparation, a new WT preparation was simultaneously made, and the mutant and WT proteins were used for binding measurements at the same time to control for variabilities in conditions from day to day. Shown in the figures are representative curves averaged from three readings from a single preparation or set of preparations (for example, WT and mutant proteins); data from multiple preparations were averaged and are summarized in Supplementary Table 1. For many experiments, only a lower bound of a Kd value was possible to estimate, owing to a lack of saturation in the binding isotherm. For such cases, a mean Kd was not determined. Source data for Figures 3–5 are available online; for Supplementary Figures 1 and 2, source data are in Supplementary Table 2.
Actin-activated ATPase assay
The human β-cardiac 2-hep and 25-hep HMMs were prepared simultaneously to minimize the effects of preparation variability. Only freshly prepared HMM-eGFP was used for this assay. After purification, the HMM was buffer-exchanged into ATPase buffer (10 mM imidazole, pH 7.5, 5 mM KCl, 1 mM DTT, and 3 mM MgCl2) with Amicon Ultracell 10-K filters. Myosin concentration was measured on the basis of eGFP absorbance and confirmed with Bradford assays. G-actin was prepared as previously described79. F-actin was prepared by extensive dialysis of G-actin into ATPase buffer to remove any residual ATP. Actin concentration was then measured on the basis of absorbance at 290 nm in a spectrophotometer, thus eliminating any contribution from the nucleotide at 280 nm.
The steady-state actin-activated ATPase activities of the 2-hep and 25-hep (phosphorylated and unphosphorylated) human β-cardiac myosins were determined with a colorimetric assay to measure inorganic phosphate production at various time points (0–30 min) from a solution containing myosin (0.01 mg ml−1), ATP, and increasing amounts of actin filaments (0–100 μM) (ref. 80). All measurements were made at 23 °C. The time-dependent rate for each actin concentration was calculated by fitting the phosphate signal as a function of time to a linear function. The slope was then converted to activity units normalized to a single myosin head. Kinetic parameters (such as kcat) were extracted from the data by fitting the activity at each actin concentration to the Michaelis–Menten equation to determine maximal activity with the curve fitting toolbox in MatLab81. The errors in the fitted values were determined with 100 bootstrap iterations82.
Statistics
Statistical significance was calculated with Student’s unpaired t test, when applicable; P values are reported in Supplementary Table 1.
Data availability
Source data files for Figures 3–5 and Supplementary Figures 1 and 2 are available online. Our model of the human β-cardiac myosin folded structure, which is complete (with all missing residues in the crystal structures used for modeling being modeled in), is available as Supplementary Data Set 1 and for download on our website http://spudlab.stanford.edu/homology-models/. Please refer to our website to access continually improving homology models of the folded-back structure. The majority of the data presented in this manuscript have been deposited in bioRxiv44. All other data are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank A. Houdusse for helpful suggestions regarding the myosin model.
We thank A. Borrayo of the laboratory of J.A.S. for technical help in maintaining virus and cell stocks. We also thank the members of NanoTemper Technologies (South San Francisco) for training us on the MST instrument and analysis. This work was funded by NIH grants GM33289 and HL117138 to J.A.S.; a Stanford Lucile Packard CHRI Postdoctoral Award (UL1 TR001085) and an American Heart Association Postdoctoral Fellowship (17POST33411070) to D.V.T.; and a Stanford Lucile Packard CHRI Postdoctoral Award (UL1 TR001085), a Stanford ChEM-H Postdocs at the Interface Award, and an American Heart Association Postdoctoral Fellowship (16POST30890005) to A.S.A.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
Molecular modeling studies were performed by J.A.S. and M.S.S. All binding experiments were performed by S.N. and D.V.T., and those involving the 2-hep HMM were performed with help from S.S.S. and K.M.R. S.N., D.V.T., and J.A.S. contributed to data analysis and interpretation. A.S.A. purified the R249Q protein and performed the MST with D.V.T. All ATPase data were performed and analyzed by A.S.A. with help from S.S.S. for reagent production. S.N., D.V.T., and J.A.S. wrote the manuscript. S.N., D.V.T., S.S.S., A.S.A., K.M.R., and J.A.S. contributed to editing of the final manuscript. S.N., D.V.T., S.S.S., A.S.A., S.S., and K.M.R. contributed to the development of new reagents.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Woodhead JL, et al. Atomic model of a myosin filament in the relaxed state. Nature. 2005;436:1195–1199. doi: 10.1038/nature03920. [DOI] [PubMed] [Google Scholar]
- 2.Alamo L, et al. Three-dimensional reconstruction of tarantula myosin filaments suggests how phosphorylation may regulate myosin activity. J Mol Biol. 2008;384:780–797. doi: 10.1016/j.jmb.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao FQ, Craig R, Woodhead JL. Head-head interaction characterizes the relaxed state of Limulus muscle myosin filaments. J Mol Biol. 2009;385:423–431. doi: 10.1016/j.jmb.2008.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto A, Sánchez F, Alamo L, Padrón R. The myosin interacting-heads motif is present in the relaxed thick filament of the striated muscle of scorpion. J Struct Biol. 2012;180:469–478. doi: 10.1016/j.jsb.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Alamo L, et al. Conserved intramolecular interactions maintain myosin interacting-heads motifs explaining tarantula muscle super-relaxed state structural basis. J Mol Biol. 2016;428:1142–1164. doi: 10.1016/j.jmb.2016.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodhead JL, Zhao FQ, Craig R. Structural basis of the relaxed state of a Ca2+-regulated myosin filament and its evolutionary implications. Proc Natl Acad Sci USA. 2013;110:8561–8566. doi: 10.1073/pnas.1218462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zoghbi ME, Woodhead JL, Moss RL, Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci USA. 2008;105:2386–2390. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Khayat HA, Kensler RW, Squire JM, Marston SB, Morris EP. Atomic model of the human cardiac muscle myosin filament. Proc Natl Acad Sci USA. 2013;110:318–323. doi: 10.1073/pnas.1212708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.González-Solá M, Al-Khayat HA, Behra M, Kensler RW. Zebrafish cardiac muscle thick filaments: isolation technique and three-dimensional structure. Biophys J. 2014;106:1671–1680. doi: 10.1016/j.bpj.2014.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nogales E, Scheres SH. Cryo-EM: a unique tool for the visualization of macromolecular complexity. Mol Cell. 2015;58:677–689. doi: 10.1016/j.molcel.2015.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Y. Single-particle cryo-EM at crystallographic resolution. Cell. 2015;161:450–457. doi: 10.1016/j.cell.2015.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai XC, McMullan G, Scheres SH. How cryo-EM is revolutionizing structural biology. Trends Biochem Sci. 2015;40:49–57. doi: 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Henderson R. Realizing the potential of electron cryo-microscopy. Q Rev Biophys. 2004;37:3–13. doi: 10.1017/s0033583504003920. [DOI] [PubMed] [Google Scholar]
- 14.Hu Z, Taylor DW, Reedy MK, Edwards RJ, Taylor KA. Structure of myosin filaments from relaxed Lethocerus flight muscle by cryo-EM at 6 Å resolution. Sci Adv. 2016;2:e1600058. doi: 10.1126/sciadv.1600058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 16.Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194:355–365. doi: 10.1083/jcb.201101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buvoli M, Hamady M, Leinwand LA, Knight R. Bioinformatics assessment of β-myosin mutations reveals myosin’s high sensitivity to mutations. Trends Cardiovasc Med. 2008;18:141–149. doi: 10.1016/j.tcm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh R, Rutland C, Thomas R, Loughna S. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology. 2010;115:49–60. doi: 10.1159/000252808. [DOI] [PubMed] [Google Scholar]
- 19.Colegrave M, Peckham M. Structural implications of β-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken) 2014;297:1670–1680. doi: 10.1002/ar.22973. [DOI] [PubMed] [Google Scholar]
- 20.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 21.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104:557–567. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 22.Seidman CE, Seidman JG. Hypertrophic cardiomyopathy. In: Scriver CR, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; 2000. pp. 5532–5452. [Google Scholar]
- 23.Tyska MJ, et al. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res. 2000;86:737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 24.Spudich JA. Hypertrophic and dilated cardiomyopathy: four decades of basic research on muscle lead to potential therapeutic approaches to these devastating genetic diseases. Biophys J. 2014;106:1236–1249. doi: 10.1016/j.bpj.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adhikari AS, et al. Early-onset hypertrophic cardiomyopathy mutations significantly increase the velocity, force, and actin-activated ATPase activity of human β-cardiac myosin. Cell Rep. 2016;17:2857–2864. doi: 10.1016/j.celrep.2016.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nag S, et al. Contractility parameters of human β-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv. 2015;1:e1500511. doi: 10.1126/sciadv.1500511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sommese RF, et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human β-cardiac myosin motor function. Proc Natl Acad Sci USA. 2013;110:12607–12612. doi: 10.1073/pnas.1309493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Homburger JR, et al. Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc Natl Acad Sci USA. 2016;113:6701–6706. doi: 10.1073/pnas.1606950113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawana M, Sarkar SS, Sutton S, Ruppel KM, Spudich JA. Biophysical properties of human β-cardiac myosin with converter mutations that cause hypertrophic cardiomyopathy. Sci Adv. 2017;3:e1601959. doi: 10.1126/sciadv.1601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kampourakis T, Irving M. Phosphorylation of myosin regulatory light chain controls myosin head conformation in cardiac muscle. J Mol Cell Cardiol. 2015;85:199–206. doi: 10.1016/j.yjmcc.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart MA, Franks-Skiba K, Chen S, Cooke R. Myosin ATP turnover rate is a mechanism involved in thermogenesis in resting skeletal muscle fibers. Proc Natl Acad Sci USA. 2010;107:430–435. doi: 10.1073/pnas.0909468107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hooijman P, Stewart MA, Cooke R. A new state of cardiac myosin with very slow ATP turnover: a potential cardioprotective mechanism in the heart. Biophys J. 2011;100:1969–1976. doi: 10.1016/j.bpj.2011.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brito R, et al. A molecular model of phosphorylation-based activation and potentiation of tarantula muscle thick filaments. J Mol Biol. 2011;414:44–61. doi: 10.1016/j.jmb.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blankenfeldt W, Thomä NH, Wray JS, Gautel M, Schlichting I. Crystal structures of human cardiac beta-myosin II S2-Delta provide insight into the functional role of the S2 subfragment. Proc Natl Acad Sci USA. 2006;103:17713–17717. doi: 10.1073/pnas.0606741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spudich JA. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem Soc Trans. 2015;43:64–72. doi: 10.1042/BST20140324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spudich JA, et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin. J Exp Biol. 2016;219:161–167. doi: 10.1242/jeb.125930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Craig R, Offer G. The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B Biol Sci. 1976;192:451–461. doi: 10.1098/rspb.1976.0023. [DOI] [PubMed] [Google Scholar]
- 39.Wendt T, Taylor D, Trybus KM, Taylor K. Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc Natl Acad Sci USA. 2001;98:4361–4366. doi: 10.1073/pnas.071051098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burgess SA, et al. Structures of smooth muscle myosin and heavy meromyosin in the folded, shutdown state. J Mol Biol. 2007;372:1165–1178. doi: 10.1016/j.jmb.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 41.Jung HS, Komatsu S, Ikebe M, Craig R. Head-head and head-tail interaction: a general mechanism for switching off myosin II activity in cells. Mol Biol Cell. 2008;19:3234–3242. doi: 10.1091/mbc.E08-02-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung HS, et al. Role of the tail in the regulated state of myosin 2. J Mol Biol. 2011;408:863–878. doi: 10.1016/j.jmb.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jung HS, et al. Conservation of the regulated structure of folded myosin 2 in species separated by at least 600 million years of independent evolution. Proc Natl Acad Sci USA. 2008;105:6022–6026. doi: 10.1073/pnas.0707846105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nag S, et al. Beyond the myosin mesa: a potential unifying hypothesis on the underlying molecular basis of hyper-contractility caused by a majority of hypertrophic cardiomyopathy mutations. 2016 Preprint at http://biorxiv.org/content/early/2016/07/24/065508/
- 45.Winkelmann DA, Forgacs E, Miller MT, Stock AM. Structural basis for drug-induced allosteric changes to human β-cardiac myosin motor activity. Nat Commun. 2015;6:7974. doi: 10.1038/ncomms8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scruggs SB, Solaro RJ. The significance of regulatory light chain phosphorylation in cardiac physiology. Arch Biochem Biophys. 2011;510:129–134. doi: 10.1016/j.abb.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toepfer C, et al. Myosin regulatory light chain (RLC) phosphorylation change as a modulator of cardiac muscle contraction in disease. J Biol Chem. 2013;288:13446–13454. doi: 10.1074/jbc.M113.455444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine RJ, Kensler RW, Yang Z, Stull JT, Sweeney HL. Myosin light chain phosphorylation affects the structure of rabbit skeletal muscle thick filaments. Biophys J. 1996;71:898–907. doi: 10.1016/S0006-3495(96)79293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trybus KM, Freyzon Y, Faust LZ, Sweeney HL. Spare the rod, spoil the regulation: necessity for a myosin rod. Proc Natl Acad Sci USA. 1997;94:48–52. doi: 10.1073/pnas.94.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo R, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell. 2014;56:298–310. doi: 10.1016/j.molcel.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo Y, Scheuermann TH, Partch CL, Tomchick DR, Gardner KH. Coiled-coil coactivators play a structural role mediating interactions in hypoxia-inducible factor heterodimerization. J Biol Chem. 2015;290:7707–7721. doi: 10.1074/jbc.M114.632786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gruen M, Gautel M. Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J Mol Biol. 1999;286:933–949. doi: 10.1006/jmbi.1998.2522. [DOI] [PubMed] [Google Scholar]
- 54.Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286:12650–12658. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaski JP, et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009;2:436–441. doi: 10.1161/CIRCGENETICS.108.821314. [DOI] [PubMed] [Google Scholar]
- 56.Morita H, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358:1899–1908. doi: 10.1056/NEJMoa075463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naber N, Cooke R, Pate E. Slow myosin ATP turnover in the super-relaxed state in tarantula muscle. J Mol Biol. 2011;411:943–950. doi: 10.1016/j.jmb.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nogara L, et al. Spectroscopic studies of the super relaxed state of skeletal muscle. PLoS One. 2016;11:e0160100. doi: 10.1371/journal.pone.0160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cremo CR, Sellers JR, Facemyer KC. Two heads are required for phosphorylation-dependent regulation of smooth muscle myosin. J Biol Chem. 1995;270:2171–2175. doi: 10.1074/jbc.270.5.2171. [DOI] [PubMed] [Google Scholar]
- 60.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–764. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Helms AS, et al. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:434–443. doi: 10.1161/CIRCGENETICS.113.000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marston S, et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- 63.Yang Y, et al. Rigor-like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure. 2007;15:553–564. doi: 10.1016/j.str.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 64.von der Ecken J, Heissler SM, Pathan-Chhatbar S, Manstein DJ, Raunser S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 2016;534:724–728. doi: 10.1038/nature18295. [DOI] [PubMed] [Google Scholar]
- 65.Behrmann E, et al. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–338. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rayment I, et al. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 67.Schröder RR, et al. Three-dimensional atomic model of F-actin decorated with Dictyostelium myosin S1. Nature. 1993;364:171–174. doi: 10.1038/364171a0. [DOI] [PubMed] [Google Scholar]
- 68.Huang J, Koide A, Makabe K, Koide S. Design of protein function leaps by directed domain interface evolution. Proc Natl Acad Sci USA. 2008;105:6578–6583. doi: 10.1073/pnas.0801097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siemankowski RF, White HD. Kinetics of the interaction between actin, ADP, and cardiac myosin-S1. J Biol Chem. 1984;259:5045–5053. [PubMed] [Google Scholar]
- 70.Rybakova IN, Greaser ML, Moss RL. Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J Biol Chem. 2011;286:2008–2016. doi: 10.1074/jbc.M110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jia W, Shaffer JF, Harris SP, Leary JA. Identification of novel protein kinase A phosphorylation sites in the M-domain of human and murine cardiac myosin binding protein-C using mass spectrometry analysis. J Proteome Res. 2010;9:1843–1853. doi: 10.1021/pr901006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 2013;41:D43–D47. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Syamaladevi DP, et al. Myosinome: a database of myosins from select eukaryotic genomes to facilitate analysis of sequence-structure-function relationships. Bioinform Biol Insights. 2012;6:247–254. doi: 10.4137/BBI.S9902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins. 2012;80:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, et al. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lovell SC, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 77.Duhr S, Braun D. Why molecules move along a temperature gradient. Proc Natl Acad Sci USA. 2006;103:19678–19682. doi: 10.1073/pnas.0603873103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Seidel SA, et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013;59:301–315. doi: 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85(Pt B 164) doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 80.Trybus KM. Biochemical studies of myosin. Methods. 2000;22:327–335. doi: 10.1006/meth.2000.1085. [DOI] [PubMed] [Google Scholar]
- 81.De La Cruz EM, Ostap EM. Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 2009;455:157–192. doi: 10.1016/S0076-6879(08)04206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Efron B. The Jackknife, the Bootstrap, and other Resampling Plans. Vol. 2. Society for Industrial and Applied Mathematics; 1982. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data files for Figures 3–5 and Supplementary Figures 1 and 2 are available online. Our model of the human β-cardiac myosin folded structure, which is complete (with all missing residues in the crystal structures used for modeling being modeled in), is available as Supplementary Data Set 1 and for download on our website http://spudlab.stanford.edu/homology-models/. Please refer to our website to access continually improving homology models of the folded-back structure. The majority of the data presented in this manuscript have been deposited in bioRxiv44. All other data are available from the corresponding author upon reasonable request.